
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceConstructing real-time, wash-free, and reiterative sensors for cell surface proteins using binding-induced dynamic DNA assembly†
Yanan
Tang
a,
Zhixin
Wang
b,
Xiaolong
Yang
a,
Junbo
Chen
bc,
Linan
Liu
d,
Weian
Zhao
d,
X. Chris
Le
b and
Feng
Li
*a
aDepartment of Chemistry, Centre for Biotechnology, Brock University, St. Catharines, L2S3A1, Canada. E-mail: fli@brocku.ca
bDepartment of Laboratory Medicine and Pathology, University of Alberta, Edmonton, T6G2G3, Canada
cAnalytical & Testing Center, Sichuan University, Chengdu, Sichuan 610064, China
dDepartment of Pharmaceutical Sciences, Sue and Bill Gross Stem Cell Research Center, Chao Family Comprehensive Cancer Center, Department of Biomedical Engineering, Edwards Lifesciences Center for Advanced Cardiovascular Technology, University of California at Irvine, Irvine, USA
First published on 8th July 2015
Abstract
Cell surface proteins are an important class of biomarkers for fundamental biological research and for disease diagnostics and treatment. In this communication, we report a universal strategy to construct sensors that can achieve rapid imaging of cell surface proteins without any separation by using binding-induced dynamic DNA assembly. As a proof-of-principle, we developed a real-time and wash-free sensor for an important breast cancer biomarker, human epidermal growth factor receptor-2 (HER2). We then demonstrated that this sensor could be used for imaging and sensing HER2 on both fixed and live breast cancer cells. Additionally, we have also incorporated toehold-mediated DNA strand displacement reactions into the HER2 sensor, which allows for reiterating (switching on/off) fluorescence signals for HER2 from breast cancer cells in real-time.
The rapid development in the field of dynamic DNA nanotechnology has greatly impacted the current practice in designing sensors and assays for biomolecules by providing novel homogeneous signal generation mechanisms.1 A unique feature of these mechanisms is the adoption of the DNA strand displacement reaction that is accelerated through a short single-stranded sticky end known as DNA toehold to induce the changes of thermodynamically stable DNA structures or assemblies.1 Many toehold-mediated DNA strand displacement strategies, such as strand displacement beacon,2 hybridization chain reactions,3 and catalytic hairpin assemblies,4 have been successfully used for analysing and imaging nucleic acid targets in solution or in living cells. To expand this powerful strand displacement strategy for protein analysis, we have previously developed the concept and strategies of binding-induced dynamic DNA assembly,5 where we harness sandwiched affinity binding complexes to accelerate DNA strand displacement rather than DNA toeholds. The success of our strategies have led to the rapid development of various sensors or assays for solution-based protein analyses.6 In this communication, we aim to further expand binding-induced dynamic DNA assembly to cell imaging applications by tailoring it into a universal strategy to construct sensors for cell surface proteins.
Cells interact and communicate with their surrounding microenvironments through a diverse range of proteins that are displayed on cell surfaces.7 Because distinct patterns of cell surface proteins are expressed among difference cell types or at different stages of the same cell, they have been frequently used as markers for identifying specific cell types or subtypes.8 Many cell surface proteins are also important biomarkers for disease diagnostics. For example, human epidermal growth factor receptor-2 (HER2) has been found to be overexpressed in approximately 20–30% of breast cancer tumours and thus becomes an important biomarker for breast cancer diagnostics and treatment.9 Similar to HER2, a large number of cell surface proteins are specific receptors for important biological processes and their expression levels are highly dynamic. Therefore, techniques that are able to rapidly monitor the expression levels of cell surface proteins will be highly valuable to increase the accuracy and throughput of cell identification and disease diagnostics, as well as to obtain enriched information on how cells respond to external stimuli, e.g., drugs, ligands, or other changes in the microenvironments. However, conventional immunostaining techniques relying on antibodies that are conjugated with fluorescent organic dyes or nanoparticles to label specific proteins of interest commonly require tedious cell fixation and washing steps to achieve sufficient signal to background ratios for cell imaging and analysis, and thus are not ideal to achieve the rapid monitoring of cell surface proteins.10 To this end, we leverage the homogeneous signal generation capability of binding-induced dynamic DNA assemblies to construct real-time, wash-free, and reiterative sensors for cell surface proteins.
