
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Dynamic behaviour of monohaptoallylpalladium species: internal coordination as a driving force in allylic alkylation chemistry†
Lan-Gui
Xie‡
a,
Viktor
Bagutski‡
b,
Davide
Audisio
c,
Larry M.
Wolf
c,
Volker
Schmidts
b,
Kathrin
Hofmann
d,
Cornelia
Wirtz
c,
Walter
Thiel
c,
Christina M.
Thiele
*b and
Nuno
Maulide
*a
aUniversity of Vienna, Institute of Organic Chemistry, Währinger Strasse 38, 1090 Vienna, Austria. E-mail: nuno.maulide@univie.ac.at
bTechnische Universität Darmstadt, Clemens Schöpf Institut für Organische Chemie und Biochemie, Alarich-Weiss-Str. 4, 64287 Darmstadt, Germany. E-mail: cthiele@thielelab.de
cMax-Planck-Institut für Kohlenforschung, Kaiser-Wilhelm-Platz 1, 45470 Mülheim an der Ruhr, Germany
dTechnische Universität Darmstadt, Eduard-Zintl-Institute, Alarich-Weiss-Str. 12, 64287 Darmstadt, Germany
First published on 6th July 2015
Abstract
Contemporary catalytic procedures involving alkylpalladium(II) have enriched the arsenal of synthetic organic chemistry. Those transformations usually rely on internal coordination through “directing groups”, carefully designed to maximize catalytic efficiency and regioselectivity. Herein, we report structural and reactivity studies of a series of internally coordinated monohaptoallylpalladium complexes. These species enable the direct spectroscopic observation and theoretical study of π–σ–π interconversion processes. They further display unusual dynamic behavior which should be of direct relevance to chemistries beyond catalytic allylic alkylation.
Introduction
Recent developments in catalysis involving alkylpalladium(II) intermediates have enabled a myriad of selective transformations in organic synthesis.1 Particularly widespread is the importance of such intermediates in C–H functionalizations of sp3 C–H bonds.2 Common in those chemistries is the requirement for pre-installed “directing groups”, which stabilize the intermediate metal centre through chelation and thus help to prevent decomposition.2Understanding how such coordinating moieties can affect not only the structure of intermediate metal complexes but also the reaction pathways available to them is a valuable endeavour that might lead to the discovery of new reactivity.
We have recently reported a palladium-catalysed diastereodivergent asymmetric allylic alkylation on cyclobutene substrates.3 In that transformation, unusually strong ligand effects were observed that led, in the presence of stabilized carbanions and depending on the ligand employed, either to the products of overall retention or overall inversion of configuration (Scheme 1).
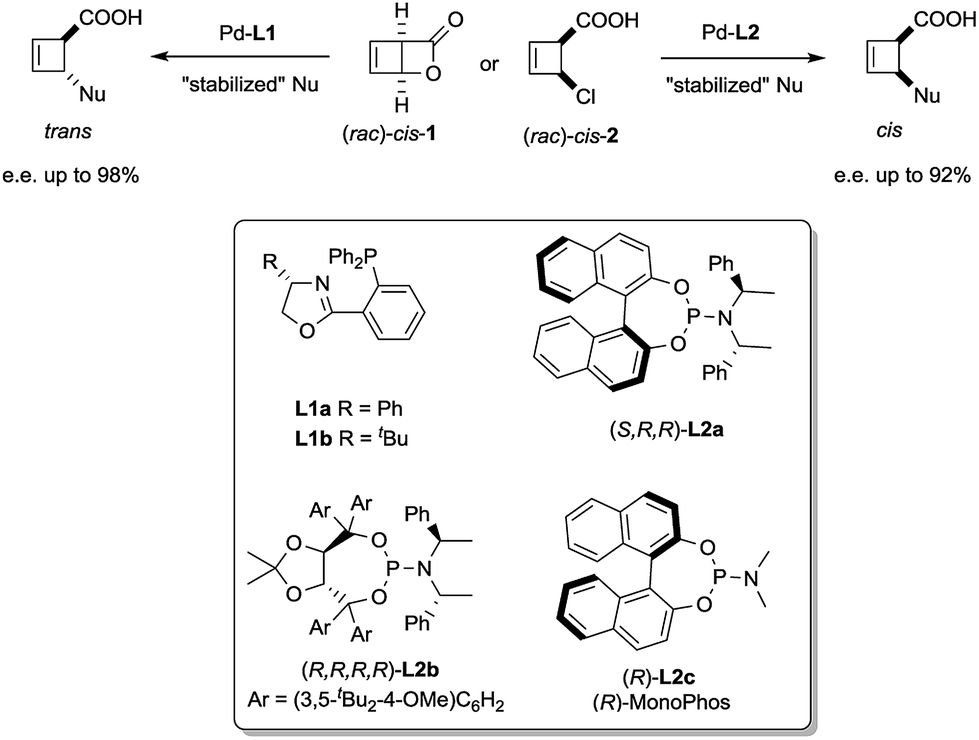 | ||
| Scheme 1 Stereoselective synthesis of trans and cis-disubstituted cyclobutenes from cis-1 and cis-2. | ||
These unusual phenomena led us to investigate the reactivity of the putative allylpalladium intermediates in more detail. Employing the bidentate ligand L1a, we eventually identified a series of η1-allyl palladium complexes, prone to very facile electrocyclic ring opening at temperatures close to r.t., as key species in the deracemization process.4 We subsequently became interested in the case of monodentate ligands such as L2a–c. Herein we present our findings on the structure of internally chelated η1-allyl palladium complexes containing those ligands, their kinetic study enabling a direct insight into metallotropic equilibria, as well as a rare showcase of the value of chelation in allylic substitution chemistry.
