
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Water oxidation catalysed by iron complex of N,N′-dimethyl-2,11-diaza[3,3](2,6)pyridinophane. Spectroscopy of iron–oxo intermediates and density functional theory calculations†
Wai-Pong
To
a,
Toby
Wai-Shan Chow
a,
Chun-Wai
Tse
a,
Xiangguo
Guan
*a,
Jie-Sheng
Huang
a and
Chi-Ming
Che
*ab
aDepartment of Chemistry and State Key Laboratory of Synthetic Chemistry, The University of Hong Kong, Pokfulam Road, Hong Kong, China. E-mail: xgguan@hku.hk; cmche@hku.hk
bHKU Shenzhen Institute of Research and Innovation, Shenzhen 518053, China
First published on 22nd July 2015
Abstract
The macrocyclic [FeIII(L1)Cl2]+ (1, L1 = N,N′-dimethyl-2,11-diaza[3,3](2,6)pyridinophane) complex is an active catalyst for the oxidation of water to oxygen using [NH4]2[CeIV(NO3)6] (CAN), NaIO4, or Oxone as the oxidant. The mechanism of 1-catalysed water oxidation was examined by spectroscopic methods and by 18O-labelling experiments, revealing that FeIV![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O and/or FeVO species are likely to be involved in the reaction. The redox behaviour of 1 and these high-valent FeO species of L1 has been examined by both cyclic voltammetry and density functional theory (DFT) calculations. In aqueous solutions, the cyclic voltammograms of 1 at different pH show a pH-dependent reversible couple (E1/2 = +0.46 V vs. SCE at pH 1) and an irreversible anodic wave (Epa = +1.18 V vs. SCE at pH 1) assigned to the FeIII/FeII couple and the FeIII to FeIV oxidation, respectively. DFT calculations showed that the E value of the half reaction involving [FeV(L1)(O)(OH)]2+/[FeIV(L1)(O)(OH2)]2+ is +1.42 V vs. SCE at pH 1. Using CAN as the oxidant at pH 1, the formation of an FeIVO reaction intermediate was suggested by ESI-MS and UV-vis absorption spectroscopic measurements, and the rate of oxygen evolution was linearly dependent on the concentrations of both 1 and CAN. Using NaIO4 or Oxone as the oxidant at pH 1, the rate of oxygen evolution was linearly dependent on the concentration of 1, and a reactive FeVO species with formula [FeV(L1)(O)2]+ generated by oxidation with NaIO4 or Oxone was suggested by ESI-MS measurements. DFT calculations revealed that [FeV(L1)(O)2]+ is capable of oxidizing water to oxygen with a reaction barrier of 15.7 kcal mol−1.
O and/or FeVO species are likely to be involved in the reaction. The redox behaviour of 1 and these high-valent FeO species of L1 has been examined by both cyclic voltammetry and density functional theory (DFT) calculations. In aqueous solutions, the cyclic voltammograms of 1 at different pH show a pH-dependent reversible couple (E1/2 = +0.46 V vs. SCE at pH 1) and an irreversible anodic wave (Epa = +1.18 V vs. SCE at pH 1) assigned to the FeIII/FeII couple and the FeIII to FeIV oxidation, respectively. DFT calculations showed that the E value of the half reaction involving [FeV(L1)(O)(OH)]2+/[FeIV(L1)(O)(OH2)]2+ is +1.42 V vs. SCE at pH 1. Using CAN as the oxidant at pH 1, the formation of an FeIVO reaction intermediate was suggested by ESI-MS and UV-vis absorption spectroscopic measurements, and the rate of oxygen evolution was linearly dependent on the concentrations of both 1 and CAN. Using NaIO4 or Oxone as the oxidant at pH 1, the rate of oxygen evolution was linearly dependent on the concentration of 1, and a reactive FeVO species with formula [FeV(L1)(O)2]+ generated by oxidation with NaIO4 or Oxone was suggested by ESI-MS measurements. DFT calculations revealed that [FeV(L1)(O)2]+ is capable of oxidizing water to oxygen with a reaction barrier of 15.7 kcal mol−1.
Introduction
Water oxidation is an energetically uphill reaction (E = +1.23 V vs. NHE at pH 0) involving the simultaneous removal of four electrons and four protons from two water molecules to give one oxygen molecule.1 Due to its importance, there have been tremendous efforts dedicated to the design of metal catalysts for water oxidation over the past decades.2 It is envisioned that mechanistic insights into the fundamental steps of this important reaction can be obtained by using structurally defined molecular catalysts.In the literature, many examples of molecular catalysts, particularly polypyridyl ruthenium(II) and organometallic iridium(III) complexes, and also complexes of 1st-row transition metals such as manganese, iron and cobalt bearing chelating N and/or O ligands, have been reported for water oxidation.2 Mechanistic studies revealed that the availability of labile coordination site(s) for the formation of metal–oxo species is crucial for a metal complex to be used to catalyse water oxidation.2 To support the formation of reactive/oxidizing metal–oxo species, ligands that bind strongly to metal ions and are resistant to oxidation are highly desirable. In this regard, macrocyclic N-donor ligands are appealing, as highly oxidizing metal–oxo complexes of both ruthenium and iron have been isolated and structurally characterized by using macrocyclic tertiary amine ligands such as 14-TMC (1,4,8,11-tetramethyl-1,4,8,11-tetraazacyclotetradecane).3
Because of the high earth abundance of iron and the recent impressive advances made in the isolation and characterization of non-heme iron–oxo complexes, notably by Nam, Que, and co-workers,4,5 there has been increasing interest in the development of new iron-based oxidation chemistry, including the oxidation of water.2 Since the first report by Bernhard, Collins, and co-workers in 2010,6a several types of water oxidation reactions catalysed by mononuclear iron complexes have been reported, including chemical oxidation with CAN (cerium ammonium nitrate [NH4]2[CeIV(NO3)6]) or NaIO4,6 photochemical oxidation,6c,e,7 electrocatalytic oxidation,8 and photoelectrochemical oxidation.9 Dinuclear iron complexes that can catalyse water oxidation with CAN or NaIO4,10 or can oxidize water to the hydroxyl radical,11 are also documented.
Mononuclear iron catalysts for water oxidation are supported by tetraanionic tetraamide ligands (TAML)6a,7,8b or monoanionic pentadentate N5 ligands,8a or bear neutral chelating N ligands,6b–g,8c such as mcp, Me2Pytacn, tpa, and bqen (Fig. 1), which allow the formation of mononuclear metal complexes with two cis labile sites (the abovementioned 14-TMC ligand forms trans complexes such as trans-[Fe(14-TMC)(OTf)2] which was found to be unreactive for catalytic water oxidation with CAN or NaIO4 (ref. 6b)). Notably, the ‘[FeII(mcp)(OTf)2] + NaIO4’ system reported by Costas, Lloret-Fillol, and co-workers formed oxygen with a turnover number (TON) of up to >1000 at pH 2.6b Oxoiron(IV) (FeIVO) intermediates supported by Me2Pytacn6b,f and bqen,6e generated from reaction of the corresponding iron catalysts with CAN in aqueous solution, were detected by UV-vis spectroscopy and electrospray-ionization mass spectrometry (ESI-MS); it was proposed that [FeIV(bqen)(O)(OH2)]2+ may be involved in O–O bond formation,6e and density functional theory (DFT) calculations on the FeII–Me2Pytacn system favoured FeIVO reactive intermediates.12b A recent report demonstrated the involvement of OFeIV–O–CeIV species in the water oxidation with CAN catalysed by [FeII(mcp)(OTf)2].13 In most cases, oxoiron(V) (FeVO) species are proposed to be the active intermediates directly responsible for O–O bond formation in water oxidation reactions,6a,b,f,7,8a as revealed by DFT calculations on water oxidation with CAN catalysed by the FeIII–TAML,12a,d FeII–Me2Pytacn,12e,f and FeII–mep12f systems. Several non-heme FeVO species, which were generated by oxidation with peracids, H2O2 or tBuOOH in organic solvents, have been reported in the literature.14 Notable examples include [FeV(TAML)(O)]−,14a,j [FeV(Me2Pytacn)(O)(OH)]2+,14d and [FeV(L)(O)(S)]3+ (L = tpa, mep; S = H2O or MeCN).14c Recently, [FeV(TAML)(O)]− (I, Fig. 1) generated by the photochemical reaction of [FeIII(TAML)(H2O)]− with ‘[RuII(bipy)3]2+ + Na2S2O8’ in 50% MeCN–borate buffer mixture, was reported to be an active intermediate in the [FeIII(TAML)(H2O)]−-catalysed photochemical water oxidation.7 The proposed FeVO intermediates with neutral chelating N ligands (II, Fig. 1) remain elusive for water oxidation reactions.
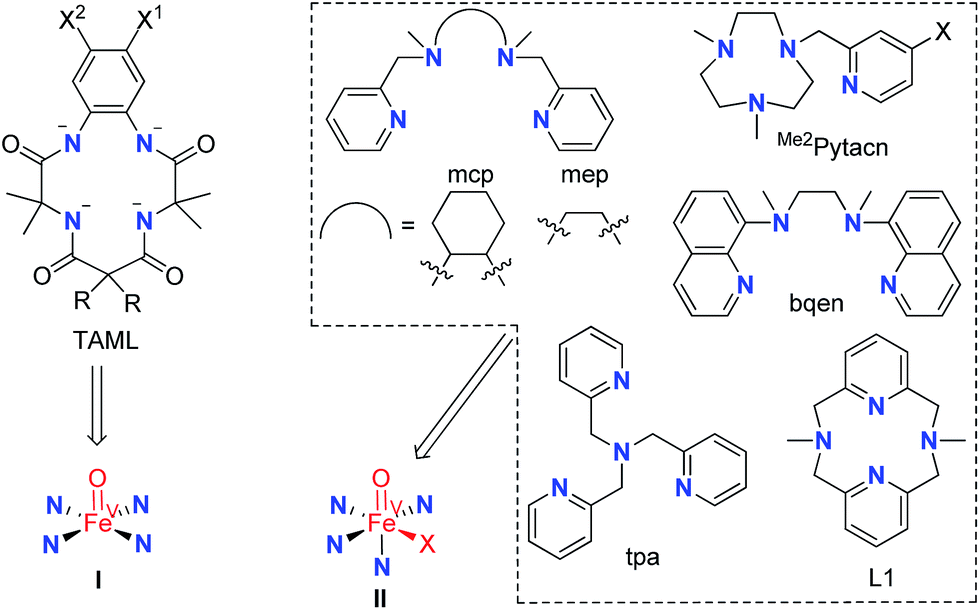 | ||
| Fig. 1 Examples of N ligands in iron complexes used as water oxidation catalysts. The corresponding proposed FeVO species are indicated. X in II stands for solvent, OH− or another ligand. | ||
N,N′-Dimethyl-2,11-diaza[3,3](2,6)pyridinophane (L1, Fig. 1),15a a neutral macrocyclic N4 ligand, is well known to form iron complexes in the cis-configuration, including [FeIII(L1)Cl2]+ (1).15b In 2010, we reported that [FeIII(L1)Cl2][FeCl4] (1·FeCl4) is an efficient catalyst for the cis-dihydroxylation of alkenes using Oxone (potassium peroxymonosulfate, 2KHSO5·KHSO4·K2SO4) as the oxidant, and the generation of an [FeV(L1)(O)2]+ intermediate in the reaction was inferred from high resolution ESI-MS analysis and DFT calculations.16a This prompted us to examine the catalytic activity of 1 toward water oxidation. It is noted that ligand L1 has also been employed for developing the oxidation chemistry of other transition metal complexes, including [OsIII(L1)(OH)(OH2)]2+, which can catalyse the cis-dihydroxylation of alkenes by H2O2via a reactive [OsV(L1)(O)(OH)]2+ intermediate,16c and [PdII(L1)(Me)2], which reacts with oxygen to generate [PdIII(L1)(Me)2(OO˙)] followed by protonation to give [PdIV(L1)(Me)2(OOH)]+.16b Hydrogen peroxide disproportionation catalysed by [Mn(L1)(H2O)2]2+ and electrochemical oxidation of water catalysed by [Mn(L1′)(H2O)2]2+ (L1′ = the N-tBu counterpart of L1) have been reported as well.17
In the present work, we report the use of [FeIII(L1)Cl2]+ (1) for water oxidation with Oxone, as well as CAN and NaIO4, under mild conditions, together with mechanistic studies by means of high-resolution ESI-MS, UV-vis absorption spectroscopy, 18O-labelling experiments, kinetic studies, cyclic voltammetry, EPR analysis, and DFT calculations. During the course of this study, Sun and co-workers communicated their findings on water oxidation with CAN catalysed by [FeII(L1)(MeCN)2]2+.6g Detailed mechanistic studies on water oxidation catalysed by Fe–L1 systems have not been reported in the literature. Also, Oxone has not been used as an oxidant in previously reported water oxidation reactions catalysed by iron complexes,6,10,11,13 despite literature reports on the manganese-catalysed oxidation of water with Oxone.2c,d The experimental studies and DFT calculations in the present work point to the generation of an FeVO species responsible for O–O bond formation in water oxidation catalysed by Fe–L1 systems.
