
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Coordination diversity in hydrogen-bonded homoleptic fluoride–alcohol complexes modulates reactivity†
Keary M.
Engle
,
Lukas
Pfeifer
,
George W.
Pidgeon
,
Guy T.
Giuffredi
,
Amber L.
Thompson
,
Robert S.
Paton
,
John M.
Brown
and
Véronique
Gouverneur
*
Chemistry Research Laboratory, Department of Chemistry, Oxford University, OX1 3TA, UK. E-mail: veronique.gouverneur@chem.ox.ac.uk; Fax: +44 (0)1865 285002
First published on 22nd June 2015
Abstract
The nucleophilic reactivity of fluoride ion is altered in the presence of hydrogen-bond donors, including alcohols. Relatively little is known about the coordination involved; to rectify this, the X-ray structures of fourteen novel fluoride–alcohol complexes with tetrabutylammonium as the counterion have been determined. The coordination number varies from two to four depending on the steric bulk of the alcohol and is closely linked to trends in reactivity. This diversity in coordination stoichiometry is unprecedented but significant, as it implies differences in the ability of the fluoride–alcohol complexes to dissociate in solution with release of a more active and/or selective fluoride source.
Introduction
Atom-economical fluorination processes are highly sought-after, especially those using readily available starting materials and inexpensive fluoride sources. The renewed interest in “F−” chemistry has also been driven by the global growth of the radiopharmaceutical industry and the increasing demand for 18F-fluoride based radiochemistry for applications in positron emission tomography.1 The fluoride salt fluorite (also called fluorspar) is an important industrial chemical for the production of hydrogen fluoride, a precursor of many fluorine-containing fine pharmaceuticals.2 In research laboratories, inexpensive anionic fluoride sources are increasingly used as an alternative to F+ reagents for transition metal-catalyzed reactions leading to C(sp2)–F and C(sp3)–F bond construction.3 Examples of metal-free catalytic nucleophilic fluorinations with fluoride are rare. A remarkable exception is the native fluorinase enzyme, with its ability to produce 5′-fluoro-5′-deoxyadenosine from fluoride in aqueous medium (Fig. 1(I)).4 This unique enzyme increases fluoride nucleophilicity within the active site through desolvation for substitution at the preactivated C-center of S-adenosylmethionine (SAM). Single-crystal X-ray diffraction studies of substrate- and product-bound structures revealed that fluoride forms two hydrogen bonds to Ser-158 when it binds in the active site.5 Subsequent substrate (SAM) binding encourages fluoride ion dehydration, thereby facilitating nucleophilic fluorination. An additional hydrogen-bonding interaction of fluoride with Thr-80 likely stabilizes the transition state of the SN2 fluorination process. This enzymatic fluorination reaction is highly significant because the use of fluoride ion for C–F bond formation is not trivial and is often met with complications. One challenge is the poor solubility of common fluoride salts in organic solvent. Moreover, fluoride is strongly basic in its unsolvated form, and solvation through hydrogen bonding typically lowers nucleophilicity. Numerous strategies have been considered to augment the scope of fluoride-based chemistry, either by diversifying the range of fluoride ion sources or by achieving controlled fluoride release in solution from neutral reagents. Our own contribution to catalytic nucleophilic fluorination processes established that the use of TBAF(t-BuOH)4 is critically important in Pd- and Ir-catalyzed fluorination of allylic p-nitrobenzoates and carbonates.6,7 This reagent is by far the most suitable fluoride source for these reactions; neither ammonium fluoride nor a range of inorganic alkali fluorides led to effective product formation (Fig. 1(IIa) and 1(IIb)). | ||
| Fig. 1 Hydrogen bonding and fluoride reactivity. | ||
These findings raise the question of how hydrogen bonding to fluoride influences reactivity. Such a study may facilitate the development of superior F− reagents by design, and inform the development of a biomimetic fluorinase catalyst capable of broad substrate tolerance, with no compromise on efficacy. The ability of fluoride to engage in hydrogen bonding has been previously evoked as a parameter that influences fluoride reactivity,8 but no detailed analysis is available on how the coordination sphere of hydrogen-bonded fluoride complexes correlates with reactivity and product distribution (SN2/E2 selectivity). In 1994, the first study examining the effect of hydrogen bonding on fluoride reactivity was disclosed by Yonezawa and co-workers, who prepared a series of hydrogen-bonded TBAF complexes from TBAF(H2O)3 using alcohol solvents as hydrogen bond donors.9 A study of their reactivity in a model SN2 reaction with benzyl bromide revealed that the reaction rate was positively correlated with the steric bulk of the alcohol (t-BuOH ≫ i-PrOH > n-BuOH ∼ n-PrOH > H2O). Kim and co-workers subsequently reported that kinetic reactivity and SN2 versus E2 selectivity were enhanced when CsF was used in the presence of bulky tertiary alcohols (e.g., t-BuOH, t-AmylOH and 3-methyl-3-pentanol) (Fig. 1(IIc)).10 In 2008, the same group published the isolation, characterization and X-ray structure of TBAF(t-BuOH)4, confirming its solid-state coordination stoichiometry and tetrahedral geometry.11 These preliminary data encouraged us to further study hydrogen-bonding interactions with fluoride as tool to rationally tune reactivity. This problem is of fundamental interest, particularly given that Nature has evolved a fluorinase enzyme capable of partially desolvating fluoride through hydrogen bonding to key residues at the active site to improve nucleophilicity, as discussed earlier. The importance of hydrogen bonding to fluoride extends to transformations other than C–F bond formation, including catalytic, stereoselective desilylation. Selected examples include the catalytic kinetic resolution of silyl-protected secondary benzylic alcohols using chiral hydroxyl-terminated polyether catalysts with potassium fluoride12 and the asymmetric acylation of silyl ketene acetals performed in the presence of a dual-function chiral thiourea organocatalyst.13 These processes are proposed to involve complexes in which fluoride is hydrogen-bonded to the polyether or thiourea catalyst.
