
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEfficient chemoenzymatic synthesis of an N-glycan isomer library†
Lei
Li‡
a,
Yunpeng
Liu‡
a,
Cheng
Ma‡
a,
Jingyao
Qu
a,
Angie D.
Calderon
a,
Baolin
Wu
b,
Na
Wei
a,
Xuan
Wang
a,
Yuxi
Guo
a,
Zhongying
Xiao
a,
Jing
Song
a,
Go
Sugiarto
c,
Yanhong
Li
c,
Hai
Yu
c,
Xi
Chen
c and
Peng George
Wang
*a
aDepartment of Chemistry and Center of Diagnostics & Therapeutics, Georgia State University, 50 Decatur St SE, Atlanta, GA 30303, USA. E-mail: pwang11@gsu.edu
bChemily, LLC, 58 Edgewood Ave NE, Atlanta, GA 30303, USA
cDepartment of Chemistry, University of California, Davis, One Shields Avenue, Davis, CA 95616, USA
First published on 23rd June 2015
Abstract
Quantification, characterization and biofunctional studies of N-glycans on proteins remain challenging tasks due to the complexity, diversity and low abundance of these glycans. The availability of structurally defined N-glycan (especially isomer) libraries is essential to help solve these tasks. We report herein an efficient chemoenzymatic strategy, namely Core Synthesis/Enzymatic Extension (CSEE), for rapid production of diverse N-glycans. Starting with 5 chemically prepared building blocks, 8 N-glycan core structures containing one or two terminal N-acetyl-D-glucosamine (GlcNAc) residue(s) were chemically synthesized via consistent use of oligosaccharyl thioethers as glycosylation donors in a convergent fragment coupling strategy. Each of these core structures was then extended to 5 to 15 N-glycan sequences by enzymatic reactions catalyzed by 4 robust glycosyltransferases. Success in synthesizing N-glycans with Neu5Gc and core-fucosylation further expanded the ability of the enzymatic extension. Meanwhile, high performance liquid chromatography with an amide column enabled rapid and efficient purification (>98% purity) of N-glycans in milligram scales. A total of 73 N-glycans (63 isomers) were successfully prepared and characterized by MS2 and NMR. In summary, the CSEE strategy provides a practical approach for “mass production” of structurally defined N-glycans, which are important standards and probes for glycoscience.
Introduction
Glycans are ubiquitous and play diverse roles in a wide range of biological processes, such as protein folding and degradation, glycoproteostasis, cell adhesion and trafficking, cell signaling, fertilization and embryogenesis, as well as pathogen recognition and immune responses.1 Abnormal cell surface glycoforms and/or glycan-profiles are usually related to diseases such as cancer and atherosclerosis. Accordingly, glycan/glycoprotein biomarkers have been developed.2 Thus, elucidating the structures and functions of glycans is essential for understanding carbohydrate related biological and pathological processes, and for developing diagnostics and therapeutics for human diseases.N-glycans found in nature possess an inherited complexity and diversity. These are mainly due to the variable and multiple connectivity of glycan building blocks (monosaccharides) and the processes by which they are assembled in biosystems. In mammalian glycomes, numerous glycan structures can be formed including branched-, regio- and stereo-isomers from only 10 common monosaccharide building blocks.3 Unlike precise template directed transcription/translation of nucleic acids/proteins, glycan structures are determined by the activities of glycosyltransferases (GTs), glycosidases, and other glycan biosynthetic enzymes, as well as the availability of donor substrates. For example, more than 30 GTs and glycosidases in the Golgi complexes of human cells are involved in processing N-glycans.4 The expression, activity, substrate specificity, and localization of each enzyme each has the potential to influence the assembly of N-glycans. It is thus understandable that N-glycans are extremely micro-heterogeneous even in one particular N-glycosylation site. For example, 58 different complex N-glycan structures were identified at one N-glycan site in mouse zona pellucida glycoprotein 3.4 As a result, despite decades of effort in developing novel approaches for glycan analysis,5 absolute quantification and characterization of complex mixtures of N-glycans remain challenging tasks. At present, the main approach for characterizing N-glycan isomers is ion-trap mass spectrometry (MS) analysis of permethylated glycans, which requires large quantities of samples, and is therefore not suitable for low abundance glycans and rare biological samples. The availability of libraries of structurally defined N-glycans (especially isomers) provides essential standards and probes for MS-based N-glycan analysis and glycan microarray studies of carbohydrate binding proteins.
Given the difficulties in separating structurally defined glycans from natural resources, chemical or chemoenzymatic approaches have been developed for the synthesis of mostly symmetric N-glycans in the last two decades.6 Among chemically synthesized N-glycans, only a few contain terminal sialic acid (Sia) due to difficulties in sialic acid chemistry,7 which were only lately overcome by enzymatic glycosylation using sialyltransferases.8 Most recently, Boons developed a strategy for chemoenzymatic synthesis of asymmetrical N-glycans, and 14 tri-antennary complex N-glycans were obtained.9 Nevertheless, only a few N-glycan structures were prepared in each report, mainly due to their complexity and diversity. A simple and robust strategy for efficient production of large numbers of N-glycan structures is still highly desirable.
Another roadblock to the rapid access of glycans in high purity is the purification strategy, which now largely relies on gel filtration chromatography (usually Sephadex G-25 or Bio-gel P2).8,9 However, even though gel filtration has been applied for decades in purifying glycans, it is time-consuming, less efficient, and may waste a significant portion of products in preparing small quantities of precious N-glycans. Therefore, a more reliable and rapid N-glycan purification approach is yet to be developed.
LewisX [LeX, Galβ1,4-(Fucα1,3-)GlcNAc] and sialyl LewisX [SLeX, Siaα2,3-Galβ1,4-(Fucα1,3-)GlcNAc] are among the most biologically significant glycan epitopes. For example LeX, also known as CD15 antigen or SSEA-1 trisaccharide, plays a role in the development of the central nervous systems of vertebrates,10 and interferes with pathogen transfer in breastfed infants.11 SLeX is a specific ligand on human leukocytes for E-, L-, and P-selectins, and has been shown to mediate leukocyte recruitment.12 SLeX (on both N- and O-glycans of glycoprotein zona pellucida) has also been shown to mediate human sperm binding during fertilization.13 In addition, LeX and SLeX are usually overexpressed on the surface of cancer cells.14 Despite its high significance, N-glycans containing these epitopes were not synthesized until recently.9 In this study, we describe an efficient Core Synthesis/Enzymatic Extension (CSEE) strategy and a HPLC based purification approach for rapid preparation of N-glycans with/without (S)LeX epitopes. In this strategy, 8 N-glycan core structures with GlcNAc residue(s) at the non-reducing terminal were first synthesized by convergent assembly of 5 building blocks. A set of robust GTs were then used to elongate these cores to yield a library of 73 N-glycans (Fig. 1 and 3). The development of a HPLC based approach using an amide column enabled rapid purification of milligrams of the chemoenzymatically synthesized N-glycans to a minimum 98% purity. In addition, MS2 analysis of selected N-glycans yielded unique fragmentation patterns that may be used for distinguishing certain isomers.