Results and discussion
Oxidative addition of Pd(dba)2 to the chloroamide (rac)-cis-3 in the presence of 2 equivalents of ligand L2a proceeded to full conversion at room temperature (Scheme 2). The resulting organometallic species 4 was assigned as a single η1-coordinated allylpalladium complex by multinuclear NMR spectroscopy. 31P-NMR spectroscopy was diagnostic for two non-equivalent proximal phosphines attached to the presumed square-planar palladium(II) centre. Replacement of L2a with the less bulky (and more amenable to detailed NMR analysis) MonoPhos ligand L2c similarly led to an intermediate of structure 5. The stability of these complexes at room temperature stands in contrast to the temperature-sensitive nature of the analogous complexes of bidentate ligand L1a.4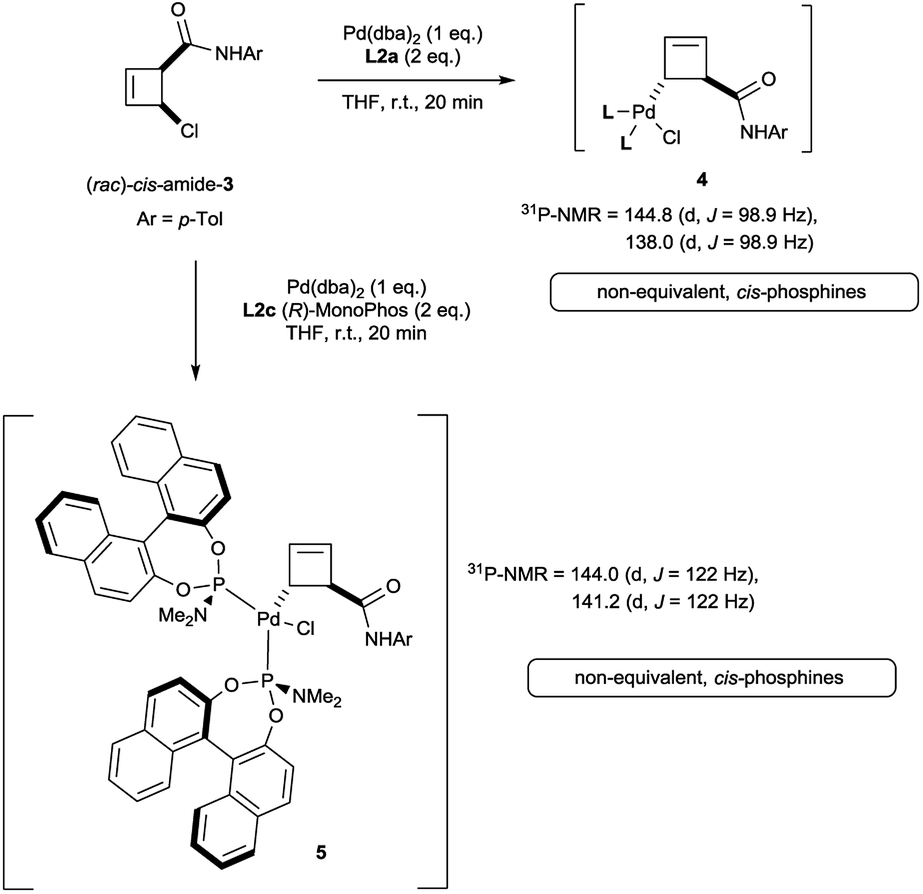 | ||
| Scheme 2 Initial results on the synthesis of allylpalladium(II) complexes of chloroamide 3. | ||
The trans-rac-bromoamide 6 (Scheme 3)5 smoothly reacts at room temperature with stoichiometric amounts of monodentate L2c and Pd(dba)2 to form two fairly stable, isomeric species 7a and 7b in 6![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :5 ratio. These compounds could be assigned as 1:1 Pd:L2c complexes with a syn relationship between the metal center and the cyclobutene substituent,6 confirming that oxidative addition occurs with inversion of configuration.7 As we will show, these are the two diastereomeric products of suprafacial allylic migration (metallotropic shift) of the Pd(L2c)Br-fragment across the cyclobutene ring. Remarkably (Scheme 3), the 13C-NMR resonance of the carbonyl carbon was shifted from δ = 167.6 ppm in starting material 6 to δ = 182.0 and 181.8 ppm in the complexes 7a and 7b. This strongly suggests internal coordination of the amide carbonyl as the fourth ligand of the tetra-coordinate sphere around palladium. Worthy of note, this mixture of isomers survived purification by silica gel column chromatography, though required an inert atmosphere.
:5 ratio. These compounds could be assigned as 1:1 Pd:L2c complexes with a syn relationship between the metal center and the cyclobutene substituent,6 confirming that oxidative addition occurs with inversion of configuration.7 As we will show, these are the two diastereomeric products of suprafacial allylic migration (metallotropic shift) of the Pd(L2c)Br-fragment across the cyclobutene ring. Remarkably (Scheme 3), the 13C-NMR resonance of the carbonyl carbon was shifted from δ = 167.6 ppm in starting material 6 to δ = 182.0 and 181.8 ppm in the complexes 7a and 7b. This strongly suggests internal coordination of the amide carbonyl as the fourth ligand of the tetra-coordinate sphere around palladium. Worthy of note, this mixture of isomers survived purification by silica gel column chromatography, though required an inert atmosphere.
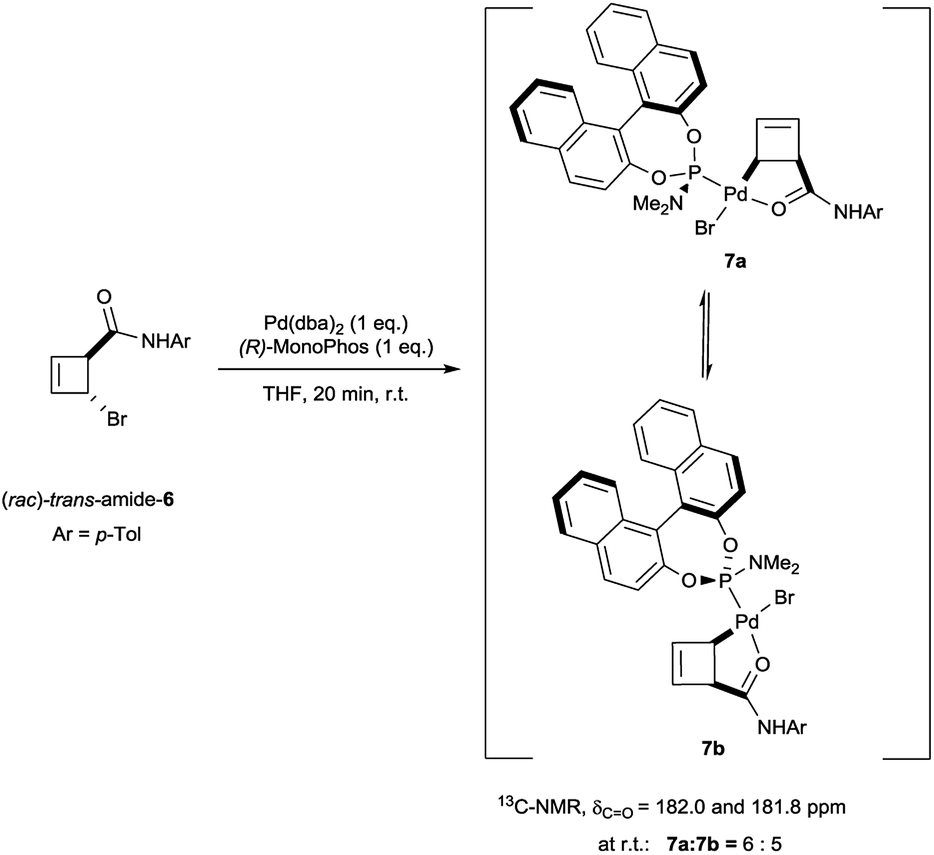 | ||
| Scheme 3 Formation of cyclobut-2-enyl η1-allyl complexes from trans-amide-6. | ||
Single crystals of 7a were obtained by crystallization of the crude mixture of 7a/7b from CH2Cl2, and investigated by X-ray diffraction.8 The resulting molecular structure (Fig. 1) confirms the syn-orientation of substituents across the cyclobutene ring. The internal distances of the four-membered ring have typical lengths for localized C–C single (1.621 to 1.660 Å) and C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C double (1.365 Å) bonds. In contrast to our prior findings,4 the internal disubstituted C–C single bond is shorter than the other two, due to the chelation from the amide carbonyl. The short distance between the Pd-centre and the amide carbonyl oxygen (bond distance 2.08 Å) further validates this assumption. These two points are likely responsible for the remarkably enhanced stability of this compound at room temperature. To the best of our knowledge, crystal structures of internally coordinated monohaptoallyl-Pd complexes are not known in spite of the obvious relevance of such compounds even beyond the realm of catalytic allylic alkylation.9 Indeed, internally coordinated species such as 7 are postulated in virtually all established catalytic, directed C–H activation procedures; interestingly, amide coordination at Pd(II) centre is most often observed or postulated to involve bonding through nitrogen9f,i rather than oxygen, as we observe in this case.