Results
Water oxidation catalysed by complex 1
At the outset, a series of iron complexes bearing N3, N4 and N5 ligands (e.g. 1,4,7-trimethyl-1,4,7-triazacyclononane, 2,2′:6′:2′′-terpyridine, L1, and 2,2′:6′,2′′:6′′,2′′′:6′′′,2′′′′-quinquepyridine) were screened for catalytic water oxidation with CAN as oxidant under different reaction conditions (Table S1 in the ESI†). We found that [FeIII(L1)Cl2][FeCl4] (1·FeCl4) exhibited the best performance, affording oxygen with TONs of up to 41 and 32 in 0.1 M HNO3 and in pure water, respectively, after 30 minutes with CAN (840 equiv.) as oxidant (Table 1, entries 1d, 1e). With NaIO4 as oxidant, the reaction in 0.1 M HNO3 afforded oxygen with TON of 12, whereas oxygen evolved with TON of only 3 from the reaction in pure water (Table 1, entries 2a and 2b). With Oxone as oxidant, under the conditions of 100 μM 1 and 84 mM Oxone in 0.1 M HNO3, oxygen was produced with TON of 89 in 30 minutes (Table 1, entry 3a); lowering the concentration of 1 to 12.5 μM increased the TON to 113 (Table 1, entry 3b), a value higher than those obtained using CAN or NaIO4 as oxidant (TONs of 93 and 44 respectively, entries 1f and 2c in Table 1). Control experiments using [Et4N][FeCl4] instead of 1·FeCl4 as the catalyst did not produce detectable amounts of oxygen for the reactions with CAN and NaIO4, and afforded oxygen with TON of only 1.2 for the reaction with Oxone, revealing that 1 is the key species accounting for the catalytic activity of 1·FeCl4. With 1·ClO4 as catalyst, similar oxygen evolution to that catalysed by 1·FeCl4 was observed (e.g. TON of O2: 40 vs. 41 with 84 mM CAN as oxidant, 11 vs. 12 with 84 mM NaIO4 as oxidant, and 91 vs. 89 with 84 mM Oxone as oxidant for reactions using 100 μM catalyst in 0.1 M HNO3).| Entry | Oxidant | [Catalyst] (μM) | [Oxidant] (mM) | Medium | TON of O2b | OEc (%) |
|---|---|---|---|---|---|---|
a All reactions were performed under argon atmosphere and in 0.1 M HNO3 or water at room temperature for 30 min.
b Determined by GC integration.
c Based on the stoichiometric ratio (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :4) of O2 formed per oxidant in 4e− oxidation; oxidant efficiency (OE) = [no. of moles of O2 formed/(no. of moles of oxidant used/n)] × 100%, where n = 4 for CAN and n = 2 for NaIO4 and Oxone. :4) of O2 formed per oxidant in 4e− oxidation; oxidant efficiency (OE) = [no. of moles of O2 formed/(no. of moles of oxidant used/n)] × 100%, where n = 4 for CAN and n = 2 for NaIO4 and Oxone.
|
||||||
| 1a | CAN | 100 | 10 | 0.1 M HNO3 | 17 | 68 |
| 1b | CAN | 100 | 20 | 0.1 M HNO3 | 19 | 38 |
| 1c | CAN | 100 | 42 | 0.1 M HNO3 | 31 | 30 |
| 1d | CAN | 100 | 84 | 0.1 M HNO3 | 41 | 20 |
| 1e | CAN | 100 | 84 | H2O | 32 | 15 |
| 1f | CAN | 12.5 | 125 | 0.1 M HNO3 | 93 | 4 |
| 1g | CAN | 12.5 | 125 | H2O | 81 | 3 |
| 2a | NaIO4 | 100 | 84 | 0.1 M HNO3 | 12 | 3 |
| 2b | NaIO4 | 100 | 84 | H2O | 3 | 1 |
| 2c | NaIO4 | 12.5 | 125 | 0.1 M HNO3 | 44 | 1 |
| 2d | NaIO4 | 12.5 | 125 | H2O | 3 | 0.1 |
| 3a | Oxone | 100 | 84 | 0.1 M HNO3 | 89 | 21 |
| 3b | Oxone | 12.5 | 125 | 0.1 M HNO3 | 113 | 2 |
The origin of the oxygen gas produced from the reaction mixture of 1-catalysed water oxidation was investigated by using a mixture of H216O and H218O (v/v = 1:1; the H218O used had 97 atom% 18O) as the medium and by analysing the gaseous product(s) using GC-MS. With CAN as oxidant, the gaseous products obtained in the first 5 minutes were a mixture of 16O2/16O18O/18O2 with a ratio of 30.0:49.6:20.4, which is close to the theoretical value of 26.5:50.0:23.5 (ref. 6b) expected for oxygen gas derived from water. In the case of NaIO4 as oxidant, similar 18O-labelling studies using H218O were not performed, because the oxygen atoms of IO4− undergo rapid exchange with those of water.18 However, the exchange of oxygen atoms between Oxone and water is sufficiently slow for useful 18O-labelling studies.19 Water oxidation with Oxone (84 mM) in a 1:1 mixture of H218O and 0.2 M HNO3 gave oxygen with a 16O2/16O18O/18O2 ratio of 68.9:29.9:1.2 in the first 5 minutes. This ratio falls within the range of (49–91.9):(7.6–39):(0.51–12) reported for water oxidation with Oxone catalysed by a MnIII(O)2MnIV complex.19
Kinetic studies
The time courses of 1-catalysed water oxidation with CAN in 0.1 M HNO3 using different concentrations of CAN (12.5–125 mM) and 1 (6.25–25 μM) were examined; the plots of oxygen evolution (measured by GC) over time are depicted in Fig. S1 and S2 in the ESI.† A linear dependence of the initial rate of oxygen evolution on both concentration of CAN and concentration of 1 was observed, as depicted in Fig. 2.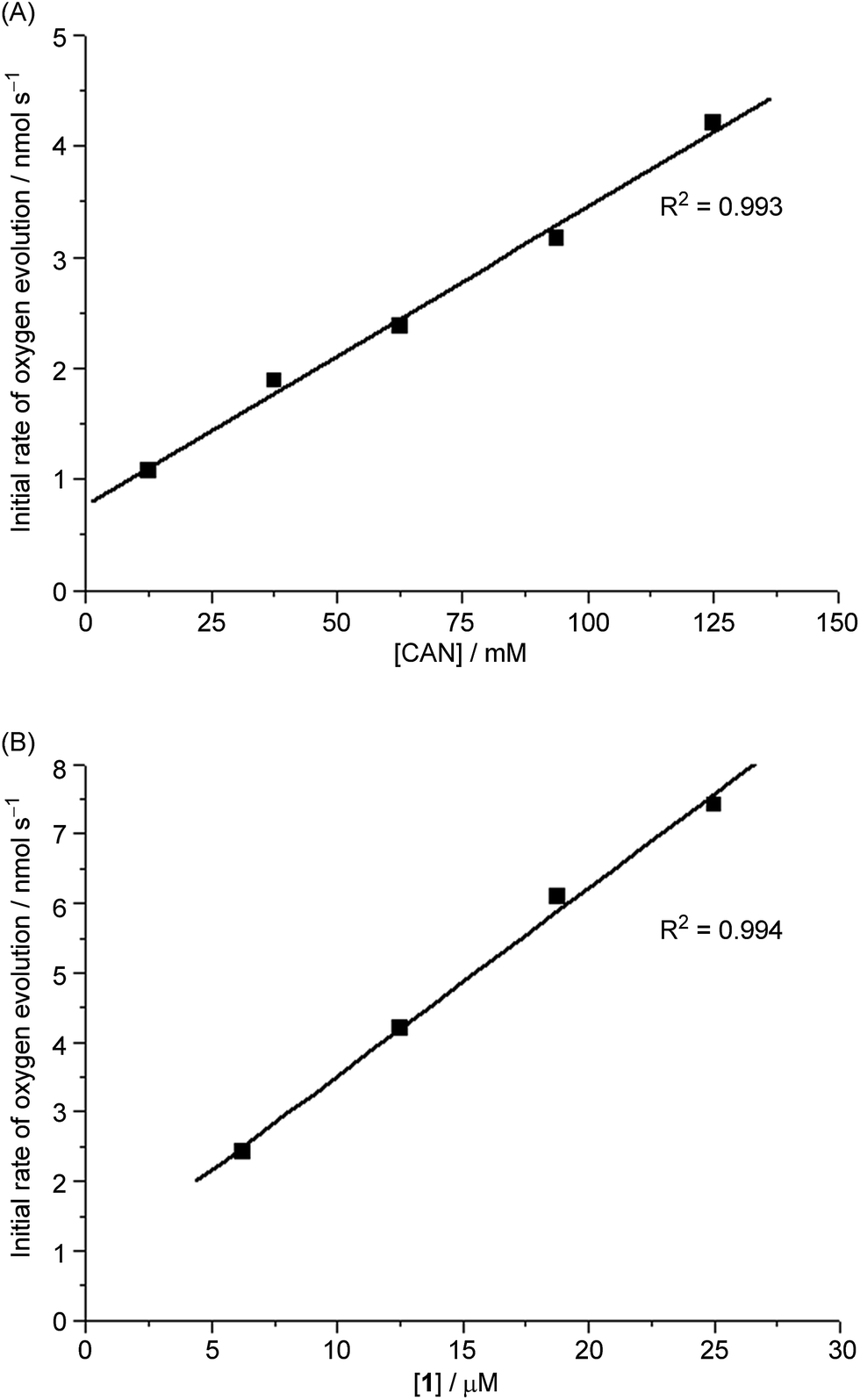 | ||
| Fig. 2 (A) Plot of initial rate of O2 evolution against different [CAN] (12.5–125 mM) at fixed [1] (12.5 μM) in 0.1 M HNO3. (B) Plot of initial rate of O2 evolution against different [1] (6.25–25.0 μM) at fixed [CAN] (125 mM) in 0.1 M HNO3. | ||
For 1-catalysed water oxidation with NaIO4, the time course (0–60 min) plots of oxygen evolution at different concentrations of NaIO4 (12.5–125 mM) and 1 (6.25–25 μM) in 0.1 M HNO3 are shown in Fig. S3 and S4,† respectively. In this case, the initial rate of oxygen evolution showed a linear dependence on the concentration of 1 but not on the concentration of NaIO4 (see Fig. S5†).
We then examined the time courses of oxygen evolution during water oxidation with Oxone (37.5–125 mM) catalysed by 1 (6.25–25 μM) in 0.1 M HNO3 (Fig. S6 and S7†). Again, a linear correlation between the initial rate of oxygen evolution and the concentration of 1 was observed, whereas the initial rate was not significantly dependent on Oxone concentration (Fig. S8†), which is analogous to the findings obtained with NaIO4 as oxidant.
High-resolution ESI-MS analysis
In our previous work, analysis of a reaction mixture of [Fe(L1)Br2]+ and Oxone (8 equiv.) in MeCN–H2O (5:1 v/v) by high-resolution ESI-MS revealed a cluster peak at m/z 356.0981, attributed to [FeV(L1)(O)2]+.16a In this work, in order to detect the reaction intermediates in the oxidation reactions of 1 with CAN and NaIO4 in aqueous solution, we performed ESI-MS measurements on the corresponding reaction mixtures.