The principle of our strategy is illustrated in Fig. 1A. The target recognition is achieved through the binding of two specific antibodies to the same target cell surface protein. Because various antibodies are readily available for most cell surface proteins, the use of antibodies as target recognition components ensures the generalizability of our strategy. Moreover, the availability of two or more antibodies binding to the same target protein has been demonstrated by the widely used proximity ligation assays.11 The signal generation component is designed based on the principle of binding-induced dynamic DNA assembly (details are described in the ESI and Fig. S1†).5 Briefly, DNA probes TB and B*C are each conjugated to one of the two antibodies that can bind to the same target but at different epitopes through a flexible poly-thymine linker. TB and B*C are designed to have 6 complementary bases, so that they cannot form a stable DNA duplex at room temperature or 37 °C. However, in the presence of the target cell surface protein, the binding of the two antibodies to the same protein brings TB and B*C to close proximity, greatly increasing their local effective concentrations. Consequently, TB and B*C hybridize to each other to form a stable TB:B*C DNA duplex. Once forming the duplex, TB:B*C triggers a subsequent toehold-mediated strand displacement reaction with a reporter DNA duplex T*C*:C, forming a binding-induced DNA three-way junction (TB:B*C:T*C*) and releasing the probe C. Because a pair of fluorophore and quencher is initially conjugated to T*C* and C, the formation of the binding-induced DNA three-way junction (TWJ) separates the fluorophore from the quencher, turning on the fluorescence for the target protein on the cell.
 | ||
| Fig. 1 (A) Schematic illustrating the principle of the real-time cell surface sensor. (B) Confocal fluorescent microscopy images of SK-BR-3 cells incubated with real-time HER2-specific DNA sensors (25 nM TB, 25 nM B*C, and 50 nM T*C*:C) and 100 nM 4′,6-diamidino-2-phenylindole (DAPI) at RT for 30 min. (C) Confocal fluorescent microscopy images of SK-BR-3 cells incubated with isotype control probes and 100 nM DAPI. Scale bars at x-axis represent 9 μm. | ||
To demonstrate the proof-of-principle, we chose HER2 as a target cell surface protein due to its important roles in breast cancer biology and treatments.6 DNA probes TB and B*C were conjugated to HER2 specific polyclonal antibodies through streptavidin–biotin interactions. The use of polyclonal antibodies here is a practical way to construct HER2 sensors, because the high binding diversity of polyclonal antibodies will ensure a portion of antibody molecules to form correct proximity pairs on each HER2 molecule and trigger subsequent formation of binding-induced DNA TWJ. We first examined DNA sensors for analyzing HER2 proteins in buffer (Fig. S2A†). Fig. S2B† shows the fluorescence signal increase of the sensor as a function of time. Within a period of 5 min, fluorescence intensities from 10 nM HER2 are readily distinguishable from the blank (green curve) that contained all reagents but not the target HER2. The rapid fluorescence increase suggests that it is possible to achieve real-time sensing using our sensor system. To test the specificity of the sensor, we also designed an isotype control (I.C. in Fig. S2B†) that was constructed the same way as the HER2 sensor except that nonspecific antibodies were used instead of HER2 specific antibodies. No fluorescence signal increase was observed from this control group, confirming the target-specificity of our sensor.
