
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTricyclic analogues of epidithiodioxopiperazine alkaloids with promising in vitro and in vivo antitumor activity†
Marcus
Baumann
a,
André P.
Dieskau
a,
Brad M.
Loertscher
a,
Mary C.
Walton
a,
Sangkil
Nam
b,
Jun
Xie
b,
David
Horne
*b and
Larry E.
Overman
*a
aDepartment of Chemistry, 1102 Natural Sciences II, University of California, Irvine, California 92697-2025, USA. E-mail: leoverma@uci.edu
bDepartment of Molecular Medicine, Beckman Research Institute of City Hope Comprehensive Cancer Center, 1500 E. Duarte Road, Duarte, California 91010, USA. E-mail: dhorne@cih.org
First published on 26th May 2015
Abstract
Epipolythiodioxopiperazine (ETP) alkaloids are structurally elaborate alkaloids that show potent antitumor activity. However, their high toxicity and demonstrated interactions with various biological receptors compromises their therapeutic potential. In an effort to mitigate these disadvantages, a short stereocontrolled construction of tricyclic analogues of epidithiodioxopiperazine alkaloids was developed. Evaluation of a small library of such structures against two invasive cancer cell lines defined initial structure–activity relationships (SAR), which identified 1,4-dioxohexahydro-6H-3,8a-epidithiopyrrolo[1,2-a]pyrazine 3c and related structures as particularly promising antitumor agents. ETP alkaloid analogue 3c exhibits low nanomolar activity against both solid and blood tumors in vitro. In addition, 3c significantly suppresses tumor growth in mouse xenograft models of melanoma and lung cancer, without obvious signs of toxicity, following either intraperitoneal (IP) or oral administration. The short synthesis of molecules in this series will enable future mechanistic and translational studies of these structurally novel and highly promising clinical antitumor candidates.
Introduction
Epipolythiodioxopiperazines (ETPs) constitute a group of structurally complex and biologically active fungal alkaloids that are characterized structurally by a transannular polysulfide unit joining carbons 3 and 6 of their 2,5-dioxopiperazine rings (Fig. 1). A broad spectrum of biological activities is associated with this family of natural products.1 Two commercially available members of this group, chaetomin (1) and chaetocin A (2), have received extensive study. Early on, chaetomin was identified as an inhibitor of HIF-1 transcriptional activity, although its toxicity prevented its further development for cancer therapy.2 Chaetocin A continues to be actively investigated as a potential cancer therapeutic, with good activity registered in vitro1,3 and in animal models1,3d,4 against blood and solid tumors. A number of molecular targets of chaetocin A have been identified, and various mechanisms contributing to its cytotoxicity are now recognized.3b–e,5 Recently, several structurally simple analogues of ETP alkaloids have been prepared and their in vitro cytotoxicity registered against a few cancer cell lines.6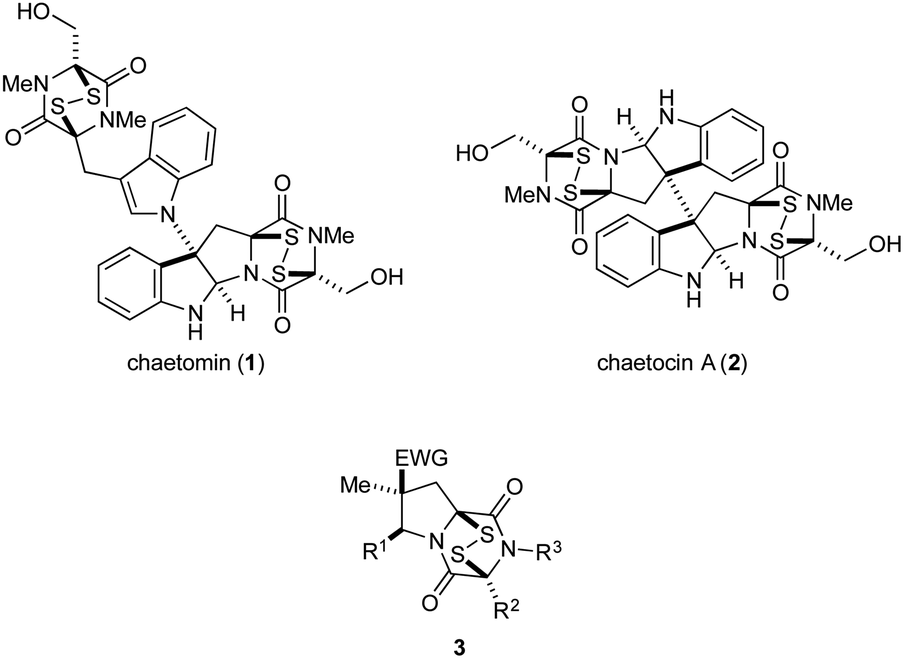 | ||
| Fig. 1 Two ETP alkaloids and structurally simplified tricyclic analogues 3. | ||
The high cytotoxicity of ETP alkaloids 1 and 2,2,5a and their promiscuous interactions with biological receptors, renders these natural products unattractive candidates for further development as anticancer agents. Nonetheless, we anticipated that less toxic and more selective antitumor agents could be found by significantly simplifying the structure of these alkaloids. As good in vitro cytotoxicity against several cancer cell lines is maintained in natural product analogues of chaetocin A that contain a single ETP fragment,6b–d we investigated 1,4-dioxohexahydro-6H-3,8a-epidithiopyrrolo[1,2-a]pyrazines 3, analogues of chaetomin and chaetocin A that retain only three of the natural products' nine or ten rings.7
We report herein an expeditious synthesis of analogues 3 that allows easy introduction of substituents at four sites. The in vitro antitumor activity of 19 molecules in this series was evaluated against invasive human prostate cancer and melanoma cell lines. IC50 values of 3c, the most potent compound in this series, were determined using nine additional cancer cell lines, and 3c was evaluated also in xenograft tumor models of human lung cancer and melanoma.
Results and discussion
Synthesis of ETP analogues 3
The general scheme used to prepare a library of 1,4-dioxohexahydro-6H-3,8a-epidithiopyrrolo[1,2-a]pyrazines 3 is outlined in Scheme 1. As the building blocks 4, 6, and 7 can be varied widely, a diversity of structures is readily prepared, most by way of only two isolated and purified intermediates (pyrrolidine 5 and dioxopiperazine 8).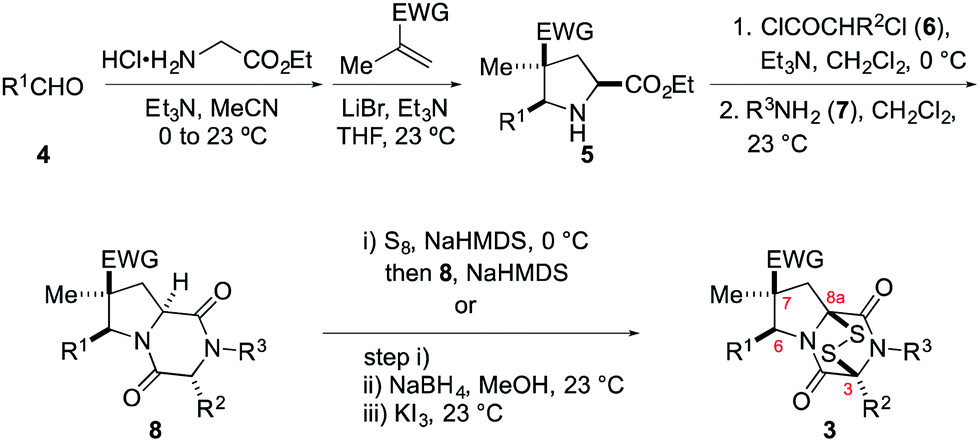 | ||
| Scheme 1 Synthesis of ETP alkaloid analogues 3 (EWG = CO2Me or CN). | ||
The synthesis begins with 1,3-dipolar cycloaddition of azomethine ylides generated in situ by 1,2-protropic shifts of glycine imines with α-methyl-substituted dipolarophiles in the presence of LiBr and Et3N.8,9 It was found that imine formation was most efficient in acetonitrile, whereas the cycloaddition was cleanest in THF. In general, this two-step process delivered the desired pyrrolidine products in useful yields (45–85%) and high purity on multi-gram scales. As expected, this cycloaddition took place with high endo stereoselectivity (>10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) when methyl methacrylate was employed as the dipolarophile. Cycloadditions employing methacrylonitrile took place with lower endo stereoselectivity, which varied considerably with the nature of the R1 substituent (see Table 1 of the ESI†). In some cases, the major endo cycloadduct 5 could be separated in good yield from the cycloadduct mixture by its lower solubility in 1:1 MeOH/CH2Cl2, although generally higher yields of this adduct were realized by purification of the crude product by flash chromatography on silica gel.