C double (1.365 Å) bonds. In contrast to our prior findings,4 the internal disubstituted C–C single bond is shorter than the other two, due to the chelation from the amide carbonyl. The short distance between the Pd-centre and the amide carbonyl oxygen (bond distance 2.08 Å) further validates this assumption. These two points are likely responsible for the remarkably enhanced stability of this compound at room temperature. To the best of our knowledge, crystal structures of internally coordinated monohaptoallyl-Pd complexes are not known in spite of the obvious relevance of such compounds even beyond the realm of catalytic allylic alkylation.9 Indeed, internally coordinated species such as 7 are postulated in virtually all established catalytic, directed C–H activation procedures; interestingly, amide coordination at Pd(II) centre is most often observed or postulated to involve bonding through nitrogen9f,i rather than oxygen, as we observe in this case.
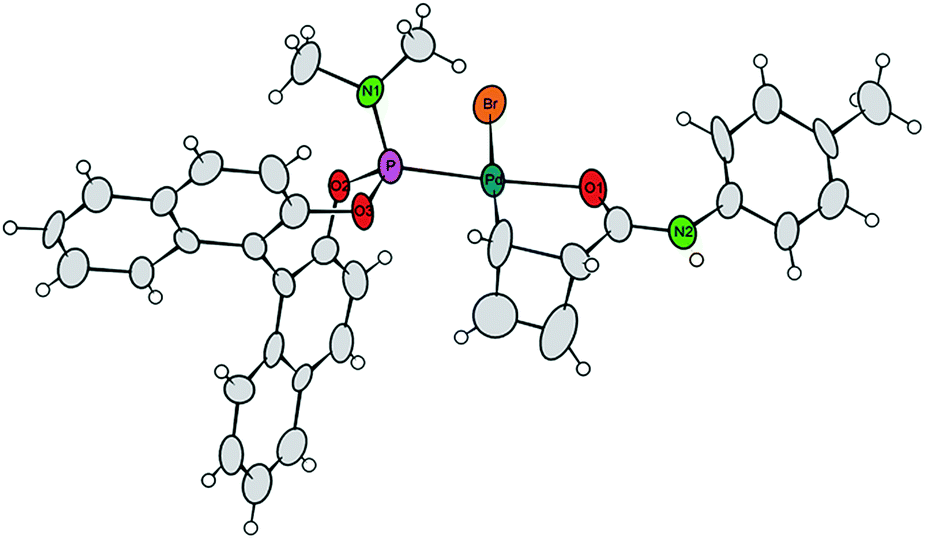 | ||
| Fig. 1 Projection of the molecular structure of the isomer 7a (two crystal water molecules are omitted for clarity). Ellipsoids of the displacement parameters are drawn at 40% probability level.8 | ||
The unexpectedly selective crystallization of only one out of the two isomers 7a/7b offers the possibility for direct observation of the metallotropic equilibrium of isomers 7a and 7b in solution.10 Additionally, solution conformations may differ from those determined in the solid state. Initial studies based on intramolecular NOEs provided useful guidance for the assignment of complexes 7a/7b as allylic rearrangement isomers,6 but reliable quantification of interatomic distances relating the ligand and the cyclobutene fragments proved difficult, as only a few, weak NOE contacts were observed in the spectra (see Fig. 2). We thus sought to introduce the mixture of complexes 7a/b into an anisotropic medium to measure residual dipolar couplings (RDC).11,12 After some experimentation we chose chemically cross-linked PDMS (polydimethylsiloxane)13 as an orienting medium. This choice yielded ω1-coupled HSQC14 spectra of excellent quality and enabled the identification of 8 C–H RDCs for each isomer.6
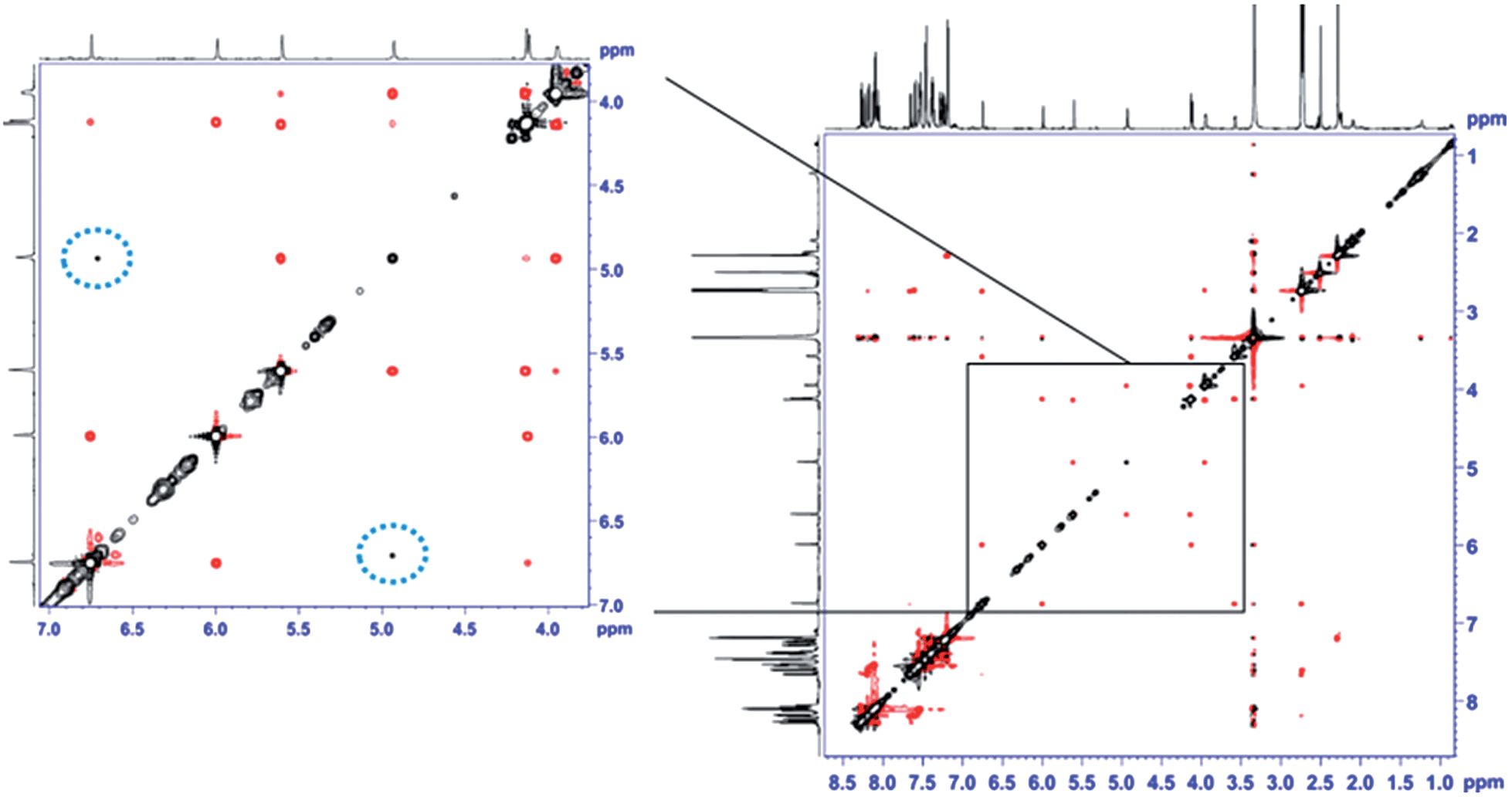 | ||
| Fig. 2 EASY-ROESY (τmix = 300 ms, 5 kHz spinlock field, 45° flip angle)17 spectrum of the isomeric mixture of 7a and 7b in DMSO-d6 at 300 K after covariance processing.18 The expansion shows the spectral region of the signals of the cyclobutene protons at a lower intensity level. NOE contacts between protons within the cyclobutene moiety of the same isomer show the opposite phase (red) as the diagonal signals (black). Cross peaks resulting from exchange of cyclobutene protons of different isomers (7a → 7b, and vice versa) show the same phase as the diagonal (circled with a dotted blue line). | ||
Structural models for the two isomers and the transitional η3-coordinated species were generated computationally by molecular modelling and subsequent geometry optimization by density functional theory (DFT) using ORCA.15 Fitting the experimental RDC data to the computed structure models of the isomers was performed with the RDC module of the hot-FCHT software package.16 Comparison of the experimental and back-calculated RDCs yielded excellent quality factors for the data assigned to the respective isomers, while all other combinations of experimental data and structure model showed a significantly worse fit.6 This validates the proposed structure models in solution.