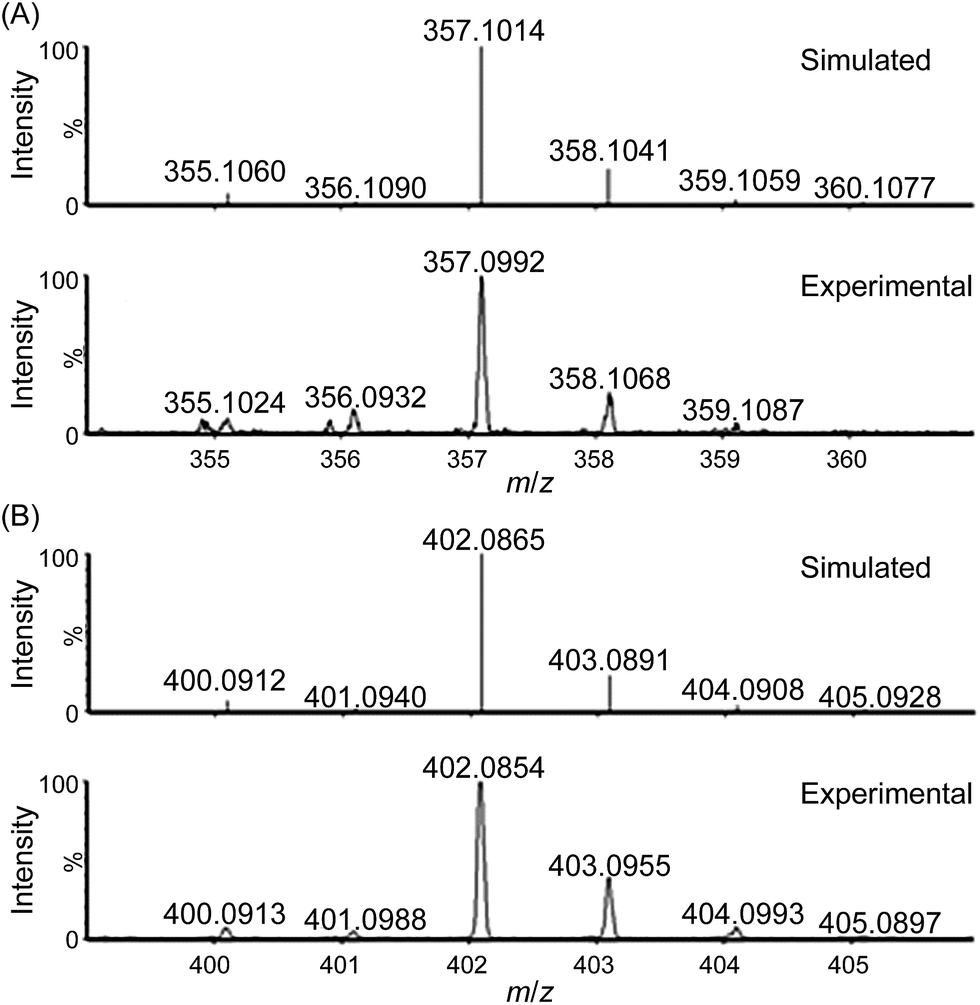 | ||
| Fig. 3 Cluster peaks at m/z 357.0992 and 402.0854 observed during high-resolution ESI-MS analysis of a reaction mixture of 1 and CAN (200 equiv.) in H2O. Simulated isotopic patterns for (A) [FeIV(L1)(O)(OH)]+ and (B) [FeIV(L1)(O)(NO3)]+ are shown. | ||
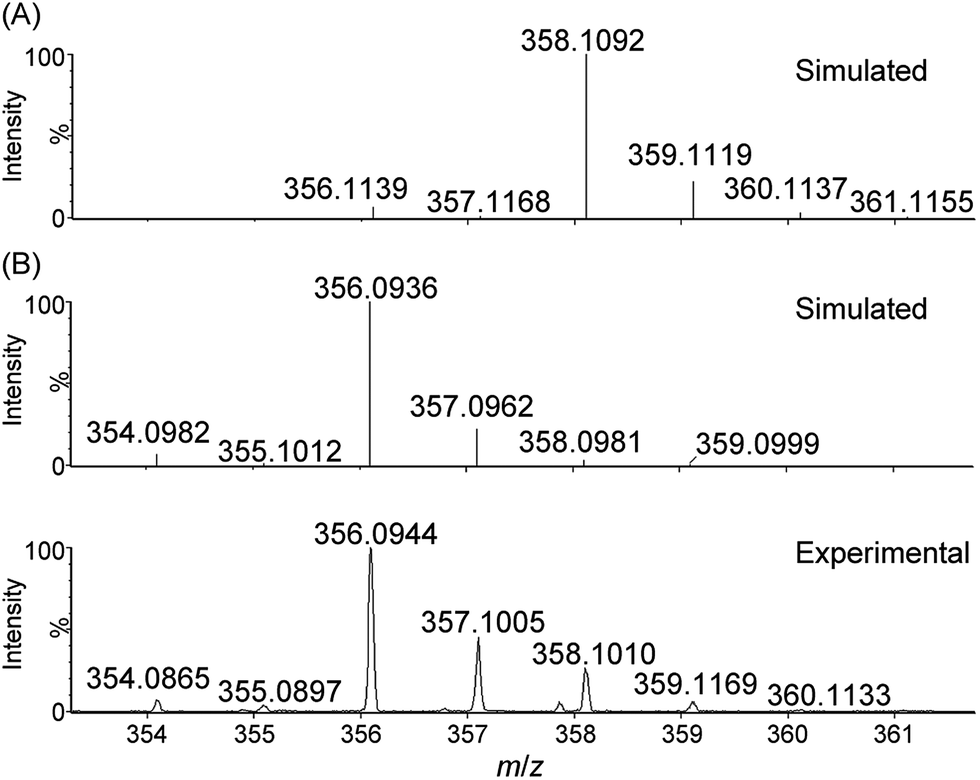 | ||
| Fig. 4 Cluster peak at m/z 356.0944 observed during high-resolution ESI-MS analysis of the reaction mixture of 1 with NaIO4 (800 equiv.) in 0.1 M HNO3. Simulated isotopic patterns for (A) [FeIII(L1)(OH)2]+ and (B) [FeV(L1)(O)2]+ are shown. | ||
UV-vis absorption spectroscopy
The reaction of 1 (1.5 mM) with 5 equiv. of CAN in 0.1 M HNO3 at room temperature was monitored by UV-vis absorption spectroscopy. This reaction immediately (within 10 seconds) generated a new species with λmax 830 nm and a shoulder near 540 nm (Fig. 5). The new species decayed rapidly with a half-life of 107 seconds (Fig. 5, inset).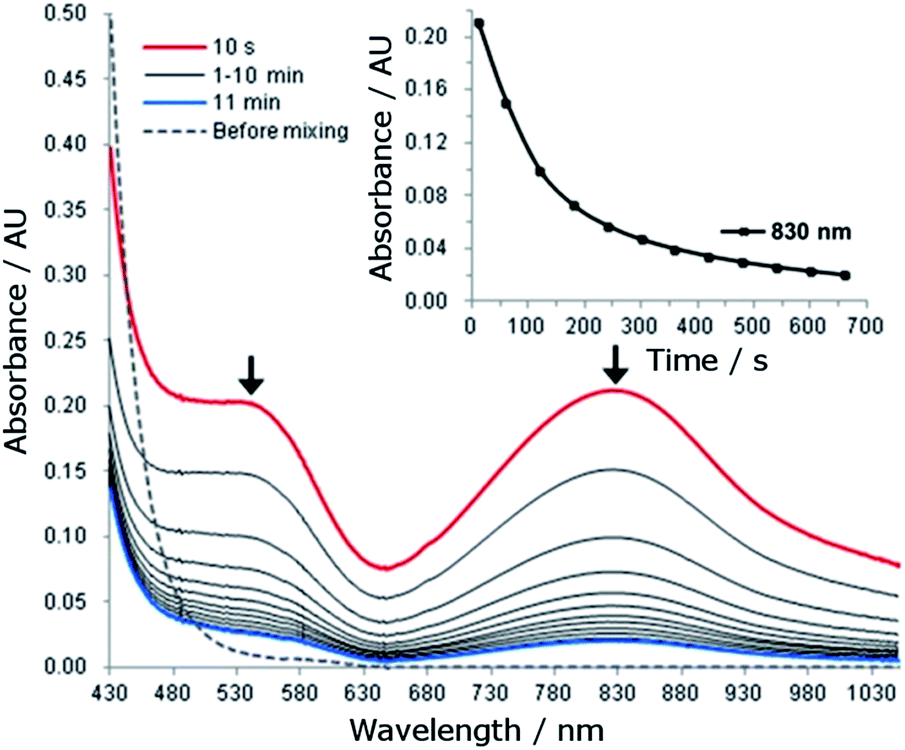 | ||
| Fig. 5 UV-vis absorption spectral changes for the reaction of 1 (1.5 mM) with CAN (7.5 mM) in 0.1 HNO3 at room temperature. Inset: time course of the decay of the absorption band at 830 nm. | ||
For the reaction of 1 (1.5 mM) with 5 equiv. of NaIO4 or Oxone in 0.1 M HNO3, UV-vis measurements revealed different phenomena from that observed for the ‘1 + CAN’ system. Upon treatment of 1 with NaIO4, a new species with λmax 830 nm and a shoulder near 540 nm was gradually formed over 10 minutes (Fig. S25†) and decayed much more slowly with a half-life of ∼40 minutes (see the inset of Fig. S26†), in contrast to the CAN counterparts (Fig. 5). In the case of Oxone, the new band at ∼830 nm was barely discernible in the UV-vis spectrum of the reaction mixture, being much weaker than that observed for the reaction with NaIO4.
EPR analysis
EPR spectroscopy was employed to examine the reaction mixture of 1-catalysed water oxidation, using NaIO4 oxidant as an example. Complex 1 is a high-spin FeIII complex with S = 5/2;15b however, the X-band EPR spectrum of 1·ClO4 in 0.1 M HNO3 recorded at 6 K did not show an appreciable signal (Fig. 6, black line), presumably due to quick relaxation. The X-band EPR spectrum of the reaction mixture obtained by mixing 1·ClO4 with NaIO4 (10 equiv.) in 0.1 M HNO3 at room temperature followed by immediate cooling to 6 K is depicted in Fig. 6 (red line). In this EPR spectrum, there are two prominent signals, one with gavg = 5.54 and the other with g ≈ 2.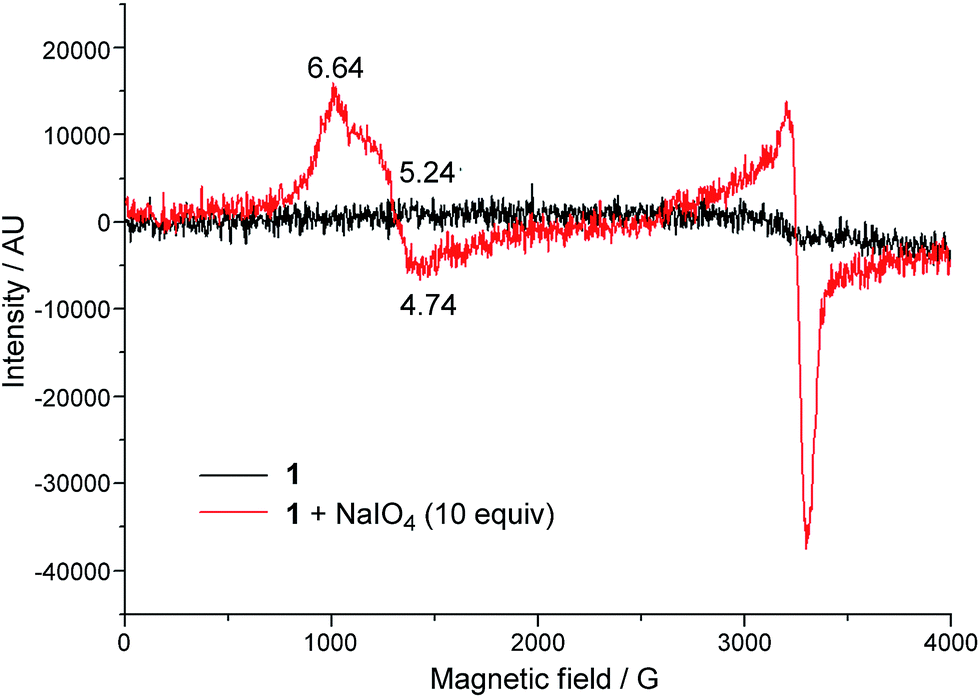 | ||
| Fig. 6 X-band EPR spectra (at 6 K) of 1·ClO4 in 0.1 M HNO3 (black line), and a reaction mixture of 1·ClO4 (1 mM) and NaIO4 (10 mM) in 0.1 M HNO3 (red line). | ||
Electrochemistry of complex 1
In order to avoid interference arising from the electrochemical reactions of the FeCl4− anion in 1·FeCl4, we used the ClO4− salt of 1 and examined its electrochemical properties in 0.1 M HNO3 at pH 1 and in buffered solutions at various pH by means of cyclic voltammetry and rotating disk voltammetry using glassy carbon as the working electrode. The cyclic voltammograms of 1·ClO4 at pH 1–6 are depicted in Fig. 7A and S27,† and show a reversible couple I at E1/2 +0.46 V (pH 1) to +0.12 V (pH 6) vs. SCE, a small irreversible oxidation wave II at Epa +1.18 V (pH 1) to +0.86 V (pH 6) vs. SCE, and the onset of a catalytic oxidation wave at +1.4 V (pH 1) to ca. +1.1 V (pH 6) vs. SCE. Both E1/2 of the reversible couple I and Epa of the irreversible wave II shift cathodically by ∼67 mV per pH unit upon increasing the pH (Fig. 7B and S28†). The magnitude of the irreversible wave II was found to be sensitive to the electrode surface, but could be reproducibly observed during both the cyclic voltammetric scans and the rotating disk voltammetric experiments. The linear scan voltammograms of 1·ClO4 at pH 1, 3, and 5 obtained from rotating disk voltammetric measurements are depicted in Fig. 7C and S29,† and that recorded at pH 1 at various rotation rates is shown in Fig. S30.† For example, at pH 3, a small but distinct current was recorded at ca. +1.05 V vs. SCE (Fig. 7C), which could be correlated to the small irreversible wave II in the corresponding cyclic voltammogram (Fig. 7A). Notably, the catalytic oxidation wave, presumably due to water oxidation, can be observed at pH 1 to 6 (Fig. 7A and S27†). The limiting current (iL) of the redox process corresponding to the reversible couple I increased linearly with the square root of the rotation rate (ω1/2). A plot of iL against ω1/2 (Levich plot) shows a straight line with R2 = 0.999 (Fig. S31†). This is indicative of a totally mass-transfer-limited condition at the electrode surface. From the slope of the Levich plot, the diffusion coefficient of 1·ClO4 under the experimental conditions was calculated to be 0.48 × 10−5 cm2 s−1.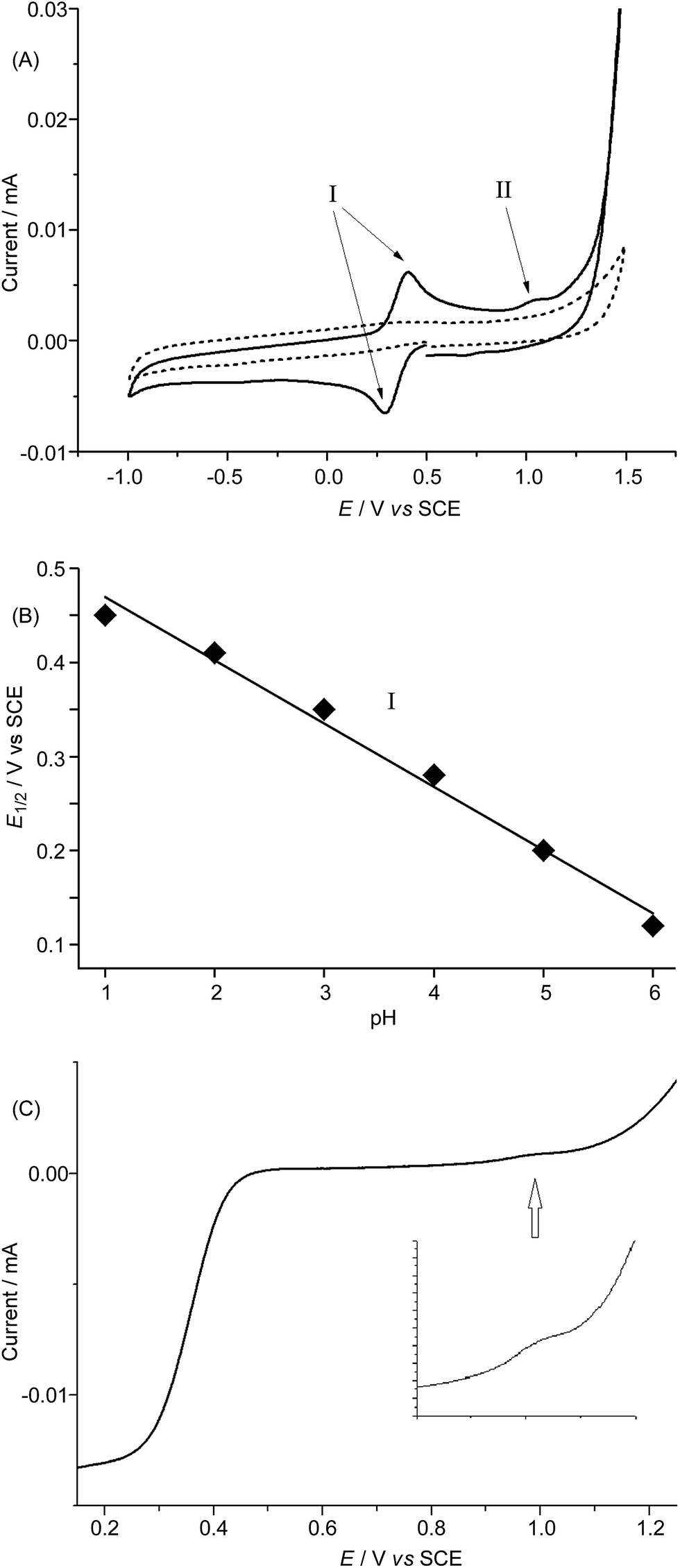 | ||
