
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Copper-catalyzed diastereoselective aerobic intramolecular dehydrogenative coupling of hydrazones via sp3 C–H functionalization†
Xuesong
Wu
a,
Mian
Wang
b,
Guangwu
Zhang
a,
Yan
Zhao
a,
Jianyi
Wang
*b and
Haibo
Ge
*a
aDepartment of Chemistry and Chemical Biology, Indiana University-Purdue University Indianapolis, Indianapolis, Indiana 46202, USA. E-mail: geh@iupui.edu
bSchool of Chemistry and Chemical Engineering, Guangxi University, Nanning 530004, Guangxi, P. R. China. E-mail: jianyiwang@gxu.edu.cn
First published on 14th July 2015
Abstract
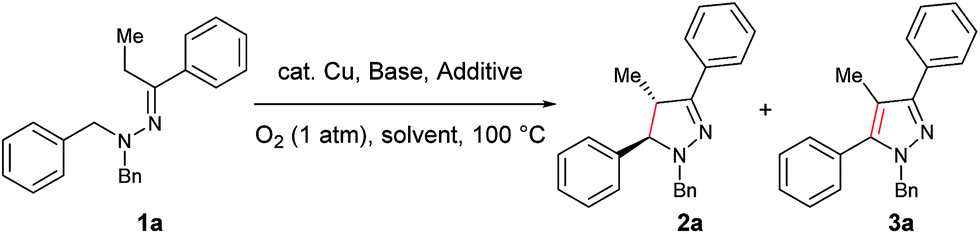
Transition metal-catalyzed cross dehydrogenative coupling is an important tool for functionalization of the α Csp3–H bond of amines. Among this reaction category, copper-catalyzed selective C–C bond formation under atmospheric O2 is of considerable research interest and significant progress has been achieved in recent years. In comparison, development of the intramolecular version of this transformation is still in its infancy. Furthermore, diastereoselective cyclization with this transformation has not been achieved. Here, we describe the highly diastereoselective intramolecular dehydrogenative cyclization of N,N-disubstituted hydrazones by a copper-catalyzed sp3 C–H bond functionalization process. The reaction protocol utilizes O2 as the oxidant and shows great functional group compatibility. Computational studies suggest that a 5-center/6-electron disrotatory cyclization mechanism is probably involved in the process for controlling the diastereoselectivity. This work represents the first example of a copper-catalyzed, direct intramolecular diastereoselective coupling reaction via an iminium ion intermediate. Additionally, it provides an environmentally friendly and atom efficient approach to access substituted pyrazolines, an important structural unit in many biologically active compounds.
Introduction
Selective carbon–carbon (C–C) bond formation is a fundamental focus in chemical research due to its importance in organic, medicinal, agricultural, and material chemistry.1 Conventional methods for the construction of C–C bonds, such as nucleophilic addition and substitution reactions, Friedel–Crafts-type reactions, and transition-metal-catalyzed or -mediated cross coupling reactions, rely primarily on prefunctionalized reactants, which somehow limit the synthetic application of these transformations.2 Additionally, stoichiometric amounts of toxic metal waste are often generated in the processes, which constitutes an environmental issue. For these reasons, transition metal-catalyzed cross coupling reactions via direct functionalization of relatively unreactive C–H bonds has been of great interest in the past two decades.3 Among this reaction class, copper-catalyzed aerobic dehydrogenative couplings via a double C–H bond functionalization process have received considerable attention in recent years due to the economical and ecological advantages.4The oxidative dimerization of terminal alkynes, the Glaser reaction reported in 1869, is the first example of copper-catalyzed aerobic dehydrogenative coupling.5 Since then, considerable efforts have been devoted in this area for selective C–C bond construction, and a variety of copper-catalyzed aerobic dehydrogenative coupling reactions have been reported via sp or sp2 C–H bond functionalization. Representative examples, which have expanded product diversity, include oxidative dimerization of phenols, naphthols and electron-deficient arenes, cross coupling of terminal alkynes with electron-deficient arenes, and intramolecular dehydrogenative cyclization of anilides.4e,g,6 Furthermore, copper-catalyzed aerobic cross dehydrogenative coupling (CDC) via an sp3 C–H bond functionalization process has also been established on tertiary amines by Miura, Li, and others.4a,e,g It has been demonstrated that a wide range of nucleophiles, such as alkynes, nitroalkanes, malonic esters, ketones, and electron-rich (hetero)arenes, are compatible coupling partners under aerobic oxidative conditions. Mechanistically, an iminium ion intermediate is generated via oxidation of the corresponding amine. This intermediate then serves as an active electrophile for the subsequent nucleophilic addition reaction.7 Despite its considerable potential, this reaction suffers from a restricted substrate scope of tertiary amines; only substituted 1,2,3,4-tetrahydroisoquinoline and N,N-dimethylaniline derivatives have been reported as suitable substrates. Furthermore, this transformation is limited to the intermolecular version, in part due to the non-selective oxidation of the amine over the internal nucleophile.8
We envisaged that the use of an enamine-type motif as an internal nucleophile could potentially overcome this drawback and extend the reaction scope to intramolecular cyclizations.9 Based on this design, copper-catalyzed aerobic intramolecular dehydrogenative cyclization of hydrazones was examined and realized in our laboratory.10,11 However, it was noted that the initial cyclized intermediate was very unstable under the reaction conditions and was rapidly aromatized to pyrazoles. Furthermore, only α-unsubstituted 1-benzyl-1-isopropyl-2-(1-phenylethylidene)hydrazines were well tolerated under the catalyst system. Additionally, aliphatic hydrazones completely failed in this process due to competitive decomposition of the starting materials. To overcome these drawbacks and thus to provide a straightforward environmentally friendly and atom efficient access to diversified pyrazoline derivatives, copper-catalyzed diastereoselective cyclization of aromatic and aliphatic hydrazones was investigated. It is noteworthy that pyrazoline is a prominent structural motif in many pharmacological compounds, and pyrazoline derivatives display a broad spectrum of biological activities including antibacterial, antidepressant, antidiabetic, antiepileptic, antihypotensive, anti-inflammatory, antimalarial, antimicrobial, antipyretic-analgesic, antituberculotic, antitumor, immunosuppression, insecticidal, muscle relaxant, psycho analeptic, and tranquilizing activities.12
Here we report detailed studies in which the copper-catalyzed sp3 C–H functionalization strategy was thoroughly examined and further explored to expand its scope of synthetic utility. To provide important mechanistic insights into various factors that control the product distribution, density functional theory (DFT) calculations were further conducted to rationalize the diastereoselectivity observed. The combined work of chemical synthesis, NMR, and computation revealed how the interplay of molecular configurations, steric effects, and the symmetry requirement of electronic structure reactivity, can be utilized to precisely control the diastereoselectivity in the products, thereby providing a valuable guidance to rational designs of the related synthetic routes.
Results and discussion
Our study commenced with copper-catalyzed dehydrogenative cyclization of 1,1-dibenzyl-2-(1-phenylpropylidene)hydrazine (1a) under atmospheric O2 (Table 1). After an initial screening of the catalyst, base, and solvent, it was found that the pyrazoline 2a was obtained in 27% yield, with only trace amount of the undesired pyrazole compound 3, by using a catalytic amount of Cu(OTf)2 and stoichiometric KI, KOAc, and DBU in NMP with O2 as the external oxidant (entry 5). Interestingly, this reaction showed high diastereoselectivity, with the anti isomer as the major product. Further optimization showed that this reaction was improved by using NMP and tAmOH as co-solvent (entry 8). It was then noticed that the reaction yield was significantly increased by extending the reaction time to 12 h from 4 h (entry 9). In addition, the replacement of DBU with DBN significantly increased the reaction rate, and the desired product was obtained in 86% yield within 4 h (entry 10). Moreover, the cyclization could also be effectively catalyzed by other CuI or CuII sources under the above modified conditions (entries 13–17).|
|
|||||||
|---|---|---|---|---|---|---|---|
| Entry | Cu source (mol%) | Base (equiv.) | Additive (equiv.) | Solvent | Yieldb (%) | d.r.c | |
| 2a | 3 | ||||||
a Reaction conditions: 1a (0.3 mmol), Cu source, base, additive, O2 (1 atm), 2 mL of solvent, 100 °C, 4 h.