:1) when methyl methacrylate was employed as the dipolarophile. Cycloadditions employing methacrylonitrile took place with lower endo stereoselectivity, which varied considerably with the nature of the R1 substituent (see Table 1 of the ESI†). In some cases, the major endo cycloadduct 5 could be separated in good yield from the cycloadduct mixture by its lower solubility in 1:1 MeOH/CH2Cl2, although generally higher yields of this adduct were realized by purification of the crude product by flash chromatography on silica gel.
The relative configuration of the cycloadducts in the nitrile series was assigned on the basis of their distinctive 1H NMR chemical shifts (Fig. 2). In the exo cycloadducts, the C4 methyl substituent was observed at a diagnostic upfield chemical shift (δ 0.95–1.05 ppm), consistent with its position within the shielding cone of the C5 aryl substituent. The chemical shift of the C5 benzylic methine hydrogen was also diagnostic, appearing upfield by ∼0.6 ppm in the endo cycloadduct.
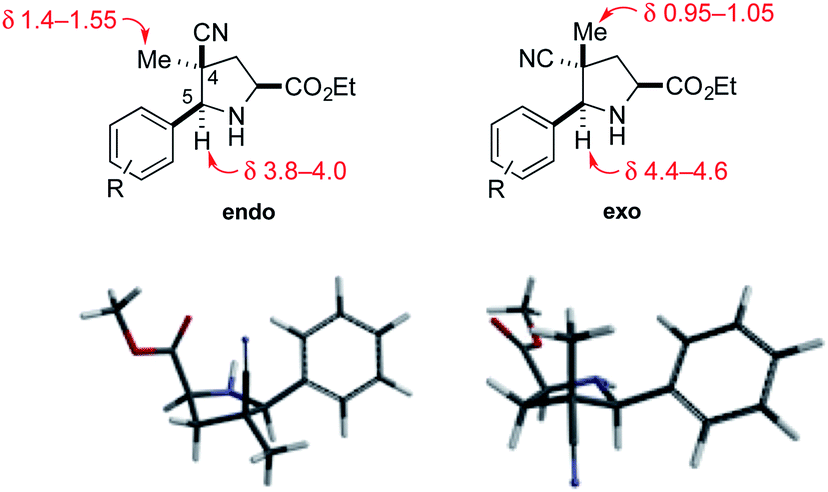 | ||
| Fig. 2 Endo and exo pyrrolidine cycloadducts: diagnostic 1H NMR signals (in CDCl3) and models of their low-energy conformations. | ||
Cycloadducts 5 could be elaborated to dioxopiperazines 8 in a number of standard ways.10 A convenient sequence is depicted in Scheme 1 whereby the amino ester 5 was first acylated with 2-chloropropionyl chloride (6, R2 = Me) and the crude α-chloroamide intermediate was coupled with a primary amine 7 to form the dioxopiperazine product. For example, the formation of dioxopiperazines 8 having R3 = Me was accomplished by stirring a biphasic mixture of the crude α-chloroamide in CH2Cl2 with 40% aqueous methylamine at room temperature for 12–16 h. In most cases, the dioxopiperazine intermediate was isolated as a crystalline solid without the need for chromatographic purification. This solid was either a single stereoisomer or a mixture of C3 epimers. In two cases, the relative configuration of the major stereoisomer was confirmed by single-crystal X-ray analysis.11
An alternate sequence that allows ready incorporation of a variety of R2 substituents for the synthesis of dioxopiperazine 8p having R2 = Bn is illustrated in Scheme 2. Coupling of cycloadduct 5c with N-Boc-phenylalanine using BOPCl gave dipeptide 9 in high yield. Standard removal of the Boc group followed by heating the product in refluxing 2-butanol/toluene (4:1) gave dioxopiperazine 10 in 70% yield as a 1:1 mixture of stereoisomers. After these products were separated on silica gel, they were individually N-methylated in high yield upon reaction with MeI and K2CO3. That products 8p were epimers at the benzyl stereocenter was established by strong 1H NOEs between the quaternary methyl substituent and the angular methine hydrogen.
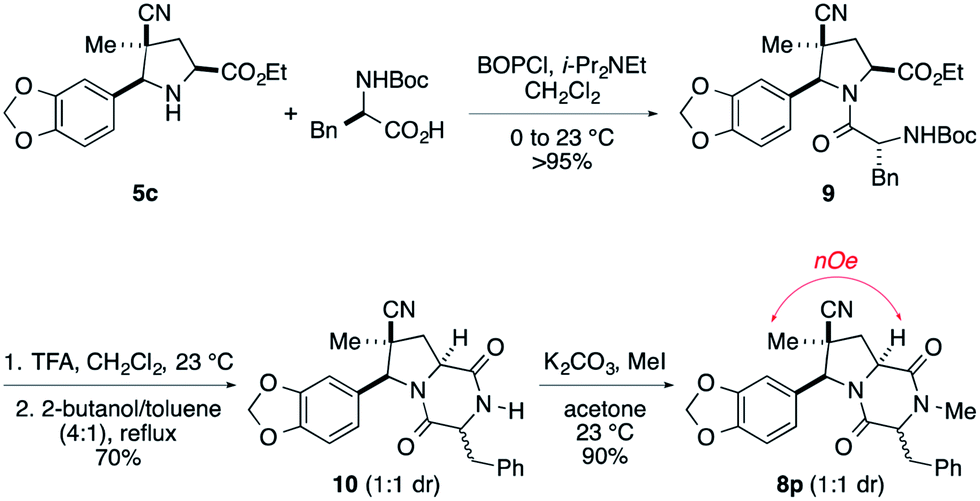 | ||
| Scheme 2 An alternate route to dioxopiperazine intermediates. | ||
We initially examined elaboration of dioxopiperazine 8c to the corresponding ETP derivatives by attempting to first selectively oxidize the methine hydrogens of the dioxopiperazine ring. The most successful of the oxidants screened was Pyr2AgMnO4, which had been employed with great success by Movassaghi for this purpose.12 This reagent cleanly hydroxylated the angular carbon; however, under the conditions we examined, hydroxylation at C3 did not proceed to full conversion before decomposition of the diol product became problematic (eqn (1)).‡
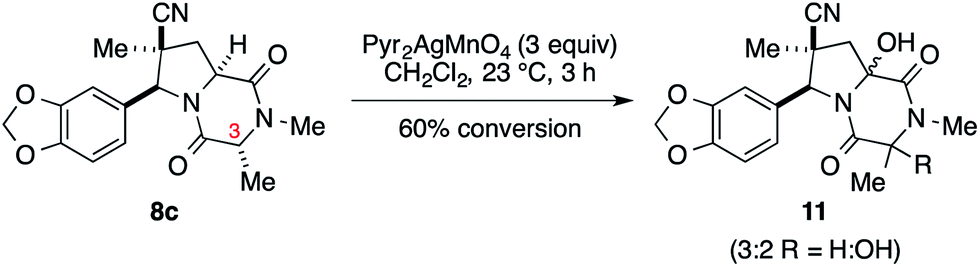 | (1) |
We turned to introduce the epidisulfide unit by base-promoted disulfenylation of the dioxopiperazine intermediates.13 In initial studies, dioxopiperazines 8 were converted to the corresponding ETP derivatives by the general procedure introduced by Nicolaou and coworkers.14 After purification by flash chromatography or preparative TLC, ETP products 3 were obtained in low to moderate yields (generally 20–30%, Scheme 1). During attempts to optimize this sequence, we discovered that the desired disulfide 3, typically contaminated with 5–20% of the corresponding trisulfide, was the predominant sulfur-containing product produced after the sulfenylation step. This result stands in contrast to the formation of largely the tetrasulfide intermediate in the base-promoted sulfenylation of nearly flat dioxopiperazines.15 As the epidithiodioxopiperazine products 3 could be separated from their trisulfide analogues by flash chromatography or preparative TLC, this operationally simple one-step procedure was generally used in our studies giving 3 in 6–46% yield (see Table 1 of the ESI†).