| Fig. 7 (A) Solid line: cyclic voltammogram of 1·ClO4 in 0.1 M acetate buffer at pH 3; dotted line: buffer background; working electrode: glassy carbon; scan rate: 100 mV s−1. (B) Redox potentials (E1/2 of reversible couple I) of 1·ClO4 in solutions of various pH. (C) Linear scan voltammogram of 1·ClO4 in 0.1 M acetate buffer at pH 3. Working electrode: rotating glassy carbon disk; rotation rate: 100 rpm; scan rate: 5 mV s−1 (inset: magnified region at approximately 1.0 V). | ||
Density functional theory calculations
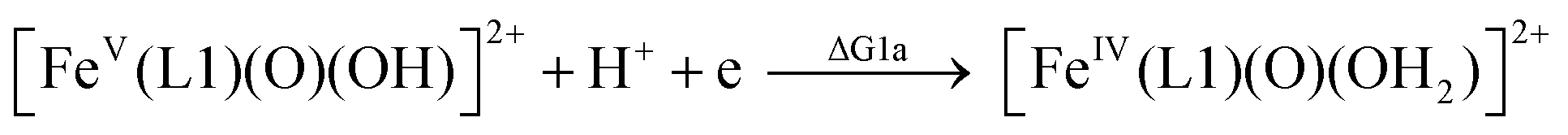 | (1a) |
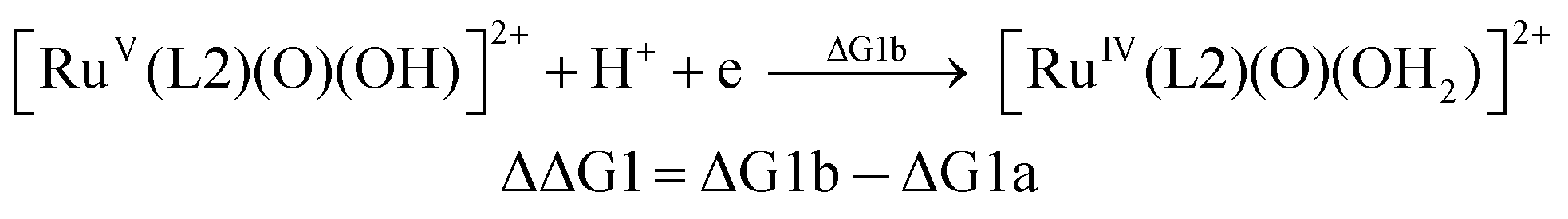 | (1b) |
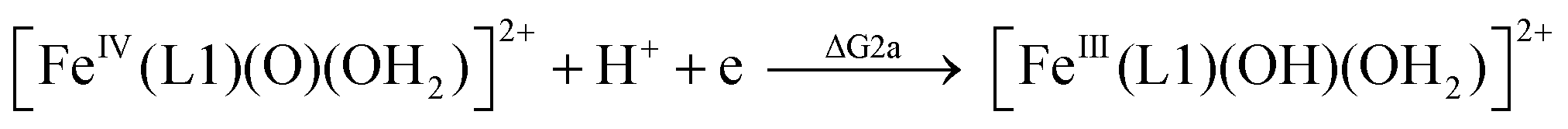 | (2a) |
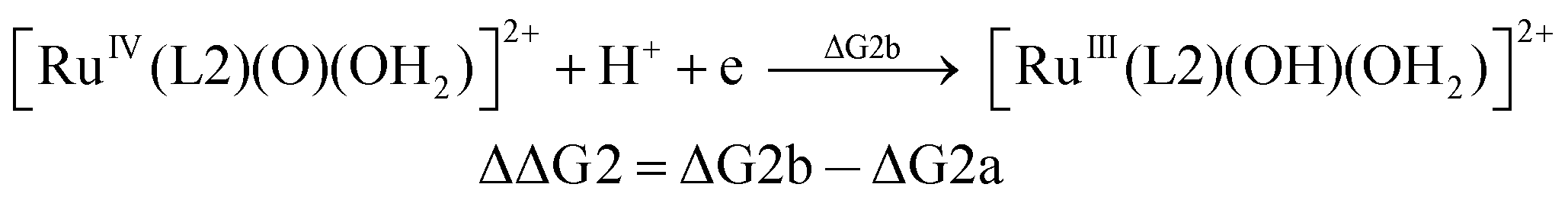 | (2b) |
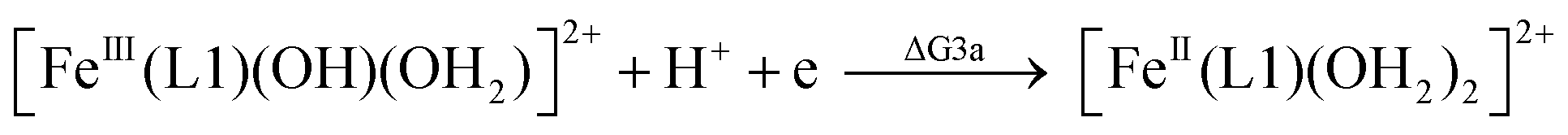 | (3a) |
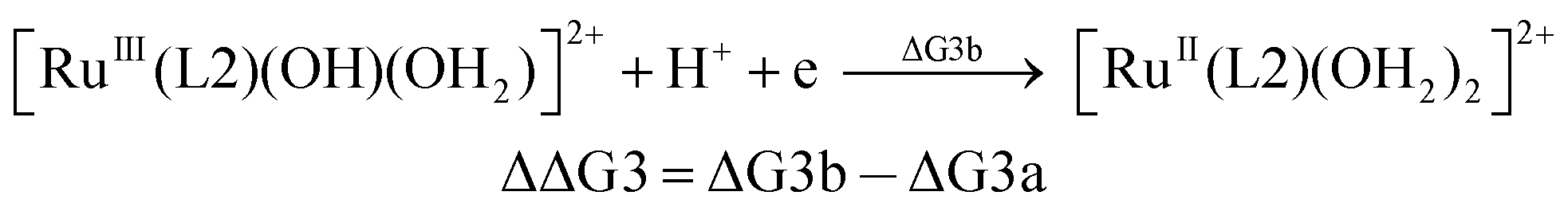 | (3b) |
From the DFT computed ΔΔG1, ΔΔG2 and ΔΔG3 values, and the experimental redox potentials of the RuV/RuIV, RuIV/RuIII and RuIII/RuII couples,20 the redox potentials of the FeV/FeIV, FeIV/FeIII and FeIII/FeII couples can be calculated as:
| E(FeV/FeIV) = E(RuV/RuIV) + ΔΔG1 |
| E(FeIV/FeIII) = E(RuIV/RuIII) + ΔΔG2 |
| E(FeIII/FeII) = E(RuIII/RuII) + ΔΔG3 |
We calculated the FeIII/FeII and FeIV/FeIII redox potentials at different pH by using three commonly used density functionals (DFs): BPW91 (pure-GGA), B3LYP (hybrid-GGA), and M06L (meta-GGA). A correlation between the experimental redox potentials of FeIII/FeII and FeIV/FeIII and those calculated using these DFs is depicted in Fig. 8 (note of caution: calculated redox potentials are thermodynamic values). It is evident that BPW91 and B3LYP led to marked-to-severe (up to ca. +0.57 V) overestimation of the E1/2 of FeIII/FeII, although B3LYP showed good performance in the prediction of the electrochemical potential of FeIV/FeIII. Only M06L gave good estimations of the electrochemical potentials of both FeIII/FeII and FeIV/FeIII at different pH with a linear fit (E(M06L) = E(expt) + 0.06, R = 0.998, SD = 0.03) between the calculated redox potentials and the experimental data. Hence, M06L was chosen for the subsequent DFT calculations in this work. In the literature, the M06L functional has been reported to show good performance in modelling water oxidation by ruthenium21 and iron12a,c complexes.
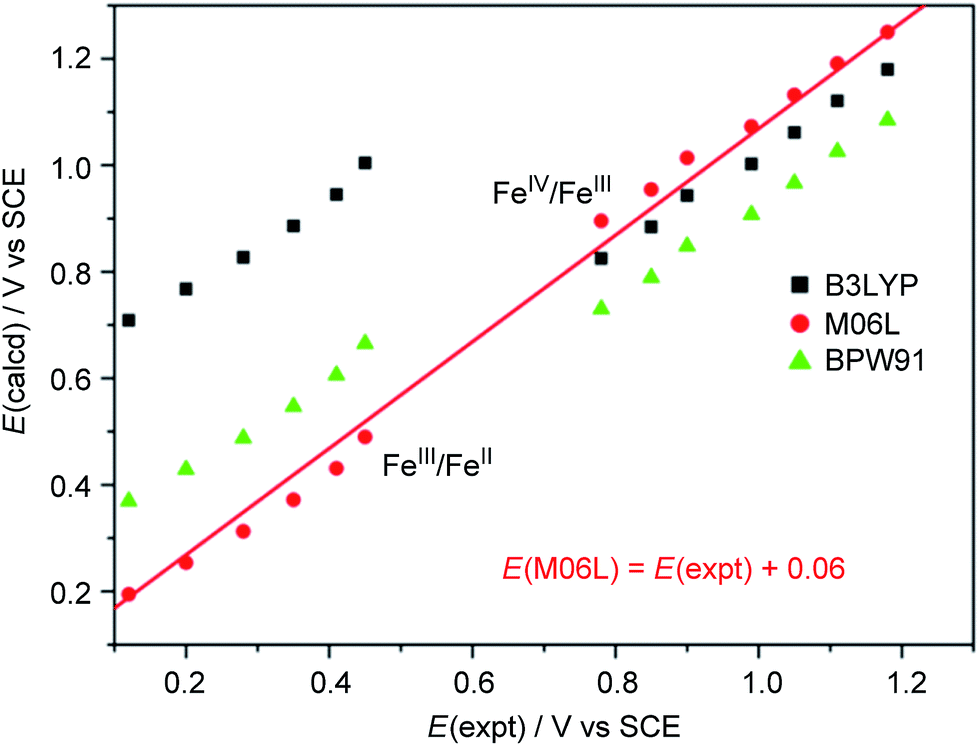 | ||
| Fig. 8 Correlation between experimental and calculated redox potentials. The E(expt) values for the FeIII/FeII and FeIV/FeIII couples were taken from Fig. 7 and S28† (see Discussion section). Note of caution: E(expt) values for FeIV/FeIII are the Epa values of the irreversible wave II. | ||
For the calculation of the pKa values, as an example, the pKa of [FeV(L1)(O)(OH)]2+ was calculated based on the following pair of isodesmic reactions (reactions (4a) and (4b)).
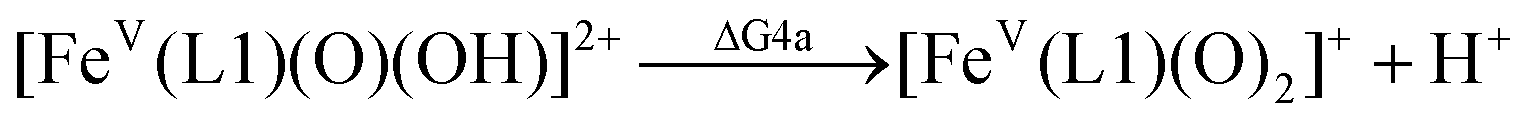 | (4a) |
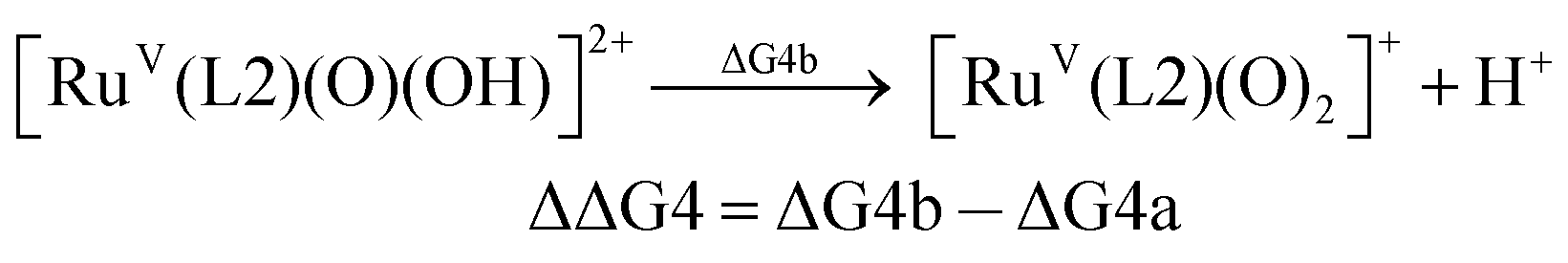 | (4b) |
| pKa([FeV(L1)(O)(OH)]2+) = pKa([RuV(L2)(O)(OH)]2+) − ΔΔG4/2.303RT |
The calculated redox potentials for various FeV/FeIV, FeIV/FeIII and FeIII/FeII couples, including those of [Fe(L1)(O)(X)]n+ (X = H2O, HO−, O2−), along with the calculated pKa values, are depicted in Scheme 1, revealing the following features: (1) the pKa of the [FeV(L1)(O)(OH)]2+/[FeV(L1)(O)2]+ equilibrium is 3.0; (2) the calculated E1/2 of [FeV(L1)(O)(OH)]2+/[FeIV(L1)(O)(OH2)]2+ (+1.42 V vs. SCE at pH 1) is comparable to the E1/2 of CeIV/CeIII (+1.38 V vs. SCE) and close to the calculated E1/2 values of [FeV(L)(O)(OH)]2+/[FeIV(L)(O)(OH2)]2+ with L = Me2Pytacn or mep (+1.70 to +1.73 V vs. SHE,12f i.e. +1.46 to +1.49 V vs. SCE); (3) the calculated E1/2 of [FeIV(L1)(O)(OH2)]2+/[FeIII(L1)(OH)(OH2)]2+ (+1.25 V vs. SCE at pH 1) matches the experimental Epa value of FeIV/FeIII (Epa = 1.18 V vs. SCE at pH 1, see Discussion section); (4) the calculated E1/2 of the FeIII/FeII couple is +0.49 V at pH 1, which matches the experimental E1/2 value for FeIII/FeII (+0.46 V vs. SCE at pH 1, see Discussion section) well; (5) the calculated E1/2 of the FeV/FeIII couple (E1/2 = +1.34 V vs. SCE at pH 1) coincides with the catalytic oxidation wave at +1.4 V observed in the cyclic voltammogram of 1 at pH 1. It is noted that there is just 170 mV difference in the calculated E1/2 values of [FeV(L1)(O)(OH)]2+/[FeIV(L1)(O)(OH2)]2+ and [FeIV(L1)(O)(OH2)]2+/[FeIII(L1)(OH)(OH2)]2+ at pH 1.
