
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A recyclable polyoxometalate-based supramolecular chemosensor for efficient detection of carbon dioxide†
Haibing
Wei
ab,
Jinlong
Zhang
a,
Nan
Shi
a,
Yang
Liu
a,
Ben
Zhang
a,
Jie
Zhang
*a and
Xinhua
Wan
*a
aBeijing National Laboratory for Molecular Sciences, Key Laboratory of Polymer Chemistry and Physics of Ministry of Education, College of Chemistry and Molecular Engineering, Peking University, Beijing 100871, China. E-mail: jz10@pku.edu.cn; xhwan@pku.edu.cn
bSchool of Chemistry and Chemical Engineering, Provincial Key Laboratory of Advanced Functional Materials and Devices, Hefei University of Technology, Hefei, Anhui 230009, China
First published on 7th September 2015
Abstract
A new type of supramolecular chemosensor based on the polyoxometalate (POM) Na9DyW10O36 (DyW10) and the block copolymer poly(ethylene oxide-b-N,N-dimethylaminoethyl methacrylate) (PEO114-b-PDMAEMA16) is reported. By taking advantage of the CO2 sensitivity of PDMAEMA blocks to protonate the neutral tertiary amino groups, CO2 can induce the electrostatic coassembly of anionic DyW10 with protonated PDMAEMA blocks, and consequently trigger the luminescence chromism of DyW10 due to the change in the microenvironment of Dy3+. The hybrid complex in dilute aqueous solution is very sensitive to CO2 content and shows rapid responsiveness in luminescence. The luminescence intensity of the DyW10/PEO-b-PDMAEMA complex increases linearly with an increasing amount of dissolved CO2, which permits the qualitative and quantitative detection of CO2. The complex solution also shows good selectivity for CO2, with good interference tolerance of CO, N2, HCl, H2O and SO2. The supramolecular chemosensor can be recycled through disassembly of the hybrid complex by simply purging with inert gases to remove CO2.
Introduction
Carbon dioxide (CO2) is a known greenhouse gas which is responsible for global climate change and also related to many human diseases, such as hypercapnia, hypocapnia and metabolic disorders, as well as being important in coalmine safety and volcanic activity.1 CO2 sensing and detecting is of great significance. For example, monitoring of dissolved CO2 in arterial blood allows a timely clinical response in the case of patients with pneumonia or acute respiratory distress syndrome.2 Nowadays, CO2 detecting and sensing methods, including electrochemical systems,3 near-infrared spectroscopic techniques,4 gas chromatography5 and optical chemosensors6 are well-established. Among these, state-of-the-art Severinghaus-type electrochemical CO2 sensors are widely used in commercial clinical blood gas analyzers, but these probes still suffer from a long response time and are only capable of detecting a relatively high CO2 concentration, because their working performance relies on diffusion and the establishment of an equilibrium between the internal pH electrode and the sample.7 Alternatively, CO2 chemosensor systems based on colorimetric and fluorimetric analysis take advantage of the outcome of chromism visible to the naked eye, which can be helpful for rapid on-site monitoring.8 A few pH indicators or pH dependent fluorescent dyes have been used to produce such a type of optical sensor.6a,8c–e However, pH-dependent organic chromatic molecules that can simultaneously meet the needs of appropiate pKa, specificity, photostability, and contrast are very limited.Supramolecular chemosensors are promising candidates to overcome the limitations of conventional optical chemosensors. Specially designed supramolecular chemosensors are fabricated by noncovalent binding between the molecular recognizer and the molecular reporter, and the sensing works by a relay mechanism:9 the recognizer detects the analyte, then communicates with the reporter by physical or chemical means, and eventually the optical signals of the reporter are switched on. This strategy integrates the complementary functions of multiple components, and is advantageous for CO2 sensing in that it greatly expands the options of chromatic molecules and improves sensitivity and stability. In addition, by taking advantage of the dynamic nature of the noncovalent binding of supramolecular systems, device recyclability can be achieved, which is crucial for low cost and environmental concerns. Tang et al.8b developed a CO2 sensor based on a fluorogen with aggregation-induced emission in dipropylamine. Besides this, supramolecular CO2 sensors are still rare and it is a challenge to elaborately construct a supramolecular system for CO2 detection with the characteristics of high sensitivity, low detection limit, specificity, photostability, and recyclability.