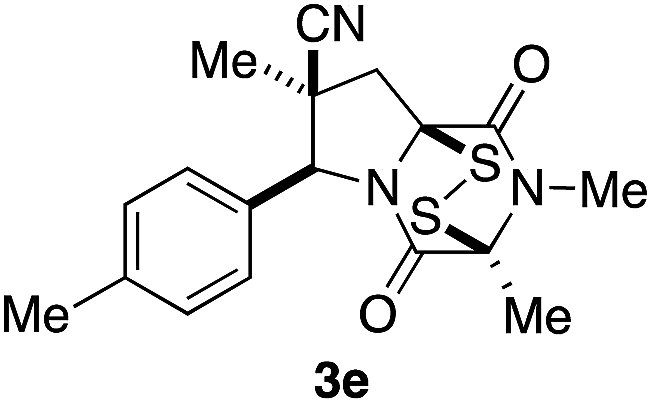
Stereoisomer 3 having the disulfide bridge on the same face as the nitrile and aryl substituents was typically produced with 5–10:1 diastereoselectivity. In two cases (3c and 3e), this product was characterized by single-crystal X-ray analysis.11 Epidisulfide stereoisomer 3e and diastereomer 12 having the disulfide bridge on the opposite face of the dioxopiperazine are easily distinguished by 1H NMR analysis. Particularly diagnostic are the chemical shifts of the C7 methylene hydrogens HA (syn to CN) and HB (anti to CN).§ As illustrated in Fig. 3 for 3e and 12, these diastereotopic hydrogens are observed at similar chemical shift in the major stereoisomer 3e, whereas in diastereomer 12 the chemical shifts of these hydrogens differ by 1.3 ppm, with HA being observed at δ 3.84 and HB at δ 2.53. The observation of the C7 methylene hydrogens at similar chemical shift (Δδ 0.3–0.4 ppm) was seen also in 1H NMR spectra of crystallographically characterized 3c, and this trend in chemical shift was used to assign the relative configuration to other ETP products 3 prepared in this study. In cases where the epidisulfide diastereomer was isolated and characterized, the chemical shift difference between its methylene hydrogens ranged from 1.1–1.4 ppm (see the ESI†). Using the X-ray model of 3e and a computational model (B3LYP/631-G*) of diastereomer 12, the chemical shifts (shown in parentheses in Fig. 3A and B) of the C7 methylene hydrogens of the two ETP stereoisomers were calculated and found to be in accord with the observed trend.¶
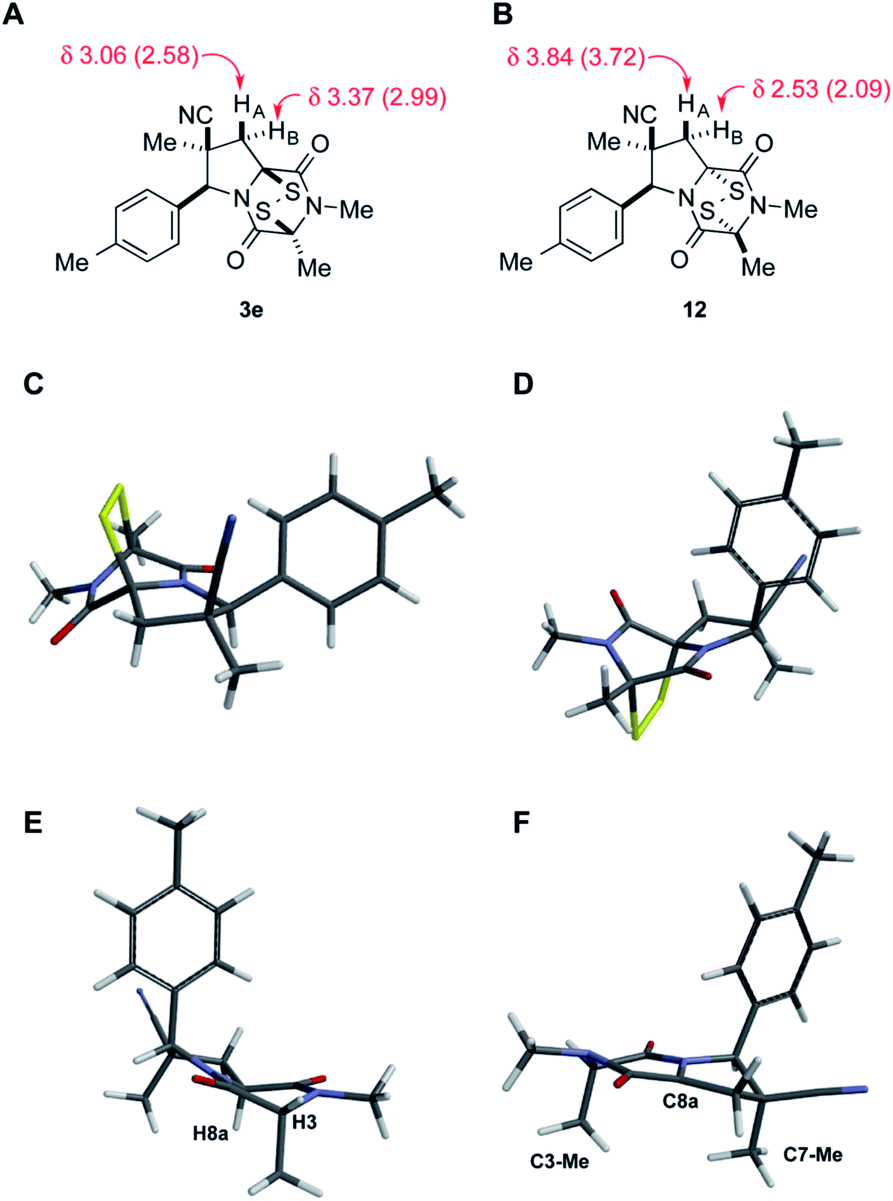 | ||
| Fig. 3 Representative data used to assign the relative configuration of the disulfide bridge in ETP products, X-ray model of dioxopiperazine 8e, and a computation model of the derived angular enolate. (A and B) show the observed (in CDCl3) and calculated (in parentheses) chemical signals of the methylene hydrogens of the pyrrolidine ring of ETP diastereomers 3e and 12. (C) is the X-ray model of 3e. (D) is the computational model (B3LYP/631-G*) of 12. (E) is the X-ray model of dioxopiperazine 8e. (F) is the predominant conformer (B3LYP/631-G*) of the angular enolate formed from dioxopiperazine 8e. | ||
The origin of diastereoselection in the preferential formation of epidisulfide stereoisomer 3 is poorly understood at this time. We hypothesize that enolization and subsequent configuration-determining sulfenylation occurs initially at the angular C8a carbon, because the expected boat conformation of the dioxopiperazine precursor positions the C3 methine hydrogen nearly coplanar with the adjacent carbonyl group (see X-ray model Fig. 3E).11 Consistent with this suggestion, treatment of dioxopiperazine 8e with 0.5, 1.0 and 1.5 equiv. of NaHMDS at room temperature, followed by quenching with D2O after 10 min resulted in preferential deuteration at C8a. The predominant conformer of the C8a enolate of 8e found by computational modeling is depicted in Fig. 3F. Sulfenylation at C8a from the bottom face (anti to CN) of this enolate would be disfavored by the “axially” oriented methyl groups at C3 and C7, whereas sulfenylation from the top face would be disfavored by the orientation of the p-tolyl substituent. Perhaps the potential of the p-tolyl substituent to minimize its steric influence by a 30–40° rotation about the aryl carbon–C6 σ-bond is responsible for the observed facial selectivity in forming the disulfide bridge.