The paucity of structural data on fluoride–alcohol complexes prompted us to examine in detail the coordination chemistry of fluoride–alcohol complexes with the aim of determining how structure correlates with reactivity. Complex formation between halide anions and alcohols has been investigated by gas-phase experimental methods14 and computational techniques.15 Compared to other halides, the fluoride ion stands out with the largest bonding enthalpies and the shortest X⋯H hydrogen-bond lengths with various hydrogen-bond donors. Detailed information on the structural properties in the solid state for complexes of fluoride with alcohols other than phenol derivatives and t-BuOH is surprisingly lacking.16 Herein, we disclose the synthesis of fourteen new fluoride–alcohol complexes and their full characterization in the solid state using single-crystal X-ray diffraction. We also present data on the relative reactivity of these fluoride ion complexes towards a model reactant, demonstrating their potential as useful fluoride reagents in organic synthesis. Many of these new complexes are easy to handle solids that are less hygroscopic than TBAF(H2O)3 and TBAF(t-BuOH)4.
Results and discussion
Overview
The alcohols in this study were chosen on the basis of varying steric bulk, in order to elicit a range of coordination geometries in the solid state. The complexes were prepared in good yields by adapting an established synthetic protocol; TBAF(H2O)3 was combined with the alcohol (1–4 eq.) in vigorously refluxing hexane for 2 h. The ensuing crude solid materials were characterized by 1H and 13C NMR, and recrystallized as appropriate to obtain single crystals suitable for X-ray diffraction studies (vide infra). Tetra-alkylammonium fluoride precursors other than TBAF(H2O)3 (e.g., TMAF and TEAF) were not studied in detail because the resulting alcohol complexes were found to be more difficult to handle and crystallize. All alcohols examined gave either tetra-, tri-, or dicoordinate fluoride–alcohol complexes, with the coordination number decreasing as the degree of branching and steric bulk of the alcohol increased. This variability in coordination stoichiometry had not been observed previously (Table 1).|
|
||||
|---|---|---|---|---|
| Entry | Alcohol | Yield | Complex | C.N.a |
| a Coordination number in (ROH)nF−. b See formulae block. c 3ROH, 1H2O. d 2(ROH)2, 1H2O. | ||||
| 1 | 1-Adamantanol 1a | 84% | 2a | 4 |
| 2 | Pentaerythritol 1b | 77% | 2b | 4 |
| 3 | Tris(hydroxymethyl)ethane 1cb | 95% | 2c | 4 |
| 4 | Neopentylglycol 1d | 89% | 2d | 4 |
| 5 | (R,R)-di-(i-Pr)-tartrate 1e | 61% | 2e | 4 |
| 6 | Mannitol derivative 1fb | 95% | 2f | 4 |
| 7 | Pinacol 1g | 93% | 2g | 4 |
| 8 | (R)-BINOL 1h | 88% | 2h | 3 |
| 9 | Cyclic hemiacetal 1ib | 89% | 2i | 3c |
| 10 | 9-Phenylfluoren-9-ol 1j | 91% | 2j | 2, 3 |
| 11 | Diphenylmethanol 1k | 91% | 2k | 3 |
| 12 | Triphenylmethanol 1l | 61% | 2l | 2 |
| 13 | Tri-(p-tolyl)-methanol 1m | 76% | 2m | 2 |
| 14 | Pyrrolidine 1nb | 77% | 2n | 2d |

|
||||
Fluoride complexes with four ROH ligands
The only closely relevant structure preceding this work is that of the tetra-alcohol complex TBAF(t-BuOH)4.10 Accordingly, the tertiary alcohol 1-adamantanol 1a, which is nearly isosteric with t-BuOH around the hydroxyl group but has distinct packing requirements, was examined. Crystallisation of the fluoride complex 2a and X-ray diffraction (see Experimental section) gave the anion structure shown in Fig. 2. Key geometric parameters describing the environment of one of the two closely similar but crystallographically inequivalent fluorides in the unit cell are given. The other symmetrically equivalent fluoride possesses F⋯O distances of 2.680(3) Å and O⋯F⋯O angles of 95.17(8)° and 117.06(5)°. These structures are in close accord with the single known homoleptic alcohol complex TBAF(t-BuOH)4 (O⋯F distance: 2.643(7) Å, O⋯F⋯O angles: 97.98(18)° and 115.50(18)°) reported by Kim and co-workers,9,10 despite the difference in steric bulk and hydrophobicity of the alcohol hydrogen-bond donors in the two cases. Structures in this class may be analysed for deviations from a formal tetrahedral structure and Td symmetry.17 Within the coordination sphere, four F⋯O distances and six O⋯F⋯O angles can be measured, as in Fig. 2. For an individual complex, the O⋯F distances generally do not vary widely and pairs of chelating diols in a 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 complex exhibit similar bite angles that constrain two of the six angle parameters.
:1 complex exhibit similar bite angles that constrain two of the six angle parameters.
 | ||
| Fig. 2 Anion formula and crystal structure together with key geometric parameters for complex 2a. | ||
The tetra-(1-adamantanol) complex 2a is the only tetracoordinate structure in this study that does not involve a chelating diol. Consider more generally the three possible coupled-pair distortions from a pure Td structure shown in Fig. 3, which represents a structure for which two angle parameters are constrained by bis-chelation and all distances from the central atom are equal. Any possible geometry may be realised by a combination of three movements of one pair, keeping the second stationary. These three modes can be identified as twist A, roll B and glide C. Using this analysis, the two independent molecules in the unit cell of complex 2a may both be fully described by simply imposing a slight C2 distortion A on the Td model so that two of the O⋯F⋯O angles become smaller than the remaining four.18
 | ||
| Fig. 3 Distortions from T4 symmetry in a bis-chelated complex with comparable distances in ligation; AC2 twist, B Out-of-plane roll, C In-plane glide. | ||
For chelating diol-based structures, the results are more diverse (Fig. 4).
 | ||
| Fig. 4 Anion formulae and crystal structures, together with key geometric parameters for the O⋯F⋯O cores of 2b in (a), 2c in (b) and 2d in (c). | ||
The three 1,3-diol based structures that were obtained form a closely related set in molecular terms but exhibit distinct coordination modes. The most clear-cut case is 2b, derived from pentaerythritol 1b. This 2:1 fluoride complex crystallized as its DCM solvate, forming a one-dimensional linear hydrogen-bonded coordination polymeric structure (Fig. 4a) The individual F(diol)2 units of the 1D ribbons experience a simple C2 twist A from an ideal orthogonal geometry, resulting in an interplanar angle between the two O⋯F⋯O units of 50.54(12)°.