b Yields and conversions are based on 1a, determined by GC/MS using diphenylketone as the internal standard.
c d.r. (anti![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :syn): determined by 1H NMR spectroscopy.
d 2:3 (v/v).
e 12 h.
f Isolated yields. :syn): determined by 1H NMR spectroscopy.
d 2:3 (v/v).
e 12 h.
f Isolated yields.
|
|||||||
| 1 | Cu(OTf)2 (10) | KOAc (1.0) | — | NMP | <5 | — | — |
| 2 | Cu(OTf)2 (10) | DBU (1.0) | — | NMP | <5 | — | — |
| 3 | Cu(OTf)2 (10) | KOAc (1.0) | KI (1.0) | NMP | 23 | — | 6.8:1 |
| 4 | Cu(OTf)2 (10) | DBU (1.0) | KI (1.0) | NMP | 10 | — | – |
| 5 | Cu(OTf)2 (10) | KOAc (0.5)/DBU (0.5) | KI (1.0) | NMP | 27 | — | 6.3:1 |
| 6 | Cu(OTf)2 (10) | KOAc (0.5)/DBU (0.5) | — | NMP | <5 | — | — |
| 7 | Cu(OTf)2 (10) | — | KI (1.0) | NMP | <5 | — | — |
| 8 | Cu(OTf)2 (10) | KOAc (0.5)/DBU (0.5) | KI (1.0) | NMP/tAmOHd | 45 | 3 | 6.0:1 |
| 9e | Cu(OTf)2 (10) | KOAc (0.5)/DBU (0.5) | KI (1.0) | NMP/tAmOHd | 81 | 4 | 6.1:1 |
| 10 | Cu(OTf) 2 (10) | KOAc (0.5)/DBN (0.5) | KI (1.0) | NMP/ t AmOH | 88 (86) | 6 |
6.8:1 |
| 11 | Cu(OTf)2 (10) | KOAc (0.5)/DBN (0.5) | KI (1.0) | t AmOH | 43 | 3 | 5.1:1 |
| 12 | — | KOAc (0.5)/DBN (0.5) | KI (1.0) | NMP/tAmOHd | 0 | — | — |
| 13 | Cu(TFA)2 (10) | KOAc (0.5)/DBN (0.5) | KI (1.0) | NMP/tAmOHd | 67 | 5 | 7.4:1 |
| 14 | Cu(OAc)2 (10) | KOAc (0.5)/DBN (0.5) | KI (1.0) | NMP/tAmOHd | 73 | 5 | 6.5:1 |
| 15 | CuI (10) | KOAc (0.5)/DBN (0.5) | KI (1.0) | NMP/tAmOHd | 62 | 4 | 6.3:1 |
| 16 | CuBr (10) | KOAc (0.5)/DBN (0.5) | KI (1.0) | NMP/tAmOHd | 60 | 4 | 6.3:1 |
| 17 | (CuOTf)2Py (5) | KOAc (0.5)/DBN (0.5) | KI (1.0) | NMP/tAmOHd | 82 | 5 | 6.7:1 |
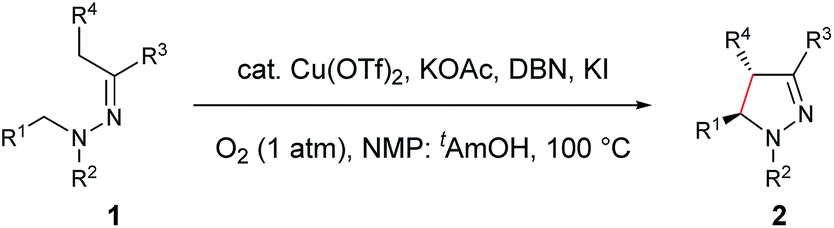
With the optimized conditions in hand, the scope of hydrazone substrates was studied (Table 2). As expected, aromatic, acyclic and cyclic aliphatic hydrazones were compatible with the oxidative reaction conditions (2a–h). Interestingly, the reaction showed a substrate-dependent diastereoselectivity pattern, and the anti-isomers of 2a and 2b were obtained as the major products while the syn-isomers of 2c–g were the major forms. It is noteworthy that the current methods for the synthesis of syn-4,5-pyrazolines rely primarily on the [3 + 2]-cyclization of a diazo compound and an olefin, which often suffers from poor regioselectivity.13 Furthermore, the N-benzyl group could also be replaced with another alkyl group (2i–n), in which the aromatic hydrazones favored the anti-isomers, and the aliphatic substrates favored the syn-isomers. Additionally, an N-phenyl substituted hydrazone yielded exclusively the syn-isomer 2o, providing an important and tunable strategy to selectively access either anti or syn pyrazolines. Moreover, 1,2,3,4-tetrahydroquinoline is also an effective substrate (2p).
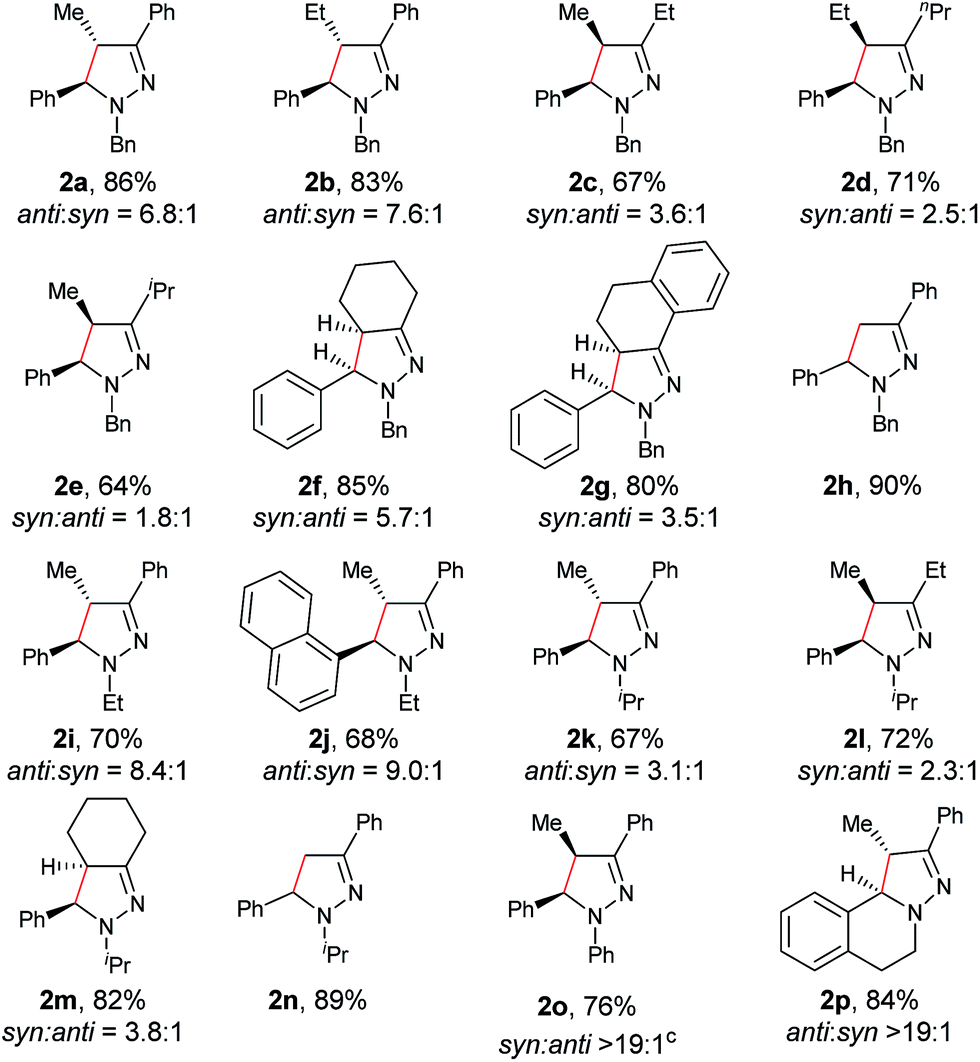
In our previous study of dehydrogenative cyclization/aromatization of 1-benzyl-1-isopropyl-2-(1-phenylethylidene)hydrazine, it has been demonstrated that both electron-donating and electron-withdrawing groups on the phenyl rings were compatible.11 It is also believed that an oxidized iminium ion intermediate is involved in this process, serving as the precursor for the cyclization. Therefore, in order to simplify the complexity of configuration of the iminium ion intermediates, and thus to better understand the outcome of diastereoselectivity, we carried out an extensive substrate scope study on N-cyclohexylidene-3,4-dihydroisoquinolin-2(1H)-amine (Table 3). For each substrate, only one isomeric iminium ion intermediate will be generated, which will undergo either 5-center/6-electron cyclization or intramolecular nucleophilic addition to provide the corresponding diastereoisomers. It is worth mentioning that extensive synthetic studies have been carried out with azomethine imines, which have resulted in a variety of applications of the products obtained.14 However, to our knowledge, there are only two reported examples which involve the direct use of a dihydroisoquinoline derivative via an oxidative sp3 C–H activation process: mercuric oxide-mediated dimerization and Rh(III)-catalyzed cycloaddition.9,15 Furthermore, systematic electronic or steric effects of the above reactions on 1,2,3,4-tetrahydroisoquinolines have barely been studied.