A summary of the ETP alkaloid analogues prepared during this investigation and the yields obtained for each of the three steps is provided in Table 1 of the ESI.† Purification by chromatography was only necessary after the 1,3-dipolar cycloaddition and disulfenylation steps, whereas purification of the 2,6-dioxopiperazines was accomplished by trituration in most cases.
Antitumor activity
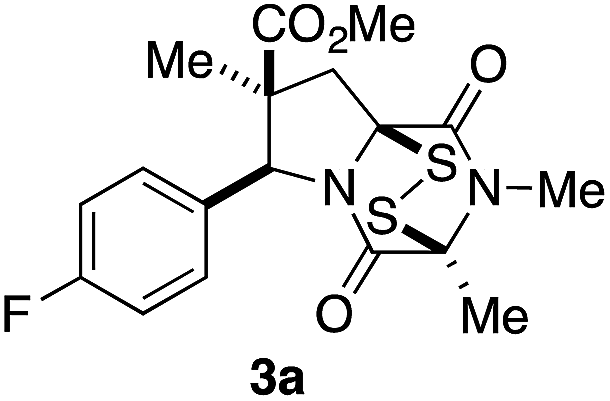
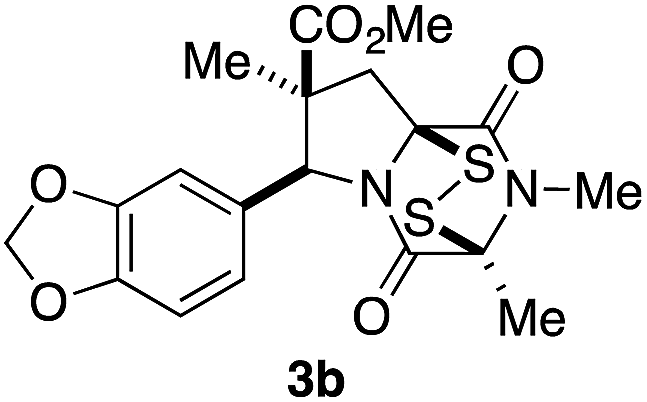
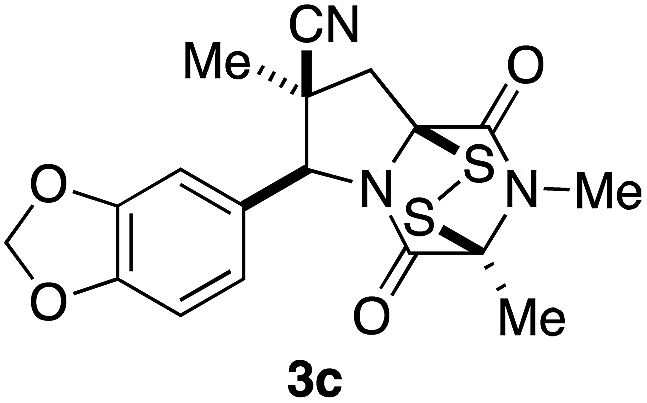
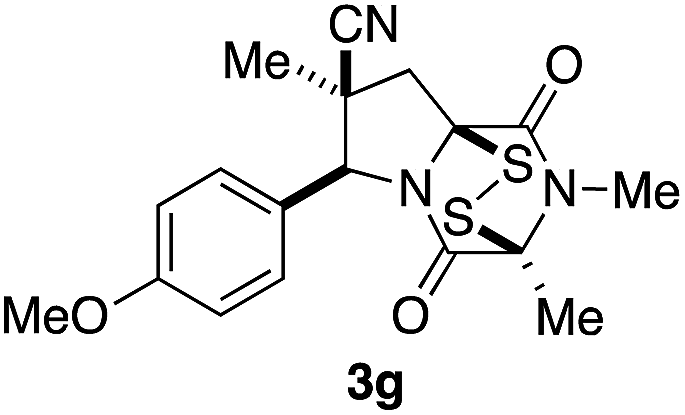
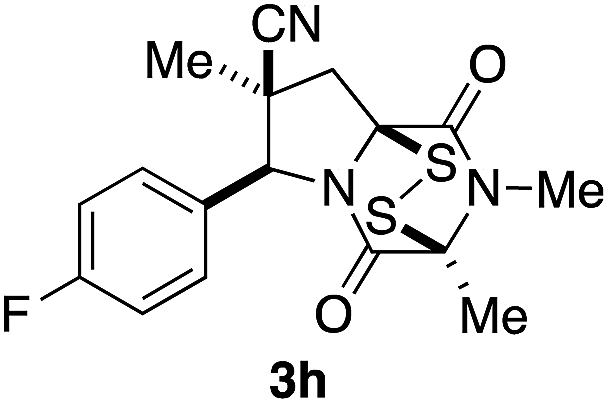
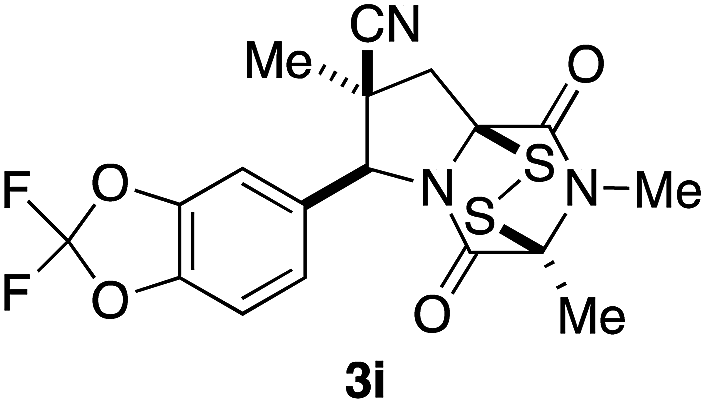
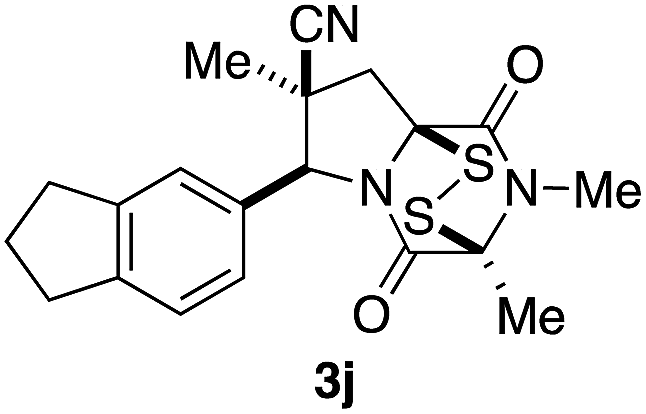
With a selection of ETP alkaloid analogues in hand, their in vitro cytotoxicity against two highly invasive cancer cell lines, DU145 (human prostate cancer) and A2058 (human melanoma) were determined (Table 1). Analogues 3a and 3b, in which the electron-withdrawing substituent in the pyrrolidine ring is a methyl ester, exhibited no observable activity (IC50 values > 5 μM). In contrast, the corresponding analogues incorporating a nitrile are considerably more cytotoxic, particularly in the methylenedioxyphenyl series as shown by comparison of 3b and 3c. Of the molecules in this series explored to date, 3c is the most potent showing IC50 values against DU145 and A2058 tumor cells in the double-digit nanomolar range that are comparable to those found for chaetocin A (2) under identical assay conditions.6d The presence of oxygen at meta and para positions of the aryl substituent is beneficial, with the absence of oxygen at the meta position (3g), at the para position (3k), or at both the meta and para positions (3f and 3j) resulting in a 4–15-fold reduction in cytotoxicity. When the electron-donating groups at C3 and C4 are replaced by weaker donors, cytotoxicity is