For the structure of complex 2c, derived from triol 1c (Fig. 4b), two hydrogen bonds to fluoride per 1c molecule were observed, with the third hydroxyl group forming an additional hydrogen bond that activates and shortens the neighbouring O–H⋯F bond. At 81.32(6)°, the plane between the pairs of O⋯F⋯O angles of the chelates is close to that of an undistorted tetrahedron. In effect however, through roll and glide motions B and C, one of the two donor –OH groups from one diol remains approximately in its tetrahedral position while the other diol has been rotated away, leading to an arrangement where one oxygen is in the O⋯F⋯O plane of the first diol ligand.
The third member of the series, 2d derived from neopentyl glycol 1d, is again distinct, possessing four different O⋯F⋯O distances, as shown in Fig. 4c. The two chelate units are close to coplanarity with an interplanar angle of 11.51(8)° but further modified by a significant contribution of roll distortion B.
Three ostensibly similar fluoride anion complexes with the same counterion thus show quite distinct geometrical parameters. The variation observed points to a structural model for which the overall lattice geometry is primarily determined by the TBAF cation and the alcohols, with fluoride ion demonstrating a capacity to fit within that structure. The O⋯F distances, however, vary only to a small extent in any given structure. Fig. 5 illustrates these tendencies for the three structures discussed above. Viewing the structures through an axis between the central C–C bonds emphasises the marked variation in fluoride ion location relative to its ligands.
 | ||
| Fig. 5 Structures (i) 2b, (ii) 2c, (iii) 2d: view along the axis linking quaternary carbons. | ||
Three analogous 1,2-diol complexes were prepared, and their crystal structures were analysed similarly. Complex 2e, derived from the enantiomerically pure hydrogen-bond donor (R,R)-di-isopropyl tartrate 1e, was crystallized as a hexane solvate. The anion in the ensuing 2:1 complex is C2-symmetric with the two O⋯F⋯O planes oriented at 60.05(7)° to one another. In this geometry the two central C–C bonds are very nearly coplanar, with the F atom close to equidistant from the carbon atoms of these bonds and just 0.1391(11) Å from their mean plane. The basic geometry of the complex is imposed by its overall C2 symmetry (Fig. 6a).
 | ||
| Fig. 6 Anion formulae and X-ray structures, together with key geometric parameters for the O⋯F⋯O cores of 2e, 2f and 2g. | ||
The related complex 2f, derived from 1,2,5,6-(R,S,S,R)-di-isopropylidene mannitol 1f, crystallized as an EtOAc solvate. This structure also possesses local C2 symmetry about the anion, and here the interplanar twist of the two O⋯F⋯O subunits is very similar to 2e at 61.03(5)°. The actual geometry, however, is quite distinct from 2e through substantial rolling distortion. When one subunit is aligned in plane, the oxygen atoms of the other subunit are respectively 0.7222(8) Å above and 1.7874(9) Å below that plane. The central C–C bonds of the two ligands are no longer co-planar (Fig. 6b).
More significant structural variation was observed in the complex 2g, derived from pinacol, where two of the four O⋯F distances are equal to one another and distinct from the remaining two. Here the twist angle between the two O⋯F⋯O subunit planes is 64.96(8)°, but all four OH ligands are now clearly confined to one coordination hemisphere (Fig. 6c). With respect to one subunit plane, the oxygen atoms of the second diol ligand are respectively 0.7417(13) Å and 2.3880(13) Å, both below that plane. If the subunit planes are created directly from the hydroxyl H⋯F⋯H positions, they still occupy a single hemisphere. In order to pursue this observation further, the corresponding tetraethylammonium and tetramethylammonium complexes were synthesized, but both resisted attempts to prepare X-ray diffraction quality crystals.