Notably, the sparse solubility of 7a in THF-d8 enabled the enrichment of an isomeric mixture of 7a/7b up to 95% in 7a, thus furnishing its clean NMR spectrum. Upon standing at room temperature, enriched 7a gradually equilibrates back to the 6:5 thermodynamic ratio of isomers. This process was too slow in THF-d8 to be quantified by EXSY. Nevertheless, in DMSO-d6 we were able to qualitatively follow this exchange process by 2D EASY-ROESY spectra (see Fig. 2).16 Using less measurement-time-consuming, selective 1D PFGSE NOE spectra, we quantified activation parameters of that equilibrium at 320 K of ΔG≠ = 21.1 kcal mol−1 for 7a → 7b and 20.7 kcal mol−1 for the reverse process.6
The mechanism for this apparent metallotropic shift was investigated computationally at the B3LYP-D3 level (Fig. 3). The predicted free energy difference between 7a and 7b (−0.3 kcal mol−1) is very small, which is consistent with the observed 6:5 equilibrium ratio of 7a/7b (corresponding to a free energy difference of −0.1 kcal mol−1). Two potential pathways were investigated for their isomerization, the first of which proceeds initially via an η1 → η3 conversion to 7a-INT1 (black path). This η3 intermediate 7a-INT1 then can undergo an η3 → η1 conversion to form 7a-INT2 followed by positional isomerization to generate 7b. Alternatively, 7a-INT1 could isomerize to 7b-INT1, accessing the second pathway, via an apparent rotation which could be facilitated by coordinating solvent in the absence of excess ligand.19 A separate pathway accessible to 7a proceeds first via positional isomerization followed by an analogous η1 → η3 → η1 shift (red path). The maximum heights of the two pathways differ by 1.7 kcal mol−1 in favor of the first pathway, which is caused by the contrasting steric effects imparted by the chiral ligand. Either pathway is expected to translate to a relatively facile equilibration of 7a and 7b at room temperature, consistent with the experimentally determined activation parameters.20
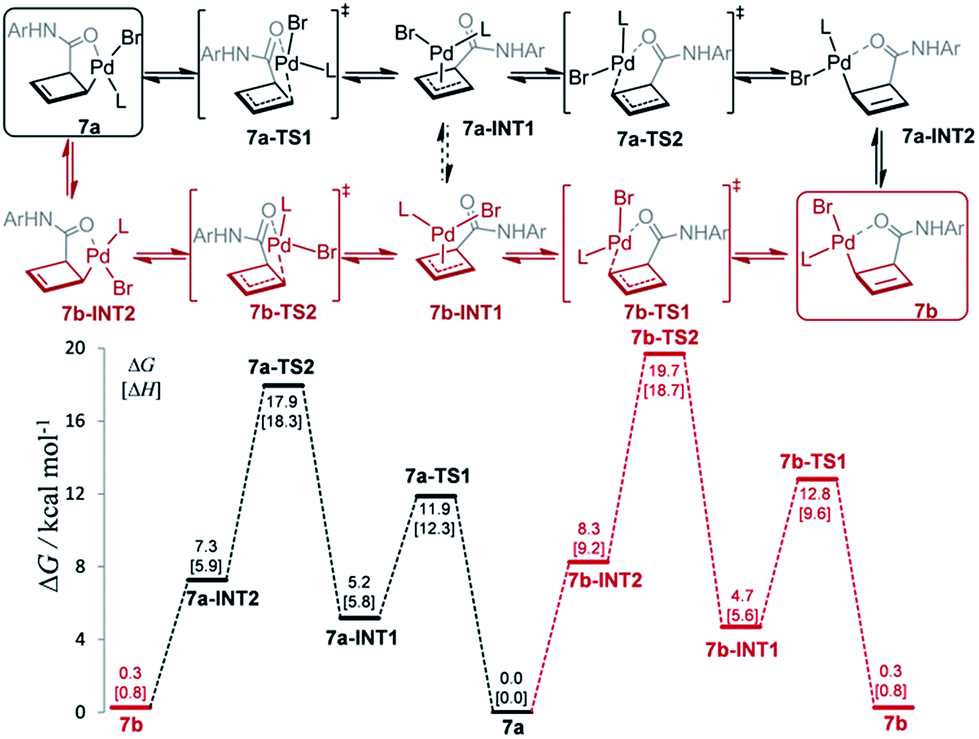 | ||
| Fig. 3 Computed Gibbs free energy profile (298.15 K) for the equilibration of 7a and 7b at the SMD(THF)-B3LYP-D3/def2-TZVP//B3LYP-D3/def2-SVP(def2-TZVP for Pd) level via two possible pathways. L = L2c. | ||
In close analogy with amide 3, the (rac)-cis-chloroacid 2 also leads to the formation of a single anti-η1-allyl palladium complex 8 upon stoichiometric combination with Pd(dba)2 and L2a (Scheme 4). As the acid 2 had proved to be an ideal electrophile for catalytic deracemization in our previous work,4 we investigated the reaction of the monohaptoallylpalladium complex 8 with a suitable nucleophile. As shown, treatment of complex 8 with sodium (2-methyl)dimethylmalonate at 0 °C yielded the cis-disubstituted cyclobutene 9 in 74% ee. This result is consistent with the observed enantioselectivity in the catalytic process employing ligand L2a,3 thus demonstrating that 8 is a catalytically active intermediate.