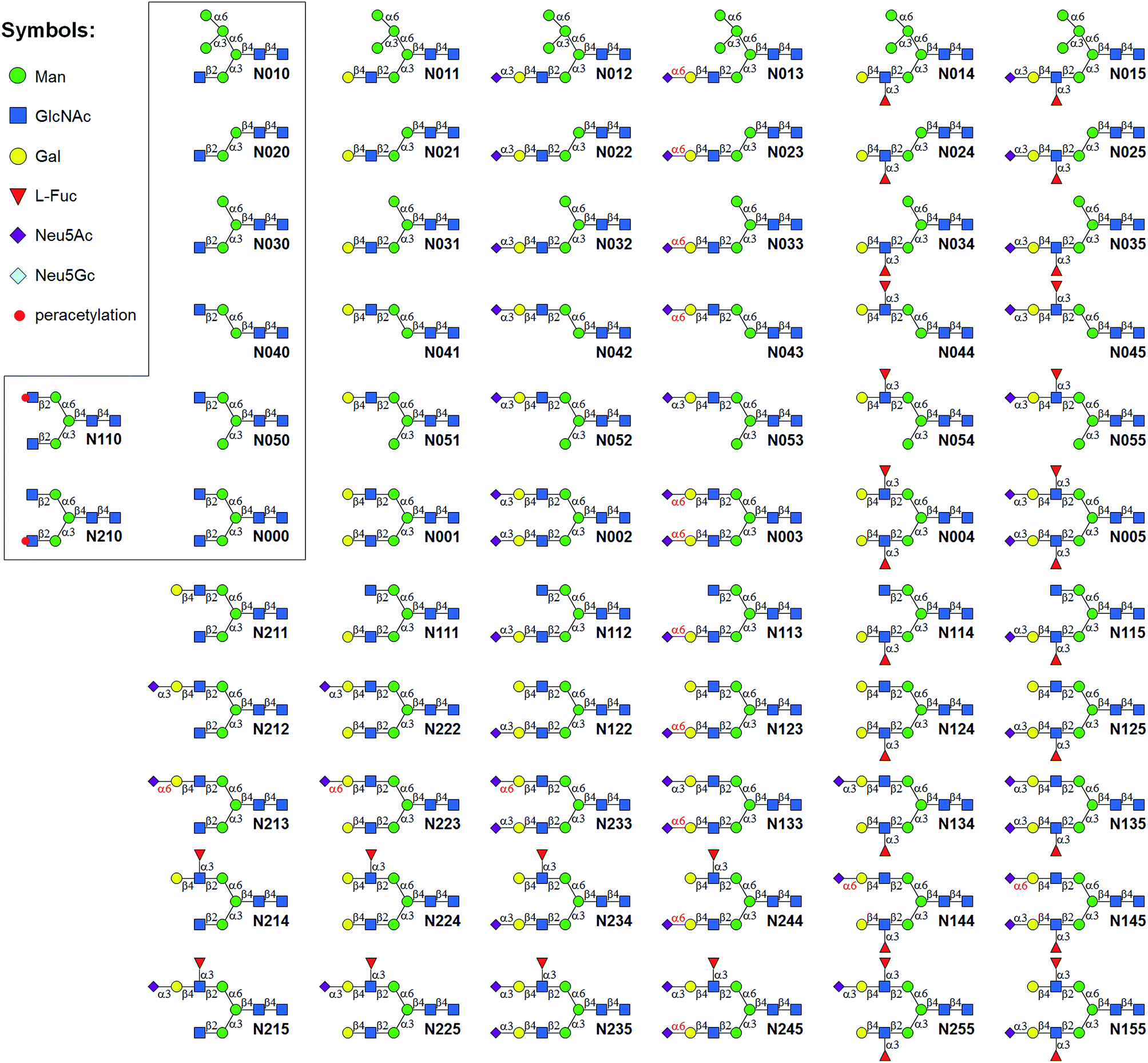 | ||
| Fig. 1 N-glycans synthesized by the Core Synthesis/Enzymatic Extension (CSEE) strategy. N-glycan core structures (boxed) were chemically synthesized. Glycans N0xx were enzymatically synthesized from corresponding cores N0x0, whereas N1xx and N2xx were enzymatically synthesized from N110 or N210. | ||
Results
Convergent core synthesis
A good amount of work had been reported in chemically synthesizing N-glycan structures. For example, Danishefsky7a,15 constructed a core pentasaccharide by glycosylating a trisaccharide with a monosaccharide thioether donor; Unverzagt16 synthesized multi-antennary complex type N-glycans with acetated Schmidt's trichloroacetimidate donor; Wong8a used oligosaccharyl fluorides as donors to produce bi-, tri- and tetra-antennary complex type N-glycans; Boons9 developed a strategy by sequential removal of the protecting groups, and then by chemical glycosylation using a diverse set of trifluoroacetimidate donors. In these cases, to prepare various glycosyl donors, a temporary anomeric protecting group was used prior to transformation into trichloroacetimidates, fluorides or trifluoroacetimidates, depending on the choice of glycosylation reaction. In this study, we developed an efficient convergent strategy that utilized oligosaccharyl thioether as a versatile donor for glycosylation, facilitating antennae assembly in just one or two glycosylation step(s) with excellent yield and good stereoselectivity.We envisaged that trisaccharide 1 (Fig. 2) containing a crucial β-mannoside would be a versatile precursor for the synthesis of the core structures, as the C4,C6-hydroxyl groups (OH) of the β-Man are protected with benzylidene and the C3-OH is unprotected to allow further chemical glycosylation. Installation of β-mannoside, the most challenging task in N-glycan synthesis, was accomplished using Crich–Kahne conditions with satisfactory yield and β-selectivity.17 The benzylidene acetal ring can be selectively opened at either C6-OH9 or C4-OH18 of the β-mannoside for further chemical assembly. In order to prepare the target N-glycans, 8 GlcNAc terminated core structures (Fig. 2) were designed. Among these, N110 and N210 were partially protected by peracetylation of the GlcNAc residue on either the α1,6Man or α1,3Man branch for the synthesis of asymmetric bi-antennary N-glycans. After enzymatic extension of the unprotected branch, the acetyl groups can be removed easily for further elongation.
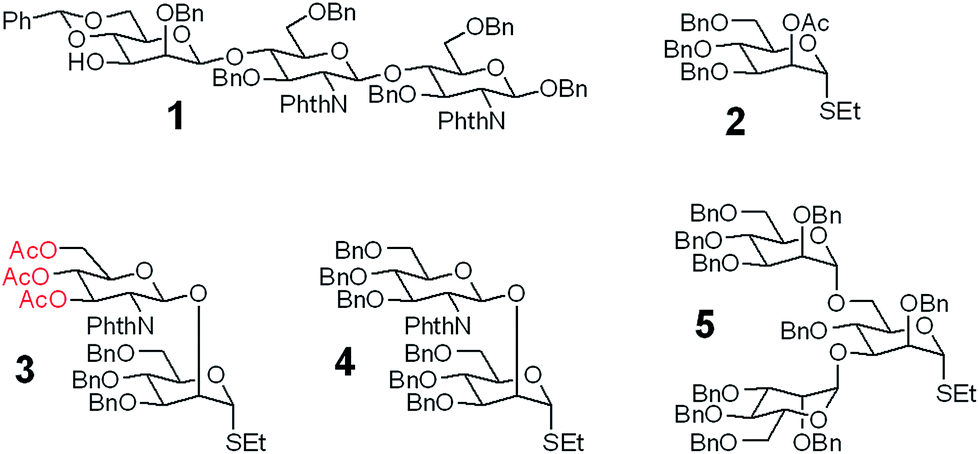 | ||
| Fig. 2 The versatile trisaccharide precursor 1 and four donor fragments (2, 3, 4, 5) for the assembly of the 8 core structures. | ||
The versatile precursor trisaccharide 1 (ref. 7b) and donor fragments 2,193,204 and 5 (ref. 21) were prepared as previously reported (ESI†). Using these building blocks, the syntheses of the 8 core structures were performed in a convergent strategy (Scheme 1). For example, to synthesize core structure N010, thioether donor 4 was first stereoselectively installed onto C3-hydroxyl of trisaccharide 1 in the presence of N-iodosuccinimide (NIS)/AgOTf with a yield of 93%. Pentasaccharide 7 was then obtained in an excellent yield (96%) by selective opening of the benzylidene ring at C6 using Et3SiH/PhBCl2. The octasaccharide 8 was assembled by stereoselective installation of 5 onto C6-hydroxyl of β-Man of acceptor 7 with a yield of 85%. The two phthalimides of 8 were then converted into acetamides, followed by the global deprotection of Bn by catalytic hydrogenolysis with Pd(OH)2/H2 in MeOH/H2O (10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1). The core structure N010 was produced in a total yield of 63% over the three steps.