Having established the HER2 sensor in solution, we further demonstrate that this sensor can be used to light up endogeneous HER2 from breast cancer cells. To achieve this goal, we chose breast cancer cell line SK-BR-3 as a model system, because of its high HER2 expression level. The ability to homogeneously stain endogenous HER2 using our DNA sensor was first tested by labelling HER2 from fixed SK-BR-3 cells. As shown in Fig. 1B, after incubating sensor components (TB, B*C, and T*C*:C) with fixed SK-BR-3 cells for 30 min, intensive fluorescence signals were found to be generated at cell surfaces through the observation under a confocal fluorescence microscope. Further comparing our sensor with an isotype control (Fig. 1C) and a non-HER2 expression bladder cancer cell T24 (Fig. S3†), we confirmed that our sensor is HER2-specific. To illustrate the function of each sensor component, we also compared the intact sensor with a number of controls shown in Fig. S4.† No fluorescence was observed in the absence of B*C (Fig. S4B†), TB (Fig. S4C†), or both (Fig. S4D†), suggesting that fluorescence can only be turned on when dynamic DNA assembly happens. We have also compared our strategy with two other standard fluorescence immunostaining strategies (Fig. S5†). Our strategy has demonstrated comparable signal-to-background ratio (S/B = 4.2 ± 0.6, Fig. S6,†vs. S/B = 3.7 ± 0.8 for FITC-labeled HER2 Ab, and 4.0 ± 0.8 for PE-labelled HER2 Ab), but is much faster and simpler due to the elimination of all washing steps during fluorescence staining.
We next examined the real-time sensing capability of our DNA sensor. To enable the real-time sensing, it is essential that both target recognition and signal transduction components of the sensor are rapid. We have previously demonstrated that near-instantaneous signal transduction can be achieved by tuning the sequences of DNA probes.5 Therefore, the affinity binding is likely to be the rate limiting step here, since the optimal DNA designs have been used. To ensure a fast binding between HER2 specific antibodies and HER2 proteins, 25 nM DNA probes TB and B*C were used instead of that in solution-based experiments (10 nM, Fig. S2†). Further increase in DNA probe concentrations will lead to nonspecific staining. In the experiment, fixed SK-BR-3 cells were first labeled with 4′,6-diamidino-2-phenylindole (DAPI) and observed under a confocal fluorescent microscope to determine a proper position for subsequent real-time staining. HER2 specific DNA sensors were then added into the same culture dish and time-lapse fluorescence imaging was carried out immediately with a 30 s interval. As shown in Fig. 2, HER2 proteins are clearly stained at cell surfaces within 2 min and optimal fluorescence signal was achieved within 5 min. Further quantitative analyses of the cell imaging data (Fig. 3) revealed that detectable fluorescence was achieved for almost all cells as early as 30 s and increased linearly as a function of time over the 5 min measurement window.
 | ||
| Fig. 2 Real-time imaging of HER2 from fixed SK-BR-3 cells using HER2-specific DNA sensors. Time-lapse confocal fluorescent microscopy images of SK-BR-3 cells were taken at a 30 s interval immediately after mixing cells with HER2-specific DNA sensors that contained 25 nM TB, 25 nM B*C, and 50 nM T*C*:C. Scale bars represent 20 μm. | ||
 | ||
| Fig. 3 Quantitative measurement of fluorescence increase from each single cell (A) and total fluorescence increase (B) as a function of time. All fluorescence measurements were carried out using ImageJ 1.47. For each cell, fluorescence = integrated density − (area of selected cell × mean fluorescence of background readings).12 | ||
Once establishing real-time cell sensing using fixed cells, we aim to achieve live cell sensing and analysis. We first attempted to use our sensors to image live cells directly from culture media. However, high background fluorescence was observed likely due to the degradation of DNA probes in the media. We later found that a brief washing with PBS buffer can effectively solve this issue. This observation makes our sensor particularly useful to analyze live cells using flow cytometry. Fig. 4 shows a typical result of the flow cytometric analysis of live SK-BR-3 cells stained using HER2 DNA sensor. After incubating live SK-BR-3 cells with HER2 sensors in PBS buffer for 10 min, fluorescence signals are readily distinguishable from negative controls (SK-BR-3 cells, no sensor). Fluorescence signals from the isotype control and the T*C*:C control (a mixture of SK-BR-3 cells and T*C*:C) are identical to the negative control. These results suggest that our sensor can be used to analyze cell surface proteins from live cells using flow cytometry.