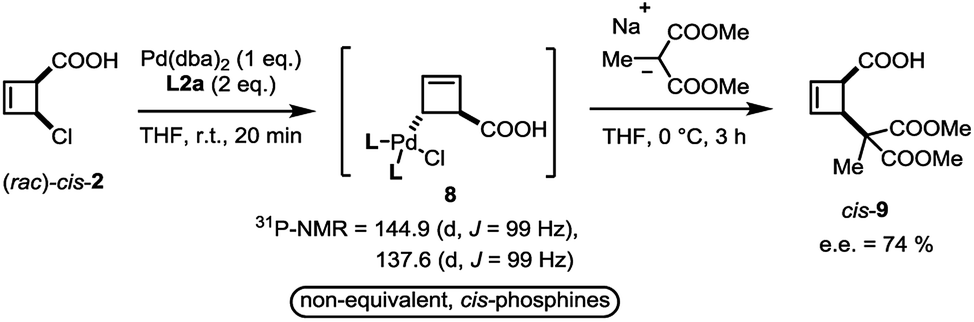 | ||
| Scheme 4 Formation of four membered ring η1-allyl complex from cis-2. | ||
To account for the observation of a single diastereomer from the cis-configured substrates amide 3 and acid 2, DFT modelling was performed on the amide complex 5 (Fig. 4). Complexes 5a and 5b were identified as the lowest-energy conformers for 5 (Scheme 2) and the diastereomer of 5 respectively.21 A key interaction common to both structures is a hydrogen bond between the chlorine and the hydrogen atoms of the amide moiety.4,22 Additionally, two CH/π interactions between the naphthyl groups of both ligands are apparent in both structures, with H-arene distances within the purview of what has been observed experimentally and computationally for this type of interaction.23 A structural feature distinguishing between the two diastereomers is the positioning of the dimethyl amino group that is located in 5b under and in close proximity to the cyclobutene ring. This repulsive contact is imposed by the combined hydrogen bond and CH/π interactions. The contact is not as repulsive in 5a, as judged from a greater separation, and thus appears to be responsible for the 2.1 kcal mol−1 preference for 5a.
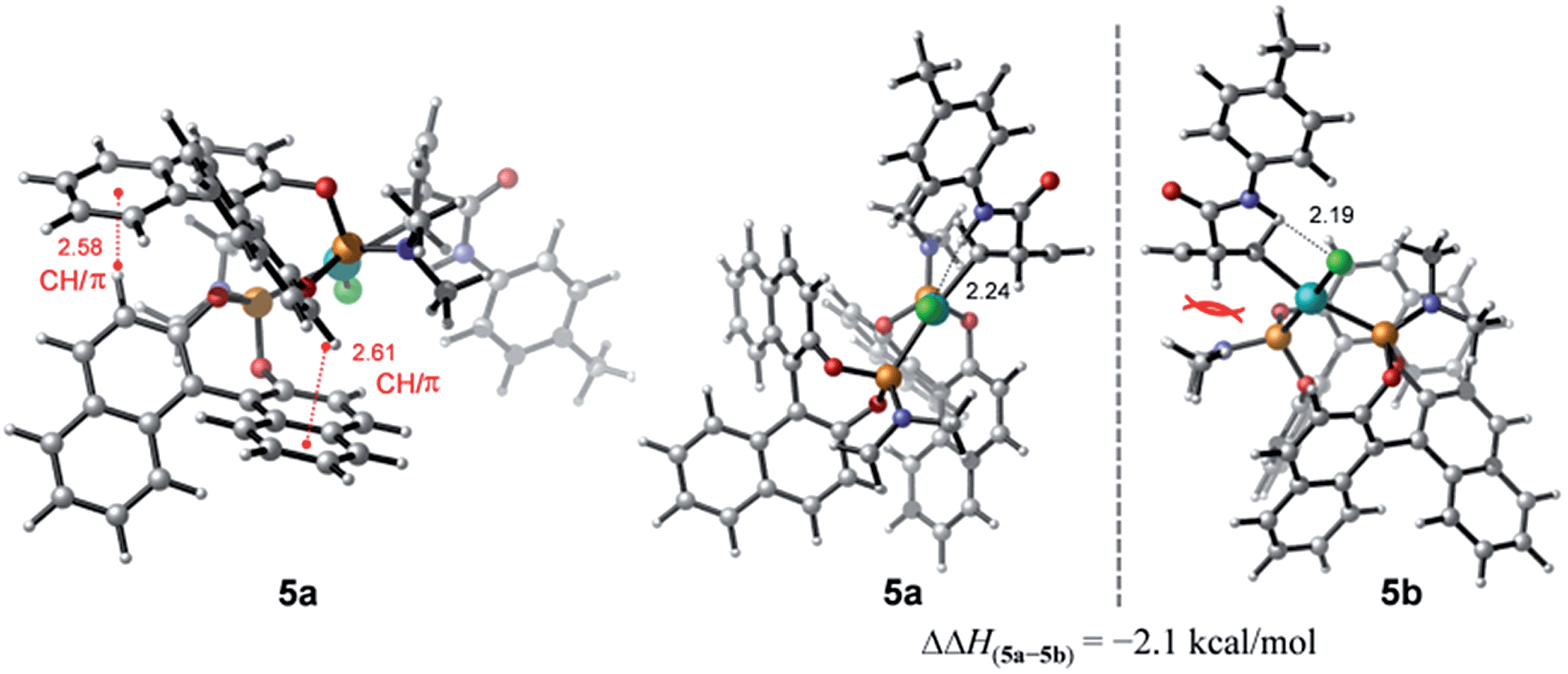 | ||
| Fig. 4 Computed diastereomers 5a and 5b and their corresponding enthalpy difference (ΔΔH) obtained at the SMD(THF)-M06-D3/def2-TZVP//TPSS-D3/def2-SVP(def2-TZVP for Cl and Pd) level. Distances are in units of Å. | ||
In contrast to this and as in the case of the bromoamide 6, subjection of the (rac)-trans-chloroamide 10 to the action of stoichiometric amounts of Pd(dba)2 and ligand L2c led to the formation of two η1-allylpalladium complexes in 6:5 ratio (Scheme 5). The spectral signature of this mixture is very similar to that of 7a/b, supporting its analogous assignment as an internally coordinated, syn-species bearing a single phosphorus ligand. Simple demonstration of this analogy was achieved by exposing both the bromo-7a/b and the chloro-11a/b complexes to the action of silver triflate. Following filtration of the corresponding silver halide, an identical mixture of diastereoisomeric cationic palladium(II) complexes 12 (7:5) was observed in solution.