Recently we have discovered hybrid supramolecular systems with the variable luminescence properties of lanthanide-containing polyoxometalates (POMs) by coacervate complexation with block polyelectrolytes.10 The lanthanide-containing polyoxometalates possess excellent photoluminescent properties, i.e. narrow emission bands, large Stokes shifts, long lifetimes and photostability, and are sensitive to ambient chemical environments.11 The dysprosium-containing POM Na9DyW10O36 (DyW10) has two characteristic emission bands, viz., 4F9/2 → 6H15/2 (blue emission, λmaxem = 476 nm) and 4F9/2 → 6H13/2 (yellow emission, λmaxem = 574 nm) transitions, and their relative intensity ratio 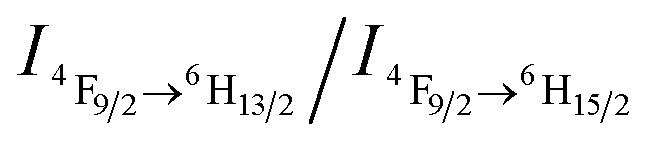 varies according to the surrounding microenvironment, which results in luminescence chromism.11a,12 Although the luminescence of lanthanide-containing POMs is not sensitive to CO2, assembly approaches can effectively tune their emission colors and intensities, and therefore their characteristics are promising when employed as supramolecular sensors.13 In the present work, we constructed a DyW10-based supramolecular chemosensor for CO2 detection and quantitation in aqueous systems with high sensitivity and specificity, rapid response and recyclability. Unlike previous CO2 chemosensors, which depend primarily on chromism of organic fluorogens at the molecular level, our supramolecular CO2 sensor is based on hybrid core–shell assemblies composed of the block copolymer poly(ethylene oxide-b-N,N-dimethylaminoethyl methacrylate) (PEO114-b-PDMAEMA16) and DyW10 in aqueous solution. By taking advantage of the CO2 sensitivity of PDMAEMA blocks to protonate the neutral tertiary amino groups,14 CO2 can induce the electrostatic coassembly of anionic DyW10 with protonated PDMAEMA blocks, and consequently trigger the luminescence chromism of DyW10 due to the change in the microenvironment of Dy3+ (Scheme 1a). The luminescence variation is closely related to the CO2 content in solution, which can be used to quantitate dissolved CO2.
varies according to the surrounding microenvironment, which results in luminescence chromism.11a,12 Although the luminescence of lanthanide-containing POMs is not sensitive to CO2, assembly approaches can effectively tune their emission colors and intensities, and therefore their characteristics are promising when employed as supramolecular sensors.13 In the present work, we constructed a DyW10-based supramolecular chemosensor for CO2 detection and quantitation in aqueous systems with high sensitivity and specificity, rapid response and recyclability. Unlike previous CO2 chemosensors, which depend primarily on chromism of organic fluorogens at the molecular level, our supramolecular CO2 sensor is based on hybrid core–shell assemblies composed of the block copolymer poly(ethylene oxide-b-N,N-dimethylaminoethyl methacrylate) (PEO114-b-PDMAEMA16) and DyW10 in aqueous solution. By taking advantage of the CO2 sensitivity of PDMAEMA blocks to protonate the neutral tertiary amino groups,14 CO2 can induce the electrostatic coassembly of anionic DyW10 with protonated PDMAEMA blocks, and consequently trigger the luminescence chromism of DyW10 due to the change in the microenvironment of Dy3+ (Scheme 1a). The luminescence variation is closely related to the CO2 content in solution, which can be used to quantitate dissolved CO2.