 | ||
| Fig. 4 Flow cytometry analysis of live SK-BR-3 cells that are labeled with HER2 specific DNA sensors. Fluorescent intensities are displayed in a hyperlog scale at x-axis. | ||
Having constructed real-time cell surface sensors, we further explored the possibility to construct reiterative sensors for switching on/off fluorescence signals of cellular HER2 in real-time. The erasable and reiterative cell imaging capabilities are highly desirable to highlight specific cellular proteins and to achieve multiplexity and logic gating.13–15 Previously, Diehl and coworkers have developed reiterative fluorescent cell imaging strategies through toehold-mediated DNA strand displacement.13 As our real-time sensor is also constructed through dynamic DNA assemblies, we propose that it is also possible to construct erasable and reiterative DNA probes by incorporating toehold-mediated DNA strand displacement components into our real-time sensor. Moreover, we aim to construct reiterative DNA sensors that are able to switch on/off fluorescence for specific cell proteins in real-time.
To achieve reiterative cell imaging using our sensor, we extended the reporter DNA duplex T*C*:C with 6 DNA nucleotides (motif E*) and the new reporter duplex is denoted as E*T*C*:C (Fig. 5A). An erasing probe ETC is designed accordingly, which is a fully complementary DNA strand to E*T*C* but labeled with a quencher. In the presence of a target cell that expresses specific cell surface proteins, DNA probes TB, B*C, and E*T*C*:C assemble into a binding-induced DNA TWJ TB:B*C:E*T*C* and turn on fluorescence signals. Once formed, this binding-induced DNA TWJ contains a sticky end (motif E*) that can serve as a DNA toehold to trigger toehold-mediated DNA strand displacement reactions. To specifically erase the fluorescence signals, erasing probe ETC is added at a concentration equal to probe E*T*C*:C, and a subsequent toehold-mediated