:1). The core structure N010 was produced in a total yield of 63% over the three steps.
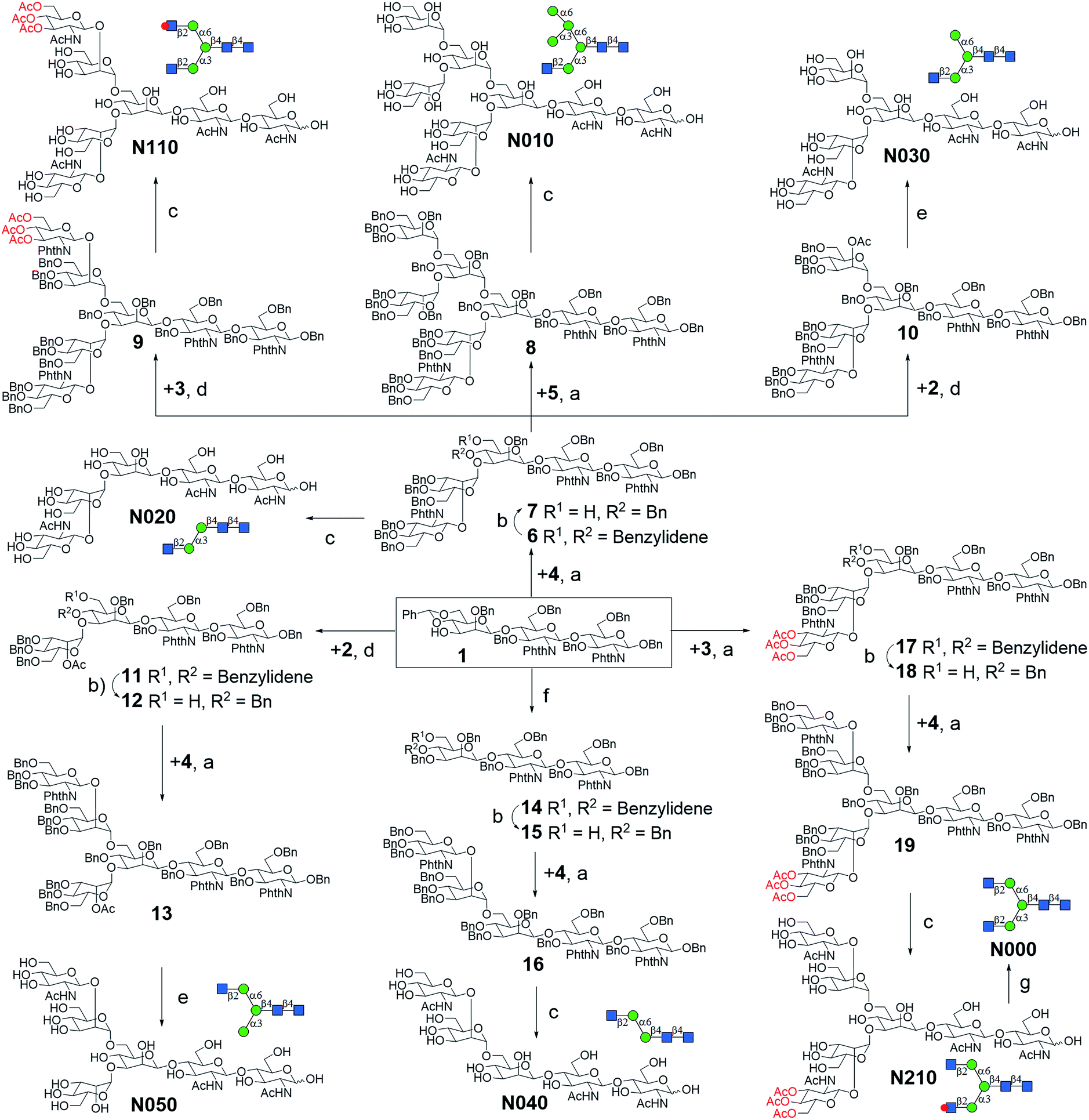 | ||
| Scheme 1 Convergent synthesis of N-glycan core structures. Reagents and conditions: (a) NIS, AgOTf, Et2O, 0 °C, 6: 93%; 8: 85%; 9: 83%; 13: 90% (α/β = 5:1); 16: 91% (α/β = 3.5:1); 17: 94%; 19: 86% (α/β = 4:1); (b) PhBCl2, Et3SiH, DCM, −78 °C, 7: 96%; 12: 92%; 15: 95%; 18: 93%; (c) (1) ethylenediamine, n-butanol, 90 °C; (2) Ac2O, pyridine, rt; (3) Pd(OH)2, H2, MeOH/H2O (10:1), over three steps, N010: 63%; N020: 67%; N040: 61%; N110: 69%; N210: 65%; (d) NIS, AgOTf, DCM, 0 °C, 10: 91%; 11: 95%; (e): (1) ethylenediamine, n-butanol, 90 °C; (2) Ac2O, pyridine, rt; (3) NaOMe, MeOH; (4) Pd(OH)2, H2, MeOH/H2O (10:1), over four steps, N030, 53%; N050: 55%; (f) BnBr, NaH, DMF, 90%; (g) 30% NH4OH:H2O (1:10), quant. | ||
Similarly, cores N000, N020, N030, N050, N110, and N210 were synthesized by first installing 2, 3 or 4 onto C3-hydroxyl of β-man of 1, followed by installation of the corresponding building blocks onto the α1,6Man branch. For the synthesis of N040, simple 3-O-benzylation and controlled reductive cleavage of the benzylidene acetal of 1 was performed to afford acceptor 15, which was then glycosylated with 4 to yield pentasaccharide 16 in 91% yield and with satisfactory stereoselectivity (α/β = 3.5:1). Compound 16 was further deprotected to yield N040 as previously described. The structures and stereochemistry of all glycosidic linkages was confirmed by NMR (ESI†).