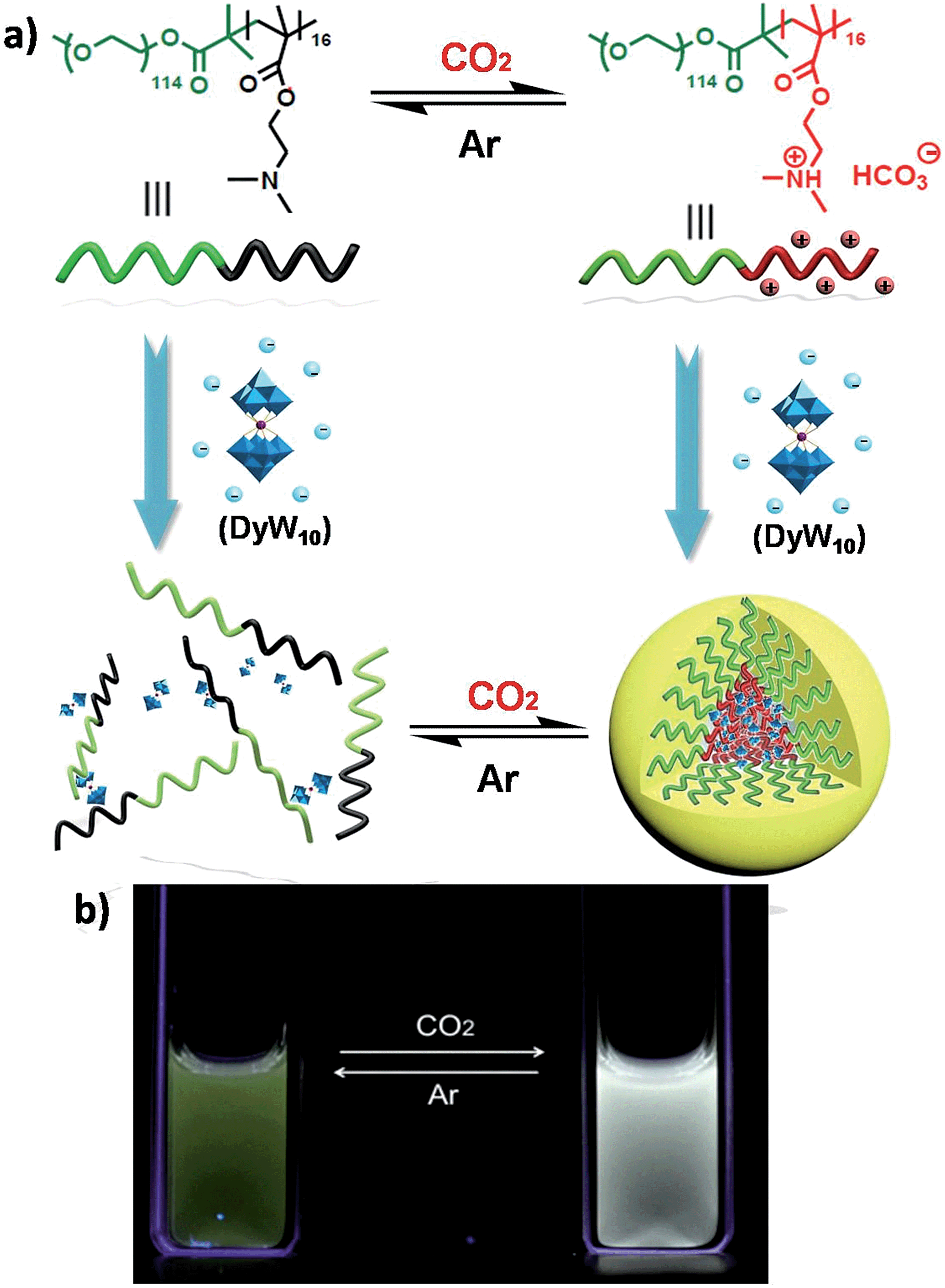 | ||
| Scheme 1 (a) Structural change of the PEO-b-PDMAEMA block copolymer and schematic representation of the reversible formation of a hybrid micelle after the reaction with CO2 in aqueous medium. (b) Photos taken under illumination with 254 nm UV light, representing the CO2-responsive luminescence chromism of the DyW10/PEO-b-PDMAEMA complex in aqueous solution before/after CO2 sensing. | ||
Results and discussion
The DyW10/PEO-b-PDMAEMA complex was prepared by the addition of PEO-b-PDMAEMA to a dilute DyW10 solution (0.2 mg mL−1, 9.8 mL), where the molar ratio of tertiary amino groups in PDMAEMA to DyW10 was set as 13.5 (the charge ratio of DyW10/PDMAEMA ∼1.0). Bubbling a small volume of CO2 gas through the solution in as short a time as <1 minute caused a striking change in the emission of DyW10 from weak green light to intense white light (Scheme 1b), while bubbling CO2 into dilute DyW10 solution for a long time did not cause a color change. The quantum yield of the DyW10/PEO-b-PDMAEMA complex increased from 0.78% to 2.10% after treatment with CO2, while the molar absorptivity was almost unchanged (∼7.0 × 103 L mol−1 cm−1 on the basis of Na9DyW10O36). As a potential CO2 sensor, the complex solution is very sensitive to dissolved CO2 content and shows rapid responsiveness. To track the luminescence variation at low contents of dissolved CO2, a certain amount of saturated CO2 aqueous solution (1.45 g L−1 at 100 kPa and 25 °C) was directly added to the hybrid complex solution. In situ PL monitoring was conducted to investigate the sensing time of the CO2, and a substantial increase in the emission intensity was observed after only 30 s. As can be seen in Fig. 1a, the luminescence intensity of the DyW10/PEO-b-PDMAEMA complex increased linearly with increasing content of dissolved CO2 with a correlation coefficient of 0.9956 (Fig. 1b). As the value of decreased with increasing CO2 concentration (Fig. S1†), the emission color gradually evolved from green to white, as could be seen with the naked eye. The detection limit of dissolved CO2 was around 1.5 mg L−1. The DyW10/PEO-b-PDMAEMA sensor responds to dissolved CO2 in the range 0–47.9 mg L−1, and the linear range can be extended by changing the initial concentration of the DyW10/PEO-b-PDMAEMA complex (Fig. S2†). Therefore, this photophysical characteristic of DyW10/PEO-b-PDMAEMA solution allows for the qualitative and quantitative detection of CO2. Moreover, upon exposure to air, atmospheric CO2 (∼300 ppm) can stimulate the luminescence variation of the DyW10/PEO-b-PDMAEMA complex system (Fig. S3†), further demonstrating its high sensitivity.
decreased with increasing CO2 concentration (Fig. S1†), the emission color gradually evolved from green to white, as could be seen with the naked eye. The detection limit of dissolved CO2 was around 1.5 mg L−1. The DyW10/PEO-b-PDMAEMA sensor responds to dissolved CO2 in the range 0–47.9 mg L−1, and the linear range can be extended by changing the initial concentration of the DyW10/PEO-b-PDMAEMA complex (Fig. S2†). Therefore, this photophysical characteristic of DyW10/PEO-b-PDMAEMA solution allows for the qualitative and quantitative detection of CO2. Moreover, upon exposure to air, atmospheric CO2 (∼300 ppm) can stimulate the luminescence variation of the DyW10/PEO-b-PDMAEMA complex system (Fig. S3†), further demonstrating its high sensitivity.