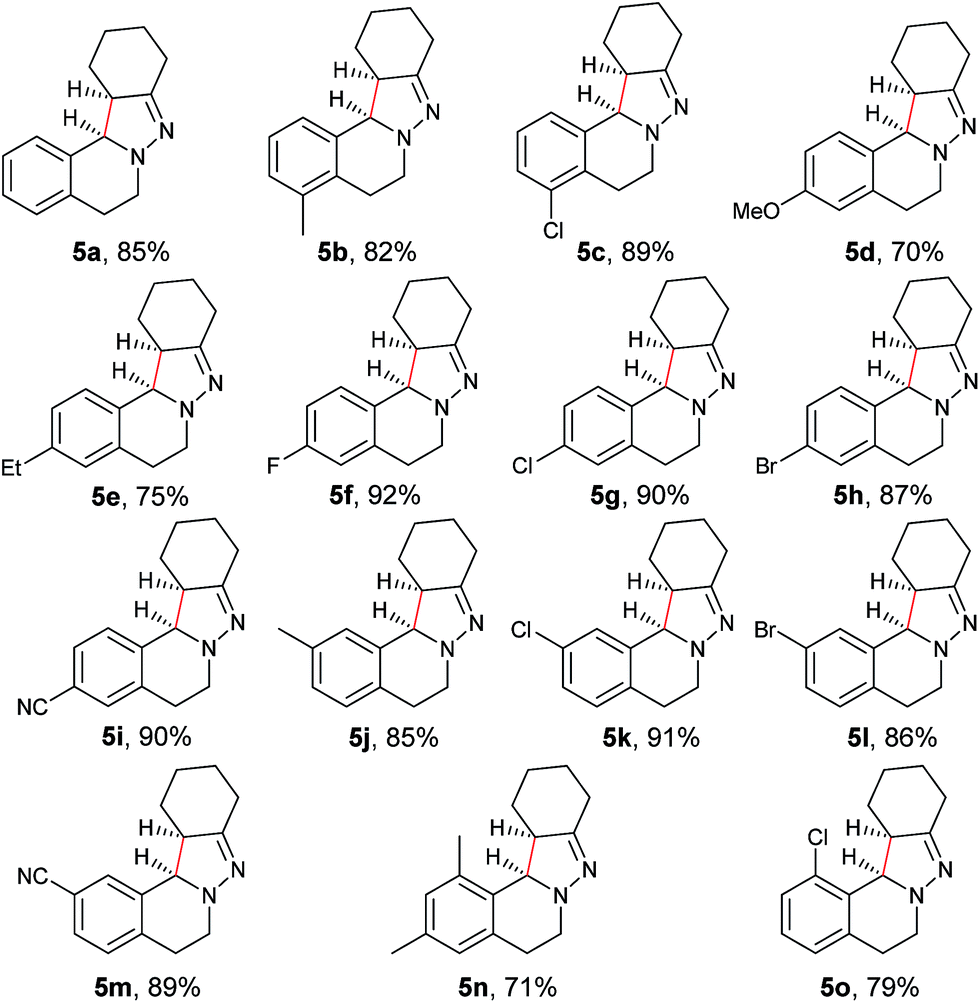
As shown in Table 3, both electron-donating and electron-withdrawing groups on the phenyl ring are compatible under the current catalytic system. Interestingly, high diastereoselectivity was observed in all cases with the syn diastereoisomers as the major products (5b–o). Generally, electron-withdrawing group-substituted substrates provided better yields compared with electron-donating substituents in the same position. Considering that an iminium ion intermediate is formed prior to cyclization, these results are not surprising since an electron-withdrawing group increases the electrophilicity of the iminium ion and thus facilitates the cyclization. Additionally, an electron-withdrawing group may retard the further oxidation of pyrazolines and thus minimize formation of the by-product pyrazoles. Furthermore, a steric effect was observed for this reaction: C8-substituted hydrazones gave lower yields of desired products compared with the C5-, C6-, or C7-substituted substrates. Finally, halogens (F, Cl, and Br) are also well tolerated, which provides the opportunity for further product manipulation.
NOESY analysis was carried out to confirm the relative configuration of the product (Fig. 1). NOEs between the aromatic proton H1 of 5a and H12a, H12b were observed. Additionally, there was no observable NOE between H1 and H13, or H14 and H12a or H12b. These results indicate that H13 and H14 have the syn relationship.
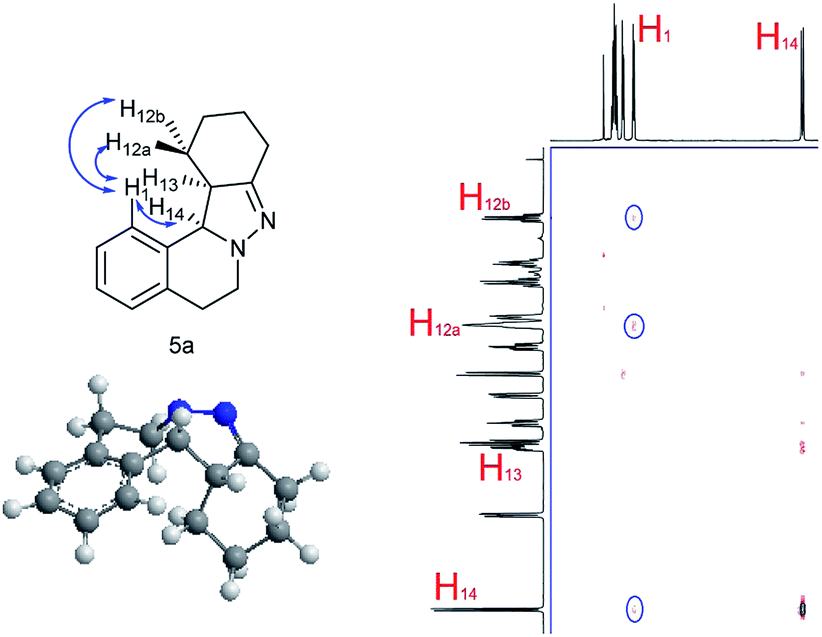 | ||
| Fig. 1 NOESY and relative configuration of 5a. | ||
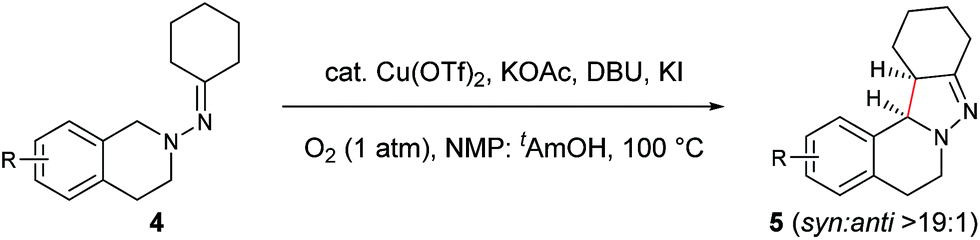
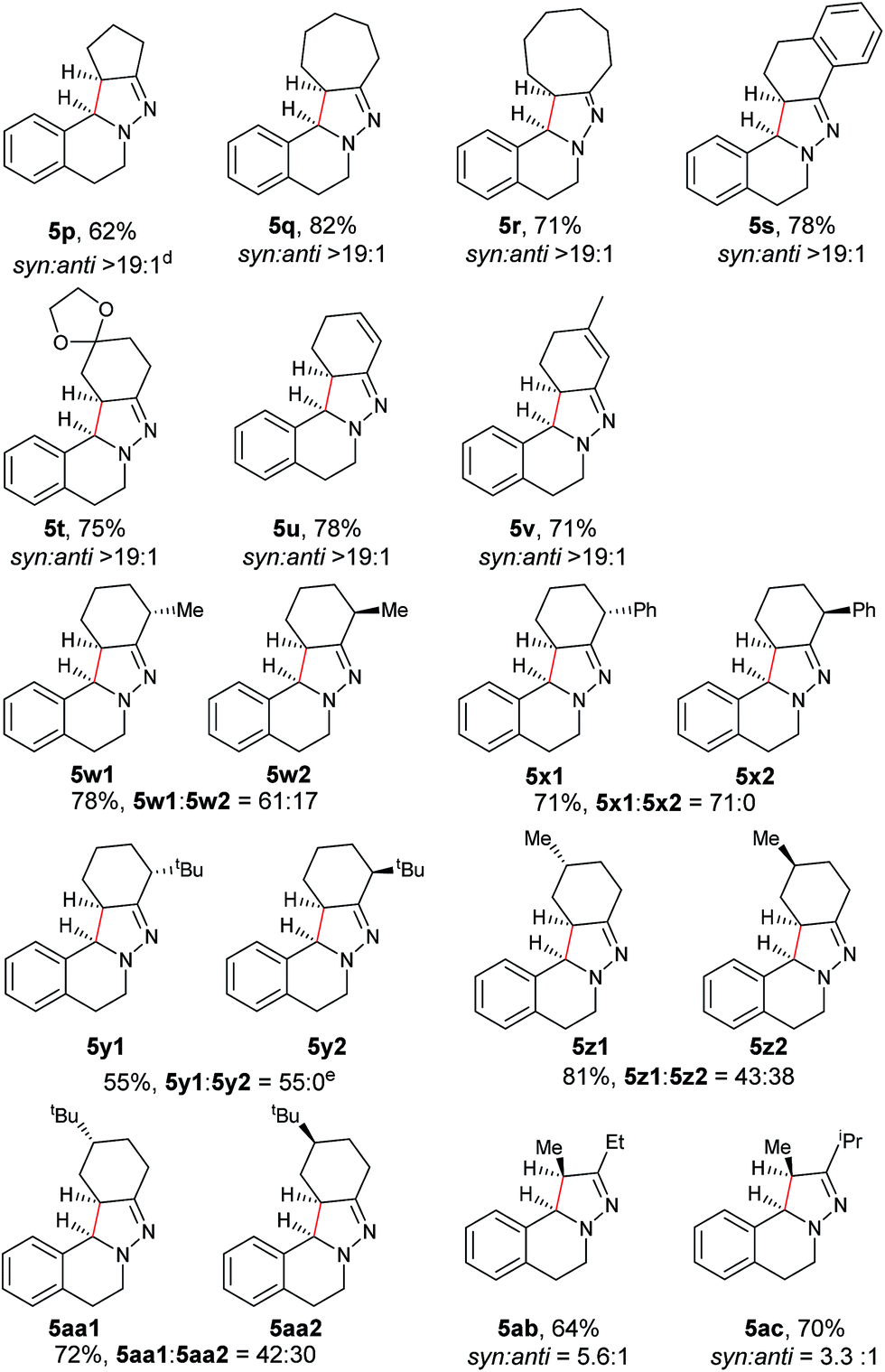
Next, an imine substrate scope study was carried out (Table 4). The reaction occurred with high diastereoselectivity on substrates with a larger ring (5q and 5r). The 5-membered ring substrate also provided a good yield of product 5p under the modified reaction conditions. Furthermore, α,β-unsaturated hydrazones are also compatible (5u and 5v), which allows for further transformation of the initial products. As expected, good to high yields of products were obtained on substrates with substituted cyclohexylidenehydrazine moieties (5s, 5t, 5w–z and 5aa). In addition, good to excellent diastereoselectivity