Enzymatic extension of N-glycans
We intended to develop an enzymatic extension strategy that could efficiently generate 6 glycans (including LeX and SLeX) starting with any terminal GlcNAc residue. To this end, several robust GTs were chosen for the proof-of-concept experiment: β1,4-galactosyltransferase from bovine milk (B4GALT1); α2,3-sialyltransferase 1 mutant E271F/R313Y from Pasteurella multocida (PmST1m) with reduced α2,3-sialidase activity;22 α2,6-sialyltransferase from Photobacterium damselae (Pd2,6ST);23 and C-terminal 66 amino acids truncated α1,3-fucosyltransferase from Helicobacter pylori (Hpα1,3FT).24 Except for the commercially available B4GALT1, all GTs were from bacteria and had high expression levels in Escherichia coli, high activity, and relatively relaxed substrate specificities. Using GlcNAc-OBn as a starting material, we validated the activities of these GTs (Scheme 2). As previously reported, the enzymes could efficiently catalyze the formation of LacNAc (Galβ1,4-GlcNAc)-, SLacNAc (Siaα2,3-Galβ1,4-GlcNAc)-, S6LacNAc (Siaα2,3-Galβ1,4-GlcNAc)-, LeX- and SLeX-OBn. For reactions catalyzed by B4GALT1, Pd2,6ST and Hpα1,3FT, no product hydrolysis was observed (analyzed by MS) even with excess enzyme and up to 24 h of incubation. However, product hydrolysis was observed for PmST1m with an excess of enzyme and an extended reaction time (>1 h); even the α2,3-sialidase activity has been reduced for 6333-fold by mutation of two amino acid residues.22 Therefore, controlling the amount of PmST1m and the reaction time is still important for the synthesis of α2,3-sialosides. The attempt to synthesize unnatural tetrasaccharide S6LeX [Siaα2,6-Galβ1,4-(Fucα1,3-)GlcNAc] failed utilizing either Hpα1,3FT or Pd2,6ST, with the formation of only a MS detectable product after 24 h of Pd2,6ST incubation (Fig. S3†). In addition, PmST1m can hardly catalyze the sialylation of LeX to form SLeX. Such information is important for designing sequential enzymatic N-glycan synthesis schemes.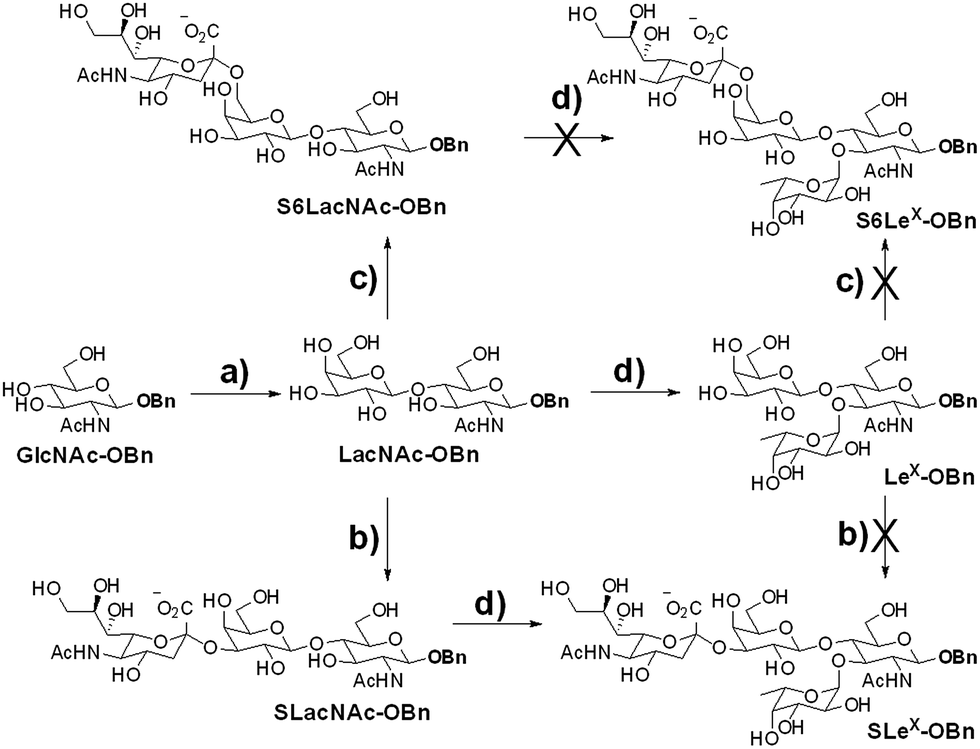 | ||
| Scheme 2 Proof-of-concept experiment for the proposed enzymatic extension strategy. Reagents and conditions: (a) UDP-Gal, Mn2+ and β1,4-galactosyltransferase from bovine milk (B4GALT1); (b) CMP-Sia and double mutant E271F/R313Y of α2,3-sialyltransferase 1 from Pasteurella multocida (PmST1m); (c) CMP-Sia and α2,6-sialyltransferase from Photobacterium damselae (Pd2,6ST); (d) GDP-Fuc, Mn2+ and C-terminal 66 amino acids truncated α1,3-fucosyltransferase from Helicobacter pylori (Hpα1,3FT). | ||
Utilizing the enzymatic extension strategy, N011–N015 were prepared starting with the chemically prepared core N010 (Fig. 3A). Firstly, in a 1.5 mL reaction system, 9 mg of N010 (4 mM) was incubated with UDP-Gal (8 mM), MnCl2 (5 mM), and B4GALT1 (20 μU per μmole acceptor). One microliter of the reaction mixture was aliquoted every hour for analysis. MS analysis showed a peak at m/z = 719.7645, corresponding to N011 [M + 2H]2+. Meanwhile, on the HPLC-ELSD (Evaporative Light Scattering Detector) profile, a new peak (TR = 16.79 min) was observed, of which the area underneath grew while that of the peak corresponding to N010 (TR = 14.86 min) became smaller. After 6 h of incubation, the reaction was freeze-quenched at −80 °C for 30 min, and condensed into 300 μl for HPLC purification using a water/acetonitrile gradient elution, yielding 9.4 mg of N011 (94% yield). The purified N011 (99% pure) was then utilized for the syntheses of N012, N013, and N014 (Fig. 3A) catalyzed by PmST1m, Pd2,6ST, and Hpα1,3FT, respectively (see ESI† for details). It is worth noting that the reaction for the synthesis of N012 was only allowed to proceed for 30 min due to the sialidase activity of PmST1m. N015 was then synthesized from N012 using Hpα1,3FT. The reaction took 20 h to achieve complete conversion (Fig. S4†). Similarly, starting with other chemically synthesized cores (N000, N020, N030, N040 and N050), N-glycans N001–N005, N021–N025, N031–N035, N041–N045 and N051–N055 were prepared in a manner analogous to that described above. All prepared N-glycans were analyzed by HPLC-ELSD, ESI/MALDI-MS, and NMR to confirm their purity and structure (ESI†).