reduced (compare 3g with 3h, and 3c with 3i). A small alkyl substituent on the nitrogen atom of dioxopiperazine ring is favored, with an n-Bu or morpholinoethyl substituent reducing cytotoxicity by an order of magnitude (compare 3c, 3m, and 3o). However, substituting methyl for ethyl (3l) or cyclopropyl (3n) at this position has only minimal effect on cytotoxicity. Methyl is favored over benzyl at the sulfur-bearing carbon of the dioxopiperazine ring, although the significant activity of benzyl analogue 3p suggests that substitution is tolerated at this site.| Compound | IC50 (μM) | |
|---|---|---|
| DU145 (prostate) | A2058 (melanoma) | |
|
>5 | >5 |
|
>5 | >5 |
|
0.10 | 0.06 |
|
0.26 | 0.24 |
|
0.59 | 0.50 |
|
0.78 | 0.87 |
|
0.42 | 0.65 |
|
2.0 | 1.6 |
|
0.71 | 0.42 |
|
0.75 | 0.50 |
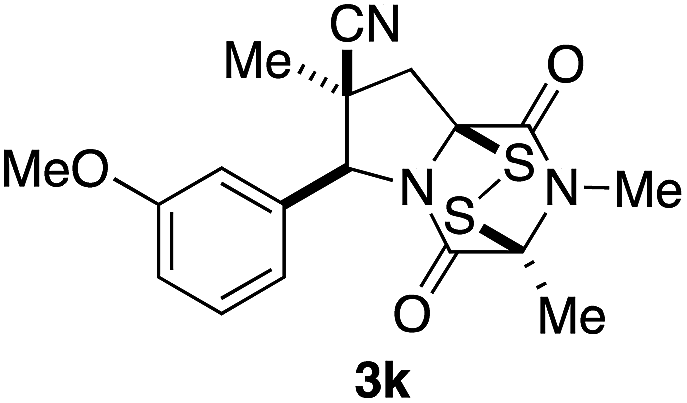
|
0.92 | 0.50 |
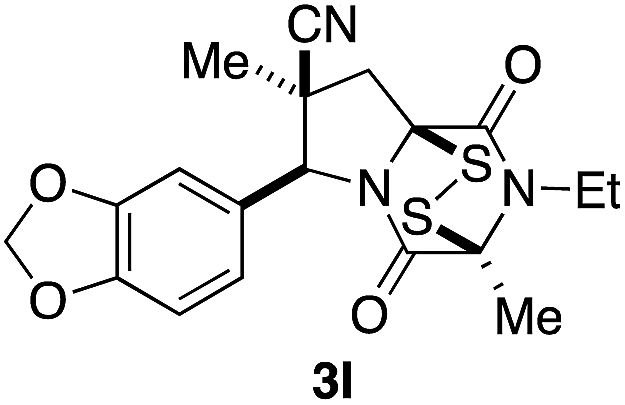
|
0.24 | 0.10 |
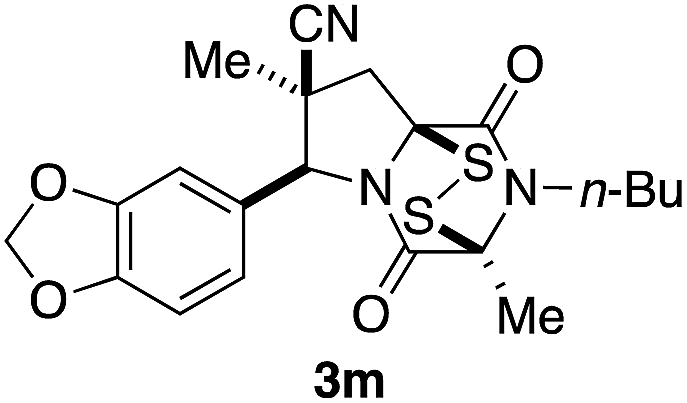
|
1.2 | 0.82 |
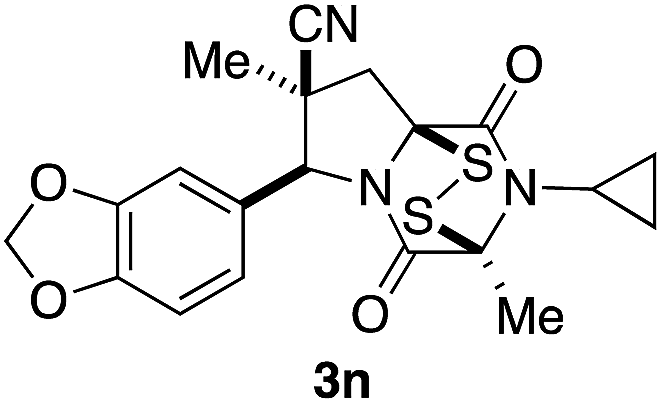
|
0.26 | 0.07 |
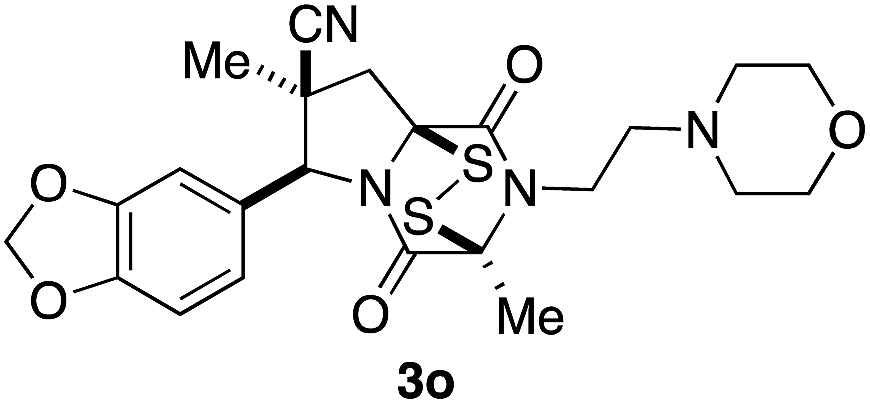
|
0.90 | 0.61 |
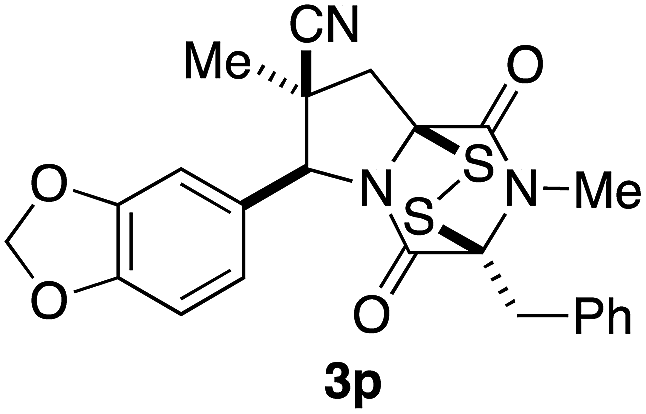
|
0.31 | 0.35 |
|
0.75 | 0.22 |
|
2.2 | 3.8 |
|
0.87 | 0.51 |
|
0.13 | 0.06 |
|
0.81 | 0.75 |
| Chaetocin A (2) | 0.073 | 0.061 |
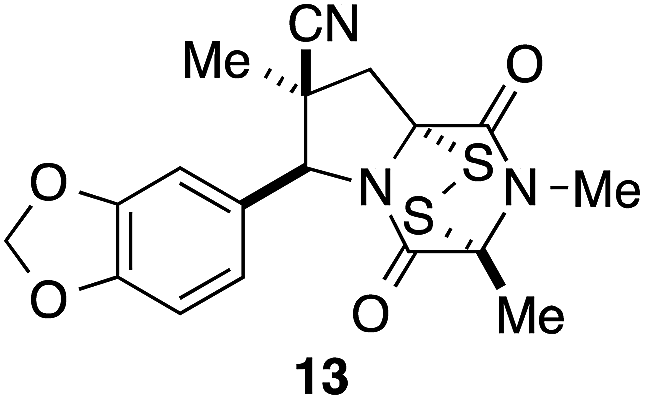
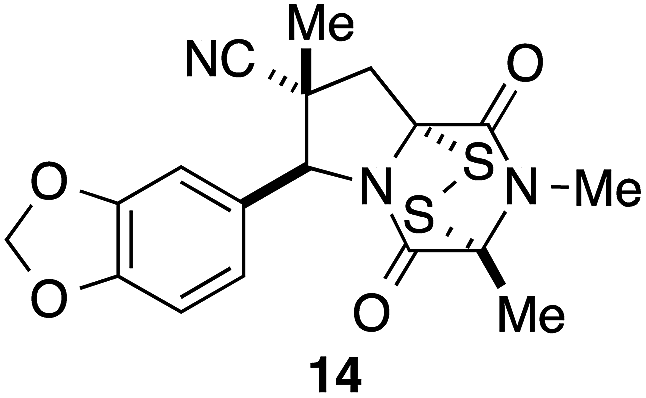
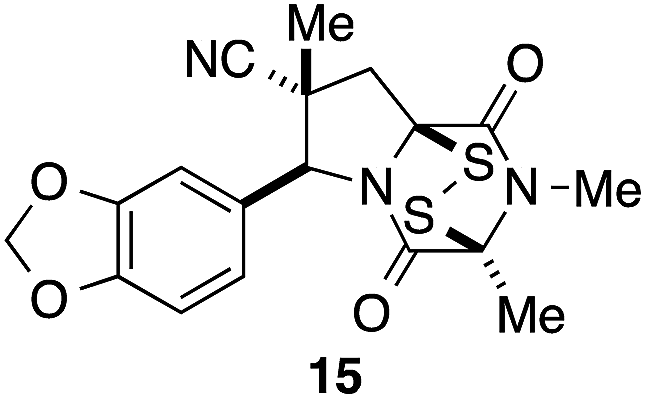
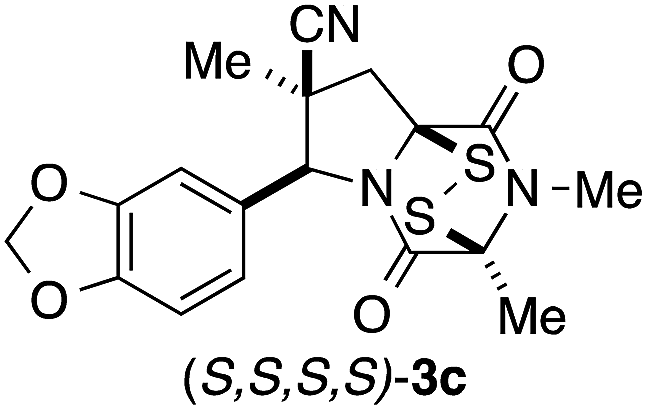
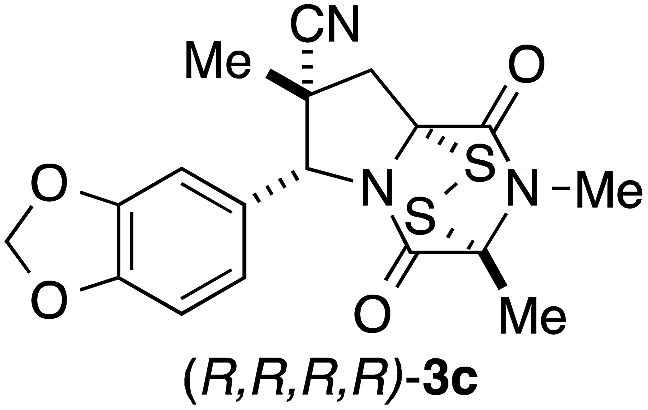
It was particularly important to establish that relative and absolute configuration of the early lead compound 3c is important for activity. Analogue 13, having the opposite relative configuration of the disulfide bridge to 3c, showed decreased potency against DU145 and A2058. Analogues 14 and 15, which are derived from the minor exo cycloadduct epi-5c, and, thus, have the nitrile substituent trans to the aryl substituent, showed weak activity only. We were able to separate the two enantiomers of 3c by enantioselective chromatography and assign their absolute configurations by a combination of CD analysis and single-crystal X-ray crystallography.