strand displacement reaction occurs between TB:B*C:E*T*C* and ETC to form a E*T*C*:ETC duplex. As a result, fluorophore labeled E*T*C* is removed from target and fluorescence is quenched within the E*T*C*:ETC duplex. In the same process, binding-induced DNA duplex TB:B*C is regenerated and fluorescence can be turned on again if reporter DNA duplex E*T*C*:C is added again.
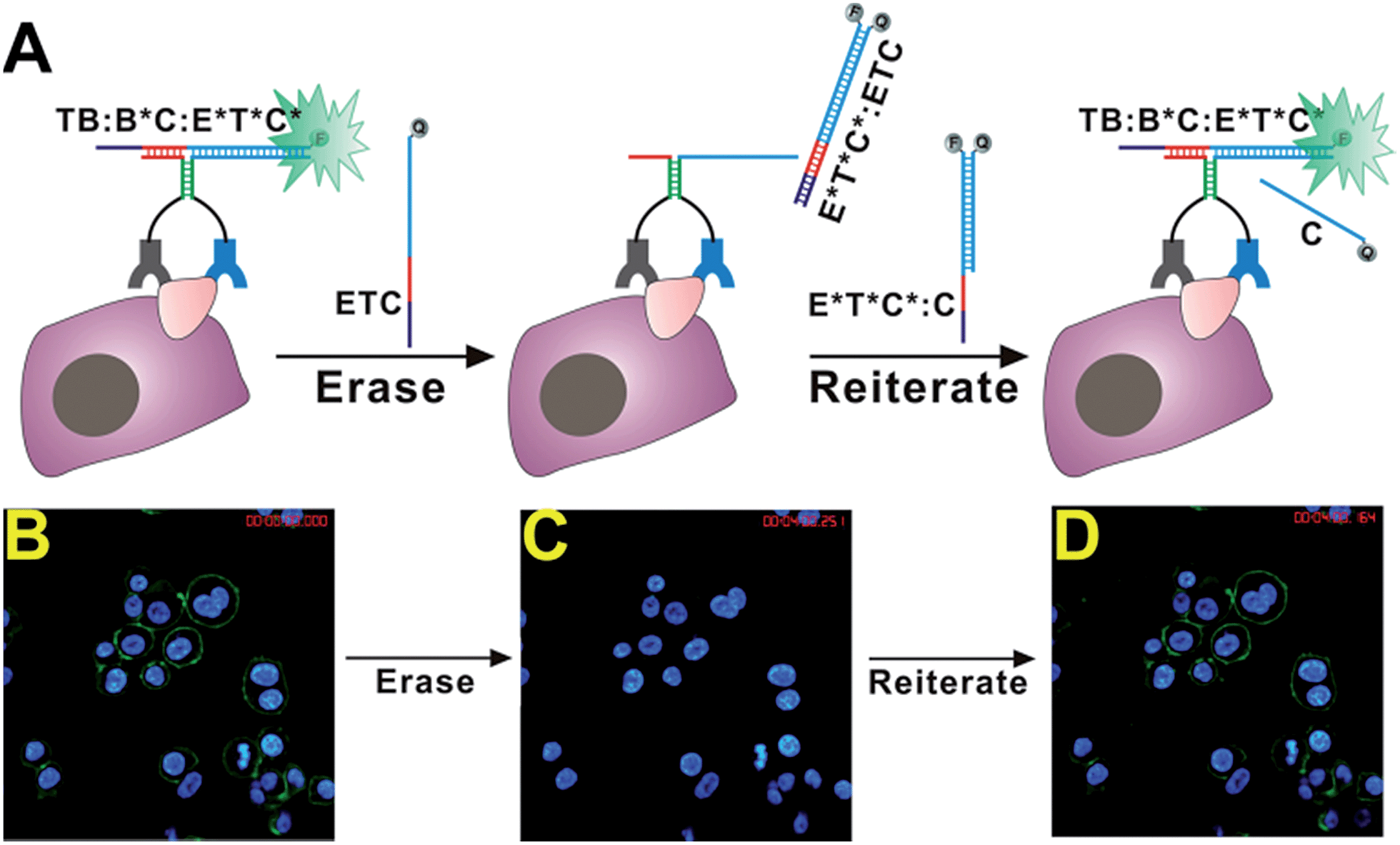 | ||
| Fig. 5 (A) Schematic illustrating the principle of real-time erasing and reiterating fluorescence for HER2 using DNA strand displacement. (B–D) Fluorescent imaging of SK-BR-3 cells before erasing fluorescence (B), after erasing fluorescence (C), and after regenerating fluorescence (D). | ||
To examine this strategy in experiments, we first labeled HER2 proteins on fixed SK-BR-3 using the HER2 sensor containing TB, B*C, and E*T*C*:C (Fig. 5B). After incubating the sensor components with cells for 5 min, erasing DNA probe ETC was added into the same culture dish and fluorescently labeled cells were monitored using the time-lapse confocal fluorescent microscopy at a 1 min interval. As shown in Fig. S7,† fluorescence signals decrease was clearly observed after incubating cells with erasing probe ETC for 1 min. The fluorescence was completely turned off within 4 min (Fig. S7† and 5C), confirming that our strategy can effectively erase fluorescent signals from cell surfaces through simple DNA strand displacement. Once turning off the fluorescence signal from cell surfaces, we then examined whether our sensor could be turned on again by adding reporter DNA duplex E*T*C*:C back into the system. As shown in Fig. S8,† immediately after adding E*T*C*:C, fluorescence on cell surfaces was regenerated in real-time (Fig. 5D). These results suggest that our sensor system is high flexible and can easily be programmed through DNA hybridizations to achieve desired functions for cell imaging and analysis.