was observed with α-substituted cyclohexylidenehydrazine, favoring the anti-products (5w–y). In comparison, γ-substituents on the cyclohexylidenehydrazine moiety did not significantly affect the relative diastereoselectivity (5z and 5aa). Additionally, linear hydrazones are also compatible with the oxidative conditions, favoring the formation of the syn-isomers. However, in this case, the ratio of diastereoselectivity was dramatically decreased. Considering that there are two possible conformers present in the iminium ion intermediate oxidized either from 5z or 5aa, the above result is not surprising.
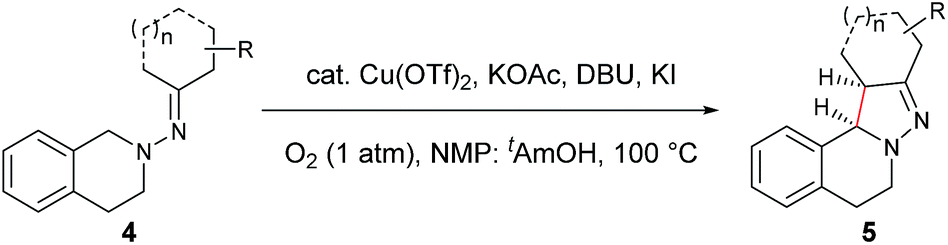
On the basis of the above results and our previous report,11 a plausible reaction mechanism is proposed (Scheme 1).4g,7 Oxidation of the amine nitrogen on 1 generates the radical cation Avia an SET process. The radical cation A is then converted into the iminium ion intermediates B1 and B2via either an oxidation/deprotonation or a homolytic cleavage process. Tautomerization of the imine moiety on B1 and B2 to enamine-type diastereoisomers C1, C2, C3 and C4, followed by subsequent nucleophilic addition provides the intermediate D1 and D2, which then give pyrazoline 2 upon deprotonation. The rationale of high syn over anti diastereoselectivity is also proposed based on the previous reports by Hoffmann and List.16,17 It is believed that the neutral nitrogen in intermediate C will behave as an sp2 hybridized atom. Therefore, the lone-pair of electrons on the nitrogen will participate in the π-conjugation together with the four π electrons in the double bonds, resulting in a 5-center/6-electron system, similar to the situation found in a pentadienyl anion. For such homoconjugated systems, the HOMO is actually a “symmetric” nonbonding orbital, which is 1,5-bonding with the preferred U planar configuration. For thermally induced electrocyclic ring closures, the symmetric HOMO requires a disrotatory mechanism, which results in the corresponding product 2. It should be mentioned that α-alkyl Cu species could potentially be formed from deprotonation of the intermediates B1/B2 under basic conditions. Intramolecular nucleophilic addition of these Cu species could also provide the desired products.