Fluoride complexes with three ROH ligands
For complexes where the alcohol is sufficiently bulky to permit just three or fewer O–H⋯F bonds to fluoride, different patterns emerge depending on the alcohol structure (Fig. 7).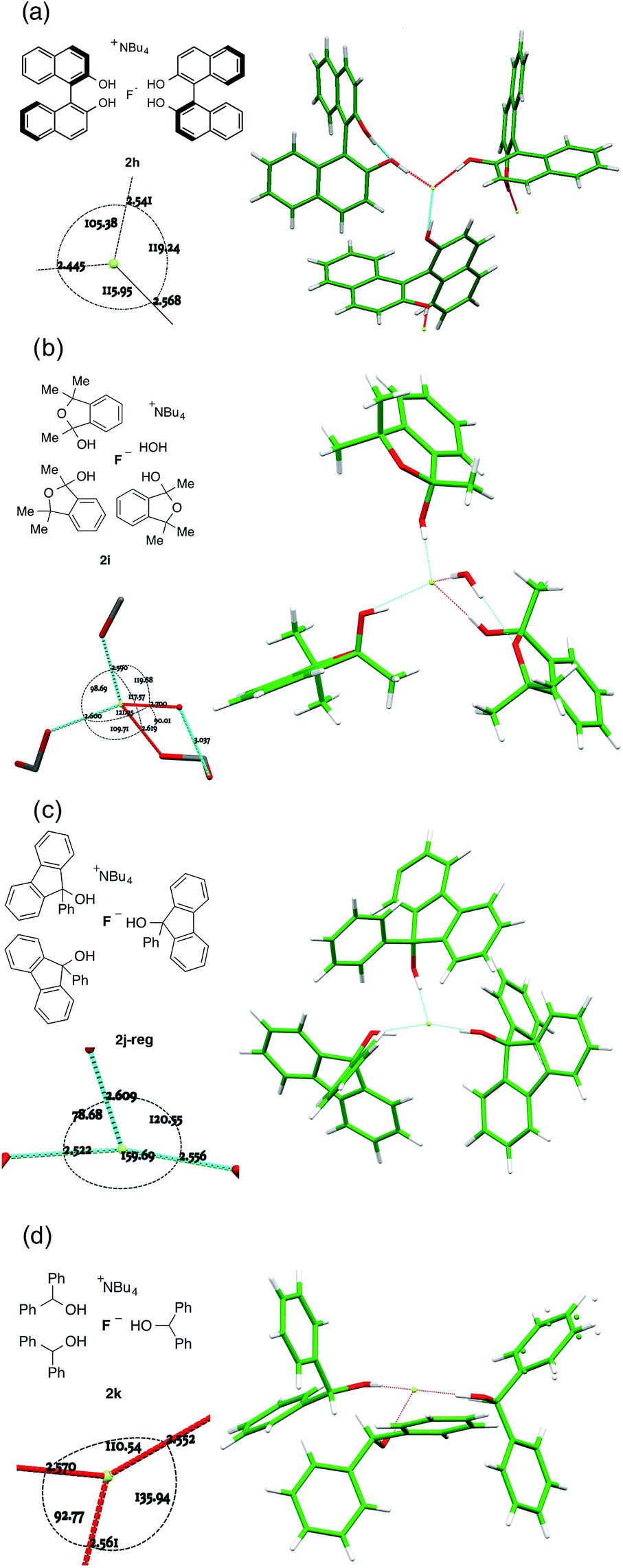 | ||
| Fig. 7 Anion formulae and X-ray structures, together with key geometric parameters for the O⋯F⋯O cores of 2h, 2i, 2j-reg and 2k. | ||
There is a tendency towards alternative bonding modes that permit higher coordination numbers.16a For the (R)-binol 2:1 complex 2h shown in Fig. 7a, three different molecules participate in bonding to a single fluoride ion as part of an extended network linked by interligand hydrogen bonding. This results in a flattened tetrahedral geometry for fluoride with an unoccupied site, where F− is 0.6501(6) Å distant from the plane described by the three alcohol oxygens. There is an ortho-aryl C–H close to the fourth apex with a C⋯F distance of 3.3398(12) Å, but it is not well directed for hydrogen bonding (C–H⋯F = 125.59(3)°).
Trimethylisobenzofuran-2-ol 1i forms complex 2i shown in Fig. 7b. The three-ligand motif is modified here by incorporation of a single ligating water molecule; the resulting structure is close to tetrahedral with all O⋯F⋯O angles between 90° and 120° and with fluoride ion 0.8942(8) Å out of the plane of the three oxygen atoms of the donor groups. The hemiacetal is chiral, although the complex crystallizes in an achiral space group. Thus, each individual anion has alternatively (R,R,S) or (S,S,R) configuration. There are two independent motifs in the crystal structure of the 9-phenylfluoren-9-ol 3:1 complex 2j, with respectively three and two donor alcohols per fluoride ion (Fig. 7c). The first 2j-reg is approximately T-shaped with O⋯F⋯O angles of 160.40(4)°, 120.00(4)° and 78.30(3)°, and the fluoride ion is just 0.1325(8) Å out of the plane of the three oxygen donor atoms; the second 2j-alt is discussed below. The 3:1 diphenylmethanol complex 2k falls into this group, with the three donor oxygens as part of a flattened tetrahedron with the fluoride 0.6483(11) Å out of plane (Fig. 7d). The remaining apex is occupied by an α-C–H bond from the TBA cation, with a C⋯F distance of 3.3331(18) Å, and a C–H⋯F angle of 161.92(4)°.
Fluoride–alcohol complexes with two ROH ligands
Dicoordinate complexes of fluoride ion are observed as the sole structural unit only in the bulky triarylmethanol complexes 2l and 2m, and as the alternative structural motif found in the unit cell of 2j (2j-alt). In the first of these (2l), the O⋯F⋯O angle is 102.94(4)°, augmented by donation from an α-C–H of the cation, for which the C⋯F distance is 3.181(12) Å; the C⋯F vector makes angles of 117.3(3)° and 133.8(3)° with the two coordinated O-atoms (Fig. 8a). There are further weak hydrogen bonds from two ortho-C–H atoms of proximal phenyl groups, where the corresponding C-atoms are 3.247(2) Å and 3.2945(18) Å distant from fluoride and the spatial orientation is favourable.19 These two phenyl rings are well ordered whilst the remaining four exhibit librational disorder. | ||
| Fig. 8 Anion formulae and X-ray structures, together with key geometric parameters for the O⋯F⋯O cores of 2l, 2m, 2j-alt. | ||
For 2m, there are two closely related alcohol-complexed anions in the crystal, and both show the same characteristic features as 2l, with an O⋯F⋯O angle of 85.21(3)° and a C⋯F distance of 3.0983(14) Å from one of the C–H groups α to nitrogen in the first crystallographically distinct equivalent cation (Fig. 8b). These parameters are respectively 87.63(3)° and 3.1207(14) Å in the otherwise similar second anion. This is the least coordinated example in the series and is also the most reactive nucleophile (vide infra).