|| The (S,S,S,S)-enantiomer of 3c (the absolute configuration depicted for all racemic ETP alkaloid analogues in this report) was found to be more active by a factor of five against DU145 cells and a factor of ten against A2058 cells compared to the (R,R,R,R)-enantiomer of 3c.
Having established that the (S,S,S,S)-enantiomer was the more active form of 3c, we developed a method to access it selectively. This was achieved by acylation of racemic pyrrolidine 5c with Fmoc-L-alanyl chloride and chromatographic separation of the diastereoisomeric products to afford enantiopure amide 16 in 45% yield (Scheme 3).** Cleavage of the Fmoc group lead to spontaneous formation of 2,6-dioxopiperazine 17 and subsequent N-methylation furnished 18. Disulfenylation of 18 proceeded in modest yield to afford enantiopure (S,S,S,S)-3c.
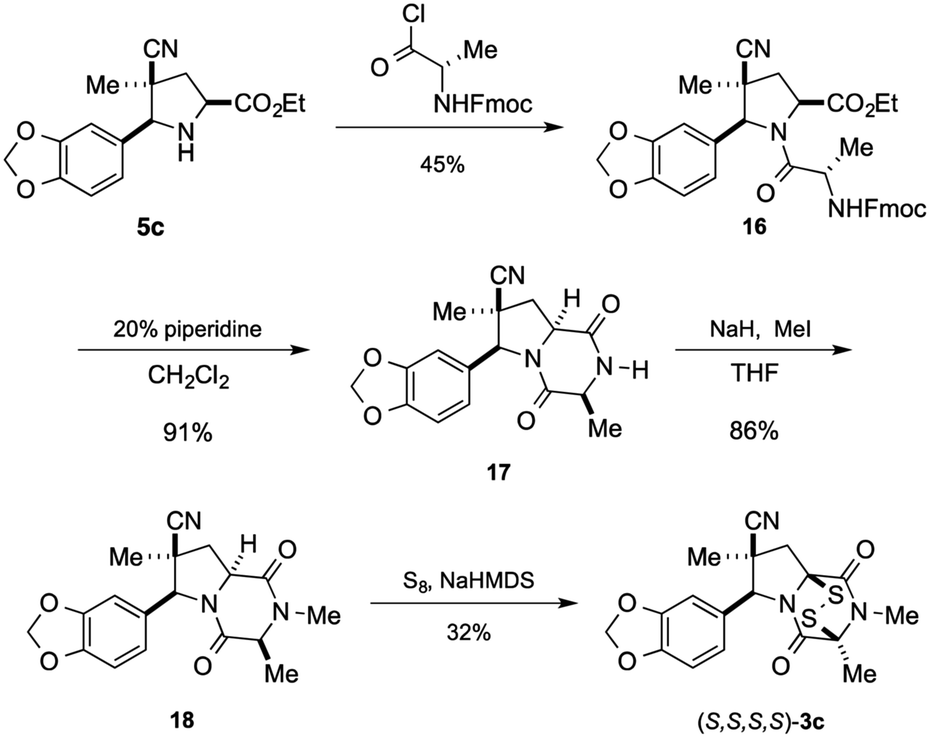 | ||
| Scheme 3 Synthesis of (S,S,S,S)-3c. | ||
The lead compound in this series, (±)-3c, was screened against eleven cancer cell lines representing eight cancer types (Table 2). Low single-digit nanomolar activity was observed for both solid (Huh-7 liver) and blood (AML) cancers. In addition, analogue (±)-3c was shown to significantly suppress tumor growth in xenograft tumor models of melanoma (Fig. 4A) and lung cancer (Fig. 4B) by intraperitoneal (IP) injection or oral administration. Consistent with suppression of tumor volumes, analogue (±)-3c slowed growth of tumor weights in both mouse xenograft models. In addition, no obvious signs of toxicity (e.g., weight loss or diarrhea) were observed in the mice during these studies.