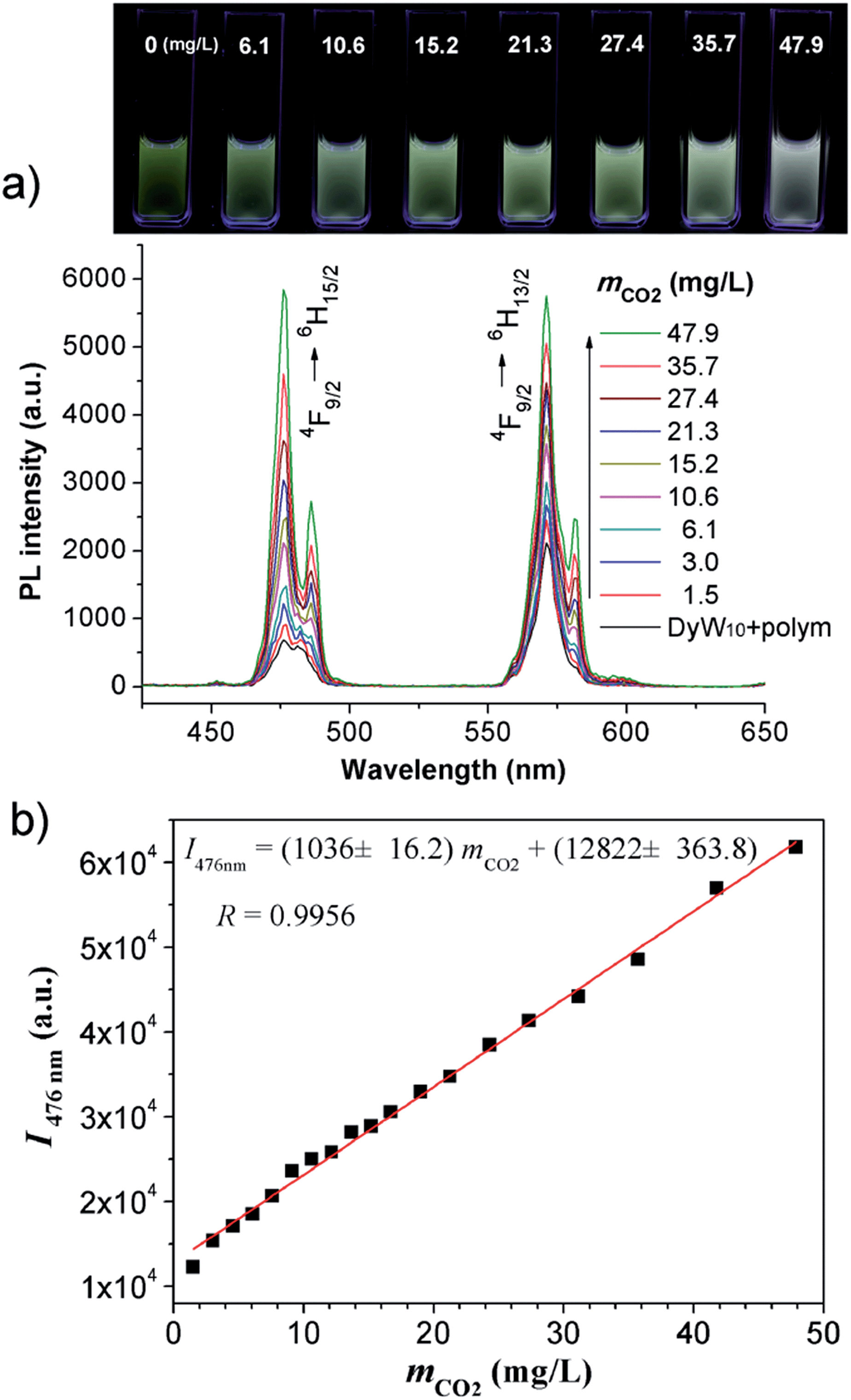 | ||
Fig. 1 (a) Variation in the PL spectra of DyW10/PEO-b-PDMAEMA hybrid complex (DyW10: 0.2 mg mL−1, 9.8 mL) in the presence of various concentrations of dissolved CO2 at 25 °C (λex = 280 nm). Insert: photograph of DyW10/PEO-b-PDMAEMA hybrid complex in aqueous solution with different concentrations of dissolved CO2 under UV illumination. (b) Plot of PL integrated intensities (blue emission, 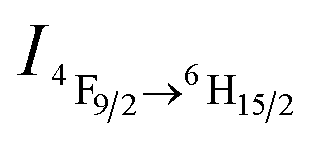 ) of the DyW10/PEO-b-PDMAEMA hybrid complex in aqueous solution as a function of dissolved CO2 concentration at 25 °C (λex = 280 nm). ) of the DyW10/PEO-b-PDMAEMA hybrid complex in aqueous solution as a function of dissolved CO2 concentration at 25 °C (λex = 280 nm). | ||
The complex solution shows good selectivity for CO2. CO, HCl, and SO2 gases were purged into the solution. CO did not change the luminescence at all (Fig. S4†), while SO2 quenched the luminescence because of the reduction of DyW10 (Fig. S5†) and HCl gas also quenched the luminescence at low pH values (Fig. S6†) owing to the decomposition of DyW10. However, after purging with a mixed gas containing 20% CO2, 70% N2, 10% O2 and 0.1% SO2, the complex solution still showed good detection of CO2 (Fig. S7†), indicating that in practical applications CO, SO2 and O2 would not affect the CO2 detection.