The second structural motif (2j-alt) in the unit cell of crystalline 2j is dicoordinate, with the third molecule of the alcohol involved in hydrogen bonding to one of the donor ligands, but not to fluoride (Fig. 8c). A far wider O⋯F⋯O angle is observed, at 151.97(5)°.
One further dicoordinate alcohol fluoride complex was characterized and provides a distinct category. Unlike the diol complexes discussed above, diol 1n forms crystals of a monohydrated anion, with the water molecule acting as an H-bond acceptor to both hydroxyl groups of a second diol. The secondary amine is not engaged in hydrogen bonding. The geometry of this second diol is almost identical to the first, such that the assembly refines as a single unit with very similar locations for the water oxygen and the fluoride atoms; the two diol ligands are distinct only in the positioning of one phenyl group. Fig. 9 shows the fluoride anion location in this complex.
 | ||
| Fig. 9 Part of the crystal structure of the 2:1 complex anion formed from 1n and the environment of fluoride ion therein. | ||
In general, the coordination number of fluoride complexes is largely determined by the steric bulk of the ligand but was never less than 2 in the series covered in this paper. In accord with the characterized crystal structures of hydrated fluoride ion,20 an optimum coordination of 4 hydrogen-bonding ligands is observed here. Computational studies suggest that water association up to hexacoordination is feasible.21 In the two published examples where the hydrated fluoride ion is unconstrained by further complexation, the structure of the complexed anion lies between tetrahedral and square planar so that only the twist distortion A from the T4 structure is involved; the O⋯F⋯O interplanar angles in those structures are respectively 35° and 37°.
The larger ligands involved in the present study elicit a far wider structural range. Whilst O⋯F, and by implication H⋯F, distances are similar for a given structure, there is a trend towards significantly shorter values with lower coordination numbers, illustrated in Fig. 10.
 | ||
| Fig. 10 The relationship between O⋯F distance in hydrogen-bonding alcohols (all examples described here), and the ROH coordination at F−. | ||
With 1,2- and 1,3-diols, the interligand angles vary widely, indicating that the geometry of the coordination sphere is far more strongly influenced by packing forces than through any predisposition to an ideal tetrahedral geometry. With low coordination numbers, there is a tendency for fluoride in these complexes to form weak C–H⋯F bonds.22 This is unambiguous for coordination of one or more α-protons of TBAF in three cases; with 2l, 2m, and 2k, the C–H⋯F angle is 160–166° and the C–F distance is between 3.10 and 3.33 Å. Other interactions involving proximal aromatic C–H protons, seen in structures of low coordination number, will contribute to the overall stability of the complex. A striking example is provided by 2l, which requires the specific orientation of two phenyl rings for optimal C–H hydrogen bonding, where the other phenyl rings in the structure are disordered.
Relative nucleophilic reactivity of ROH fluoride complexes
In the original studies of TBAF(t-BuOH)4 as a fluorinating agent, Kim and co-workers examined displacement reactions of 3a and 3b.9,10 The bromide was less selective than the mesylate and gave mixtures of the alkene 5 and fluoride 4 in which the latter predominated. The conditions used in this prior work provided a basis for systematic examination of several of the compounds characterized by X-ray diffraction as described above, as controlled sources of fluoride ion acting as nucleophile. The results show a range of reactivity of >100 fold on variation of the hydrogen-bond donor alcohol, as shown in Table 2.|
|
||||||
|---|---|---|---|---|---|---|
| Entry | Complex | C.N.a | M | k 2, M−1 s−1 | k 2 (rel) | [4]/[5] |
| a Coordination no., in (ROH)aF−. b (ROH)aF, M. c NB low (ROH)aF concn. d With 4× xs. ROH. e In C7H8. | ||||||
| 1 | 2m | 2 | 0.50 | 0.0251 | 4.1 | 2.1 |
| 2 | 2m | 2 | 0.13 | 0.0870 | 14 | 1.03 |
| 3 | 2m | 2 | 0.05 | 0.123 | 20 | 0.74 |
| 4 | 2m | 2 | 0.50 | 0.0142 | 2.3 | 3.6 |
| 5 | 2j | 2, 3 | 0.50 | 0.0032 | 0.52 | 3.5 |
| 6 | TBAF(H2O)3 | 3 | 0.50 | 0.0061 | 1.0 | 1.6 |
| 7 | TBAF(H2O)3e | 3 | 0.50 | 0.0075 | 1.2 | 4.2 |
| 8 | TBAF(t-BuOH)4 | 4 | 0.50 | 0.0130 | 2.1 | 2.0 |
| 9 | 2g | 4 | 0.50 | 0.0021 | 0.35 | 2.8 |
| 10 | 2d | 4 | 0.50 | 0.0004 | 0.066 | 3.1 |
| 11 | 2c | 4 | 0.50 | 0.0002 | 0.037 | 4.2 |
Taking first the reaction using complex 2m (entry 1), reaction is rapid and the decline in [3b] follows a 2nd order decay over the first 600 s, subsequently reacting more slowly. The product is partitioned between SN2 and E2 pathways, with the dominance of the former increasing slightly over time. Running the same reaction at higher dilution of both components demonstrates dramatic changes that increase reactivity and decrease SN2 selectivity (entries 2, 3). This is consistent with partial or complete dissociation of the L2F− complex to give more reactive LF−, or free F− that becomes kinetically dominant at low concentration. Carrying out reaction with 1 M excess alcohol 1m present (entry 4) gives a slower rate but substantially higher SN2 selectivity. As a representative of the (ROH)3F− class, the 9-phenylfluoren-9-ol derived complex 2j reacts 8 times more slowly than 2m and gives a lower proportion of product by the E2 pathway (entry 5).23
Commercial TBAF(H2O)3 was used as a benchmark of reactivity (entry 6). In CH3CN the reaction is relatively unselective between SN2 and E2 pathways, but occurs with higher SN2 selectivity in toluene (entry 7). Surprisingly, the t-BuOH complex that proved so useful in allylic fluorination,6,7 proved relatively unselective under these conditions (entry 8).