| Cancer type | Cell line | IC50 (nM) |
|---|---|---|
| Prostate | DU145 | 130 |
| Melanoma | A2058 | 100 |
| Ovarian | SKOV3 | 90 |
| Breast | HCC38 | 75 |
| AML | MV4-11 | 1.8 |
| Lung | A549 | 100 |
| Liver | Huh-7 | 3.3 |
| HepG2 | 14 | |
| Pancreatic | SU.86.86 | 86 |
| BxPC3 | 210 | |
| Panc1 | 824 |
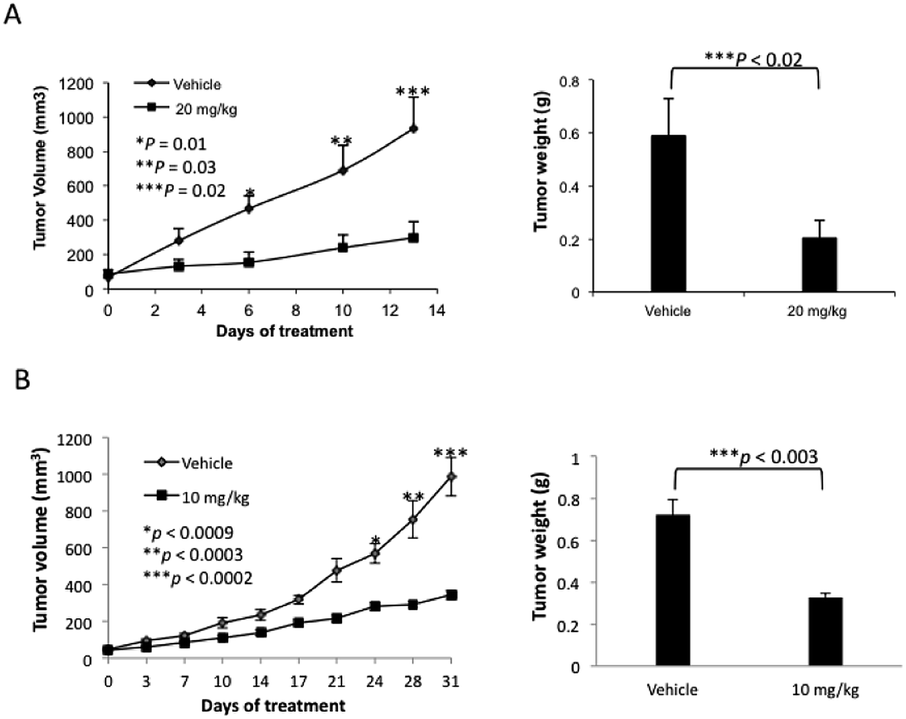 | ||
| Fig. 4 Agent (±)-3c suppresses tumor growth in xenografts of A2058 human melanoma (A) and A549 human non-small lung cancer cells (B). (A) A2058 human melanoma cells (3 × 106) were subcutaneously injected into the flanks of 5–6 weeks old athymic female nude mice. When palpable tumor sizes reached approximately 100 mm3, (±)-3c was administered by IP injection at 20 mg kg−1 once daily for 13 days. (B) A549 human non-small lung cancer cells (5 × 106) were subcutaneously injected into the flanks of 5–6 weeks old female NSG mice. When palpable tumor sizes reached approximately 50 mm3, (±)-3c was orally administered at 10 mg kg−1 once daily for 31 days. | ||
Conclusion
A short sequence, typically involving the isolation and purification of only two intermediates, was developed for preparing 1,4-dioxohexahydro-6H-3,8a-epidithiopyrrolo[1,2-a]pyrazines 3. The sequence begins with the endo-stereoselective dipolar cycloaddition of glycine imines of aryl aldehydes with methyl methacrylate or methacrylonitrile. The pyrrolidine ester products 5 are converted in standard ways to dioxopiperazines 8, which are transformed with modest stereoselectivity to ETP alkaloid analogues 3 by reaction with NaHMDS and S8. In contrast to other applications of this procedure for sulfenylation,15 epidisulfide products are formed predominantly in this step.The racemic 1,4-dioxohexahydro-6H-3,8a-epidithiopyrrolo[1,2-a]pyrazines 3 prepared in this study, which are analogues of polycyclic ETP alkaloids such as chaetomin (1) and chaetocin A (2), were screened initially against two invasive human cancer cell lines: DU145 (prostate cancer) and A2058 (melanoma). Most of these analogues showed IC50 values of <1 μM. The highest cytotoxicity against these cell lines was seen with analogues 3 containing a nitrile substituent, an electron-rich aromatic ring, and two methyl substituents in the dioxopiperazine ring, with analogue 3c emerging as the lead compound in this series. Stereoisomers of 3c (13, 14, and 15) showed reduced activity, and separation of the enantiomers of 3c demonstrated that cytotoxicity resided largely in the (S,S,S,S)-enantiomer. In screening against an additional nine cancer cell lines, analogue (±)-3c showed low single-digit nanomolar activity against both blood (AML) and solid (Huh-7 liver) cancers. Of particular significance, analogue (±)-3c showed promising in vivo activity in xenograft models of human melanoma and lung cancer by both IP and oral administration. These studies establish 1,4-dioxohexahydro-6H-3,8a-epidithiopyrrolo[1,2-a]pyrazine 3c and related structures as promising clinical candidates for cancer chemotherapy.
Acknowledgements
Partial financial support for chemical synthesis investigations was provided by the National Institute of General Medical Sciences of NIH (R01GM-30859 to L.E.O.). Biological investigations at the City of Hope were supported in part by NIH P30-CA22572 (to D.H.) and the Drug Discovery and Structural Biology Core facility. Support from the A. Gary Anderson Family Foundation and Panda Charitable Foundation (to D.H.) is gratefully acknowledged. M.B. thanks the Alexander von Humboldt Foundation for a Feodor Lynen Postdoctoral Fellowship, A.P.D. the German Academic Exchange Service (DAAD) for a postdoctoral fellowship, B.M.L. the National Cancer Institute for partial support from postdoctoral fellowship (1F32CA180741), and M.C.W. the National Science Foundation for a graduate fellowship (DGE-1321846). NMR and mass spectra were determined at UC Irvine using instruments purchased with the assistance of NSF and NIH shared instrumentation grants. The authors thank Dr Joseph Ziller and Dr John Greaves, Department of Chemistry, UC Irvine, for their assistance with X-ray and mass spectrometric analyses.Notes and references
- For reviews, see: (a) P. Waring, R. D. Eichner and A. Müllbacher, Med. Res. Rev., 1988, 8, 499 CrossRef CAS PubMed; (b) D. M. Vigushin, N. Mirsaidi, G. Brooke, C. Sun, P. Pace, L. Inman, C. J. Moody and R. C. Coombes, Med. Oncol., 2004, 21, 21 CrossRef CAS PubMed; (c) D. M. Gardiner, P. Waring and B. J. Howlett, Microbiology, 2005, 151, 1021 CrossRef CAS PubMed; (d) C.-S. Jiang and Y.-W. Guo, Mini-Rev. Med. Chem., 2011, 11, 728 CrossRef CAS; (e) E. Iwasa, Y. Hamashima and M. Sodeoka, Isr. J. Chem., 2011, 51, 420 CrossRef CAS PubMed; (f) C.-S. Jiang, W. E. G. Müller, H. C. Schröder and Y.-W. Guo, Chem. Rev., 2012, 112, 2179 CrossRef CAS PubMed.
- B. Onnis, A. Rapisarda and G. Melillo, J. Cell. Mol. Med., 2009, 13, 2780 CrossRef CAS PubMed.
- (a) T. Saito, Y. Suzuki, K. Koyama, S. Natori, Y. Iitaka and T. Kinoshita, Chem. Pharm. Bull., 1988, 36, 1942 CrossRef CAS; (b) K. M. Cook, S. T. Hilton, J. Mecinović, W. B. Motherwell, W. D. Figg and C. J. Schofield, J. Biol. Chem., 2009, 284, 26831 CrossRef CAS PubMed; (c) Y. Teng, K. Iuchi, E. Iwasa, S. Fujishiro, Y. Hamashima, K. Dodo and M. Sodeoka, Bioorg. Med. Chem. Lett., 2010, 20, 5085 CrossRef CAS PubMed; (d) C. R. Isham, J. D. Tibodeau, A. R. Bossou, J. R. Merchan and K. C. Bible, Br. J. Cancer, 2012, 106, 314 CrossRef CAS PubMed; (e) H. T. Tran, H. N. Kim, I. K. Lee, T. N. Nguyen-Pham, J. S. Ahn, Y. K. Kim, J. J. Lee, K. S. Park, H. Kook and H. J. Kim, J. Korean Med. Sci., 2013, 28, 237 CrossRef CAS PubMed.