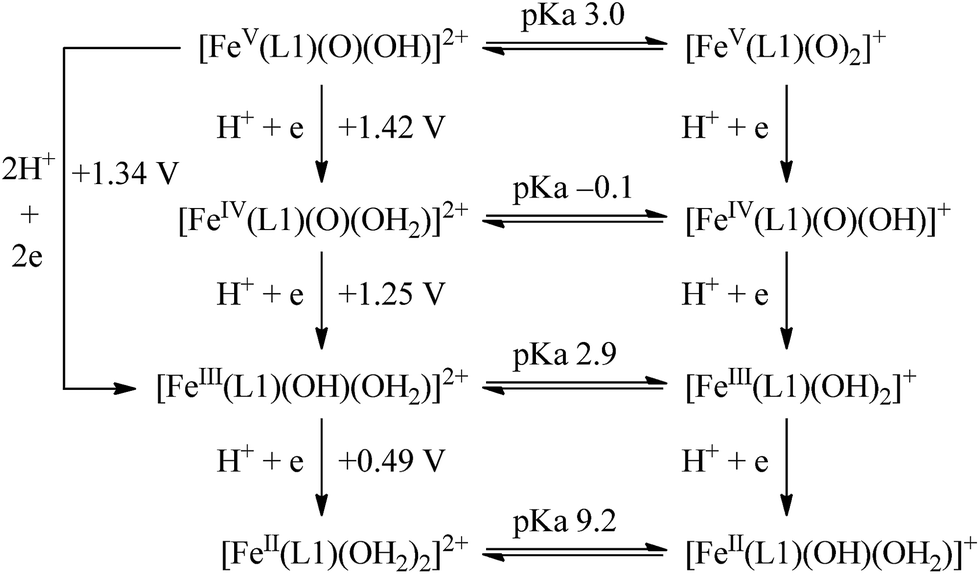 | ||
| Scheme 1 Calculated reduction potentials for FeV/FeIV, FeIV/FeIII and FeIII/FeII, together with calculated pKa values, based on the M06L functional. | ||
O species has previously been predicted by DFT calculations for [FeV(Me2Pytacn)(O)(OH)]2+.12f,14d The computed FeO distance in [FeV(L1)(O)2]+ in the quartet state (4FeV) is 1.614 Å, which is shorter than that in the sextet state (6FeV, 1.695 Å) due to the antibonding character of the d1 and d2 orbitals (Fig. 9, right). Similar FeO distances were reported for [FeV(L1)(O)2]+ (1.613–1.638 Å), computed at the B3LYP/6-31(G) (lanl2dz) level in our previous work, and for [FeV(Me2Pytacn)(O)(OH)]2+ (1.63 Å)12f and low-spin [FeV(TAML)(O)]− (1.60 Å).14a The spin density plot for [FeV(L1)(O)2]+ calculated in this work is shown in Fig. 9 (left). Apart from the spin density on Fe of 2.3, each oxo group has considerable spin density of 0.27. The MO diagram of [FeV(L1)(O)2]+ (Fig. 9, right) reveals a d3 configuration in accordance with the assignment of the Fe(V) oxidation state.
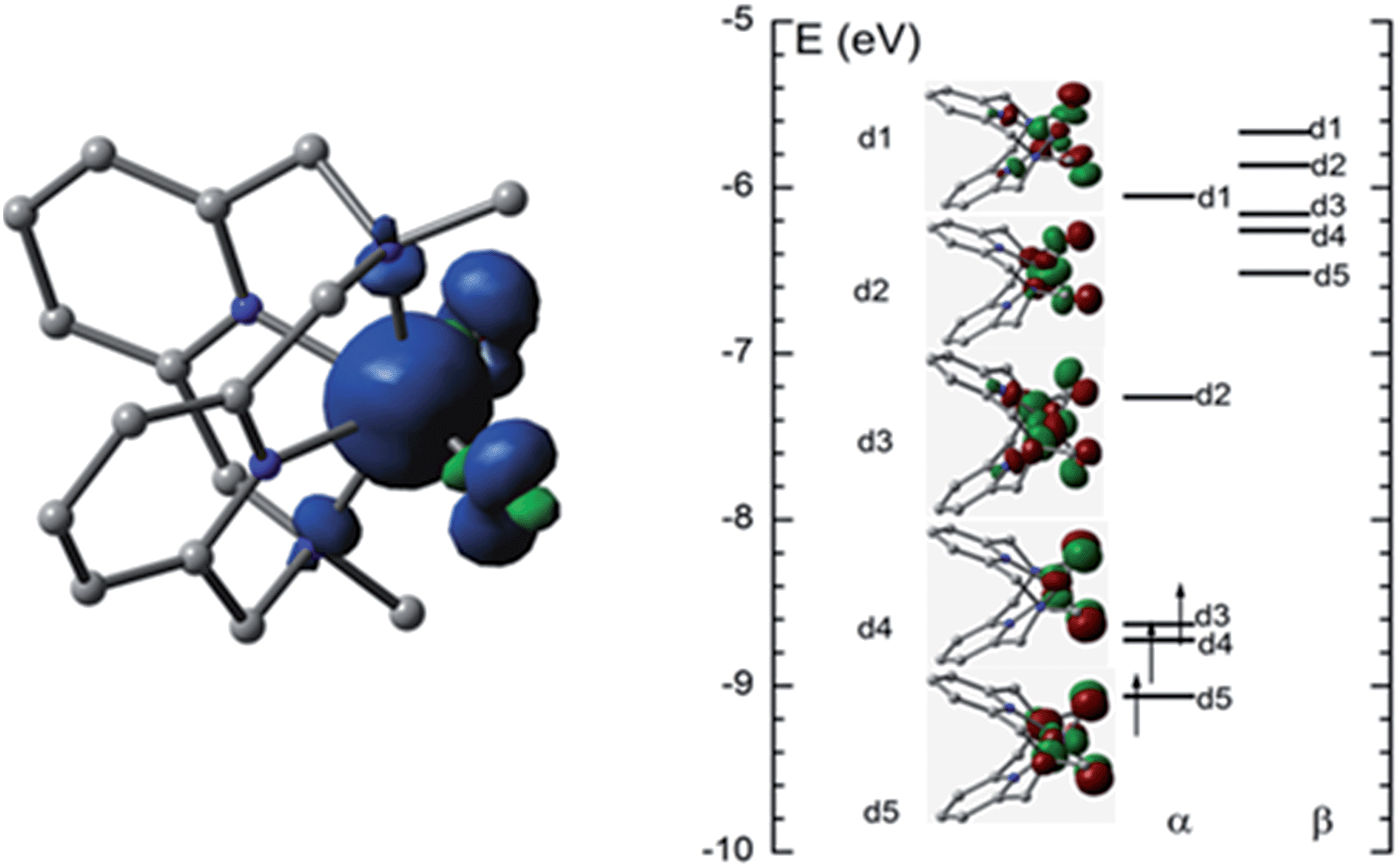 | ||
| Fig. 9 Spin density plot (left) and MO diagram (right) of [FeV(L1)(O)2]+. | ||
O distance of [FeIV(L1)(O)(OH)]+ (5FeIV) in the ground state is 1.632 Å, very close to the experimental FeO distance in [FeIV(N4Py)(O)]2+ (1.639(5) Å).5aFig. 10 (left) shows the spin density plot calculated in this work for [FeIV(L1)(O)(OH)]+. Fe has the largest spin density of 3.1; the oxo and hydroxyl groups have spin densities of 0.59 and 0.18, respectively. The MO diagram of [FeIV(L1)(O)(OH)]+ (Fig. 10, right) reveals a d4 configuration, which is consistent with the assignment of the Fe(IV) oxidation state.
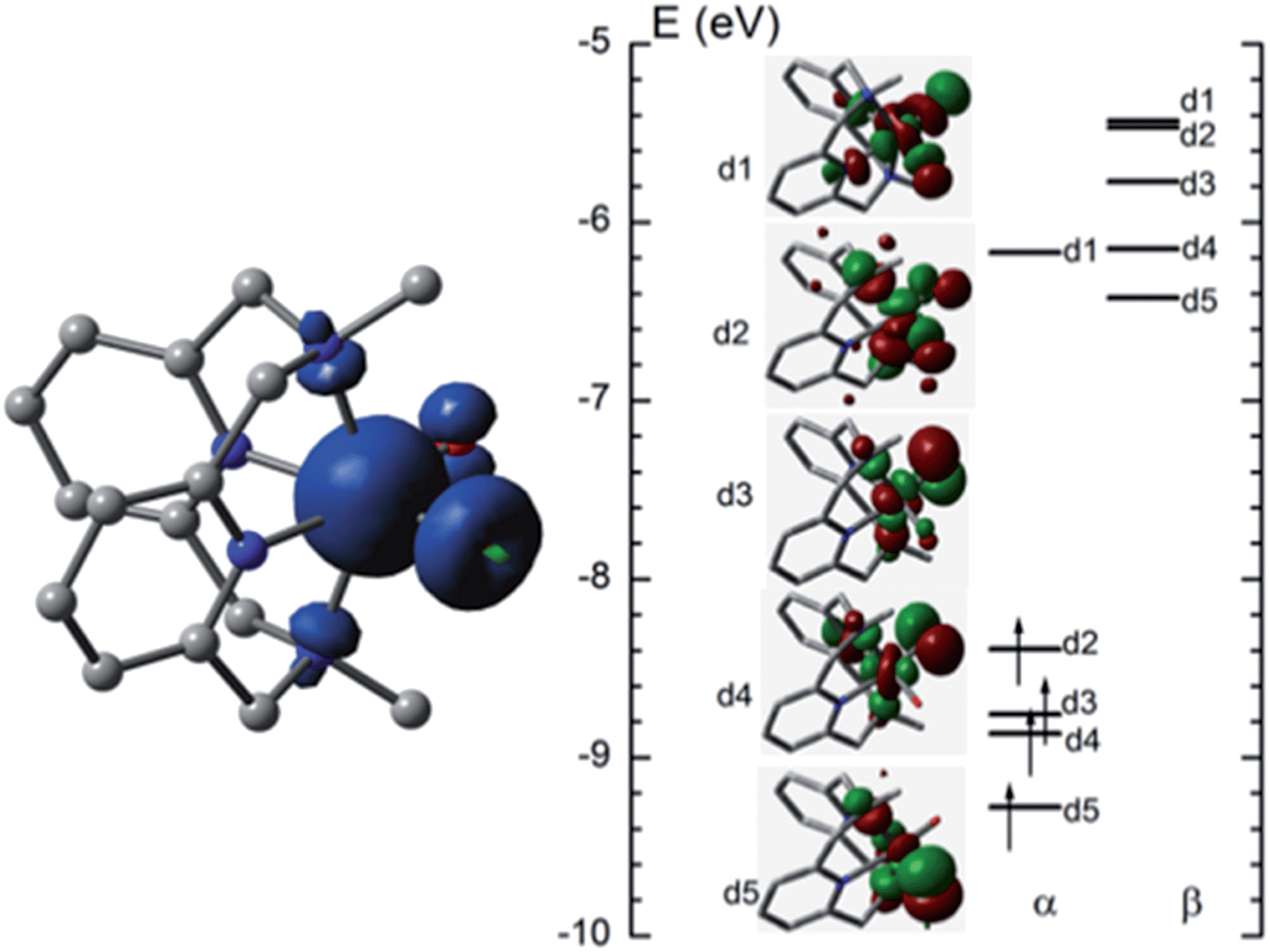 | ||
| Fig. 10 Spin density plot (left) and MO diagram (right) of [FeIV(L1)(O)(OH)]+. | ||
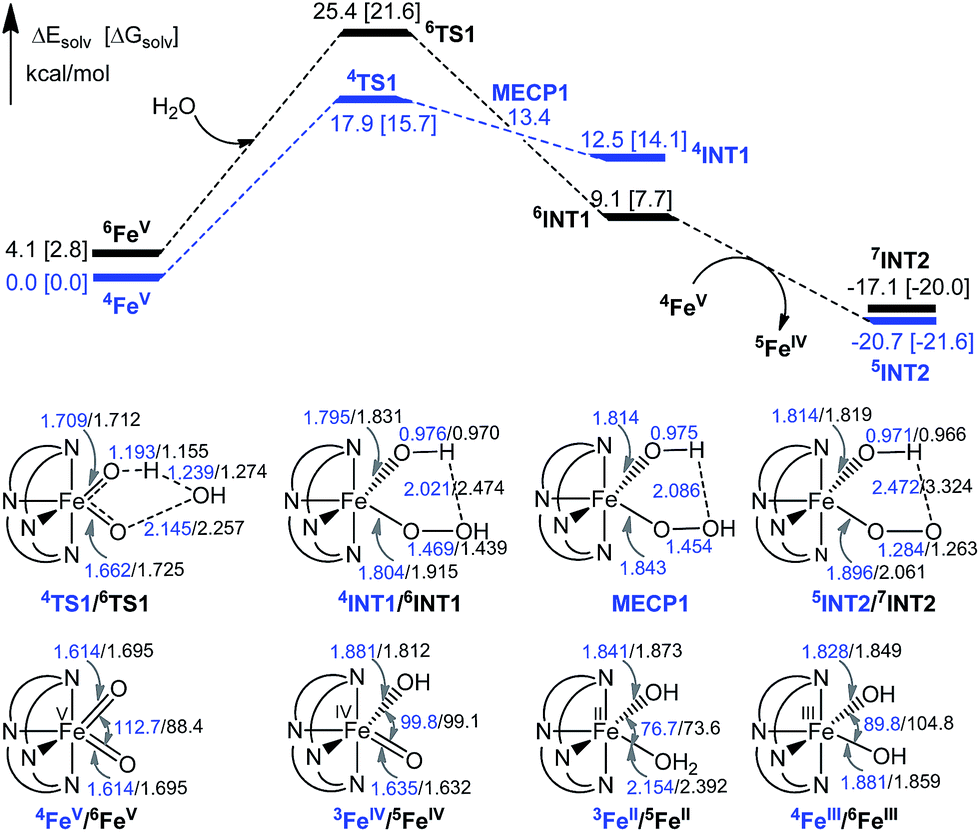 | ||
| Fig. 11 Potential energy surfaces (in kcal mol−1), key structural parameters (in Å and degrees) and relative stabilities (in kcal mol−1) of the stationary points. The energy of the MECP point (13.4 kcal mol−1) is the average energy of the quartet and sextet states in water. | ||
The 6INT1 complex can subsequently be oxidized by [FeV(L1)(O)2]+ (4FeV) to give the superoxo complex [FeIII(L1)(OO˙)(OH)]+ (7INT2/5INT2). Meanwhile, 4FeV is reduced to [FeIV(L1)(O)(OH)]+ (5FeIV). The subsequent reaction steps include exchange of an oxygen molecule by a water molecule and the release of 3O2/1O2 (reaction (5)). The search for the transition state has not been successful. A barrier of 8.8 kcal mol−1 for the release of O2 from [TAML–FeIV–OO˙]− was reported by Crammer and co-workers.12a After release of O2, complex [FeII(L1)(OH2)(OH)]+ (5FeII) with a quintet state ground state is formed; this complex can further comproportionate with [FeIV(L1)(O)(OH)]+ (5FeIV) to regenerate two [FeIII(L1)(OH)2]+ (6FeIII) molecules.