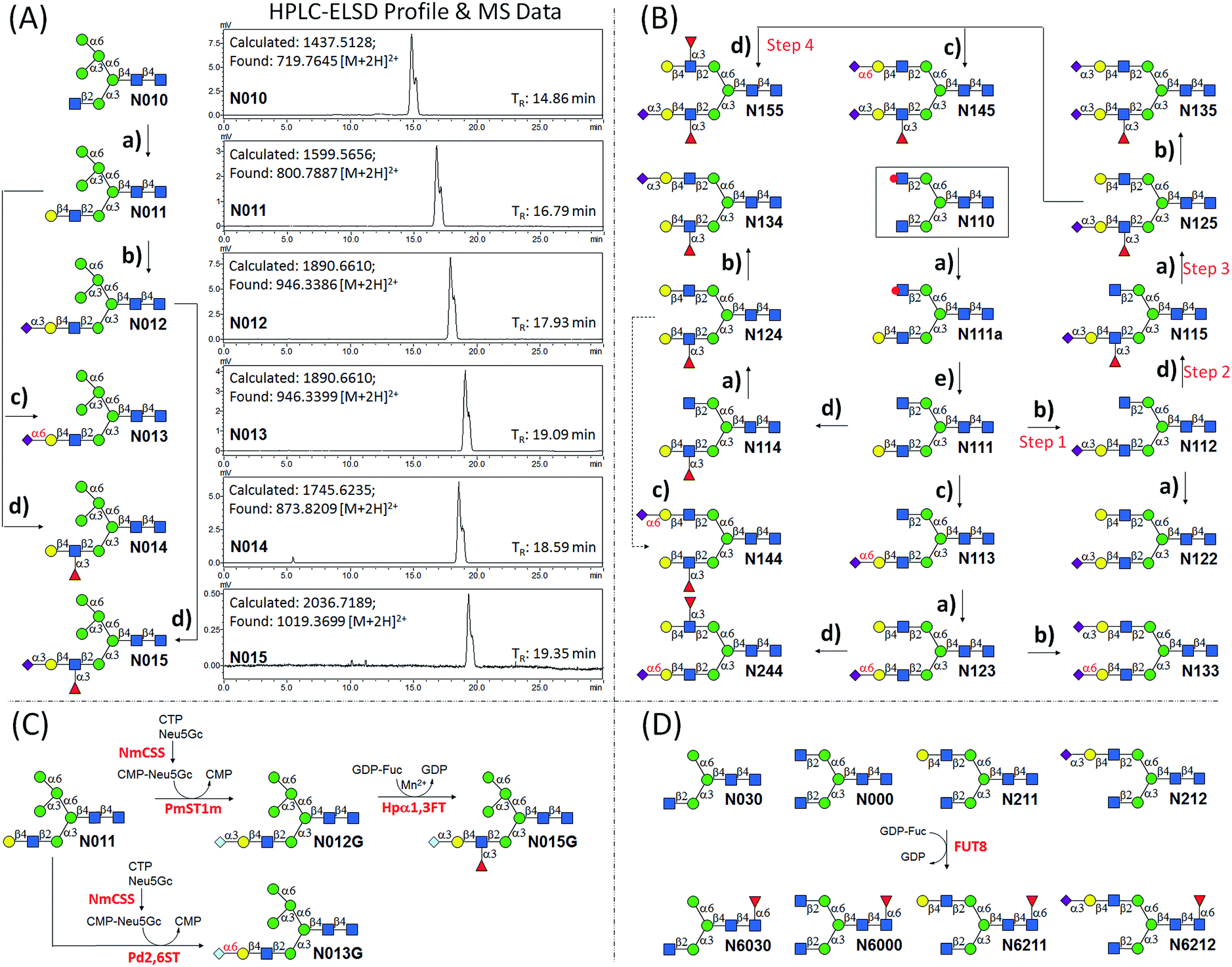 | ||
| Fig. 3 Enzymatic extension of N-glycans. (a) B4GALT1, UDP-Gal, Mn2+; (b) PmST1m, CMP-Neu5Ac; (c) Pd2,6ST, CMP-Neu5Ac; (d) Hpα1,3FT, GDP-Fuc, Mn2+; (e) 30% ammonium hydroxide:H2O (1:10), 6 h. PmST1m, Pasteurella multocida α2,3-sialyltransferase 1 mutant E271F/R313Y; Pd2,6ST, Photobacterium damselae α2,6-sialyltransferase; NmCSS, Neisseria meningitidis CMP-sialic acid synthetase; Hpα1,3FT, c-terminal 66 amino acids truncated Helicobacter pylori α1,3-fucosyltransferase; FUT8, human α1,6-fucosyltransferase. Each N-glycan was purified to minimum 98% purity by HPLC with a semi-preparative amide column (10 × 250 mm). HPLC analyses of purified N-glycans were performed using an analytical amide column (4.6 × 250 mm) under a gradient condition (solution A: 100 mM ammonium formate, pH 3.4; solution B: acetonitrile; flow rate: 1 mL min−1; B%: 65–50% within 25 min) monitored by ELSD. The adjacent two peaks in the HPLC profiles correspond to α and β anomers of N-glycans. MS data for purified N-glycans were obtained by ESI-MS. | ||
The syntheses of asymmetric bi-antennary N-glycans N1xx and N2xx (Fig. 1) were carried out by enzymatic extension of the unprotected antenna first and then the other. The synthesis of N1xx is illustrated in Fig. 3B. Firstly, Gal was added by B4GALT1 to the GlcNAc residue in the α1,3Man branch of N110 to form N110a, galactosylation on the α1,6Man branch was avoided by peracetylation of the corresponding GlcNAc residue. It should be noted that partial de-acetylation was observed when the reaction was incubated for over 12 h. After HPLC purification, N110a was de-acetylated using 30% of ammonium hydroxide:H2O (1:10) to afford N111, which was then used as a substrate for synthesizing the other N1xx glycans in a strictly controlled sequential manner. For example, to obtain N155, the α1,3Man branch was first extended by PmST1m (Step 1) and Hpα1,3FT (Step 2) to yield N115, the α1,6Man branch was then extended by B4GALT1 (Step 3) and Hpα1,3FT (Step 4) (Fig. 3B). Such synthetic routes were designed according to the substrate specificities of GTs to avoid undesirable glycosylation. Particularly, N144 was not designed to be synthesized from N124 to avoid potential sialylation on the α1,3Man branch by Pd2,6ST. Instead, N-glycan N244 was synthesized from N123 catalyzed by Hpα1,3FT (Fig. 3B). Similarly, N-glycans N2xx and N144 were synthesized from N210 (Fig. S5†).
N-Glycolylneuraminic acid (Neu5Gc), often found in mammalian glycans, is another common sialic acid molecule besides N-acetylneuraminic acid (Neu5Ac).25 Even though human cells cannot produce Neu5Gc because of the inactivation of the gene encoding CMP-Neu5Ac hydroxylase,26 it is frequently detected in glycans of cancer cells, probably due to metabolic incorporation from Neu5Gc-containing structures in the diet.25 Previously, the two sialyltransferases (PmST1, Pd2,6ST) used in the enzymatic extension were shown to be extremely promiscuous towards sugar donors, and were applied in an efficient synthesis of a number of sialosides and their derivatives.23,27 To further expand the current library, N-glycans with the Neu5Gc residue (N012G and N013G) were synthesized via a one-pot two-enzyme system (Fig. 3C). In detail, for the synthesis of N012G, 3 mM of N011 was incubated with 5 mM of Neu5Gc and cytidine 5′-triphosphate (CTP), 5 μg mL−1 of PmST1m, and excessive amounts of CMP-Sia synthetase (NmCSS). After 30 min of incubation at 37 °C (94% conversion as detected by HPLC), the reaction mixture was concentrated and subjected to HPLC purification. The synthesis of N013G was achieved by simply replacing PmST1m with Pd2,6ST. Surprisingly, it was found that the incorporation of a Neu5Gc residue resulted in a longer retention time shift (>1 min) on the amide column compared to the Neu5Ac-counterpart (N012, TR = 17.93 min; N012G, TR = 19.13 min; N013, TR = 19.09 min; N013G, TR = 20.16 min) (ESI†). Fucosylation of N012G (to generate N015G) was shown to be as efficient as that of N012, indicating that Hpα1,3FT can tolerate substrates with Neu5Gc. Theoretically, another set of 57 N-glycans can be easily synthesized by simply replacing Neu5Ac of the glycans in Fig. 1 with Neu5Gc.