Furthermore, the sensor can be recycled by purging with inert gas to degas CO2. As shown in Fig. 2a, the emission spectrum of the complex solution after CO2 treatment displays two intense blue and yellow bands, with an 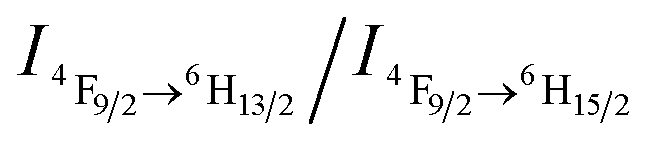 value of ∼0.92. After purging with Ar to degas CO2, the intensity strikingly decreased to the initial value before CO2 treatment.
value of ∼0.92. After purging with Ar to degas CO2, the intensity strikingly decreased to the initial value before CO2 treatment. 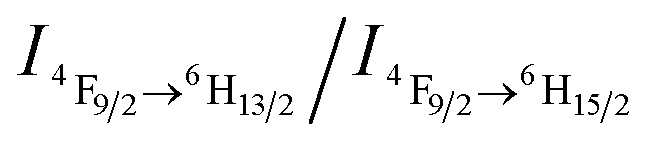 also increased to ∼2.22, and correspondingly the emissive color recovered from strong white to the original weak green. Aside from Ar, the luminescence can be restored by purging with N2 or simply heating, although the heating treatment takes longer (Fig. S8 and S9†). Unlike Ar and N2, compressed air is inefficient (Fig. S10†), which may be attributed to the fact that the air contains a small amount of CO2 (∼300 ppm). On the other hand, the complex solution in both states is very stable in an airtight environment, with no distinct changes after standing at room temperature for at least 30 h (Fig. S11†). Both the luminescence intensity and emission color of the DyW10/PEO-b-PDMAEMA complex can be reversibly switched by alternating CO2/Ar treatment for at least five cycles (Fig. 2b), which endows this complex system with the merit of recyclability, making it a more affordable CO2 detector.
also increased to ∼2.22, and correspondingly the emissive color recovered from strong white to the original weak green. Aside from Ar, the luminescence can be restored by purging with N2 or simply heating, although the heating treatment takes longer (Fig. S8 and S9†). Unlike Ar and N2, compressed air is inefficient (Fig. S10†), which may be attributed to the fact that the air contains a small amount of CO2 (∼300 ppm). On the other hand, the complex solution in both states is very stable in an airtight environment, with no distinct changes after standing at room temperature for at least 30 h (Fig. S11†). Both the luminescence intensity and emission color of the DyW10/PEO-b-PDMAEMA complex can be reversibly switched by alternating CO2/Ar treatment for at least five cycles (Fig. 2b), which endows this complex system with the merit of recyclability, making it a more affordable CO2 detector.
 | ||
| Fig. 2 (a) Emission spectra (λex = 280 nm) of DyW10/PEO-b-PDMAEMA coassembly in water before and after CO2 treatment. (b) Reversible switching of the luminescence intensity and chromism of the DyW10/PEO-b-PDMAEMA solution by alternating CO2/Ar treatment. | ||
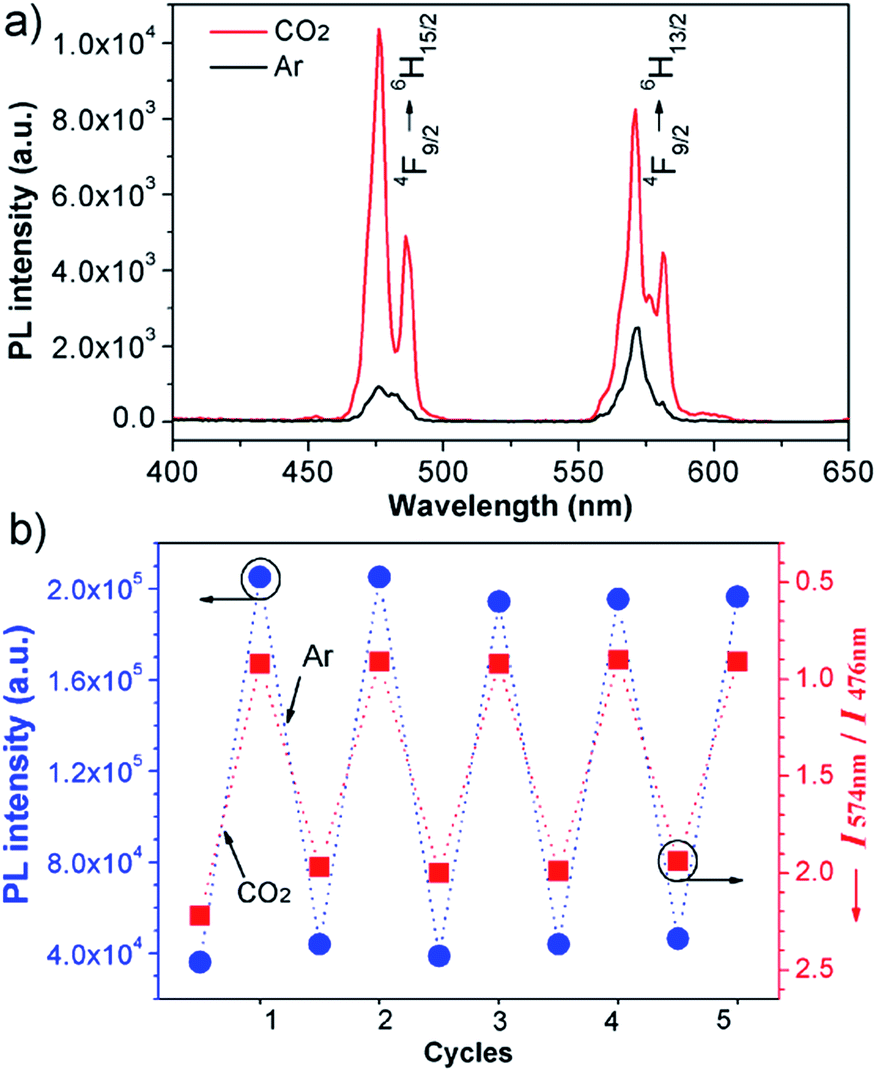
To verify the CO2-responsive assembly behavior of DyW10/PEO-b-PDMAEMA, we used in situ small angle X-ray scattering (SAXS), transmission electron microscopy (TEM), and 1H NMR spectroscopy. The scattering intensity I(q) of the DyW10/copolymer complex at low q values became stronger after purging CO2 into the complex solution (Fig. 3a), supporting the formation of large assembled aggregates. The corresponding pair-distance distribution function p(r) (Fig. 3b) was deduced by using Generalized Indirect Fourier Transform (GIFT) analysis,15 and the result shows that the scattering objects have a globular, almost spherical shape with an average diameter