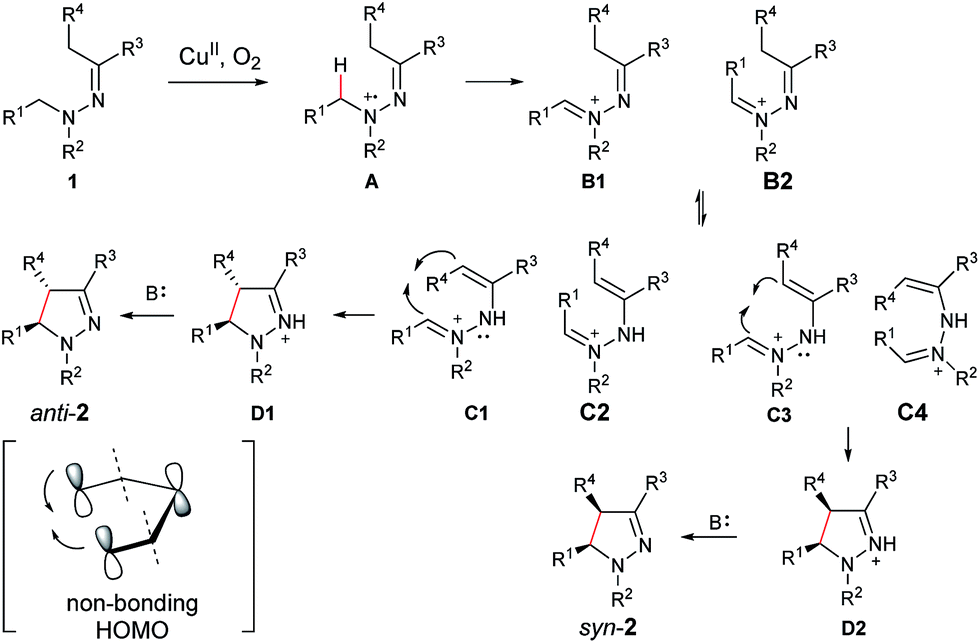 | ||
| Scheme 1 Proposed reaction mechanism. | ||
Mechanistic studies with computation
Toward molecular-level understanding of diastereoselective 5-center/6-electron cyclization mechanism displayed in these copper-based C–H functionalization reactions, we performed density functional theory (DFT) investigations on five representative systems (2a, 2f, 2p, 5a, and 5ab).Computational methods
All the calculations were carried out for the five systems by using Gaussian 09 program suites.18 The Kohn–Sham density functional theory (DFT) was solved with the B3LYP functional,19 and the 6-31G+(d,p) basis sets were selected. The key stationary species (B, C, D) related to diastereoselective cyclization in Scheme 1 of these five systems were fully optimized in the gas phase. The transition states (TS) from C → D, which seems to be the most relevant step in determining the diastereoselectivity of the products, were especially emphasized, and the transition states from B → C are outside the scope of this work and thus were excluded. The influence of the solvent environment on the reaction was evaluated by using the polarizable continuum models (PCM) method20 with the gas-phase optimized geometries. The molecular cavity was treated using the united atom Hartree–Fock (UAHF) parameterization. Since N-methylpyrrolidone (ε = 32.2) and 2-methyl-2-butanol (ε = 5.78) are not available in Gaussian 09, we chose ethanol as the solvent (ε = 24.55). Frequency analysis was carried out to verify the optimized geometry as minima or transition state, and to obtain free energy of each species. The intrinsic reaction coordinate (IRC)21 calculation was applied to judge whether the transition state connects the reactants and the products.In all systems, the symbols used to represent the related species are: hydrazine (1), the iminium ion (B), the enamine-type intermediate (C) and the charged pyrazoline product (D). For 2f, 2p, 5a, and 5ab, C1/C2 represents the enamine-type intermediates with trans/cis conformation for the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C double bond; D1/D2 represent the syn/anti charged pyrazoline intermediates. For 2a, B1/B2 is the iminium ion with trans/cis conformation for the CN+ double bond; D1/D2 are the anti/syn charged pyrazoline intermediates. Given the conformational combinations of the CN+ and CC double bonds, four enamine-type intermediates are considered for this system, which are referred to as C1 (trans, cis), C2 (cis, trans), C3 (trans, trans) and C4 (cis, cis), respectively. The transition state of a disrotatory/conrotatory reaction is specified by “a”/“b”, for example, TS2a/TS2b is the transition state of the disrotatory/conrotatory reaction of C2. The proposed mechanism, optimized geometries, some free energy profiles, and original data are shown in ESI.†
C double bond; D1/D2 represent the syn/anti charged pyrazoline intermediates. For 2a, B1/B2 is the iminium ion with trans/cis conformation for the CN+ double bond; D1/D2 are the anti/syn charged pyrazoline intermediates. Given the conformational combinations of the CN+ and CC double bonds, four enamine-type intermediates are considered for this system, which are referred to as C1 (trans, cis), C2 (cis, trans), C3 (trans, trans) and C4 (cis, cis), respectively. The transition state of a disrotatory/conrotatory reaction is specified by “a”/“b”, for example, TS2a/TS2b is the transition state of the disrotatory/conrotatory reaction of C2. The proposed mechanism, optimized geometries, some free energy profiles, and original data are shown in ESI.†
Computational results for the formation of 5a
Based on the complexity of these systems, the computational results of 5a is first represented here. The charge of the carbon atom in the CN+ bond is −0.769e in C1, −0.609e in TS1a, and −0.466e in TS1b, respectively. The charge of the terminal carbon atom in the CC bond is 0.916e in C1, 0.679e in TS1a, and 0.729e in TS1b, respectively. As shown in Fig. S1,† the dihedral angles D(CN+–N–C) and D(N+–N–CC) of the pyrazoline ring in TS1a are 22.1° and −19.6°, respectively. The CN+ and CC bonds form a pseudo-plane with the dihedral angle D(CN+⋯CC) to be 3.1°, and the neutral nitrogen atom is a little out of that plane. For TS1b, the values of D(CN+–N–C), D(N+–N–CC) and D(CN+⋯CC) are 35.0°, 7.5°, and 40.2°, respectively, implying that the pyrazoline ring in TS1b is twisted and it looks like a helical folding conformation. Clearly, compared with the CN+–N–CC in TS1b, the CN+–N–CC in TS1a is closer to a coplanar conformation. The distance R(CN+⋯CC) between the two terminal carbon atoms of the pyrazoline ring is 2.25 Å in TS1a and 2.15 Å in TS1b. These show that the C → D reaction is a 5-center/6-electron cyclization constituted by CN+–NH–CC, although this system does not completely satisfy the Hückel rule (not typically coplanar).
In the free energy profiles (Fig. 2), C is less stable than its tautomer B by 10.9 kcal mol−1. The reaction is endothermic by 1.9 kcal mol−1 for C → D1 process, and exothermic by 4.3 kcal mol−1 for C → D2 process. Even though the conformation of the cyclohexane is chair in D1 and twist boat in D2, D2 (anti) shows more stability than D1 (syn). The reason may be that the two bulky substituents are on the same side of the pyrazoline ring in D1, while the two substituents are on the different side of the pyrazoline ring in D2, causing the steric repulsion being stronger in D1 than in D2. However, the active free energy generating D1 is 1.6 kcal mol−1 lower than that generating D2, indicating that C prefers to undergo disrotatory process (TS1a) to form D1, rather than conrotatory process (TS1b) to afford D2. This may be because the CN+–N–CC (5-center/6-electron) in TS1a is more coplanar and more satisfies the Hückel rule compared with that in TS1b. These are in good agreement with the diastereoselectivity observed in the copper-based C–H functionalization reactions (syn-D1 is major product).
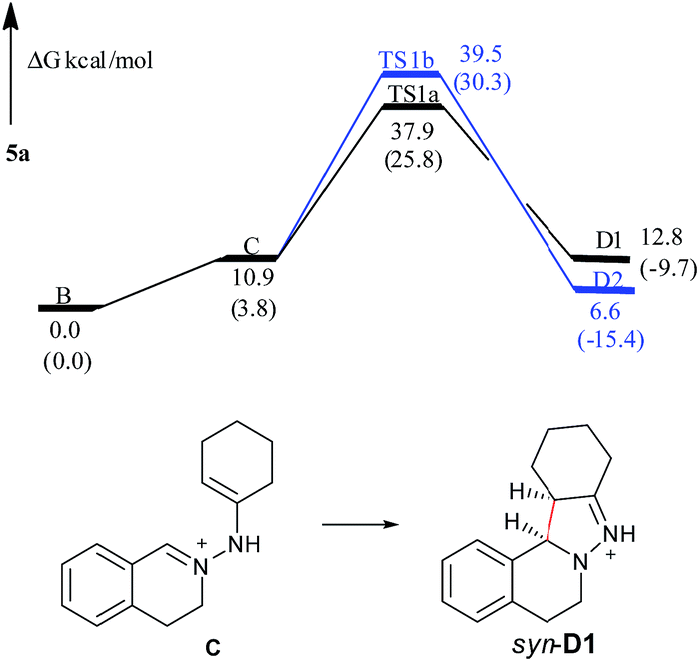 | ||
| Fig. 2 Free energy profiles of the reaction generating 5a in the gas phase. Free energies in the solution phase are given in parentheses. | ||
To better understand this point, HOMOs of TS1a and TS1b were compared. As shown in Fig. 3, the under lobe of the orbital of the C atom in the CN+ bond overlaps with the under lobe of the orbital of the terminal C atom in the CC bond, and CN+–NH constitutes a delocalized system in TS1a. In TS1b, the under lobe of the orbital of the C atom in the CN+ bond overlaps with the upper lobe of the orbital of the terminal C atom in the CC bond. However, the delocalized orbitals of the N–NH bond do not overlap with the C atom in the CN+ bond, generating an orbital nodal surface between the C atom and the N atoms. These demonstrate that TS1b is less stable and more difficult to cross than TS1a, supporting that the intermediate C tends to undergo a disrotatory process to form D1, rather than a conrotatory process to afford D2.