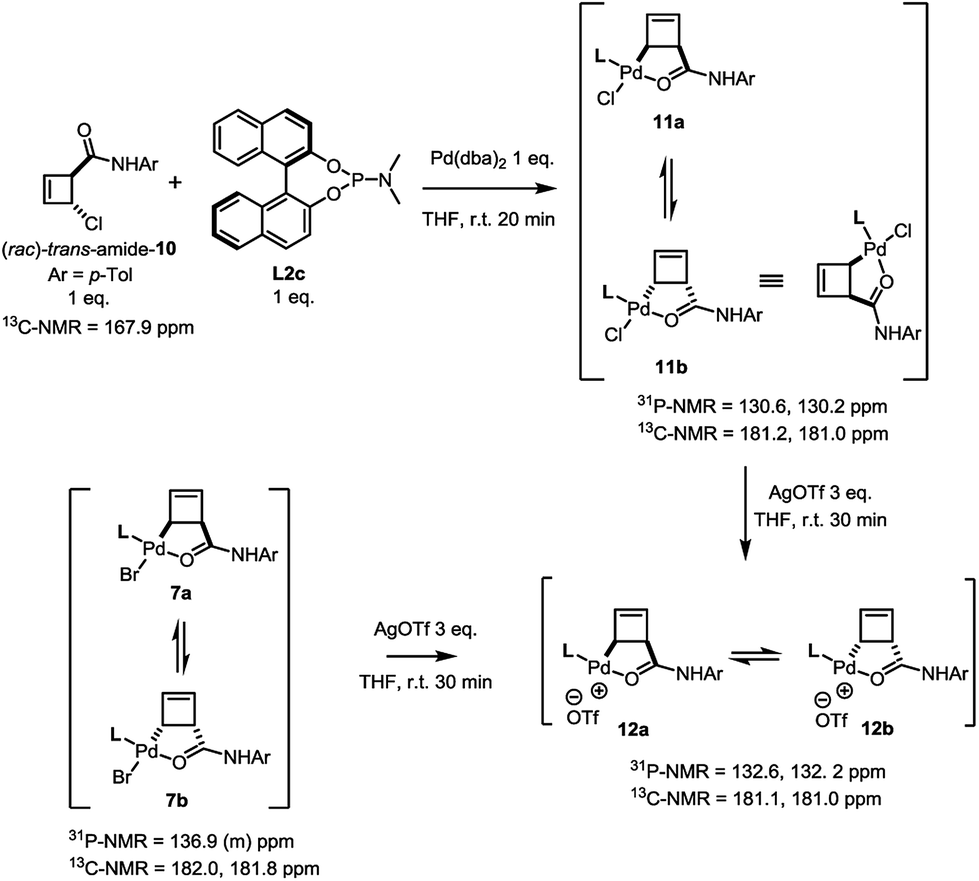 | ||
| Scheme 5 Demonstration of homology between halide complexes 7a/b and 11a/b through reaction with AgOTf. | ||
Treatment of the diastereomerically pure anti-palladium complex 5 with silver triflate also led to precipitation of silver chloride and formation of a new organometallic species (Scheme 6). Much to our surprise, this was exactly the same mixture of syn-, internally chelated diastereoisomeric palladium complexes 12 that had been obtained by halide abstraction from the syn-complexes 7 and 11! This unexpected result suggests the existence of a facile pathway for facial exchange of palladium within the cyclobutene framework. That this type of facial exchange could be triggered by ligand removal from the coordination sphere is, to the best of our knowledge, unprecedented.
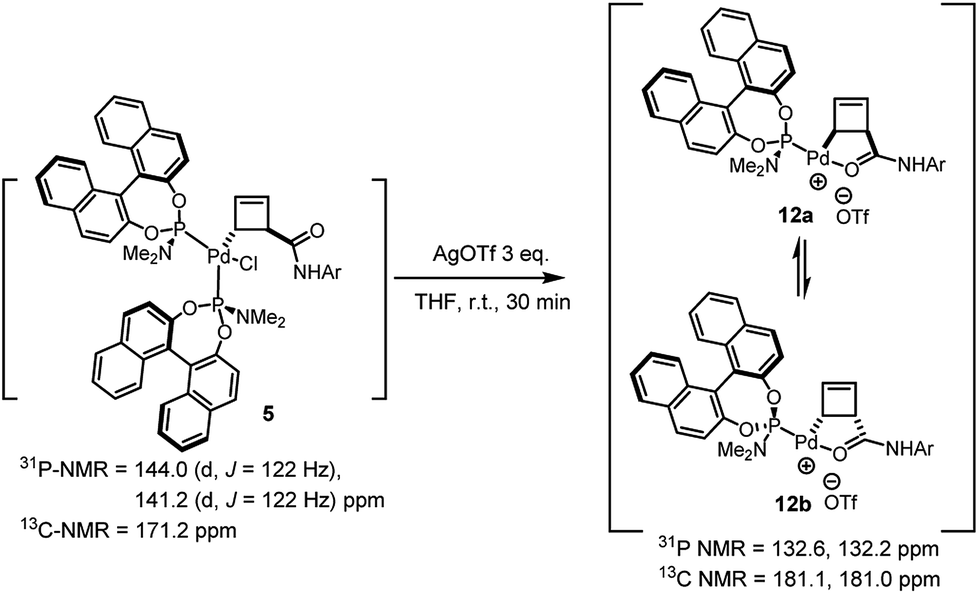 | ||
| Scheme 6 Reaction of anti-complex 5 with AgOTf. | ||
Mechanisms for such a process have been proposed in the literature and typically involve bimolecular metal displacement.24 Further studies were conducted to shed light on this reaction (5 → 12a/b, Table 1), at first focusing on the concentration of reactants. A qualitatively striking change in the time required to reach full conversion to 12a/b (from 45 min to 6 h) was observed, when the concentration of anti-complex 5 was lowered from 7.1 × 10−2 M to 1.4 × 10−2 M in THF-d8.6 Conversely, the addition of Pd(0) in the form of Pd(dba)2 accelerated the entire process, leading to full conversion in less than 5 min. Furthermore, the inhibition of conversion by addition of free ligand L2c suggests that a bimolecular process, which critically relies on the metal coordination sphere and is not promoted by nucleophilic displacement by a phosphorus centre, could be operative.20 It moreover becomes apparent that the thermodynamic value of internal coordination is remarkably high in these systems.