- (a) C. R. Isham, J. D. Tibodeau, W. Jin, R. Xu, M. M. Timm and K. C. Bible, Blood, 2007, 109, 2579 CrossRef CAS PubMed; (b) K. C. Bible, C. R. Isham, R. Xu and J. D. Tibodeau, Methods and Compositions for Treating Cancer, PCT Int. Appl., 2008 Search PubMed, WO 2008112014 A1 20080918 (c) H. Chaib, A. Nebbioso, T. Prebet, R. Castellano, S. Garbit, A. Restouin, N. Vey, L. Altucci and Y. Collette, Leukemia, 2012, 26, 662 CrossRef CAS PubMed; (d) K. M. Reece, E. D. Richardson, K. M. Cook, T. J. Campbell, S. T. Pisle, A. J. Holly, D. J. Venzon, D. J. Liewehr, C. H. Chau, D. K. Price and W. D. Figg, Mol. Cancer, 2014, 13, 91 CrossRef PubMed; (e) T. Chiba, T. Saito, K. Yuki, Y. Zen, S. Koide, N. Kanogawa, T. Motoyama, S. Ogasawara, E. Suzuki, Y. Ooka, A. Tawada, M. Otsuka, M. Miyazaki, A. Iwama and O. Yokosuka, Int. J. Cancer, 2015, 136, 289 CrossRef CAS PubMed.
- (a) D. Greiner, T. Bonaldi, R. Eskeland, E. Roemer and A. Imhof, Nat. Chem. Biol., 2005, 1, 143 CrossRef CAS PubMed; (b) J. D. Tibodeau, L. M. Benson, C. R. Isham, W. G. Owen and K. C. Bible, Antioxid. Redox Signaling, 2009, 11, 1097 CrossRef CAS PubMed; (c) E. Iwasa, Y. Hamashima, S. Fujishiro, E. Higuchi, A. Ito, M. Yoshida and M. Sodeoka, J. Am. Chem. Soc., 2010, 132, 4078 CrossRef CAS PubMed; (d) M. Sodeoka, K. Dodo, Y. Teng, K. Iuchi, Y. Hamashima, E. Iwasa and S. Fujishiro, Pure Appl. Chem., 2012, 84, 1369 CrossRef CAS; (e) D. Illner, R. Zinner, V. Handtke, J. Rouquette, H. Strickfaden, C. Lanctôt, M. Conrad, A. Seiler, A. Imhof, T. Cremer and M. Cremer, Exp. Cell Res., 2010, 316, 1662 CrossRef CAS PubMed; (f) A. Lakshmikuttyamma, S. A. Scott, J. F. DeCoteau and C. R. Geyer, Oncogene, 2010, 29, 576 CrossRef CAS PubMed; (g) Y.-M. Lee, J.-H. Lim, H. Yoon, Y.-S. Chun and J.-W. Park, Hepatology, 2011, 53, 171 CrossRef CAS PubMed; (h) C. Dong, Y. Wu, Y. Wang, C. Wang, T. Kang, P. G. Rychahou, Y.-I. Chi, B. M. Evers and B. P. Zhou, Oncogene, 2013, 32, 1351 CrossRef CAS PubMed; (i) Y. Yokoyama, M. Hieda, Y. Nishioka, A. Matsumoto, S. Higashi, H. Kimura, H. Yamamoto, M. Mori, S. Matsuura and N. Matsuura, Cancer Sci., 2013, 104, 889 CrossRef CAS PubMed; (j) F. L. Cherblanc, K. L. Chapman, J. Reid, A. J. Borg, S. Sundriyal, L. Alcazar-Fuoli, E. Bignell, M. Demetriades, C. J. Schofield, P. A. DiMaggio Jr, R. Brown and M. J. Fuchter, J. Med. Chem., 2013, 56, 8616 CrossRef CAS PubMed; (k) D. Dixit, R. Ghildiyal, N. P. Anto and E. Sen, Cell Death Dis., 2014, 5, e1212 CrossRef CAS PubMed; (l) M.-C. Lee, Y.-Y. Kuo, W.-C. Chou, H.-A. Hou, M. Hsiao and H.-F. Tien, J. Leukocyte Biol., 2014, 95, 105 CrossRef PubMed.
- (a) K. M. Block, H. Wang, L. Z. Szabó, N. W. Polaske, L. K. Henchey, R. Dubey, S. Kushal, C. F. László, J. Makhoul, Z. Song, E. J. Meuillet and B. Z. Olenyuk, J. Am. Chem. Soc., 2009, 131, 18078 CrossRef CAS PubMed; (b) S. Fujishiro, K. Dodo, E. Iwasa, Y. Teng, Y. Sohtome, Y. Hamashima, A. Ito, M. Yoshida and M. Sodeoka, Bioorg. Med. Chem. Lett., 2013, 23, 733 CrossRef CAS PubMed; (c) N. Boyer, K. C. Morrison, J. Kim, P. J. Hergenrother and M. Movassaghi, Chem. Sci., 2013, 4, 1646 RSC; (d) J. E. DeLorbe, D. Horne, R. Jove, S. M. Mennen, S. Nam, F.-L. Zhang and L. E. Overman, J. Am. Chem. Soc., 2013, 135, 4117 CrossRef CAS PubMed; (e) R. Dubey, M. D. Levin, L. Z. Szabo, C. F. Laszlo, S. Kushal, J. B. Singh, P. Oh, J. E. Schnitzer and B. Z. Olenyuk, J. Am. Chem. Soc., 2013, 135, 4537 CrossRef CAS PubMed.
- L. E. Overman, M. Baumann, S. Nam, D. Horne, R. Jove, J. Xie and C. Kowolik, Preparation of Epipolythiodioxopiperazine ETP Derivatives for Treatment of Cancer, PCT Int. Appl., 2014 Search PubMed WO 2014066435 A1.
- R. Grigg, J. Kemp and W. J. Warnock, J. Chem. Soc., Perkin Trans. 1, 1987, 2275 RSC.
- O. Tsuge, S. Kanemasa and M. Yoshioka, J. Org. Chem., 1988, 53, 1384 CrossRef CAS.
- A. D. Borthwick, Chem. Rev., 2012, 112, 3641 CrossRef CAS PubMed.
- X-ray coordinates were deposited with the Cambridge Crystallographic Data Centre: 3c: 1061873, 3e: 1061871, 8e: 1061870; and 8f: 1061869.
- (a) J. Kim, J. A. Ashenhurst and M. Movassaghi, Science, 2009, 324, 238 CrossRef CAS PubMed; (b) J. Kim and M. Movassaghi, J. Am. Chem. Soc., 2010, 132, 14376 CrossRef CAS PubMed.
- E. Öhler, H. Poisel, F. Tataruch and U. Schmidt, Chem. Ber., 1972, 105, 635 CrossRef PubMed.
- K. C. Nicolaou, D. Giguère, S. Totokotsopoulos and Y.-P. Sun, Angew. Chem., Int. Ed., 2012, 51, 728 CrossRef CAS PubMed.
- (a) J. A. Codelli, A. L. A. Puchlopek and S. E. Reisman, J. Am. Chem. Soc., 2012, 134, 1930 CrossRef CAS PubMed; (b) The formation of small amounts of the disulfide and trisulfide products in addition to the predominant tetrasulfide has been noted, see: K. C. Nicolaou, M. Lu, S. Totokotsopoulos, P. Heretsch, D. Giguère, Y.-P. Sun, D. Sarlah, T. H. Nguyen, I. C. Wolf, D. F. Smee, C. W. Day, S. Bopp and E. A. Winzeler, J. Am. Chem. Soc., 2012, 134, 17320 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Table summarizing yields and diastereoselectivity in the synthesis of ETP alkaloid analogues 3, experimental procedures, and characterization data for all new compounds. CCDC 1061869–1061873. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5sc01536g |
| ‡ At 60% conversion of the cycloadduct, mass spectrometric analysis indicated that a 3:2 ratio of mono to dihydroxylated products was present. |
| § The relative configuration of the C7 hydrogens was established by 1H NMR NOE studies. |
| ¶ Conformer populations were generated by molecular mechanics and low energy conformations were optimized by DFT calculations at the B3LYP/631-G* level. Calculations were done using Spartan 14 (Wavefunction, Inc.). |
| || See ESI for details. |
| ** The yield for the other diastereomer was 42%. |
| This journal is © The Royal Society of Chemistry 2015 |