Core-fucosylated N-glycans were widely found in mammalian glycoproteins and are particularly abundant in brain tissues.28 The alteration of core-fucosylation was proven to be associated with human cancers, chronic hepatitis, etc.29 Thus, the ability to prepare homogenous core-fucosylated N-glycans was believed to be important. The N-glycans prepared above are perfect substrates for specificity studies of α1,6-fucosyltransferase (FUT8), the sole enzyme responsible for the core-fucosyaltion of N-glycans, and for preparing a core-fucosylated N-glycan library. Specifically, 4 N-glycans with an identical α1,3Man branch but a different α1,6Man branch were selected for FUT8-catalyzed core-fucosylation (Fig. 3D). The results showed that FUT8 was highly active in using all 4 N-glycans as acceptors. Corresponding core-fucosylated N-glycans (N6030, N6000, N6211, N6212) (0.5–1 mg each) were synthesized accordingly. Further substrate specificity studies showed that FUT8 may have stricter requirement for structures on the α1,3Man branch than that of the α1,6Man branch (detailed study is ongoing).
A HPLC based approach for rapid access to pure N-glycans
Gel filtration (Bio-gel P2, 1 × 110 cm) was first applied in the purification of synthesized N-glycans. Taking the separation of N001 (synthesized from N000 by a B4GALT1 catalyzed reaction) as an example, MS of the P2 purified product showed a major peak at m/z = 821.2992 corresponding to N001 [M + 2H]2+, and a minor peak at m/z = 740.2735 corresponding to N001 minus a Gal residue [M + 2H]2+, which possibly comes from incomplete galactosylation of N000. This result again indicated that gel filtration is inefficient for purifying N-glycans to high purity, especially when incomplete glycosylation has occurred. As a consequence, previously reported N-glycan syntheses have usually employed excessive enzymes and long incubation times to push GT-catalyzed reactions towards completion.9Hydrophilic interaction liquid chromatography (HILIC) provides a rapid and effective strategy for separating small polar compounds, and has been used extensively in glycan analysis.30 In these cases, N-glycans from biological samples have usually been fluorescently labeled via reductive amination and then detected on a picomole scale by UPLC-HILIC. However, HILIC has not been applied to milligram scale N-glycan purification. Using an analytical HILIC column (XBridge BEH amide column, 5 μm, 4.6 mm × 250 mm, waters) under a gradient running condition (solvent A: 100 mM ammonium formate, pH 3.4; solvent B: acetonitrile; flow rate: 1 mL min−1; B%: 70–50% within 50 min), the abovementioned Bio-gel P2-purified products were analyzed. Four peaks were observed in the HPLC profile using an evaporative light scattering detector (Fig. S1†). Peaks 1 (TR = 21.68 min) and 2 (TR = 22.16 min) were next to each other and partially overlapped. The same observation was found for peaks 3 (TR = 24.51 min) and 4 (TR = 25.04 min). These peaks were collected in a parallel run monitored at A210 nm and subjected to MS analysis. The same m/z values were observed for peaks 3 (821.2997) and 4 (821.2991) (Fig. S1†), implying that both peaks represented N-glycan N001 [M + 2H]2+, possibly for α and β anomers, which is common for free glycans due to the process of mutarotation in water. This was confirmed by 1H NMR analysis that showed chemical shifts of both α and β anomer protons (ESI†). Similarly, peaks 1 and 2 represented α and β anomers of N001 minus a Gal residue [M + 2H]2+.
These results encouraged us to purify N001 using a semi-reparative HILIC column (10 × 250 mm). Under a similar gradient running condition (solvent A: 100 mM ammonium formate; solvent B: acetonitrile; flow rate: 4 mL min−1; B%: 70–50% within 50 min; monitored at A210 nm), 10.5 mg of N001 was separated by 3 injections (Fig. S2†) with a purity of higher than 98% as analyzed by HPLC-ELSD (ESI†). Different solvent combinations were later tested for N-glycan purification (Fig. S6†). The results showed that 100 mM ammonium formate/acetonitrile gradient elution gave the best separation of all N-glycans tested. In addition, N-glycans without Sia residues were separated to a similar level using water/acetonitrile gradient elution, whereas sialylated N-glycans were eluted rapidly (TR < 3 min). Furthermore, it was found that a shorter running time with a narrower B% gradient (65–50% in 25 min) was able to achieve a similarly good separation level. Such running conditions were applied to separate enzymatically synthesized N-glycan to 98% purity (ESI†).
Under a standard running condition (solvent A: 100 mM ammonium formate; solvent B: acetonitrile; flow rate: 1 mL min−1; B%: 65–50% within 25 min), all purified N-glycans were analyzed by HPLC-ELSD (ESI†). It was found that when different sugar residues were added to N-glycans, the retention time shifts of peaks on HPLC chromatograms generally decreased in the following order: Neu5Gcα2,3 with Fucα1,3 > Neu5Gcα2,6 > Neu5Acα2,3 with Fucα1,3 > Neu5Gcα2,3 > Neu5Acα2,6 > Galβ1,4 > Fucα1,3 > Neu5Acα2,3 > Fucα1,6. For example, the retention times of N015G, N013G, N015, N012G, N013, N014, N012 are 20.66, 20.16, 19.39, 19.13, 19.09, 18.59, and 17.93 min respectively. Such regularity may be found useful in HILIC-based profiling and identification of N-glycans.