| [FeIII(L1)(OO˙)(OH)]+ + OH2 → [FeII(L1)(OH2)(OH)]+ + O2 | (5) |
Discussion
The main objective of this work is to gain insight into the mechanism of water oxidation by iron complexes of tetradentate macrocyclic ligands with a cis non-planar coordination geometry. The macrocyclic N4 ligand L1 (which is well known to form iron complexes in the cis configuration15b) was chosen, as our previous work had suggested the involvement of in situ generated high-valent FeO species of L1, including [FeV(L1)(O)2]+, in the cis-dihydroxylation of alkenes with Oxone catalysed by [FeIII(L1)Cl2]+ (1),16a and FeVO species were proposed to be the reactive intermediates in iron-catalysed water oxidation reactions reported in the literature.6a,b Also, it is envisaged that the strong donor strength of L1 would stabilize iron–oxo species against demetallation in solution.
During optimization of the reaction conditions, it was noted that although the turnover number of oxygen increased with oxidant concentration, the oxidant efficiency (defined in note c of Table 1) decreased from 68% to 20% (Table 1, entries 1a–1d). Such a significant decrease in oxidant efficiency is attributed to partial decomposition/deactivation of the catalyst under strongly oxidizing reaction conditions.23 Similar findings were observed when NaIO4 or Oxone was used as oxidant.
Unlike the ‘[Fe(bqen)(OTf)2] + CAN’ system (which can oxidize water to O2 in non-buffered aqueous solution but not in 0.1 M HNO3 solution),6e the ‘1 + CAN’ system produced oxygen with comparable TONs in water and in 0.1 M HNO3 (entries 1d and 1e in Table 1), consistent with the similar ESI mass spectra obtained under the two conditions (Fig. S13†). This reflects the remarkable stability of 1 toward ligand dissociation under acidic conditions, like other iron complexes such as [FeII(mcp)(OTf)2]6b and [FeII(L1)(MeCN)2]2+ (ref. 6g) used as water oxidation catalysts at pH 1. The 1-catalysed water oxidation was sensitive to the oxidant, with the TON of oxygen evolved under the same conditions (in 0.1 M HNO3) following the order Oxone > CAN > NaIO4 (113 vs. 93 vs. 44, see Table 1, entries 1f, 2c and 3b). According to the 16O2/16O18O/18O2 ratio (68.9:29.9:1.2) obtained in the 18O-labelling studies, the oxygen evolved in the ‘1 + Oxone’ system originated not only from Oxone but also from water (see the possible pathways of oxygen evolution, along with the corresponding calculated 16O2/16O18O/18O2 ratios, depicted in Scheme S1†), indicating the involvement of water oxidation in the reaction.
Considering the excellent catalytic ability of [FeII(mcp)(OTf)2] for water oxidation, as reported by Costas, Lloret-Fillol and co-workers,6b we compared the activity of catalysts [FeII(mcp)Cl2]6b and [FeIII(L1)Cl2]+ (1) for water oxidation. Under our reaction conditions with catalyst/oxidant = 1:840, the amounts of oxygen formed in [FeII(mcp)Cl2]-catalysed water oxidation in 0.1 M HNO3 with CAN and NaIO4 as oxidants were approximately 1.4 times (TON = 56) and 2.9 times (TON = 35) the amounts formed in the 1-catalysed reactions, respectively. Changing the catalyst/oxidant ratio from 1:840 to 1:10000 led to an increase in the TON for oxygen formation from 41 to 93 for ‘1 + CAN’, from 56 to 177 for ‘[FeII(mcp)Cl2] + CAN’, from 12 to 44 for ‘1 + NaIO4’, and from 35 to 114 for ‘[FeII(mcp)Cl2] + NaIO4’. These results showed that [FeII(mcp)Cl2] is more active than 1 in catalysing water oxidation.
Iron oxide nanoparticles, which can be generated from simple iron salts or iron complexes at high pH,6c have been shown to be efficient catalysts for water oxidation.6c,24 For the 1-catalysed water oxidation with CAN, NaIO4, or Oxone in 0.1 M HNO3 (pH 1), the possibility that the observed catalytic activity of 1 is due to Fe2O3 nanoparticles generated in situ could be excluded, as in acidic solution either formation of Fe2O3 nanoparticles would be prevented or any Fe2O3 nanoparticles formed would be converted to Fe3+(aq) ions. It has been reported that Fe3+(aq) ions are not active in catalysing water oxidation with CAN at low pH.6c
Electrochemical studies of ruthenium-based molecular water oxidation catalysts often revealed strong catalytic currents due to water oxidation occurring at potentials corresponding to the generation of oxidizing Ru–oxo complexes.25 In general, electrochemical oxidation of Ru–OH2 to RuO species would be kinetically slow as a result of deprotonation of Ru–OH2 prior to oxidation. Hence the working electrode is well documented to play a crucial role in the electrochemical reversibility of the RuO/Ru–OH2 couple(s). Glassy carbon or pyrolytic carbon is commonly used as a working electrode for electrochemical studies of RuO complexes in aqueous solutions. While there have been numerous reports on the electrochemistry of RuO/Ru–OH2 couples in aqueous solution, to the best of our knowledge, related studies on the electrochemical oxidation of Fe–OH2 to FeO species in aqueous solution are sparse.26
The cyclic voltammetric and rotating disk voltammetric measurements of 1 at various pH provide useful information on the electrochemical potentials of the iron–oxo species of L1. As seen in Fig. 7B and S28,† the plots of the redox potentials against pH (from 1 to 6) for both couple I and wave II show negative slopes, and the redox potential decreases by ∼67 mV per pH unit, lending support to the two oxidation processes which are due to one electron and one proton transfer reactions. These findings, together with the experimental data obtained from rotating disk voltammetry (Fig. 7C and S29†) and the results of the DFT calculations (Fig. 8 and Scheme 1), support the assignment of couple I to the [FeIII(L1)(OH)(OH2)]2+/[FeII(L1)(OH2)2]2+ redox couple and the assignment of wave II to the oxidation of [FeIII(L1)(OH)(OH2)]2+ to [FeIV(L1)(O)(OH2)]2+. Despite the low scan rate of 5 mV s−1 for the rotating disk voltammetry, the magnitude of the current recorded for the [FeIV(L1)(O)(OH2)]2+/[FeIII(L1)(OH)(OH2)]2+ oxidation wave is not the same as that recorded for the [FeIII(L1)(OH)(OH2)]2+/[FeII(L1)(OH2)2]2+ redox process. As the oxidation of FeIII–OH to an FeIVO species involves the deprotonation of an Fe–OH unit, the electrochemical oxidation would be kinetically slow and highly sensitive to the electrode surface. Indeed, the electrochemical oxidation of Ru–OH2/Ru–OH to a RuO species is well documented to be significantly affected by the nature/surface of the working electrode.27 At pH 1 to 6, the difference between the observed redox potentials for wave II (FeIV/FeIII) and couple I (FeIII/FeII) is about 700 mV. For a ruthenium complex (also with cis vacant sites) supported by the mcp ligand,28 the difference between the redox potentials of the [RuIV(mcp)(O)(OH2)]2+/[RuIII(mcp)(OH)(OH2)]2+ and [RuIII(mcp)(OH)(OH2)]2+/[RuII(mcp)(OH2)2]2+ couples is 550 mV, which is 150 mV smaller than that between the iron analogues and can be attributed to the stronger π-bond formed in the RuIVO moiety. There is a previous report on the observation of a pH-dependent reversible couple assigned as a proton-coupled electron transfer FeIV/FeIII couple for [FeIV(N4Py)(O)]2+ (N4Py = N,N-bis(2-pyridylmethyl)-bis(2-pyridyl)methylamine) in buffered aqueous solution (pH 1.5–4) with an E1/2 value of +0.41 V vs. SCE at pH 4 (the E1/2 value anodically shifted upon decreasing the pH with a shift of 55 mV per pH unit).29 However, the FeIII/FeII couple was not reported for [FeIV(N4py)(O)]2+. The E1/2 value of +0.41 V for the [FeIV(N4py)(O)]2+/[FeIII(N4py)(OH)]2+ couple in aqueous solution at pH 4 is unexpectedly low. Should this be the case, the corresponding FeIII/FeII couple would have to occur at a potential less than 0.0 V vs. SCE based on the DFT calculations in this work. Our DFT calculations using the M06L functional gave redox potentials of +1.21 V vs. SCE for the [FeIV(N4py)(O)]2+/[FeIII(N4py)(OH)]2+ couple and +0.3 V vs. SCE for the [FeIII(N4py)(OH)]2+/[FeII(N4py)(OH2)]2+ couple at pH 1. The observation of a catalytic wave for 1 (Fig. 7A and S27†) beyond the irreversible wave II (assigned to FeIV/FeIII) at, for example, Epa +1.18 V vs. SCE (pH 1), which is comparable to the DFT-calculated potential of +1.25 V vs. SCE (pH 1) for the [FeIV(L1)(O)(OH2)]2+/[FeIII(L1)(OH)(OH2)]2+ couple, corroborates the finding that 1 can catalyse water oxidation through iron–oxo species at oxidation states beyond FeIV.
ESI-MS analysis of a solution of 1 in water (Fig. S9–S12†), which revealed a major cluster peak assigned to [FeIII(L1)(OH)2]+, suggests rapid exchange of the Cl− ligands of 1 with solvent. The new signals, generated upon addition of CAN, at m/z 357.0992, 402.0854, and 448.0815 can be attributed to [FeIV(L1)(O)(OH)]+ (calcd m/z 357.1014, Fig. 3), [FeIV(L1)(O)(NO3)]+ (calcd m/z 402.0865, Fig. 3) and [FeIII(L1)(NO3)2]+ (calcd m/z 448.0794, Fig. S14†), respectively, based on their m/z values and isotopic patterns, together with the 18O-labelling experiments which revealed new signals with m/z 361.1107 and 404.0927 attributable to [FeIV(L1)(18O)(18OH)]+ (calcd m/z 361.1099, Fig. S17†) and [FeIV(L1)(18O)(N16O3)]+ (calcd m/z 404.0907, Fig. S19†), respectively. Species [FeIV(L1)(18O)(16OH)]+ and [FeIV(L1)(16O)(18OH)]+ (calcd m/z 359.1057) were not detected in the experiment, indicating that the oxygen atoms of the hydroxide and oxo ligands in [FeIV(L1)(O)(OH)]+ come from water and not from CAN.
In the UV-vis absorption spectra of the reaction mixture of 1 with CAN (Fig. 5), the new band at λmax 830 nm is tentatively assigned to an FeIVO species of L1, with reference to the characteristic bands of FeIVO species (ranging from 700–850 nm) reported in the literature.30 This assignment is in line with the detection of species formulated as [FeIV(L1)(O)(OH)]+ and [FeIV(L1)(O)(NO3)]+ by ESI-MS. We further examined the intensity of the cluster peak at m/z 357.0992, attributed to [FeIV(L1)(O)(OH)]+, at different reaction times. The ion count of this signal decreased with reaction time, as depicted in Fig. S32,† which correlates with the decay of the absorption band at λmax 830 nm assigned to the FeIVO species in the UV-vis spectra shown in Fig. 5. FeIVO species of chelating N-donor ligands have been reported to be reasonably stable, with half-lives generally ranging from 2 to 60 hours.30 Unlike the [FeIV(mcp)(O)(H2O)]2+ species (generated in situ from [FeII(mcp)(OTf)2] and CAN in aqueous solution), which could persist under catalytic conditions with a half-life of 2.4 hours,6b the FeIVO species of L1 decayed with a half-life of 107 seconds under these conditions (Fig. 5). Such a fast decay may suggest that either it is taking part in a further reaction, or it is decomposing rapidly.