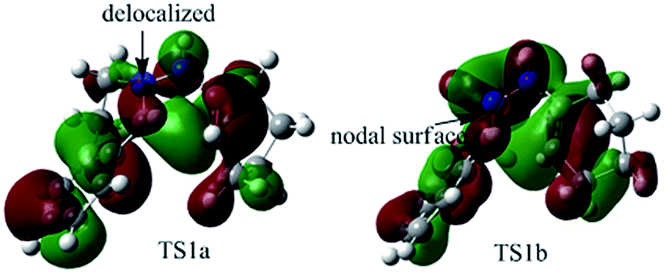 | ||
| Fig. 3 HOMOs of TS1a and TS1b generating 5a. | ||
The presence of a solvent can obviously improve the reaction process (the C → D1/D2 processes are exothermic and the active free energy barriers are easier to overcome, shown in parentheses in Fig. 2), but the overall diastereoselective tendency is the same as that captured from gas-phase calculations. Such a huge improvement of the reaction is mainly attributed to the solvation of ions.
Computational results for the formation of 2a
The charge of the carbon atom in the CN+ bond is −0.464e in C1, −0.321e in C2, −0.313e in C3, −0.579e in C4, −1.407e in TS1a, −0.388e in TS1b, −0.983e in TS2a and −0.656e in TS2b, respectively. The charge of the terminal carbon atom in the CC bond is separately 0.139, 0.337, 0.641, −0.077, 0.131, 0.297, 0.042 and −0.161e for the corresponding species. Seen from Fig. S2,† the dihedral angles D(CN+–N–C) and D(N+–N–CC) of the pyrazoline ring in TS1a are 30.0° and −33.8°, respectively. The CN+ and CC bonds form a pseudo-plane with the dihedral angle D(CN+⋯CC) of −2.4°, and the neutral nitrogen atom is a little out of that plane. The corresponding values are separately −44.1°, 4.3° and −37.7° in TS1b, also suggesting that the pyrazoline ring in TS1b is a pseudo-plane. However, this pyrazoline ring in TS1b is slightly apart from the coplanar conformation compared with that in TS1a. In TS2a, the values of D(CN+–N–C), D(N+–N–CC) and D(CN+⋯CC) are separately 37.6°, 23.3° and 56.5°, implying that the pyrazoline ring is twisted and it looks like a helical folding conformation. The corresponding values are separately −29.2, −18.9° and −43.7° in TS2b, showing that the pyrazoline ring in TS2b is twisted with the opposite direction, but the twist with the opposite direction would increase the steric hindrance between the methyl group and the benzyl group. The distance R(CN+⋯CC) between the two terminal carbon atoms of the pyrazoline ring is 2.36 Å in TS1a, 2.32 Å in TS1b, 2.34 Å in TS2a, and 2.34 Å in TS2b. These also demonstrate that the CN+–NH–CC constitutes a 5-center/6-electron system, but not a typical Hückel system (not completely coplanar).
As shown in Fig. 4, C1 and C2 are more stable than C3 and C4, supporting that C1 and C2 are major intermediate compared with C3 and C4. The anti product D1 is more stable than the syn product D2 in the gas phase, which is also attributed to the steric repulsion between the two bulky substituents on the same side of the pyrazoline ring in D2 compared with D1. TS1a is −3.3 kcal mol−1 relative to TS1b, and TS2a is −3.1 kcal mol−1 relative to TS2b, showing that C tends to undergo a disrotatory (TS1a/TS2a) process to give the product, rather than a conrotatory process (TS1b/TS2b) to afford the product. The reason may be that the CN+–N–CC in TS1a is closer to the Hückel system compared with that in TS1b (5-center/6-electron and coplanar conformation), and the steric hindrance between the methyl group and the benzyl group in TS2a is smaller than that in TS2b. Compared with C3 and C4, C1 and C2 overcome less of a free energy barrier, determining the diastereoselectivity of this reaction. Similar to 5a, the presence of a solvent can obviously improve the reaction process (the C → D1/D2 processes are exothermic and the active energy barriers become easier to overcome), but the overall diastereoselective trend is the same as that captured from gas-phase calculations. The main reason for such a huge improvement of the reaction is also the solvation of ions.
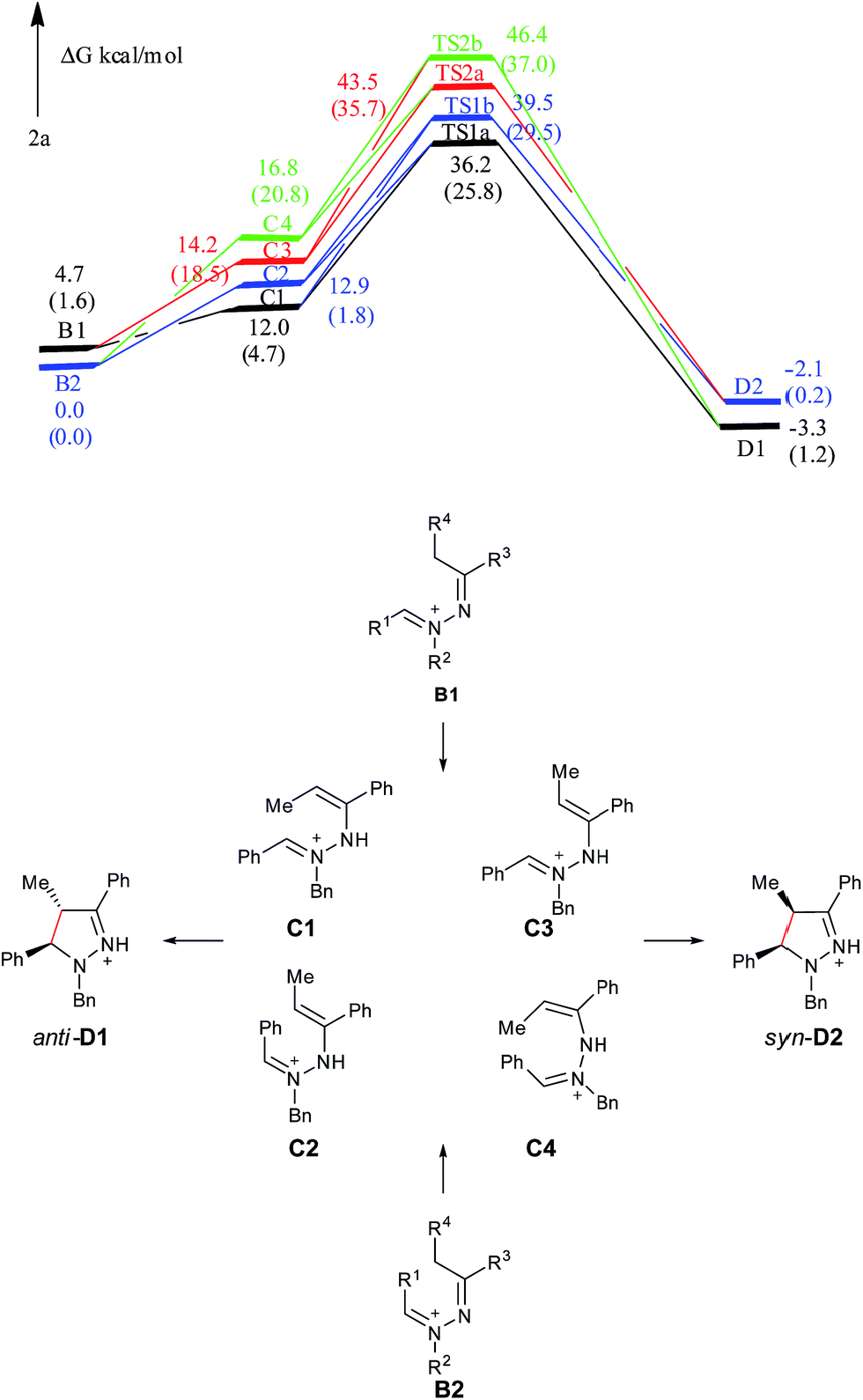 | ||
| Fig. 4 Free energy profiles of the reaction generating 2a in the gas phase. Free energies in the solution phase are given in parentheses. | ||
To better understand this point, HOMOs of TS1a, TS1b, TS2a and TS2b were compared. Seen from Fig. 5, the under lobe of the orbital of the C atom in the CN+ bond overlaps with the under lobe of the orbital of the terminal C atom in the CC bond, and CN+–NH–CC and phenyl group constitute a delocalized system in TS1a. In TS1b the under lobe of orbital of C atom in CN+ bond overlaps with the under lobe of the orbital of the terminal C atom in the CC bond, but the delocalized system only consists of CN+–NH and phenyl groups. The delocalized system in TS1b is smaller than TS1a, suggesting that TS1b is less stable and more difficult to overcome than TS1a. Compared with the orbital in TS2a, the CN+–NH–CC in TS2b does not constitute a complete delocalized orbital, and two orbital nodal surfaces are found between the C and N+–NH, and N+–NH and CC bonds, also suggesting that TS2b is less stable and more difficult to cross than TS2a. Therefore, C1 (C2) goes through a disrotatory process to form D1 (D2), instead of a conrotatory process to afford D2 (D1).