MS2 analysis of selected N-glycan isomers
The structurally well-defined N-glycans were subjected to MS2 analysis, forming a specific spectral library (see ESI† data set). The library may be used to readily differentiate N-glycan isomers in biological samples. For example, glycan structural isomers of α2,3 and α2,6Neu5Ac terminated N-glycans can be distinguished by their MS2 spectra. N235 and N245 are a pair of isomers with only a difference on the sialyl linkage of the terminal Neu5Ac residue on the α1,3Man branch. In the MS2 spectrum of N235, a base peak at m/z = 1713.59 is gained from losing Neu5Ac, Gal, and GlcNAc (break between GlcNAc and Man residues of α1,3Man branch), whereas the same ion in the MS2 spectrum of N245 is significantly weaker (Fig. 4). It is thus presumed that the glycosidic bond between GlcNAc and Man in N235 is easier to break than that in N245. The same phenomena were also observed in other Neu5Acα2,3, Neu5Acα2,6 isomer pairs, such as N042 and N043, N212 and N213, N222 and N223. Therefore, the signal intensity of fragmentation ions can be reliable evidence in distinguishing linkage patterns between Gal and Neu5Ac residues. Interestingly, this phenomenon was only observed for the structures on the α1,3Man branch, for example, while changing the linkage on the α1,3Man branch (isomers N002 and N133, or N003 and N233) resulted in similar changes in the spectra as described above, no significant changes were observed between N002 and N233 or N003 and N133, which differ only on the α1,6Man branch (Fig. S8†). This phenomenon is interesting and yet hard to comprehend. Several other unique MS2 fragmentation patterns were also found and may be used in distinguishing isomers in the future.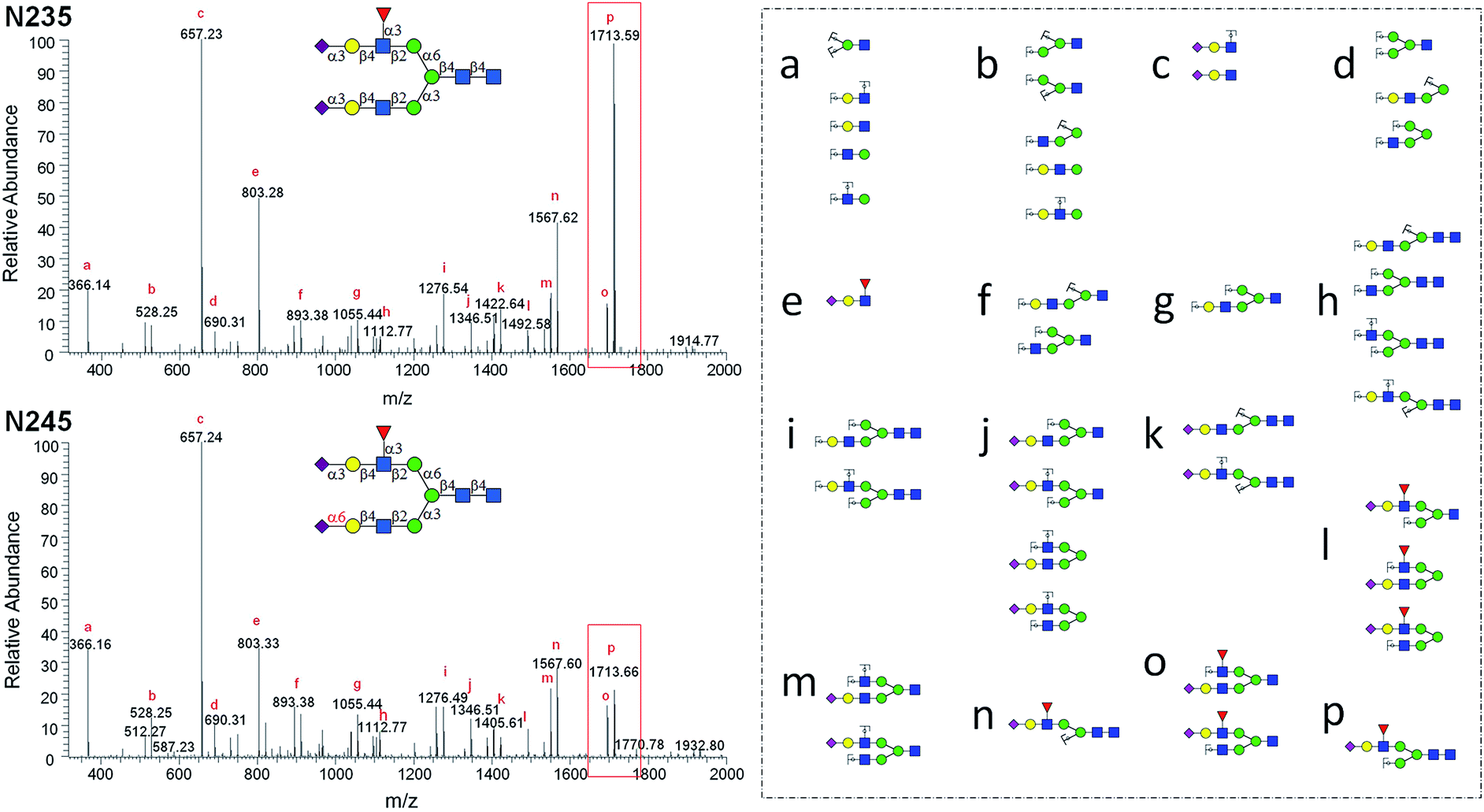 | ||
| Fig. 4 MS2 analysis of N-glycan isomers N235 and N245. | ||
Discussion
Given the diversity and micro-heterogeneous nature of complex glycans, as well as the significant roles they play in biological processes, libraries of structurally well-defined glycans are in urgent demand for studies in glycobiology and glycomedicine. Decades of effort has been devoted to developing chemical methodologies and enzymatic approaches to synthesize N-glycans, however, none were able to (cost-)efficiently generate large numbers of N-glycans. This is mainly due to the facts that: (1) the chemical methodologies developed so far are not cost-effective nor rapid enough to prepare numerous complex N-glycans. Generally, each methodology has been developed specifically for certain N-glycan structures, and is thus not suitable for efficient synthesis of other complex N-glycans. (2) The GTs applied in the enzymatic or chemoenzymatic synthesis approaches were mostly from eukaryotes,8a,9,31 which are typically hard to access and exhibit narrow substrate specificity towards glycan acceptors. Most recently, several N- and N-glycans with multisialylated poly-N-acetyllactosamine extensions were successfully synthesized using a single bacterial α2,6-sialyltransferase (Pd2,6ST).8b This revealed the power of bacterial GTs in diversifying glycans. (3) A highly efficient and rapid N-glycan purification approach is lacking. So far the only reported approach is gel-filtration, which takes many hours to separate each target, and is not able to separate complex glycans with only one monosaccharide difference.This work found answers to the above three obstacles. Firstly, a highly efficient strategy was developed based on the consistent use of oligosaccharyl thioether for the convergent installation of branched GlcNAc-terminated antennae to achieve high stereoselectivity with excellent yields. This approach minimized synthetic steps and maximized yield, and proceeded very efficiently with fewer glycosyl donor (1.3 equivalents) and mild conditions (at 0 °C). Notably, when the (Ac3)GlcNAcβ1,2-Man disaccharide thioether 3 and Bn-GlcNAcβ1,2-Man disaccharide thioether 4 were used as donors, installations on 3-OH of trisaccharide 1 were achieved with excellent yield and high stereoselectivity. We were also able to install the Man3 thioether donor 5 on the 6-OH of the β-Man in good yield and with high stereoselectivity, as seen before in our previous report.18,32 Using this strategy, 8 N-glycan core structures with 5–8 monosaccharide residues were convergently synthesized. We expect this strategy would allow us to prepare more N-glycans with various glycoforms for enzymatic extension.
Secondly, a general enzymatic extension strategy is developed that can extend any GlcNAc terminated glycans to 5 more glycans (including LeX and SLeX) using B4GALT1 and three robust bacterial GTs. Such a strategy enabled the generation of 5–15 more N-glycans from each chemically synthesized core structure. During the synthesis of these N-glycans, each of the GTs was tested towards 10 to 21 N-glycan acceptors. For example, PmST1m showed comparably high activities towards N001, N011, N021, N031, N041, N051, N111, N123, N124, N125, N211, N223, N224 and N225 (which share a common Galβ1,4-GlcNAc motif), and efficiently catalyzed the formation of the corresponding α2,3sialylted N-glycans. In addition, the successful synthesis of Neu5Gc terminated N-glycan N012G indicated that PmST1m is also promiscuous towards sugar donors. Furthermore, a substrate specificity study revealed that Hpα1,3FT can well accept various N-glycans terminated with LacNAc or Siaa2,3LacNAc (ESI, Table S1†). Similarly relaxed substrate specificities were also found for B4GALT1 and Pd2,6ST towards various N-glycan acceptors. These results clearly indicate that: (a) the 4 robust GTs only recognize the adjacent one or two monosaccharide residues in the glycosylation reactions, and thus have a great potential to extend various N-glycans; (b) the promiscuity of the bacterial GTs towards the sugar donors is not affected by acceptors, no matter if simple oligosaccharides23,27,33 or complex N-glycans were used, and thus have a great potential to synthesize N-glycan derivatives.