of ∼12 nm. The TEM image of DyW10/PEO-b-PDMAEMA assemblies after CO2 treatment displays spherical micelles with an average diameter of 10 nm with a narrow distribution (Fig. 3c), in good agreement with the SAXS results, while before CO2 treatment the TEM image displays a lesser amount of small irregular aggregates (Fig. 3d). To probe the PDMAEMA segments participating in the formation of a micellar core with the macroanionic DyW10, 1H NMR spectroscopy was utilized to characterize the signal changes of the PDMAEMA segments in D2O solution (Fig. 3e). As expected, compared to the characteristic signals of PDMAEMA at chemical shifts ∼2.38, 2.81 and 4.15 ppm recorded for the initial solution, the PDMAEMA signals disappeared completely after CO2 treatment, indicating that almost all PDMAEMA segments participate in the formation of the coacervate core. Furthermore, the 1H NMR signals of the PDMAEMA blocks can be restored upon treatment with Ar. All the above results support the co-assembly of DyW10 and PEO-b-PDMAEMA after CO2 treatment into dense spherical micelles, and the fact that the assembly/disassembly can be reversibly switched by alternating CO2/Ar treatment.
 | ||
| Fig. 3 Characterization of the morphology of the DyW10/PEO-b-PDMAEMA coassemblies before and after CO2 treatment. (a) The SAXS pattern obtained for DyW10/PEO-b-PDMAEMA and (b) the corresponding distance distribution, p(r), after treatment with CO2; TEM images of DyW10/PEO-b-PDMAEMA coassemblies after CO2 (c) and Ar (d) treatment; (e) partial 1H NMR spectra in D2O recorded for the DyW10/PEO-b-PDMAEMA complex, followed by purging with CO2 gas, and then degassing CO2 with Ar. | ||
To explore the microenvironment variations of the luminophore DyW10 before and after purging with CO2, further examination of the decay lifetimes of DyW10/PEO-b-PDMAEMA coassemblies was carried out (Fig. S12†). It is known that the emission of Dy3+ is highly dependent on coordinated water, due to radiationless deactivation of the 4F9/2 excited state through weak coupling with the vibrational states of the high-frequency OH oscillators in the water ligands.16 In the initial state without CO2, three lifetimes could be identified: τ1 ≈ 4.0 μs (f1 = 0.366), τ2 ≈ 17.3 μs (f2 = 0.201), and τ3 ≈ 59.9 μs (f3 = 0.433) (fi denotes the fractional contribution to the total fluorescence decay; detailed fitting methods and results are available in Table S1†).17 As the lifetime is correlated to the water molecules coordinated to the Dy3+ ion, the number of water ligands qH2O can be estimated to be about 6.2, 1.2, and 0.16, respectively,18 demonstrating that there are a considerable number of water molecules coordinated to Dy3+ in the initial state (detailed calculation of qH2O is given in the ESI†). In comparison, after addition of CO2, there is only a single long decay lifetime of ∼57.5 μs with a qH2O value of 0.18, which is comparable to that of DyW10 crystals (τ ≈ 58.2 μs, qH2O ∼0.17), indicating there are almost no water molecules coordinated to the Dy3+ ion. This result further suggests that after CO2 treatment DyW10 is located in a relatively hydrophobic environment in the complex core of spherical micelles, where the cationic PDMAEMA segments have strong enough electrostatic affinity to the anionic DyW10 to replace the water ligands.