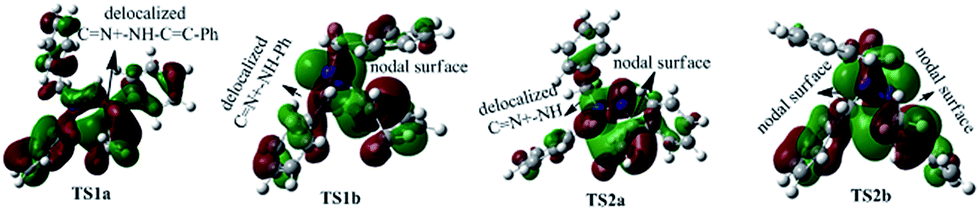 | ||
| Fig. 5 HOMOs of TS1a, TS1b, TS2a and TS2b generating 2a. | ||
Furthermore, computational studies of three more representative substrates were also carried out to account for the diastereoselective outcome (Scheme 2). For 2f, two enamine-type iminium ion intermediates are formed in the cyclization process, resulting in two different diastereoisomers. Based on the calculation (see ESI†), the (E)-isomer is more stable, resulting in syn-2f as the major product via a 5-center/6-electron disrotatory cyclization process. In the case of 2p, the intermediate with the methyl and phenyl groups on the opposite side is the major isomer, and thus the anti-isomer becomes the major product. For 5ab, the intermediate with two alkyl groups on the same side becomes the major isomers, and thus the syn-5ab is the major product. Compared with 5ab, the diastereoselectivity in 5ac is lower because the ratio of the major isomer is decreased due to the stronger steric effect by the isopropyl group. The combined results from experimental and computational studies suggest that the diastereoselectivity of products is possibly determined by the relative stability of the enamine-type iminium ion intermediates via a 5-center/6-electron disrotatory cyclization process.
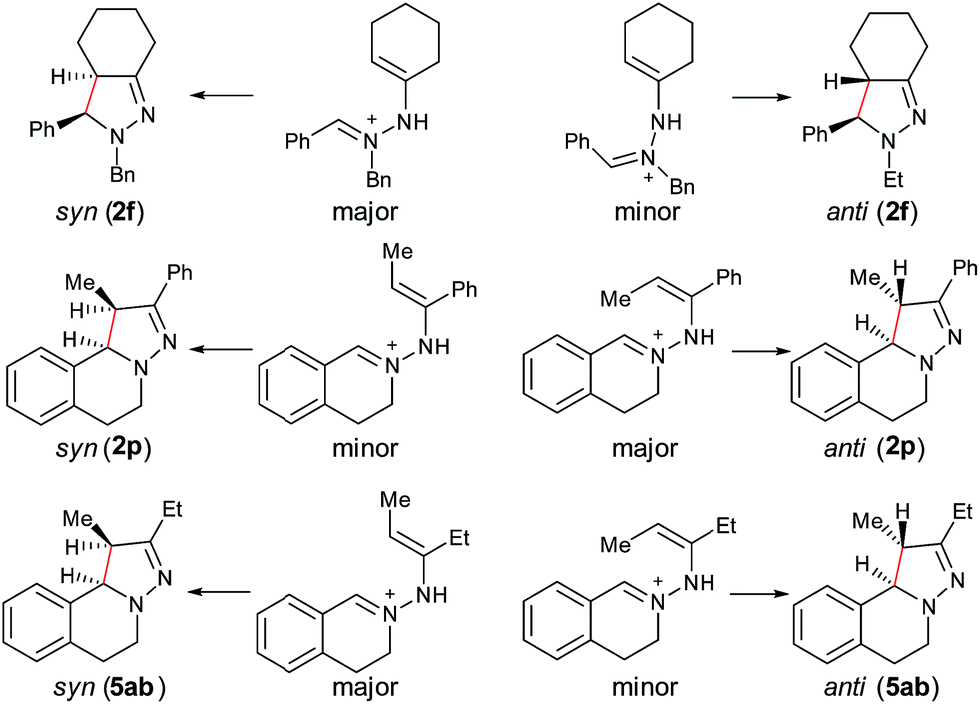 | ||
| Scheme 2 Interpretation of stereoselectivity of 2f, 2p, and 5ab. | ||
Conclusions
In summary, a highly diastereoselective aerobic intramolecular dehydrogenative cyclization of N,N-disubstituted hydrazones with good functional group tolerance was developed using a copper-catalyzed double sp3 C–H bond functionalization process. The observed diastereoselectivity was rationalized by a 5-center/6-electron disrotatory cyclization mechanism, which was supported by computational studies. As the first example of the copper-catalyzed aerobic diastereoselective intramolecular cyclization of amines via an iminium ion intermediate, this transformation provides a straightforward, environmentally friendly and atom efficient access to pyrazolines with up to five fused-ring systems. The enantioselective version of this transformation is currently under study in our laboratory.Abbreviations
| CCR2 | CC chemokine receptor 2 |
| CCL2 | CC chemokine ligand 2 |
| CCR5 | CC chemokine receptor 5 |
| TLC | Thin layer chromatography |
Acknowledgements
The authors gratefully acknowledge Indiana University-Purdue University Indianapolis and NSF CHE-1350541 for financial support. We also would like to thank Dr Yan Zhou and Professor Jingzhi Pu for helpful discussions of this manuscript. Additionally, Jianyi Wang would like to thank the National Natural Science Foundation of China (No. 21262004) for financial support.Notes and references
- E. J. Corey and X. M. Cheng, The Logic of Chemical Synthesis, John Wiley & Sons, New York, USA, 1989 Search PubMed.
- Metal Catalyzed Cross-Coupling Reactions, ed. A. de Meijere and F. Diederich, Wiley-VCH, Weinheim, Germany, 2nd edn, 2004 Search PubMed.