| Concentration (10−2 M) | Additive | Conversion time (min) |
|---|---|---|
| a No facial exchange observed. Complete decomposition in 250 min. | ||
| 7.1 | AgOTf, 3.0 eq. | 45 |
| 1.4 | AgOTf, 3.0 eq. | 360 |
| 1.4 | Pd(dba)2, 4.0 eq. and AgOTf, 3.0 eq. | <5 |
| 1.4 | L2c, 8.0 eq. and AgOTf, 3.0 eq. | —a |
| 1.4 | AgOTf, 11 eq. | 20 |
| 1.4 | AgBF4, 8.0 eq. and AgOTf, 3.0 eq. | 10 |
We thus returned to X-ray and NMR measurements of complex 7 in search of indications for aggregation to support the proposed bimolecular process. Standing at 2.575 Å, the distance between the bromine atom of one molecule in the unit cell and the N–H moiety of the next molecule is shorter than expected and suggestive of an intermolecular H-bond.6 However this might also be explained by packing effects in the solid state, which is why we investigated self-diffusion coefficients and the concentration dependence of chemical shifts in solution state NMR spectroscopy. Unfortunately, no conclusive results concerning aggregation could be obtained from the diffusion ordered spectroscopy (DOSY) spectrum of complex 7a/b in THF solution (data not shown). However, both 1H and 31P resonances show pronounced differences in 0.1 M and 0.01 M solution, clearly pointing towards aggregation playing a role in solution.6,25
Conclusion
In summary, we have identified structural features of novel internally coordinated, monohaptoallylpalladium(II) species and directly investigated their dynamic behaviour in solution. The rare possibility to observe the two limiting η1-allylpalladium intermediates of an asymmetric allylic alkylation process allowed us to propose a mechanism for their interconversion (based on an η1 → η3 → η1 isomerisation) and to obtain support by combined DFT and NMR studies (in solution) and X-ray analysis. The interplay between structure and reactivity of these species as well as the direct observation of their unusually facile isomerisation behaviour should be of direct relevance to chemistries beyond catalytic allylic alkylation, given the current prominence of chelation-directed catalytic C–H activation methodologies.Acknowledgements
Support from the Max-Planck-Institut für Kohlenforschung, the University of Vienna and the ERC (StG FLATOUT to N. M. and RDC@catalysis to C. M. T.) is gratefully acknowledged.Notes and references
- (a) E. Negishi, Handbook of Organopalladium Chemistry for Organic Synthesis, Wiley, New York, 2002 Search PubMed; (b) J. Tsuji, Palladium Reagents and Catalysts: New Perspectives for the 21st Century, John Wiley & Sons, Ltd, 2004 Search PubMed; (c) B. M. Trost and M. L. Crawley, Chem. Rev., 2003, 103, 2921–2944 CrossRef CAS PubMed; (d) R. I. McDonald, G. Liu and S. S. Stahl, Chem. Rev., 2011, 111, 2981–3019 CrossRef CAS PubMed; (e) N. Selander and K. J. Szabó, Chem. Rev., 2011, 111, 2048–2076 CrossRef CAS PubMed; (f) Y. Deng, A. K. Persson and J.-E. Bäckvall, Chem.–Eur. J., 2012, 18, 11498–11523 CrossRef CAS PubMed; (g) G. Zeni and R. C. Larock, Chem. Rev., 2006, 106, 4644–4680 CrossRef CAS PubMed.
- For selected reviews, see: (a) R. Giri, B.-F. Shi, K. M. Engle, N. Maugel and J.-Q. Yu, Chem. Soc. Rev., 2009, 38, 3242–3272 RSC; (b) K. M. Engle, T.-S. Mei, M. Wasa and J.-Q. Yu, Acc. Chem. Res., 2012, 45, 788–802 CrossRef CAS PubMed; (c) N. Nithiy, D. Rosa and A. Orellana, Synthesis, 2013, 45, 3199–3210 CrossRef CAS PubMed; (d) G. Rouquet and N. Chatani, Angew. Chem., Int. Ed., 2013, 52, 11726–11743 CrossRef CAS PubMed; (e) J. Wencel-Delord, T. Dröge, F. Liu and F. Glorius, Chem. Soc. Rev., 2011, 40, 4740–4761 RSC; (f) T. Newhouse and P. S. Baran, Angew. Chem., Int. Ed., 2011, 50, 3362–3374 CrossRef CAS PubMed; (g) O. Daugulis, H.-Q. Do and D. Shabashov, Acc. Chem. Res., 2009, 42, 1074–1086 CrossRef CAS PubMed; (h) T. W. Lyons and M. S. Sanford, Chem. Rev., 2010, 110, 1147–1169 CrossRef CAS PubMed; (i) L. Ackermann, Chem. Rev., 2011, 111, 1315–1345 CrossRef CAS PubMed.
- (a) M. Luparia, M. T. Oliveira, D. Audisio, F. Frébault, R. Goddard and N. Maulide, Angew. Chem., Int. Ed., 2011, 50, 12631–12635 CrossRef CAS PubMed; (b) D. Audisio, M. Luparia, M. T. Oliveira, D. Klütt and N. Maulide, Angew. Chem., Int. Ed., 2012, 51, 7314–7317 CrossRef CAS PubMed.