Given the instant generation of an FeIVO species upon oxidation of 1 with CAN in aqueous solution, as supported by ESI-MS and UV-vis analysis, together with the absence of an induction period for oxygen evolution, an FeIVO species is suggested to be involved in water oxidation by the ‘1 + CAN’ system. Kinetic studies revealed a linear dependence of the initial rate of oxygen evolution on the concentrations of both CAN and 1 (Fig. 2; increasing [CAN] to 125 mM lowered the pH to ∼0.6, and such pH variation alone accounted for ∼18% of the rate increase depicted in Fig. 2A), which suggests that a key step of the reaction involves an iron species, presumably [FeIV(L1)(O)(OH)]+, and one equivalent of CAN. One of the possibilities is the oxidation of [FeIV(L1)(O)(OH)]+ by CAN to give FeVO species, such as [FeV(L1)(O)(OH)]2+ or [FeV(L1)(O)2]+, and another possibility is the reaction of [FeIV(L1)(O)(OH)]+ with CAN to form an OFeIV–O–CeIV species similar to that recently reported for the ‘[FeII(mcp)(OTf)2] + CAN’ system,13 although FeVO and OFeIV–O–CeIV species have not been clearly detected in ESI-MS analysis of the reaction mixture of 1 with CAN in H2O.
For water oxidation by the ‘1 + NaIO4’ system, the new signals at m/z 356.0944 (major) and 373.0940 (minor), revealed by ESI-MS analysis of a mixture of 1 and NaIO4 in 0.1 M HNO3 (Fig. 4 and S20–S22†), could be assigned to [FeV(L1)(O)2]+ (calcd m/z 356.0936) and [FeIII(L1)(OO˙)(OH)]+ (calcd m/z 373.0963), respectively. The assignment of [FeV(L1)(O)2]+ is supported by 18O-labelling studies, revealing a shift of the signal at m/z 356.0944 to m/z 360.1042 attributable to [FeV(L1)(18O)2]+ (calcd m/z 360.1021) upon changing the reaction medium to H218O (Fig. S24†). Since IO4− can undergo rapid oxygen exchange with water,18 the oxo ligands of [FeV(L1)(18O)2]+ could come from H218O directly and/or from 18O-incorporated IO4−. Changing the reaction medium from 0.1 M HNO3 to pure water reduced the intensity of the signal assigned to [FeV(L1)(O)2]+; this observation is in line with the smaller TON of oxygen produced in pure water than in 0.1 M HNO3 (entries 2b vs. 2a, and 2d vs. 2c, Table 1).
The DFT calculations on [FeV(L1)(O)2]+ in this work and on [FeV(Me2Pytacn)(O)(OH)]2+ in the literature12f,14d revealed that the FeVO species supported by these neutral chelating N ligands all adopt a quartet ground state (S = 3/2), different from the doublet ground state (S = 1/2) of [FeV(TAML)(O)]− complexes, which bear tetraanionic tetraamide ligands and have been detected by EPR spectroscopy.7,14a,j To further test the validity of the DFT method used in this work, we performed DFT calculations on [FeV(TAML)(O)]− using the M06L functional, which revealed a doublet ground state for this species, consistent with previous DFT calculations reported in the literature.12a,14a To the best of our knowledge, no FeVO species with S = 3/2 ground state has been characterized by EPR spectroscopy. The X-band EPR spectrum of the reaction mixture of 1 with NaIO4 in 0.1 M HNO3 (Fig. 6) is dominated by two signals corresponding to S = 5/2 and S = 1/2 states; the former could be attributed to a high-spin Fe(III) species whereas the latter probably arose from decomposed NaIO4, as a similar S = 1/2 signal was observed in the X-band EPR spectrum of a solution of NaIO4 in 0.1 M HNO3 recorded at 7 K (Fig. S33†). EPR signals of S = 3/2 Fe complexes are rather close to, or considerably overlap with, those of S = 5/2 ones.31 The prominent broad S = 5/2 signal in Fig. 6 would therefore render it difficult to clearly confirm the presence of the [FeV(L1)(O)2]+ species by EPR since the signal of this S = 3/2 FeVO species, which is likely to have a low concentration, could easily be masked by the S = 5/2 signal.
Generation of [FeV(L1)(O)2]+ from the oxidation of 1 with NaIO4 resembles the generation of [FeV(L1)(O)2]+ from Oxone;16a both NaIO4 and Oxone are typically two-electron oxidants and can directly oxidize Fe(III) to Fe(V). UV-vis spectroscopy revealed that the reaction of 1 with NaIO4 or Oxone is different from the reaction of 1 with CAN, as exemplified by the immediate formation of a new band at λmax 830 nm attributable to an FeIVO species in the ‘1 + CAN’ system (Fig. 5) but gradual formation of a much weaker band at λmax 830 nm over 10 minutes in the ‘1 + NaIO4’ system (Fig. S25†). Also, for both the ‘1 + NaIO4’ and ‘1 + Oxone’ systems, kinetic studies revealed that the initial rate of oxygen evolution showed a linear dependence on the concentration of 1 but was relatively insensitive to the concentration of NaIO4 (Fig. S5†) or Oxone (Fig. S8†). This supports exclusion of the possibility of an “oxo–oxo coupling” reaction between two FeO species.
On the basis of the above findings, a mechanism involving FeIVO and/or FeVO intermediate(s) for the 1-catalysed water oxidation with CAN or NaIO4 is proposed as depicted in Scheme 2. For the corresponding reaction with Oxone, a mechanism similar to that of the reaction with NaIO4 could be proposed. The iron–oxo species depicted in Scheme 2 are [FeIV(L1)(O)(OH)]+ and [FeV(L1)(O)2]+ based on ESI-MS analysis; their protonated forms [FeIV(L1)(O)(OH2)]2+ and [FeV(L1)(O)(OH)]2+ could be involved as well (note also the computed pKa values shown in Scheme 1), but are not included in Scheme 2. Similar to [FeV(L1)(O)2]+, [FeV(L1)(O)(OH)]2+ also adopts a quartet ground state, with the doublet state being ∼15 kcal mol−1 higher in energy, according to DFT calculations. The possible involvement of the FeVO reactive species in the reaction is supported by the reasonably low reaction barrier for the oxidation of water by [FeV(L1)(O)2]+ of 15.7 kcal mol−1 obtained by the DFT calculations (Fig. 11). For [FeV(L1)(O)(OH)]2+, the reaction barrier for O–O bond formation with water was calculated to be 21.0 kcal mol−1 (Fig. S34†), which is 5.3 kcal mol−1 higher than that calculated for [FeV(L1)(O)2]+. The FeVO species is suggested to be attacked by water molecules, assisted by hydrogen-bond interactions, leading to the formation of an O–O bond, analogous to the O–O bond formation in water oxidation by MnVO,32 but the possibility of the involvement of FeIVO6e or OFeIV–O–CeIV species13 in the O–O bond formation could not be excluded, considering, for example, the small difference between the DFT-calculated redox potentials of +1.25 V for [FeIV(L1)(O)(OH2)]2+/[FeIII(L1)(OH)(OH2)]2+ and +1.42 V for [FeV(L1)(O)(OH)]2+/[FeIV(L1)(O)(OH2)]2+ at pH 1. The resulting iron(III)–peroxo intermediate in Scheme 2 is subsequently oxidized by the sacrificial oxidant (CAN, NaIO4, or Oxone) to give [FeIII(L1)(OO˙)(OH)]+ (DFT-calculated potential for [FeIII(L1)(OO˙)(OH)]+/[FeIII(L1)(OOH)(OH)]+: +0.64 V vs. SCE), the presence of which is supported by ESI-MS analysis (Fig. S20†). Extrusion of oxygen is achieved upon the substitution of [FeIII(L1)(OO˙)(OH)]+ with water; the resulting [FeII(L1)(OH)(OH2)]+ is oxidized by the sacrificial oxidant (CAN, NaIO4, or Oxone) to regenerate the catalyst [FeIII(L1)(OH)2]+ (DFT-calculated potential for [FeIII(L1)(OH)2]+/[FeII(L1)(OH)(OH2)]+: +0.26 V vs. SCE).
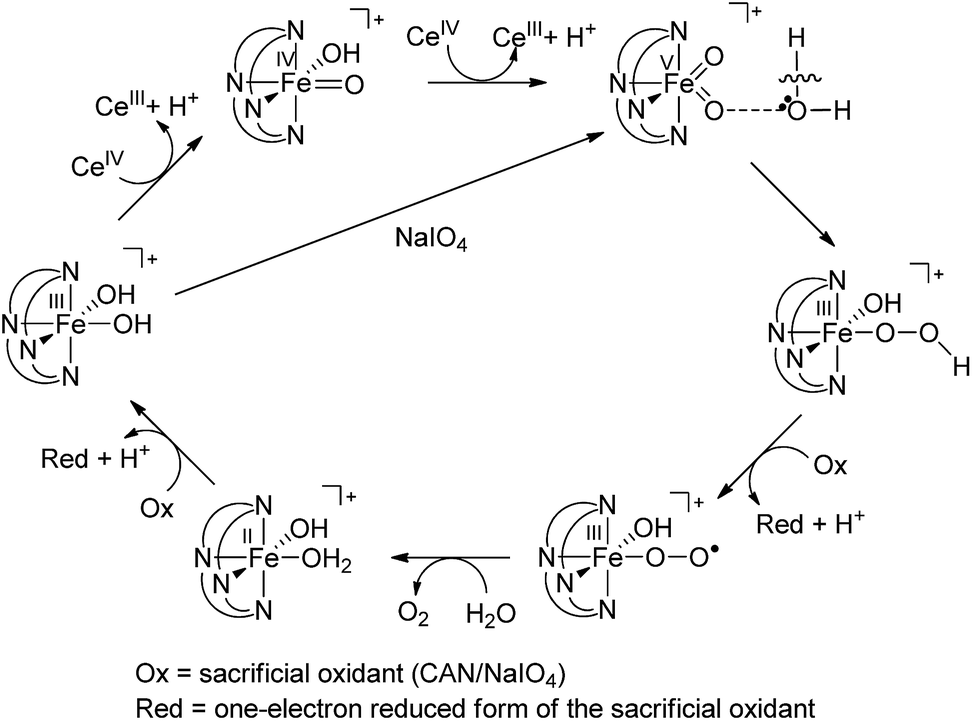 | ||
| Scheme 2 Proposed pathways for 1-catalysed water oxidation with CAN or NaIO4. | ||
Conclusions
A mononuclear iron(III) complex bearing a macrocyclic N4 diazapyridinophane ligand catalyses the oxidation of water to oxygen with NaIO4 or Oxone, as well as CAN, as the oxidant in acidic and/or neutral aqueous media. Studies using kinetic measurements, high-resolution ESI-MS, UV-vis absorption spectroscopy, 18O-labelling experiments, cyclic voltammetry, and DFT calculations lend support to the possible involvement of high-valent iron(IV)–oxo species such as [FeIV(L1)(O)(OH)]+ and/or iron(V)–oxo species such as [FeV(L1)(O)2]+, or their protonated forms [FeIV(L1)(O)(OH2)]2+ and/or [FeV(L1)(O)(OH)]2+, in the 1-catalysed water oxidation reactions.Acknowledgements
This work was supported by the Hong Kong Research Grants Council (HKU 700813), the National Key Basic Research Program of China (No. 2013CB834802), the University Grants Committee of the Hong Kong Special Administrative Region (AoE/P-03/08), and the CAS-Croucher Foundation Funding Scheme for Joint Laboratories. We thank Prof. Hung Kay Lee for assistance with EPR measurements.Notes and references
- (a) C. Tommos and G. T. Babcock, Acc. Chem. Res., 1998, 31, 18 CrossRef CAS; (b) M. H. V. Huynh and T. J. Meyer, Chem. Rev., 2007, 107, 5004 CrossRef CAS PubMed; (c) R. Eisenberg and H. B. Gray, Inorg. Chem., 2008, 47, 1697 CrossRef CAS PubMed.
- (a) D. G. H. Hetterscheid and J. N. H. Reek, Angew. Chem., Int. Ed., 2012, 51, 9740 CrossRef CAS PubMed; (b) D. J. Wasylenko, R. D. Palmer and C. P. Berlinguette, Chem. Commun., 2013, 49, 218 RSC; (c) A. Singh and L. Spiccia, Coord. Chem. Rev., 2013, 257, 2607 CrossRef CAS PubMed; (d) M. D. Kärkäs, O. Verho, E. V. Johnston and B. Ǻkermark, Chem. Rev., 2014, 114, 11863 Search PubMed.
- (a) T. C. W. Mak, C.-M. Che and K.-Y. Wong, J. Chem. Soc., Chem. Commun., 1985, 986 RSC; (b) C.-M. Che, T.-F. Lai and K.-Y. Wong, Inorg. Chem., 1987, 26, 2289 CrossRef CAS; (c) J.-U. Rohde, J.-H. In, M. H. Lim, W. W. Brennessel, M. R. Bukowski, A. Stubna, E. Münck, W. Nam and L. Que Jr., Science, 2003, 299, 1037 CrossRef CAS PubMed.
- (a) J. Hohenberger, K. Ray and K. Meyer, Nat. Commun., 2012, 3, 720 CrossRef PubMed; (b) A. R. McDonald and L. Que Jr., Coord. Chem. Rev., 2013, 257, 414 CrossRef CAS PubMed; (c) K. P. Bryliakov and E. P. Talsi, Coord. Chem. Rev., 2014, 276, 73 CrossRef CAS PubMed; (d) K. Ray, F. F. Pfaff, B. Wang and W. Nam, J. Am. Chem. Soc., 2014, 136, 13942 CrossRef CAS PubMed; (e) W. Nam, Y.-M. Lee and S. Fukuzumi, Acc. Chem. Res., 2014, 47, 1146 CrossRef CAS PubMed.