What follows is a proposed mechanism of how CO2 triggers the luminescence chromism. In the initial DyW10/PEO-b-PDMAEMA dilute solution, the degree of protonation of the PDMAEMA blocks is estimated to be ∼61% based on the initial pH value ∼7.20 of the DyW10/PEO-b-PDMAEMA solution (see ESI†). The water molecules coordinated to Dy3+ are only partially replaced by PDMAEMA segments through electrostatic interactions, which in fact lowers the D4d symmetry of DyW10 in the solid to C4v, because the water molecules cannot lie exactly in the reflection plane of the alternating S8 axis. The 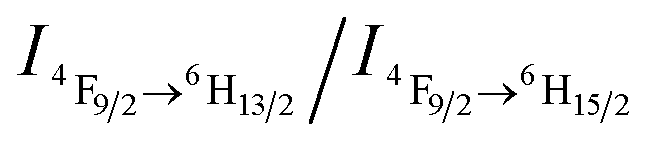 value ∼2.22 as a probe of Dy3+ symmetry in ambient microenvironments also demonstrates that DyW10 is located in a relatively asymmetrical microenvironment. In the presence of CO2, the N,N-dimethylaminoethyl tertiary amino groups of PEO-b-PDMAEMA are almost completely converted to positively-charged ammonium bicarbonates (Scheme 1), and the PDMAEMA block is almost fully positively charged with δ ∼ 99.8% as estimated from the pH value ∼4.80. Consequently, the electrostatic interactions between the cationic copolymer and macroanionic DyW10 are greatly enhanced and drive their co-assembly into dense spherical micelles consisting of a hydrophobic PDMAEMA/DyW10 complex core stabilized by a corona of neutral hydrophilic PEO blocks. As DyW10 is located in the dense core of the micelles, its bound water molecules are almost completely replaced by the protonated PDMAEMA segments, and thus the symmetric microenvironment of DyW10 is improved as evidenced by a decrease in the
value ∼2.22 as a probe of Dy3+ symmetry in ambient microenvironments also demonstrates that DyW10 is located in a relatively asymmetrical microenvironment. In the presence of CO2, the N,N-dimethylaminoethyl tertiary amino groups of PEO-b-PDMAEMA are almost completely converted to positively-charged ammonium bicarbonates (Scheme 1), and the PDMAEMA block is almost fully positively charged with δ ∼ 99.8% as estimated from the pH value ∼4.80. Consequently, the electrostatic interactions between the cationic copolymer and macroanionic DyW10 are greatly enhanced and drive their co-assembly into dense spherical micelles consisting of a hydrophobic PDMAEMA/DyW10 complex core stabilized by a corona of neutral hydrophilic PEO blocks. As DyW10 is located in the dense core of the micelles, its bound water molecules are almost completely replaced by the protonated PDMAEMA segments, and thus the symmetric microenvironment of DyW10 is improved as evidenced by a decrease in the 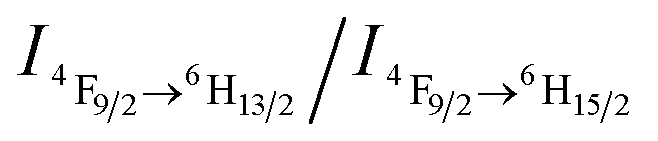 value to 0.92, which is comparable to that of DyW10 crystals. As a result, the luminescence intensity is greatly enhanced and the chromism from green to white occurs. On purging with Ar to remove CO2, the spherical micelles can disassemble because of the partial deprotonation of PDMAEMA, and consequently the complex solution can be used for recyclable CO2 sensing. Its performance does not decline with the number of cycles, because the CO2/Ar switching does not cause any salt accumulation or contamination which would destroy the electrostatic assemblies.
value to 0.92, which is comparable to that of DyW10 crystals. As a result, the luminescence intensity is greatly enhanced and the chromism from green to white occurs. On purging with Ar to remove CO2, the spherical micelles can disassemble because of the partial deprotonation of PDMAEMA, and consequently the complex solution can be used for recyclable CO2 sensing. Its performance does not decline with the number of cycles, because the CO2/Ar switching does not cause any salt accumulation or contamination which would destroy the electrostatic assemblies.
Conclusions
In summary, we have demonstrated a novel supramolecular assay for fluorimetric sensing of carbon dioxide based on a POM/copolymer hybrid complex. PDMAEMA blocks could be protonated by CO2 leading to electrostatic co-assembly with DyW10, and consequently the white emission of DyW10 is switched on. The fluorimetric characteristics of DyW10/PEO-b-PDMAEMA coassemblies permit the detection of CO2 with the merits of simplicity, sensitivity, specificity, interference tolerance, and recyclability. Furthermore, our findings may pave the way to the elaborate design of smart supramolecular materials with complementary functional components.Acknowledgements
We would like to thank Prof. Jinying Yuan at Tsinghua University for helpful discussions. This work was supported by the National Natural Science Foundation of China (21322404; 51373001; 21404030), and the Natural Science Foundation of Beijing Municipality (No. 2122024).Notes and references
- (a) D. Leaf, H. J. H. Verolme and W. F. Hunt, Environ. Int., 2003, 29, 303 CrossRef; (b) J. J. Chen and G. B. Pike, J. Cereb. Blood Flow Metab., 2010, 30, 1094 CrossRef CAS PubMed; (c) H. J. Adrogue and N. E. Madias, J. Am. Soc. Nephrol., 2010, 21, 920 CrossRef CAS PubMed.
- (a) W. Jin, J. Jiang, Y. Song and C. Bai, Respir. Physiol. Neurobiol., 2012, 180, 141 CrossRef CAS PubMed; (b) J. Zosel, W. Oelssner, M. Decker, G. Gerlach and U. Guth, Meas. Sci. Technol., 2011, 22, 072001 CrossRef.
- (a) M. E. Lopez, Anal. Chem., 1984, 56, 2360 CrossRef CAS; (b) H. Suzuki, H. Arakawa, S. Sasaki and I. Karube, Anal. Chem., 1999, 71, 1737 CrossRef CAS PubMed.
- L. Joly, F. Gibert, B. Grouiez, A. Grossel, B. Parvitte, G. Durry and V. Zeninari, J. Quant. Spectrosc. Radiat. Transfer, 2008, 109, 426 CrossRef CAS PubMed.