Conclusions
In conclusion, we have successfully developed a novel sensing strategy that is able to image and analyze specific cell surface proteins in real-time. This newly developed sensing/imaging strategy is highly versatile, and thus can potentially be applied to any target cell protein of interest by simply changing a pair of specific antibodies. The rapid and wash-free target recognition and signal transduction capability makes this strategy desirable to image and analyze both fixed and live cells in real-time. This feature can be particularly useful to be integrated with current cell isolation techniques to facilitate the rapid detection of circulating tumor cells from patient blood samples. Efforts to optimize sensor designs to achieve real-time imaging of live cells directly from culture media or complicated sample matrix (e.g., blood samples) are currently underway. The homogeneous, isothermal, and enzyme-free signal generation capability also makes our sensor a potentially useful tool for in vivo imaging or sensing applications. Recently, a number of structure-switching aptamer sensors have been successfully applied to in vivo imaging of cancer cells or cytokines.16 Because our sensor also makes use of DNA as structural scaffold and signal transduction component, it is possible for our sensor to bypass challenges arisen from complicated sample matrix, e.g., diffusion limit in viscous matrix, chemical stability, and eventually achieve the goal of in vivo sensing and imaging. The ability to real-timely erase and reiterate fluorescence labels of specific cellular proteins can potentially be used to design better highlightable and multiplexed imaging strategies. Owing aforementioned special features, we anticipate that our cell sensing/imaging strategy will find wide applications both in fundamental research and in personalized disease diagnostics.Acknowledgements
We thank the Natural Sciences and Engineering Research Council of Canada and the Brock University Start-Up Fund for financial support.References
- H. Zhang, F. Li, B. Dever, X. F. Li and X. C. le, Chem. Rev., 2013, 113, 2812–2841 CrossRef CAS PubMed; D. Y. Zhang and G. Seelig, Nat. Chem., 2011, 3, 103–113 CrossRef PubMed; C. Jung and A. D. Ellington, Acc. Chem. Res., 2014, 47, 1825–1835 CrossRef PubMed.
- D. Y. Zhang, S. X. Chen and P. Yin, Nat. Chem., 2012, 4, 208–214 CrossRef CAS PubMed.
- R. M. Dirks and N. A. Pierce, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 15274–15278 CrossRef CAS PubMed; H. M. Choi, V. A. Beck and N. A. Pierce, ACS Nano, 2014, 8, 4284–4294 CrossRef PubMed; Z. Wu, G. Q. Liu, X. L. Yang and J. H. Jiang, J. Am. Chem. Soc., 2015, 137, 6829–6836 CrossRef PubMed.
- P. Yin, H. M. T. Choi, C. R. Calvert and N. A. Pierce, Nature, 2008, 451, 318–322 CrossRef CAS PubMed; B. Li, A. D. Ellington and X. Chen, Nucleic Acids Res., 2011, 39, e110 CrossRef PubMed; Y. S. Jiang, B. Li, J. N. Milligan, S. Bhadra and A. D. Ellington, J. Am. Chem. Soc., 2013, 135, 7430–7433 CrossRef PubMed; C. Wu, S. Cansiz and L. Zhang, et al. , J. Am. Chem. Soc., 2015, 137, 4900–4903 CrossRef PubMed.
- F. Li, H. Zhang, Z. Wang, X. Li, X. F. Li and X. C. le, J. Am. Chem. Soc., 2013, 135, 2443–2446 CrossRef CAS PubMed; F. Li, H. Zhang, C. Lai, X. F. Li and X. C. le, Angew. Chem., Int. Ed., 2012, 51, 9317–9320 CrossRef PubMed; H. Zhang, F. Li, B. Dever, C. Wang, X. F. Li and X. C. le, Angew. Chem., Int. Ed., 2013, 52, 10698–106705 CrossRef PubMed; F. Li, Y. Lin and X. C. le, Anal. Chem., 2013, 85, 10835–10841 CrossRef PubMed.