Interesting contrasts were observed by using fluoride-chelating diols (entries 9–11). With the pinacol complex 2g, the rate and selectivity are comparable to 2j. For the two 1,3-diol complexes 2d and 2c the rates are considerably lower, and the slower 2c provides the highest SN2 selectivity observed in the series. Inspection of the crystal structure of 2g shows that the O–C–C–O units are gauche with dihedral angles of −68.0(2)° and −70.5(2)°, similar to the preferred tGg′ ground state of free pinacol derived by spectroscopy and QM.24 For neopentyl glycol 1d, the preferred C2 symmetric GG conformation of the chelating unit,25 is maintained in the X-ray structures, as preferred in the free diols. Hence there is no evidence of additional strain caused by complexation in either 1,2-diol or 1,3-diol fluoride anion complexes. The main structural difference between the 1,2- and 1,3-diol complexes lies in the chelate angle O⋯F⋯O that defines H-bonding, which is 68.24(5)° and 69.11(5)° for the two independent pinacol units in 2g, contrasting with 78.62(4)° and 79.12(5)° for the typical 1,3-diol complex 2d. If the wider angle in the 1,3-diol case is associated with greater stability, then the 1,2-diol complex will dissociate one pinacol more easily and hence create an active nucleophilic entity more readily. This is consistent with the observed 5–10 fold higher reactivity of 2g compared with 2c or 2d.
Overall, there is a correlation between the rates of displacement and the SN2/E2 selectivity. The clear trend towards reduced SN2 selectivity with increasing rate can be seen in Fig. 11. Stronger complexation of fluoride ion is observed with ureas, and this leads to significantly slower rates of substitution with 3b and higher selectivity towards formation of product 4.26
 | ||
Fig. 11 Correlation between rate and SN2/E2 selectivity in the reactions of 3b with TBAF(ROH)n, 70 °C, CH3CN. The additional points represent entry 4  and entry 7 and entry 7  , Table 2. , Table 2. | ||
Conclusions
From the large number of studies on nucleophilic fluorination, it appears that the nature of the fluoride reagent is critical for a particular transformation to succeed; the reasons why one fluoride source is superior to another are more often unknown. As a result, an empirical approach that involves the systematic screen of commercially available F− reagents is typically undertaken when developing nucleophilic fluorination processes. This work provides new information on the coordination chemistry and relative reactivity of a range of novel fluoride–alcohol complexes; some key findings are listed below.(a) The synthesis and characterization by single-crystal X-ray diffraction of fourteen fluoride–alcohol complexes derived from alcohols, 1,2-diols, 1,3-diols, triols and tetraols demonstrate that tetra-, tri-, or dicoordinate fluoride–alcohol complexes can be formed. This variability in coordination stoichiometry had not been observed previously.
(b) For alcohols, the coordination number to fluoride varies from two to four, and decreases as the degree of branching and steric bulk of the alcohol is increased.
(c) Complexes with lower coordination number tend to have shorter O⋯F (and therefore shorter H⋯F) distances.
(d) Complexes derived from 1,2- and 1,3-diols display a range of interligand angles; this suggests that the packing forces imposed by the ligand are more influential than the inclination of fluoride to form complexes of tetrahedral geometry. The complex derived from pentaerythrol is unique forming a linear polymeric structure with an interplanar angle between the O⋯F⋯O units of 50.54(12)°.
(e) The structural features in the solid state of hydrogen bonded fluoride–alcohol complexes provide insight into the ability of these complexes to dissociate in solution; such dissociation releases a more active fluoride source that influences rate and SN2/E2 selectivity. For fluoride complexes derived from chelating 1,2- and 1,3-diols, the ability to dissociate to give an active nucleophilic entity depends on the chelate O⋯F⋯O angle that defines hydrogen bonding since this angle influences complex stability.
(f) In solution at high dilution, the fluoride complexes LnF− partially or completely dissociate; as a result, reactivity increases but SN2 versus E2 selectivity decreases.
(g) Many complexes reported here form crystalline solids that are easy to handle and are less hygroscopic than TBAF(H2O)3 and TBAF(t-BuOH)4.
This work has demonstrated that fluoride–alcohol complexes display structural diversity in the solid state; this key observation implies that there will be significant variabilities on the ability of these complexes to dissociate in solution. This observation underscores the importance of structural analysis in the solid state combined with kinetic studies as a platform to understand fluoride reactivity. Ongoing work, applying experimental and computational methods, focuses on the examination of a larger range of small-molecule hydrogen-bond donors to activate inexpensive and widely available sources of fluoride for applications in synthesis, catalysis and [18F] radiochemistry.27–33
Experimental
For the preparation of TBAF–alcohol complexes, a flask was charged with TBAF(H2O)3 (1.0 eq.), and the alcohol (1.0–4.0 eq.) was added under an atmosphere of N2. Hexane was added, and the mixture was refluxed for 2 h, during which time droplets of water formed on the inside walls of the condenser, before letting it cool to RT. The solid products were collected by filtration, washed with hexane and dried under high vacuum, giving the desired complexes, which were used without further purification. Products were stored under an atmosphere of N2. Single-crystals suitable for X-ray analysis were obtained by recrystallization from THF, EtOAc or DCM by reducing solubility in a saturated solution through slow mixing with hexanes using a layering or vapour diffusion technique. See the ESI† for details regarding individual compounds.Low temperature (150 K) single-crystal X-ray diffraction data,27 were collected using either a Nonius Kappa CCD diffractometer or an Oxford Diffraction (Agilent) SuperNova A diffractometer and reduced using the appropriate instrument manufacturer supplied software.28 Structures were solved using either SIR92,29 or SuperFlip,30 and refined using full-matrix least-squares refinement with CRYSTALS.31 In the case of 2m, there was a small amount of diffuse residual electron density believed to be disordered solvent. This was modelled using PLATON/SQUEEZE,32 within CRYSTALS. On refinement of 2g, there was a poor agreement between the observed and calculated structure factor amplitudes. Examination of the data and model using ROTAX,33 suggested the crystal was a pseudo-merohedral twin that was included in the refinement. For further details see the full crystallographic data (in CIF format) which are available as ESI.†
Acknowledgements
The authors thank the Skaggs-Oxford Scholarship Program and NSF GRFP (predoctoral fellowships to K. M. E.), the European Union (FP7-PEOPLE-2012-ITN-RADIOMI-316882 to L.P.), and the Berrow Foundation (scholarship to G.T.G.) for generous funding. V.G. holds a Royal Society Wolfson Research Merit Award.Notes and references
- (a) M. E. Phelps, Proc. Natl. Acad. Sci. U. S. A., 2000, 97, 9226–9233 CrossRef CAS; (b) M. Tredwell and V. Gouverneur, Angew. Chem., Int. Ed., 2012, 51, 11426–11437 CrossRef CAS PubMed.