- (a) E. J. Klinker, J. Kaizer, W. W. Brennessel, N. L. Woodrum, C. J. Cramer and L. Que Jr., Angew. Chem., Int. Ed., 2005, 44, 3690 CrossRef CAS PubMed; (b) J. Yoon, S. A. Wilson, Y. K. Jang, M. S. Seo, K. Nehru, B. Hedman, K. O. Hodgson, E. Bill, E. I. Solomon and W. Nam, Angew. Chem., Int. Ed., 2009, 48, 1257 CrossRef CAS PubMed; (c) J. England, Y. Guo, E. R. Farquhar, V. G. Young Jr., E. Münck and L. Que Jr., J. Am. Chem. Soc., 2010, 132, 8635 CrossRef CAS PubMed; (d) D. C. Lacy, R. Gupta, K. L. Stone, J. Greaves, J. W. Ziller, M. P. Hendrich and A. S. Borovik, J. Am. Chem. Soc., 2010, 132, 12188 CrossRef CAS PubMed; (e) S. Fukuzumi, Y. Morimoto, H. Kotani, P. Naumov, Y.-M. Lee and W. Nam, Nat. Chem., 2010, 2, 756 CrossRef CAS PubMed; (f) H. Kotani, T. Suenobu, Y.-M. Lee, W. Nam and S. Fukuzumi, J. Am. Chem. Soc., 2011, 133, 3249 CrossRef CAS PubMed; (g) S. Hong, Y.-M. Lee, K.-B. Cho, K. Sundaravel, J. Cho, M. J. Kirn, W. Shin and W. Nam, J. Am. Chem. Soc., 2011, 133, 11876 CrossRef CAS PubMed; (h) S. A. Wilson, J. Chen, S. Hong, Y.-M. Lee, M. Clémancey, R. Garcia-Serres, T. Nomura, T. Ogura, J.-M. Latour, B. Hedman, K. O. Hodgson, W. Nam and E. I. Solomon, J. Am. Chem. Soc., 2012, 134, 11791 CrossRef CAS PubMed; (i) Y. Nishida, Y.-M. Lee, W. Nam and S. Fukuzumi, J. Am. Chem. Soc., 2014, 136, 8042 CrossRef CAS PubMed.
- (a) W. C. Ellis, M. D. McDaniel, S. Bernhard and T. J. Collins, J. Am. Chem. Soc., 2010, 132, 10990 CrossRef CAS PubMed; (b) J. Lloret Fillol, Z. Codolà, I. Garcia-Bosch, L. Gómez, J. J. Pla and M. Costas, Nat. Chem., 2011, 3, 807 CrossRef PubMed; (c) G. Chen, L. Chen, S.-M. Ng, W.-L. Man and T.-C. Lau, Angew. Chem., Int. Ed., 2013, 52, 1789 CrossRef CAS PubMed; (d) W. A. Hoffert, M. T. Mock, A. M. Appel and J. Y. Yang, Eur. J. Inorg. Chem., 2013, 3846 CrossRef CAS PubMed; (e) D. Hong, S. Mandal, Y. Yamada, Y.-M. Lee, W. Nam, A. Llobet and S. Fukuzumi, Inorg. Chem., 2013, 52, 9522 CrossRef CAS PubMed; (f) Z. Codolà, I. Garcia-Bosch, F. Acuna-Parés, I. Prat, J. M. Luis, M. Costas and J. Lloret-Fillol, Chem. – Eur. J., 2013, 19, 8042 CrossRef PubMed; (g) B. Zhang, F. Li, F. Yu, H. Cui, X. Zhou, H. Li, Y. Wang and L. Sun, Chem. – Asian J., 2014, 9, 1515 CrossRef CAS PubMed.
- C. Panda, J. Debgupta, D. D. Díaz, K. K. Singh, S. S. Gupta and B. B. Dhar, J. Am. Chem. Soc., 2014, 136, 12273 CrossRef CAS PubMed.
- (a) M. K. Coggins, M.-T. Zhang, A. K. Vannucci, C. J. Dares and T. J. Meyer, J. Am. Chem. Soc., 2014, 136, 5531 CrossRef CAS PubMed; (b) E. L. Demeter, S. L. Hilburg, N. R. Washburn, T. J. Collins and J. R. Kitchin, J. Am. Chem. Soc., 2014, 136, 5603 CrossRef CAS PubMed; (c) Z.-Q. Wang, Z.-C. Wang, S. Zhan and J.-S. Ye, Appl. Catal., A, 2015, 490, 128 CrossRef CAS PubMed.
- B. M. Klepser and B. M. Bartlett, J. Am. Chem. Soc., 2014, 136, 1694 CrossRef CAS PubMed.
- (a) M. M. Najafpour, A. N. Moghaddam, D. J. Sedigh and M. Holyńska, Catal. Sci. Technol., 2014, 4, 30 RSC; (b) A. R. Parent, T. Nakazono, S. Lin, S. Utsunomiya and K. Sakai, Dalton Trans., 2014, 43, 12501 RSC.
- D. Wang and L. Que Jr., Chem. Commun., 2013, 49, 10682 RSC.
- (a) M. Z. Ertem, L. Gagliardi and C. J. Cramer, Chem. Sci., 2012, 3, 1293 RSC; (b) E. E. Kasapbasi and M.-H. Whangbo, Inorg. Chem., 2012, 51, 10850 CrossRef CAS PubMed; (c) A. Poater, Catal. Commun., 2014, 44, 2 CrossRef CAS PubMed; (d) R.-Z. Liao, X.-C. Li and P. E. M. Siegbahn, Eur. J. Inorg. Chem., 2014, 728 CrossRef CAS PubMed; (e) F. Acuña-Parés, M. Costas, J. M. Luis and J. Lloret-Fillol, Inorg. Chem., 2014, 53, 5474 CrossRef PubMed; (f) F. Acuña-Parés, Z. Codolà, M. Costas, J. M. Luis and J. Lloret-Fillol, Chem. – Eur. J., 2014, 20, 5696 CrossRef PubMed.
- Z. Codolà, L. Gómez, S. T. Kleespies, L. Que Jr., M. Costas and J. Lloret-Fillol, Nat. Commun., 2015, 6, 5865 CrossRef PubMed.
- (a) F. T. de Oliveira, A. Chanda, D. Banerjee, X. Shan, S. Mondal, L. Que Jr., E. L. Bominaar, E. Münck and T. J. Collins, Science, 2007, 315, 835 CrossRef PubMed; (b) S. H. Lee, J. H. Han, H. Kwak, S. J. Lee, E. Y. Lee, H. J. Kim, J. H. Lee, C. Bae, S. N. Lee, Y. Kim and C. Kim, Chem. – Eur. J., 2007, 13, 9393 CrossRef CAS PubMed; (c) O. Y. Lyakin, K. P. Bryliakov, G. J. P. Britovsek and E. P. Talsi, J. Am. Chem. Soc., 2009, 131, 10798 CrossRef CAS PubMed; (d) I. Prat, J. S. Mathieson, M. Güell, X. Ribas, J. M. Luis, L. Cronin and M. Costas, Nat. Chem., 2011, 3, 788 CrossRef CAS PubMed; (e) O. Y. Lyakin, K. P. Bryliakov and E. P. Talsi, Inorg. Chem., 2011, 50, 5526 CrossRef CAS PubMed; (f) O. Y. Lyakin, I. Prat, K. P. Bryliakov, M. Costas and E. P. Talsi, Catal. Commun., 2012, 29, 105 CrossRef CAS PubMed; (g) O. Y. Lyakin, R. V. Ottenbacher, K. P. Bryliakov and E. P. Talsi, ACS Catal., 2012, 2, 1196 CrossRef CAS; (h) Y. Hitomi, K. Arakawa, T. Funabiki and M. Kodera, Angew. Chem., Int. Ed., 2012, 51, 3448 CrossRef CAS PubMed; (i) K. M. Van Heuvelen, A. T. Fiedler, X. Shan, R. F. De Hont, K. K. Meier, E. L. Bominaar, E. Münck and L. Que Jr., Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 11933 CrossRef CAS PubMed; (j) M. Ghosh, K. K. Singh, C. Panda, A. Weitz, M. P. Hendrich, T. J. Collins, B. B. Dhar and S. Sen Gupta, J. Am. Chem. Soc., 2014, 136, 9524 CrossRef CAS PubMed.
- (a) F. Bottino, M. D. Grazia, P. Finocchiaro, F. R. Fronczek, A. Mamo and S. Pappalardo, J. Org. Chem., 1988, 53, 3521 CrossRef CAS; (b) W. O. Koch, V. Schünemann, M. Gerdan, A. X. Trautwein and H.-J. Krüger, Chem. – Eur. J., 1998, 4, 686 CrossRef CAS.
- (a) T. W.-S. Chow, E. L.-M. Wong, Z. Guo, Y. Liu, J.-S. Huang and C.-M. Che, J. Am. Chem. Soc., 2010, 132, 13229 CrossRef CAS PubMed; (b) F. Tang, Y. Zhang, N. P. Rath and L. M. Mirica, Organometallics, 2012, 31, 6690 CrossRef CAS; (c) H. Sugimoto, K. Ashikari and S. Itoh, Chem. – Asian J., 2013, 8, 2154 CrossRef CAS PubMed.
- W.-T. Lee, S. B. Muñoz III, D. A. Dickie and J. M. Smith, Angew. Chem., Int. Ed., 2014, 53, 9856 CrossRef CAS PubMed.
- A. R. Parent, R. H. Crabtree and G. W. Brudvig, Chem. Soc. Rev., 2013, 42, 2247 RSC.
- J. Limburg, J. S. Vrettos, H. Chen, J. C. de Paula, R. H. Crabtree and G. W. Brudvig, J. Am. Chem. Soc., 2001, 123, 423 CrossRef CAS PubMed.
- (a) C.-K. Li, C.-M. Che, W.-F. Tong, W.-T. Tang, K.-Y. Wong and T.-F. Lai, J. Chem. Soc., Dalton Trans., 1992, 2109 RSC; (b) S. K. W. Yau, C.-M. Che and T.-C. Lau, J. Chem. Soc., Dalton Trans., 2002, 2697 RSC.
- (a) F. Bozoglian, S. Romain, M. Z. Ertem, T. K. Todorova, C. Sens, J. Mola, M. Rodríguez, I. Romero, J. Benet-Buchholz, X. Fontrodona, C. J. Cramer, L. Gagliardi and A. Llobet, J. Am. Chem. Soc., 2009, 131, 15176 CrossRef CAS PubMed; (b) X. Guan, S. L.-F. Chan and C.-M. Che, Chem. – Asian J., 2013, 8, 2046 CrossRef CAS PubMed.
- (a) J. N. Harvey, M. Aschi, H. Schwarz and W. Koch, Theor. Chem. Acc., 1998, 99, 95 CrossRef CAS; (b) J. N. Harvey and M. Aschi, Phys. Chem. Chem. Phys., 1999, 1, 5555 RSC.
- B. Limburg, E. Bouwman and S. Bonnet, Coord. Chem. Rev., 2012, 256, 1451 CrossRef CAS PubMed.
- (a) M. J. Katz, S. C. Riha, N. C. Jeong, A. B. F. Martinson, O. K. Farha and J. T. Hupp, Coord. Chem. Rev., 2012, 256, 2521 CrossRef CAS PubMed; (b) F. E. Osterloh, Chem. Soc. Rev., 2013, 42, 2294 RSC.
- (a) J. J. Concepcion, J. W. Jurss, J. L. Templeton and T. J. Meyer, J. Am. Chem. Soc., 2008, 130, 16462 CrossRef CAS; (b) J. J. Concepcion, J. W. Jurss, M. R. Norris, Z. Chen, J. L. Templeton and T. J. Meyer, Inorg. Chem., 2010, 49, 1277 CrossRef CAS PubMed; (c) D. J. Wasylenko, C. Ganesamoorthy, M. A. Henderson, B. D. Koivisto, H. D. Osthoff and C. P. Berlinguette, J. Am. Chem. Soc., 2010, 132, 16094 CrossRef CAS PubMed; (d) Y. M. Badiei, D. E. Polyansky, J. T. Muckerman, D. J. Szalda, R. Haberdar, R. Zong, R. P. Thummel and E. Fujita, Inorg. Chem., 2013, 52, 8845 CrossRef CAS PubMed.
- D.-L. Popescu, M. Vrabel, A. Brausam, P. Madsen, G. Lente, I. Fabian, A. D. Ryabov, R. van Eldik and T. J. Collins, Inorg. Chem., 2010, 49, 11439 CrossRef CAS PubMed.
- (a) G. E. Cabaniss, A. A. Diamantis, W. R. Murphy Jr., R. W. Linton and T. J. Meyer, J. Am. Chem. Soc., 1985, 107, 1845 CrossRef CAS; (b) C.-M. Che, K.-Y. Wong and F.-C. Anson, J. Electroanal. Chem., 1987, 226, 211 CrossRef CAS.
- Y. Wing-Ping, PhD Thesis, The University of Hong Kong, 2004 Search PubMed.
- D. Wang, M. Zhang, P. Buhlmann and L. Que Jr., J. Am. Chem. Soc., 2010, 132, 7638 CrossRef CAS PubMed.
- L. Que Jr., Acc. Chem. Res., 2007, 40, 493 CrossRef PubMed.
- Y. Ohgo, Y. Chiba, D. Hashizume, H. Uekusa, T. Ozeki and M. Nakamura, Chem. Commun., 2006, 1935 RSC.
- Y. Gao, T. Åkermark, J. Liu, L. Sun and B. Åkermark, J. Am. Chem. Soc., 2009, 131, 8726 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental section, Table S1, Fig. S1–S34, Scheme S1 and computational details. See DOI: 10.1039/c5sc01680k |
| This journal is © The Royal Society of Chemistry 2015 |