- (a) D. Audisio, G. Gopakumar, L.-G. Xie, L. G. Alves, C. Wirtz, A. M. Martins, W. Thiel, C. Farès and N. Maulide, Angew. Chem., Int. Ed., 2013, 52, 6313–6316 CrossRef CAS PubMed. For a cyclobutenyl palladium complex in η3-coordination mode, see: (b) P. J. Ridgwell, P. M. Bailey, S. N. Wetherell, E. A. Kelley and P. M. Maitlis, J. Chem. Soc., Dalton Trans., 1982, 999–1004 RSC.
- C. Souris, F. Frébault, A. Patel, D. Audisio, K. N. Houk and N. Maulide, Org. Lett., 2013, 15, 3242–3245 CrossRef CAS PubMed.
- See ESI for further details.†.
- B. M. Trost and D. L. van Vranken, Chem. Rev., 1996, 96, 395–422 CrossRef CAS PubMed.
- CCDC 991087 contains the supplementary crystallographic information for this paper.†.
- Selected examples: (a) L. D. Tran and O. Daugulis, Angew. Chem., Int. Ed., 2012, 51, 5188–5191 CrossRef CAS PubMed; (b) D. Shabashov and O. Daugulis, J. Am. Chem. Soc., 2010, 132, 3965–3972 CrossRef CAS PubMed; (c) B.-F. Shi, N. Maugel, Y.-H. Zhang and J.-Q. Yu, Angew. Chem., Int. Ed., 2008, 47, 4882–4886 CrossRef CAS PubMed; (d) T. M. Figg, M. Wasa, J.-Q. Yu and D. G. Musaev, J. Am. Chem. Soc., 2013, 135, 14206–14214 CrossRef CAS PubMed; (e) B. V. S. Reddy, L. R. Reddy and E. J. Corey, Org. Lett., 2006, 8, 3391–3394 CrossRef CAS PubMed; (f) N. Rodríguez, J. A. Romero-Revilla, M. A. Fernández-Ibánez and J. C. Carretero, Chem. Sci., 2013, 4, 175–179 RSC; (g) Y. Feng and G. Chen, Angew. Chem., Int. Ed., 2010, 49, 958–961 CrossRef CAS PubMed; (h) W. R. Gutekunst and P. S. Baran, J. Am. Chem. Soc., 2011, 133, 19076–19079 CrossRef CAS PubMed; (i) R. Giri, N. Maugel, B. M. Foxman and J.-Q. Yu, Organometallics, 2008, 27, 1667–1670 CrossRef CAS.
- Virtually all the crystalline samples picked up from different crops were consistent with isomer 7a only. This implies that, in the crystalline phase, the complex 7 is predominantly comprised of isomer 7a.
- B. Böttcher, V. Schmidts, J. A. Raskatov and C. M. Thiele, Angew. Chem., Int. Ed., 2010, 49, 205–209 CrossRef PubMed.
- General Reviews on RDCs: (a) B. Böttcher and C. M. Thiele, in eMagRes, John Wiley & Sons, Ltd, 2012 Search PubMed; (b) R. R. Gil, Angew. Chem., Int. Ed., 2011, 50, 7222–7224 CrossRef CAS PubMed; (c) G. Kummerlöwe and B. Luy, Annu. Rep. NMR Spectrosc., 2009, 68, 193–230 CrossRef.
- The details for the preparation of the PDMS sticks used will be the subject of a further report. For seminal work on the use of cross-linked PDMS for RDC measurements, see: J. C. Freudenberger, P. Spiteller, R. Bauer, H. Kessler and B. Luy, J. Am. Chem. Soc., 2004, 126, 14690–14691 CrossRef CAS PubMed.
- C. M. Thiele and W. Bermel, J. Magn. Reson., 2012, 216, 134–143 CrossRef CAS PubMed.
- F. Neese, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2012, 2, 73–78 CrossRef CAS PubMed.
- (a) R. Berger, C. Fischer and M. Klessinger, J. Phys. Chem. A, 1998, 102, 7157–7167 CrossRef CAS; (b) V. Schmidts, PhD thesis, Technische Universität Darmstadt, Darmstadt, Germany, 2013.
- C. M. Thiele, K. Petzold and J. Schleucher, Chem.–Eur. J., 2009, 15, 254–260 CrossRef PubMed.
- R. Brüschweiler and F. Zhang, J. Chem. Phys., 2004, 120, 5253–5260 CrossRef PubMed.
- C. Johansson, G. C. Lloyd-Jones and P.-O. Norrby, Tetrahedron: Asymmetry, 2010, 21, 1585–1592 CrossRef CAS PubMed.
- A dissociative pathway involving phosphine dissociation prior to isomerization was also considered on account of the potential assistance by the amide carbonyl. This pathway is computed to be considerably higher in energy (ΔG≠diss = 32.4 kcal mol−1) and is thus included in the ESI.†.
- See the ESI† for a discussion of results for additional conformers with and without dispersion corrections.
- A. Aullón, D. Bellamy, L. Brammer, E. A. Bruton and A. G. Orpen, Chem. Commun., 1998, 653–654 RSC.
- (a) E. Hartmann and R. M. Gschwind, Angew. Chem., Int. Ed., 2013, 52, 2350–2354 CrossRef CAS PubMed; (b) R. K. Castellano, F. Diederich and E. A. Meyer, Angew. Chem., Int. Ed., 2003, 42, 1210–1250 CrossRef PubMed; (c) M. O. Sinnokrot and C. D. Sherrill, J. Am. Chem. Soc., 2004, 126, 7690–7697 CrossRef CAS PubMed; (d) E. C. Lee, B. H. Hong, J. Y. Lee, J. C. Kim, D. Kim, Y. K. Kim, P. Tarakeshwar and K. S. Kim, J. Am. Chem. Soc., 2005, 127, 4530–4537 CrossRef CAS PubMed; (e) E. C. Lee, D. Kim, P. Jurečka, P. Tarakeshwar, P. Hobza and K. S. Kim, J. Phys. Chem. A, 2007, 111, 3446–3457 CrossRef CAS PubMed.
- (a) K. L. Granberg and J.-E. Bäckvall, J. Am. Chem. Soc., 1992, 114, 6858–6863 CrossRef CAS; (b) J.-E. Bäckvall, J. O. Vågberg, C. Zercher, J. P. Genêt and A. Denis, J. Org. Chem., 1987, 52, 5430–5435 CrossRef; (c) M. T. Oliveira, D. Audisio, S. Niyomchon and N. Maulide, ChemCatChem, 2013, 5, 1239–1247 CrossRef CAS PubMed.
- A. Mitra, P. J. Seaton, R. A. Assarpour and T. Williamson, Tetrahedron, 1998, 54, 15489–15498 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental procedures, X-ray crystallographic data, characterization data, chiral chromatographic analyses, and computational details. CCDC 991087. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5sc01867f |
| ‡ L. X. and V. B. contributed equally. |
| This journal is © The Royal Society of Chemistry 2015 |