- J. Aigueperse, P. Mollard, D. Devilliers, M. Chemla, R. Faron, R. Romano and J.-P. Cuer, Fluorine Compounds, Inorganic, Ullman's Encyclopedia of Industrial Chemistry, Wiley-VCH, Weinheim, 2005, p. 307 Search PubMed.
- (a) T. Furuya, A. S. Kamlet and T. Ritter, Nature, 2011, 473, 470–477 CrossRef CAS PubMed; (b) C. Hollingworth and V. Gouverneur, Chem. Commun., 2012, 48, 2929–2942 RSC; (c) M. G. Campbell and T. Ritter, Chem. Rev., 2015, 115, 612–633 CrossRef CAS PubMed.
- D. O'Hagan, C. Schaffrath, S. L. Cobb, J. T. G. Hamilton and C. D. Murphy, Nature, 2002, 416, 279 CrossRef PubMed.
- (a) C. Dong, F. Huang, H. Deng and C. Schaffrath, Nature, 2004, 427, 561–565 CrossRef CAS PubMed; (b) H. Deng and D. O'Hagan, Curr. Opin. Chem. Biol., 2008, 12, 582–592 CrossRef CAS PubMed; (c) D. O'Hagan, J. Fluorine Chem., 2006, 127, 1479–1483 CrossRef PubMed; (d) D. O'Hagan and H. Deng, Chem. Rev., 2015, 115, 634–649 CrossRef PubMed.
- C. Hollingworth, A. Hazari, M. N. Hopkinson, M. Tredwell, E. Benedetto, M. Huiban, A. D. Gee, J. M. Brown and V. Gouverneur, Angew. Chem., Int. Ed., 2011, 50, 2613–2617 CrossRef CAS PubMed.
- E. Benedetto, M. Tredwell, C. Hollingworth, T. Khotavivattana, J. M. Brown and V. Gouverneur, Chem. Sci., 2013, 4, 89–96 RSC.
- (a) J. Emsley, Chem. Soc. Rev., 1980, 9, 91–124 RSC; (b) D. J. Adams and J. H. Clark, Chem. Soc. Rev., 1999, 28, 225–231 RSC.
- T. Yonezawa, Y. Sakamoto and K. Nogawa, Preparation of Tetrabutylammonium Fluoride–Alcohol Adducts as Fluorination Agents, Jpn. Kokai Tokkyo Koho, 1994, JP 06316551 A Search PubMed.
- (a) D. W. Kim, H.-J. Jeong, S. T. Lim, M.-H. Sohn, J. A. Katzenellenbogen and D. Y. Chi, J. Org. Chem., 2008, 73, 957–962 CrossRef CAS PubMed; (b) D. W. Kim, D.-S. Ahn, Y.-H. Oh, S. Lee, H. S. Kil, S. J. Oh, S. J. Lee, J. S. Kim, J. S. Ryu, D. H. Moon and D. Y. Chi, J. Am. Chem. Soc., 2006, 128, 16394–16397 CrossRef CAS PubMed; (c) D. W. Kim, H.-J. Jeong, S. T. Lim and M.-H. Sohn, Tetrahedron Lett., 2010, 51, 432–434 CrossRef CAS PubMed.
- D. W. Kim, H.-J. Jeong, S. T. Lim and M.-H. Sohn, Angew. Chem., Int. Ed., 2008, 47, 8404–8406 CrossRef CAS PubMed.
- H. Yan, H. B. Jang, J.-W. Lee, H. K. Kim, S. W. Lee, J. W. Yang and C. E. Song, Angew. Chem., Int. Ed., 2010, 49, 8915–8917 CrossRef CAS PubMed.
- J. A. Birrell, J.-N. Desrosiers and E. N. Jacobsen, J. Am. Chem. Soc., 2011, 133, 13872–13875 CrossRef CAS PubMed.
- (a) V. F. DeTuri, M. A. Su and K. M. Ervin, J. Phys. Chem. A, 1999, 103, 1468–1479 CrossRef CAS; (b) V. F. DeTuri and K. M. Ervin, J. Phys. Chem. A, 1999, 103, 6911–6920 CrossRef CAS.
- B. Bogdanov and T. B. McMahon, J. Phys. Chem. A, 2000, 104, 7871–7880 CrossRef CAS.
- (a) C.-H. Chen and M. K. Leung, Tetrahedron, 2011, 67, 3924–3935 CrossRef CAS PubMed; (b) E. R. Libra and M. J. Scott, Chem. Commun., 2006, 1485–1487 RSC; (c) P. Anzenbacher, Jr., K. Jursíkova, V. M. Lynch, P. A. Gale and J. L. Sessler, J. Am. Chem. Soc., 1999, 121, 11020–11021 CrossRef.