- (a) B. Han, X. Jiang, X. Hou and C. Zheng, Anal. Chem., 2014, 86, 936 CrossRef CAS PubMed; (b) V. M. Vorotyntsev, G. M. Mochalov and I. V. Baranova, J. Anal. Chem., 2013, 68, 152 CrossRef CAS.
- (a) Q. Xu, S. Lee, Y. Cho, M. H. Kim, J. Bouffard and J. Yoon, J. Am. Chem. Soc., 2013, 135, 17751 CrossRef CAS PubMed; (b) Y. Ma, H. Xu, Y. Zeng, C.-L. Ho, C.-H. Chui, Q. Zhao, W. Huang and W.-Y. Wong, J. Mater. Chem. C, 2015, 3, 66 RSC.
- (a) Y. Ma and L.-Y. L. Yung, Anal. Chem., 2014, 86, 2429 CrossRef CAS PubMed; (b) J. J. Gassensmith, J. Y. Kim, J. M. Holcroft, O. K. Farha, J. F. Stoddart, J. T. Hupp and N. C. Jeong, J. Am. Chem. Soc., 2014, 136, 8277 CrossRef CAS PubMed; (c) E. Climent, A. Agostini, M. E. Moragues, R. Martinez-Manez, F. Sancenon, T. Pardo and M. D. Marcos, Chem.–Eur. J., 2013, 19, 17301 CrossRef CAS PubMed.
- (a) Z. Guo, N. R. Song, J. H. Moon, M. Kim, E. J. Jun, J. Choi, J. Y. Lee, C. W. Bielawski, J. L. Sessler and J. Yoon, J. Am. Chem. Soc., 2012, 134, 17846 CrossRef CAS PubMed; (b) Y. Liu, Y. Tang, N. N. Barashkov, I. S. Irgibaeva, J. W. Y. Lam, R. Hu, D. Birimzhanova, Y. Yu and B. Z. Tang, J. Am. Chem. Soc., 2010, 132, 13951 CrossRef CAS PubMed; (c) L. Q. Xu, B. Zhang, M. Sun, L. Hong, K.-G. Neoh, E.-T. Kang and G. D. Fu, J. Mater. Chem. A, 2013, 1, 1207 RSC; (d) T. A. Darwish, R. A. Evans, M. James and T. L. Hanley, Chem.–Eur. J., 2011, 17, 11399 CrossRef CAS PubMed; (e) J. Guo, N. Wang, J. Wu, Q. Ye, C. Zhang, X.-H. Xing and J. Yuan, J. Mater. Chem. B, 2014, 2, 437 RSC; (f) T. Tian, X. Chen, H. Li, Y. Wang, L. Guo and L. Jiang, Analyst, 2013, 138, 991 RSC.
- C. M. Rudzinski, A. M. Young and D. G. Nocera, J. Am. Chem. Soc., 2002, 124, 1723 CrossRef CAS PubMed.
- (a) J. Zhang, Y. Liu, Y. Li, H. Zhao and X. Wan, Angew. Chem., Int. Ed., 2012, 51, 4598 CrossRef CAS PubMed; (b) H. Wei, S. Du, Y. Liu, H. Zhao, C. Chen, Z. Li, J. Lin, Y. Zhang, J. Zhang and X. Wan, Chem. Commun., 2014, 50, 1447 RSC; (c) Y. Liu, H. Zhao, H. Wei, N. Shi, J. Zhang and X. Wan, J. Inorg. Organomet. Polym. Mater., 2015, 25, 126 CrossRef CAS.
- (a) K. Sawada and T. Yamase, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 2002, 58, i149 Search PubMed; (b) K. Binnemans, Chem. Rev., 2009, 109, 4283 CrossRef CAS PubMed; (c) T. Yamase, Chem. Rev., 1998, 98, 307 CrossRef CAS PubMed; (d) R. Ballardini, Q. G. Mulazzani, M. Venturi, F. Bolletta and V. Balzani, Inorg. Chem., 1984, 23, 300 CrossRef CAS.
- C. H. Liang, L. G. Teoh, K. T. Liu and Y. S. Chang, J. Alloys Compd., 2012, 517, 9 CrossRef CAS PubMed.
- H. Wei, N. Shi, J. Zhang, Y. Guan, J. Zhang and X. Wan, Chem. Commun., 2014, 50, 9333 RSC.
- D. Han, X. Tong, O. Boissiere and Y. Zhao, ACS Macro Lett., 2012, 1, 57 CrossRef CAS.
- (a) D. Lof, M. Tomsic, O. Glatter, G. Fritz-Popovski and K. Schillen, J. Phys. Chem. B, 2009, 113, 5478 CrossRef PubMed; (b) B. Weyerich, J. Brunner-Popela and O. Glatter, J. Appl. Crystallogr., 1999, 32, 197 CrossRef CAS.
- G. Stein and E. Wurzberg, J. Chem. Phys., 1975, 62, 208 CrossRef CAS PubMed.
- J. R. Lakowicz, Principles of Fluorescence Spectroscopy, Springer, Berlin, 3rd edn, 2006, p. 101 Search PubMed.
- C. D. Hall and N. W. Sharpe, J. Photochem. Photobiol., A, 1990, 52, 363 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5sc02020d |
| This journal is © The Royal Society of Chemistry 2015 |