- C. Zong, J. Wu, M. Liu, F. Yan and H. Ju, Chem. Sci., 2015, 6, 2602–2607 RSC; K. Ren, J. Wu, H. Ju and F. Yan, Anal. Chem., 2015, 87, 1694–1700 CrossRef CAS PubMed; B. Deng, J. Chen and H. Zhang, Anal. Chem., 2014, 86, 7009–7016 CrossRef PubMed; K. Ren, J. Wu, F. Yan and H. Ju, Sci. Rep., 2014, 4, 4360 Search PubMed; L. Zhang, K. Zhang, G. Liu, M. Liu, Y. Liu and J. Li, Anal. Chem., 2015, 87, 5677–5682 CrossRef PubMed.
- M. D. Mager, V. LaPointe and M. M. Stevens, Nat. Chem., 2011, 3, 582–589 CrossRef CAS PubMed; M. M. Ali, D. K. Kang, M. Fu, J. M. Karp and W. Zhao, Wiley Interdiscip. Rev.: Nanomed. Nanobiotechnol., 2012, 4, 547–561 CrossRef PubMed.
- F. J. Lv, R. S. Tuan, K. M. Cheung and V. Y. Leung, Stem Cells, 2014, 32, 1408–1419 CrossRef CAS PubMed; I. C. Ho, T. S. Tai and S. Y. Pai, Nat. Rev. Immunol., 2009, 9, 125–135 CrossRef PubMed.
- Z. Mitri, T. Constantine and R. O'Regan, Chemother. Res. Pract., 2012, 2012, 743193 Search PubMed.
- I. D. Odell and D. J. Cook, J. Invest. Dermatol., 2013, 133, e4 CrossRef CAS PubMed.
- M. Gullberg, S. M. Gustafsdottir, E. Schallmeiner, J. Jarvius, M. Bjarnegard, C. Betsholtz, U. Landegren and S. Fredriksson, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 8420–8424 CrossRef CAS PubMed.
- A. Burgess, S. Vigneron, E. Brioudes, J. C. Labbé, T. Lorca and A. Castro, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 12564–12569 CrossRef CAS PubMed.
- R. M. Schweller, J. Zimak, D. Y. Duose, A. A. Qutub, W. N. Hittelman and M. R. Diehl, Angew. Chem., Int. Ed., 2012, 51, 9292–9296 CrossRef CAS PubMed.
- M. You, G. Zhu, T. Chen, M. J. Donovan and W. Tan, J. Am. Chem. Soc., 2015, 137, 667–674 CrossRef CAS PubMed.
- M. Rudchenko, S. Taylor, P. Pallavi, A. Dechkovskaia, S. Khan, V. P. Butler, S. Rudchenko and M. N. Stojanovic, Nat. Nanotechnol., 2013, 8, 580–586 CrossRef CAS PubMed.
- H. Shi, X. He, K. Wang, X. Wu, X. Ye, Q. Guo, W. Tan, Z. Qing, X. Yang and B. Zhou, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 3900–3905 CrossRef CAS PubMed; W. Zhao, S. Schafer, J. Choi, Y. J. Yamanaka, M. L. Lombardi, S. Bose, A. L. Carlson, J. A. Phillips, W. Teo, I. A. Droujinine, C. Cui, R. K. Jain, J. Lammerding, J. C. Love, C. P. Lin, D. Sarkar, R. Karnik and J. M. Karp, Nat. Nanotechnol., 2011, 6, 524–531 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5sc01870f |
| This journal is © The Royal Society of Chemistry 2015 |