- For selected recent reviews, see: (a) S. Cacchi and G. Fabrizi, Chem. Rev., 2005, 105, 2873 CrossRef CAS PubMed; (b) K. Godula and D. Sames, Science, 2006, 312, 67 CrossRef CAS PubMed; (c) L.-X. Yin and J. Liebscher, Chem. Rev., 2007, 107, 133 CrossRef CAS PubMed; (d) D. Alberico, M. E. Scott and M. Lautens, Chem. Rev., 2007, 107, 174 CrossRef CAS PubMed; (e) E. M. Beccalli, G. Broggini, M. Martinelli and S. Sottocornola, Chem. Rev., 2007, 107, 5318 CrossRef CAS PubMed; (f) M. Catellani, E. Motti and N. Della Cá, Acc. Chem. Res., 2008, 41, 1512 CrossRef CAS PubMed; (g) G. C. Fu, Acc. Chem. Res., 2008, 41, 1555 CrossRef CAS PubMed; (h) X. Chen, K. M. Engle, D.-H. Wang and J.-Q. Yu, Angew. Chem., Int. Ed., 2009, 48, 5094 CrossRef CAS PubMed; (i) T. W. Lyons and M. S. Sanford, Chem. Rev., 2010, 110, 1147 CrossRef CAS PubMed; (j) S. de Ornellas, T. E. Storr, T. J. Williams, C. G. Baumann and I. J. S. Fairlamb, Curr. Org. Synth., 2011, 8, 79 CrossRef CAS; (k) C.-L. Sun, B.-J. Li and Z.-J. Shi, Chem. Rev., 2011, 111, 1293 CrossRef CAS PubMed; (l) L. McMurray, F. O'Hara and M. J. Gaunt, Chem. Soc. Rev., 2011, 40, 1885 RSC; (m) T. C. Boorman and I. Larrosa, Chem. Soc. Rev., 2011, 40, 1910 RSC; (n) T. Newhouse and P. S. Baran, Angew. Chem., Int. Ed., 2011, 50, 3362 CrossRef CAS PubMed; (o) J. Wencel-Delord, T. Droge, F. Liu and F. Glorius, Chem. Soc. Rev., 2011, 40, 4740 RSC; (p) S. H. Cho, J. Y. Kim, J. Kwak and S. Chang, Chem. Soc. Rev., 2011, 40, 5068 RSC.
- For selected recent reviews, see: (a) C.-J. Li and Z. Li, Pure Appl. Chem., 2006, 78, 935 CAS; (b) C.-J. Li, Acc. Chem. Res., 2009, 42, 335 CrossRef CAS PubMed; (c) W.-J. Yoo and C.-J. Li, Top. Curr. Chem., 2010, 292, 281 CrossRef; (d) C. S. Yeung and V. M. Dong, Chem. Rev., 2011, 111, 1215 CrossRef CAS PubMed; (e) A. E. Wendlandt, A. M. Suess and S. S. Stahl, Angew. Chem., Int. Ed., 2011, 50, 11062 CrossRef CAS PubMed; (f) C. Liu, H. Zhang, W. Shi and A. Lei, Chem. Rev., 2011, 111, 1780 CrossRef CAS PubMed; (g) Z.-Z. Shi, C. Zhang, C. Tang and N. Jiao, Chem. Soc. Rev., 2012, 41, 3381 RSC; (h) C. Zhang, C. Tang and N. Jiao, Chem. Soc. Rev., 2012, 41, 3464 RSC; (i) S. A. Girard, T. Knauber and C.-J. Li, Angew. Chem., Int. Ed., 2014, 53, 74 CrossRef CAS PubMed.
- C. Glaser, Chem. Ber., 1869, 2, 422 CrossRef PubMed.
- P. Siemsen, R. C. Livingston and F. Diederich, Angew. Chem., Int. Ed., 2000, 39, 2632 CrossRef CAS.
- (a) E. Boess, D. Sureshkumar, A. Sud, C. Wirtz, C. Farès and M. Klussmann, J. Am. Chem. Soc., 2011, 133, 8106 CrossRef CAS PubMed; (b) E. Boess, C. Schmitz and M. Klussmann, J. Am. Chem. Soc., 2012, 134, 5317 CrossRef CAS PubMed.
- (a) L. Zhao and C.-J. Li, Angew. Chem., Int. Ed., 2008, 47, 7075 CrossRef CAS PubMed; (b) L. Zhao, O. Baslé and C.-J. Li, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 4106 CrossRef CAS PubMed; (c) J. Xie and Z.-Z. Huang, Angew. Chem., Int. Ed., 2010, 49, 10181 CrossRef CAS PubMed; (d) G. Zhang, Y. Zhang and R. Wang, Angew. Chem., Int. Ed., 2011, 50, 10429 CrossRef CAS PubMed; (e) J.-C. Wu, R.-J. Song, Z.-Q. Wang, X.-C. Huang, Y.-X. Xie and J.-H. Li, Angew. Chem., Int. Ed., 2012, 51, 3453 CrossRef CAS PubMed.
- R. Grigg, F. Heaney, J. Idle and A. Somasunderam, Tetrahedron Lett., 1990, 31, 2767 CrossRef CAS.
- G. Zhang, J. Miao, Y. Zhao and H. Ge, Angew. Chem., Int. Ed., 2012, 51, 8318 CrossRef CAS PubMed.
- G. Zhang, Y. Zhao and H. Ge, Angew. Chem., Int. Ed., 2013, 52, 2559 CrossRef CAS PubMed.
- For selected recent reviews, see: (a) S. Kumar, S. Bawa, S. Drabu, R. Kumar and H. Gupta, Recent Pat. Anti-Infect. Drug Discovery, 2009, 4, 154 CrossRef CAS; (b) M. A. Rahman and A. A. Siddiqui, Int. J. Pharm. Sci. Drug Res., 2010, 2, 165 CAS; (c) X.-H. Liu, B.-F. Ruan, J. Li, F.-H. Chen, B.-A. Song, H.-L. Zhu, P. S. Bhadury and J. Zhao, Mini-Rev. Med. Chem., 2011, 11, 771 CrossRef CAS.
- (a) L. C. Behr, R. Fuso and C. H. Jarboe, Pyrazoles, Pyrazolines, Pyrazolidines, Indazoles, and Condensed Rings, The Chemistry of Heterocyclic Compounds 22, Interscience Publishers, New York, USA, 1967 Search PubMed; (b) S. U. Tekale, V. B. Jadhav, R. L. Magar, C. S. Patil, R. D. Ingle, S. R. Bembalka and Y. B. Vibhute, in Bioactive Heterocycles: Synthesis and Biological Evaluation, ed. K. L. Ameta, R. P. Pawar and A. J. Domb, Nova Science Publishers, Inc., New York, 2013, p. 259 Search PubMed.
- For selected recent reviews, see: (a) J. G. Schantl, in Science of Synthesis, ed. P. A. Georg, Thieme Verlag, Stuttgart, New York, 2004; vol. 27, p. 731 Search PubMed; (b) J. Svete, ARKIVOC, 2006, 35 CAS; (c) J. G. Schantl, in Advances in Heterocyclic Chemistry, Elsevier Inc., San Diego, 2010, vol. 99, p. 185 Search PubMed; (d) H. Suga and K. Itoh, Methods and Applications of Cycloaddition Reactions in Organic Syntheses, John Wiley & Sons, New York, 2014, p. 175 Search PubMed.
- B. F. Powell, C. G. Overberger and J.-P. Anselme, J. Heterocycl. Chem., 1983, 20, 121 CrossRef CAS PubMed.
- R. Hoffman and R. A. Oloson, J. Am. Chem. Soc., 1966, 88, 943 CrossRef.
- S. Muller and B. List, Angew. Chem., Int. Ed., 2009, 48, 9975 CrossRef PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, T. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Revision C. 01, Gaussian, Inc., Wallingford, CT, 2010 Search PubMed.
- (a) A. D. Becke, Phys. Rev. A, 1988, 38, 3098 CrossRef CAS; (b) C. Lee, W. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785 CrossRef CAS.
- S. Miertuš, E. Scrocco and J. Tomasi, Chem. Phys., 1981, 55, 117 CrossRef.
- K. Fukui, Acc. Chem. Res., 1981, 14, 363 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details including characterization data, copies of 1H, 13C NMR and NOESY spectra. See DOI: 10.1039/c5sc01736j |
| This journal is © The Royal Society of Chemistry 2015 |