- cf. similar concerns in coordination chemistry: (a) S. Alvarez, P. Alemany, D. Casanova, J. Cirera, M. Llunell and D. Avnir, Coord. Chem. Rev., 2005, 249, 1693–1708 CrossRef CAS PubMed; (b) S. Alvarez, B. Menjon, A. Falceto, D. Casanova and P. Alemany, Inorg. Chem., 2014, 53, 12151–12163 CrossRef CAS PubMed; (c) J. Cirera, P. Alemany and S. Alvarez, Chem.–Eur. J., 2004, 10, 190–207 CrossRef CAS PubMed.
- In several cases the results arising from analysis of the coordination sphere based on O⋯F distances were checked by a parallel analysis treating the hydrogen-bonding H⋯F distances directly. The conclusions were not altered.
- G. R. Desiraju and T. Steiner, The Weak Hydrogen Bond in Structural Chemistry and Biology, Oxford University Press, New York, 1999 Search PubMed.
- (a) Q.-Q. Wang, V. W. Day and K. Bowman-James, Angew. Chem., Int. Ed., 2012, 51, 2119–2123 CrossRef CAS PubMed; (b) A. Kornath, F. Neumann and H. Oberhammer, Inorg. Chem., 2003, 42, 2894–2901 CrossRef CAS PubMed; (c) M. Arunachalam and P. Ghosh, Chem. Commun., 2009, 5389–5391 RSC; (d) S. J. Brooks, P. A. Gale and M. E. Light, Chem. Commun., 2006, 4344–4346 RSC; (e) W. J. McLean and G. A. Jeffrey, J. Chem. Phys., 1967, 47, 414–417 CrossRef CAS PubMed.
- (a) J. D. C. Craig and M. H. Brooker, J. Solution Chem., 2000, 29, 879–888 CrossRef CAS; (b) J. Baik, J. Kim, D. Majumdar and K. S. Kim, J. Chem. Phys., 1999, 110, 9116–9127 CrossRef CAS PubMed; (c) J. Kim, H. M. Lee, S. B. Suh, D. Majumdar and K. S. Kim, J. Chem. Phys., 2000, 113, 5259–5272 CrossRef CAS PubMed; (d) M. A. Vincent and I. H. Hillier, Chem. Commun., 2005, 5902–5903 RSC.
- (a) T. Steiner and G. R. Desiraju, Chem. Commun., 1998, 891–892 RSC; (b) E. Arunan, G. R. Desiraju, R. A. Klein, J. Sadlej, S. Scheiner, I. Alkorta, D. C. Clary, R. H. Crabtree, J. J. Dannenberg, P. Hobza, H. G. Kjaergaard, A. C. Legon, B. Mennucci and D. J. Nesbitt, Pure Appl. Chem., 2011, 83, 1637–1641 CAS.
- The original paper (ref. 10) indicated a higher extent of substitution with RF:alkene = 3.76 with TBAF(t-BuOH)4. We note, however, that the relevant data point from entry 8 falls on the trend line in Fig. 11.
- M. Dahlqvist, M. Hotokka and M. Räsänen, Chem. Phys., 1998, 229, 137–147 CrossRef CAS.
- B. Granzow, P. Klaeboe and V. Sablinskas, J. Mol. Struct., 1995, 349, 153–156 CrossRef CAS.
- L. Pfeifer, G. W. Pidgeon, K. M. Engle, J. M. Brown and V. Gouverneur, to be published.
- J. Cosier and A. M. Glazer, J. Appl. Crystallogr., 1986, 19, 105–107 CrossRef CAS.
- Z. Otwinowski and W. Minor, in Methods in Enzymology, ed. C. W. Carter, Jr. and R. M. Sweet, Academic Press, New York, 1997, vol. 276, pp. 307–326 Search PubMed.
- A. Altomare, G. Cascarano, C. Giacovazzo, A. Guagliardi, M. C. Burla, G. Polidori and M. Camalli, J. Appl. Crystallogr., 1994, 27, 435 Search PubMed.
- L. Palatinus and G. Chapuis, J. Appl. Crystallogr., 2007, 40, 786–790 CrossRef CAS.
- (a) P. W. Betteridge, J. R. Carruthers, R. I. Cooper, K. Prout and D. J. Watkin, J. Appl. Crystallogr., 2003, 36, 1487 CrossRef CAS; (b) R. I. Cooper, A. L. Thompson and D. J. Watkin, J. Appl. Crystallogr., 2010, 43, 1100–1107 CrossRef CAS; (c) P. Parois, R. I. Cooper and A. L. Thompson, Chem. Cent. J., 2015, 9, 30 CrossRef PubMed; (d) A. L. Thompson and D. J. Watkin, J. Appl. Crystallogr., 2011, 44, 1017–1022 CrossRef CAS.
- (a) A. L. Spek, J. Appl. Crystallogr., 2003, 36, 7–13 CrossRef CAS; (b) P. van der Sluis and A. L. Spek, Acta Crystallogr., Sect. A: Found. Crystallogr., 1990, 46, 194–201 CrossRef.
- R. I. Cooper, R. O. Gould, S. Parsons and D. J. Watkin, J. Appl. Crystallogr., 2002, 35, 168–174 CAS.
- For references on the ability of organofluorine compounds to engage in hydrogen bonding, see: (a) J. D. Dunitz and R. Taylor, Chem.–Eur. J., 2006, 3, 89–98 CrossRef PubMed; (b) J. A. K. Howard, V. J. Hoy, D. O'Hagan and G. T. Smith, Tetrahedron, 1996, 52, 12613–12622 CrossRef CAS; (c) P. A. Champagne, Y. Benhassine, J. Desroches and J.-F. Paquin, Angew. Chem., Int. Ed., 2014, 53, 13835–13839 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Detailed procedures for the synthesis of fluoride–alcohol complexes, 1H and 13C NMR spectra, crystal structures and reaction kinetics. CCDC 1401765–1401778. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5sc01812a |
| This journal is © The Royal Society of Chemistry 2015 |