Thirdly, instead of generally used gel-filtration, each N-glycan was purified to >98% by HPLC utilizing a HILIC column on milligram scales (up to 4 mg per run). This HPLC-based approach could well separate complex N-glycans with only one monosaccharide difference, and takes only 30 min per injection.
Among the synthesized structures, only a few (e.g.N011, N001, N002, N003, N6000) were previously synthesized via chemical7b,15c,34 or chemoenzymatic approaches.8b,31,35 This library covers a number of low molecular weight N-glycans which have or have not been identified,36 including most common hybrid and bi-antennary complex types. More importantly, this work represents the first report preparing high pure N-glycan isomers. This N-glycan library contains 21 groups of isomers (Fig. S7†) (2 to 6 distinct structures in each group), e.g. glycans N125, N134, N144, N225, N234 and N244 are isomers with the same molecular weight of 2077.7455. These groups of isomers are valuable standards that may be applied in absolute quantification and structural identification of N-glycans by MS.
Conclusions
We have successfully developed a Core Synthesis/Enzymatic Extension (CSEE) strategy for efficient synthesis of structurally defined N-glycans and a HPLC-based approach for rapid purification of these compounds. The combination of CSEE and HPLC purification allowed rapid access to 0.5–2 mg of 73 homogenous N-glycans in high purities (>98%), including 63 isomers (21 groups). These N-glycans are valuable materials for glycan analysis and bioactivity evaluation. In this work, oligosaccharyl thioether was used as a chemical glycosylation donor for the convergent installation of branched GlcNAc-terminated antennae, this general and efficient approach produced 8 N-glycan core structures with high stereoselectivity and excellent overall yields. This work also demonstrated that any GlcNAc terminated glycans can be enzymatically extended to 5 or more longer glycans (including LeX and SLeX) using several robust glycosyltransferases. Moreover, this work showed that complex N-glycans are best purified using HPLC utilizing a HILIC column. In summary, the CSEE strategy described here provides a practical approach for rapid production of structurally defined N-glycans, and has the potential to become a general approach to solve the complexity and diversity of glycomes. This may mark the beginning of “mass production” of glycomes.Author contribution
L. L., Y. L., C. M. and P. G. W. designed experiments, analyzed data and wrote the manuscript. L. L. performed enzymatic synthesis, purification and HPLC identification of glycans; Y. L., B. W., and Z. X. performed chemical synthesis and NMR analysis; C. M., J. Q. and N. W. performed MS analysis. All other authors expressed and purified enzymes.Competing financial interests
The authors declare no competing financial interests.Acknowledgements
This work was supported by National Institute of Health (R01GM085267 to P. G. W., R01HD065122 to X. C.) and Federal funds from the National Cancer Institute, National Institutes of Health, Department of Health and Human Services, under Contract No. HHSN261201300040C.References
- A. Varki and J. B. Lowe, in Essentials of Glycobiology, ed. A. Varki, R. D. Cummings, J. D. Esko, H. H. Freeze, P. Stanley, C. R. Bertozzi, G. W. Hart and M. E. Etzler, Cold Spring Harbor, New York, 2nd edn, 2009, ch. 6 Search PubMed.
- B. Adamczyk, T. Tharmalingam and P. M. Rudd, Biochim. Biophys. Acta, 2012, 1820, 1347 CrossRef CAS PubMed.
- C. R. Bertozzi and D. Rabuka, in Essentials of Glycobiology, ed. A. Varki, R. D. Cummings, J. D. Esko, H. H. Freeze, P. Stanley, C. R. Bertozzi, G. W. Hart and M. E. Etzler, Cold Spring Harbor, New York, 2nd edn, 2009, ch. 2 Search PubMed.
- P. Stanley, H. Schachter and N. Taniguchi, in Essentials of Glycobiology, ed. A. Varki, R. D. Cummings, J. D. Esko, H. H. Freeze, P. Stanley, C. R. Bertozzi, G. W. Hart and M. E. Etzler, Cold Spring Harbor, New York, 2nd edn, 2009, ch. 8 Search PubMed.
- M. J. Kailemia, L. R. Ruhaak, C. B. Lebrilla and I. J. Amster, Anal. Chem., 2014, 86, 196 CrossRef CAS PubMed.
- (a) T. J. Boltje, T. Buskas and G. J. Boons, Nat. Chem., 2009, 1, 611 CrossRef CAS PubMed; (b) C. Unverzagt and Y. Kajihara, Chem. Soc. Rev., 2013, 42, 4408 RSC.
- (a) P. Wang, J. Zhu, Y. Yuan and S. J. Danishefsky, J. Am. Chem. Soc., 2009, 131, 16669 CrossRef CAS PubMed; (b) M. A. Walczak and S. J. Danishefsky, J. Am. Chem. Soc., 2012, 134, 16430 CrossRef CAS PubMed.
- (a) S. S. Shivatare, S. H. Chang, T. I. Tsai, C. T. Ren, H. Y. Chuang, L. Hsu, C. W. Lin, S. T. Li, C. Y. Wu and C. H. Wong, J. Am. Chem. Soc., 2013, 135, 15382 CrossRef CAS PubMed; (b) C. M. Nycholat, W. Peng, R. McBride, A. Antonopoulos, R. P. de Vries, Z. Polonskaya, M. G. Finn, A. Dell, S. M. Haslam and J. C. Paulson, J. Am. Chem. Soc., 2013, 135, 18280 CrossRef CAS PubMed.
- Z. Wang, Z. S. Chinoy, S. G. Ambre, W. Peng, R. McBride, R. P. de Vries, J. Glushka, J. C. Paulson and G. J. Boons, Science, 2013, 341, 379 CrossRef CAS PubMed.
- C. Brito, L. Danglot, T. Galli and J. Costa, Brain Res., 2009, 1287, 39 CrossRef CAS PubMed.
- M. A. Naarding, I. S. Ludwig, F. Groot, B. Berkhout, T. B. Geijtenbeek, G. Pollakis and W. A. Paxton, J. Clin. Invest., 2005, 115, 3256 CrossRef CAS PubMed.
- (a) J. B. Lowe, L. M. Stoolman, R. P. Nair, R. D. Larsen, T. L. Berhend and R. M. Marks, Cell, 1990, 63, 475 CrossRef CAS; (b) M. L. Phillips, E. Nudelman, F. C. Gaeta, M. Perez, A. K. Singhal, S. Hakomori and J. C. Paulson, Science, 1990, 250, 1130 CAS.
- P. C. Pang, P. C. Chiu, C. L. Lee, L. Y. Chang, M. Panico, H. R. Morris, S. M. Haslam, K. H. Khoo, G. F. Clark, W. S. Yeung and A. Dell, Science, 2011, 333, 1761 CrossRef CAS PubMed.
- R. D. Astronomo and D. R. Burton, Nat. Rev. Drug Discovery, 2010, 9, 308 CrossRef CAS PubMed.
- (a) G. Ragupathi, D. M. Coltart, L. J. Williams, F. Koide, E. Kagan, J. Allen, C. Harris, P. W. Glunz, P. O. Livingston and S. J. Danishefsky, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 13699 CrossRef CAS PubMed; (b) V. Y. Dudkin, J. S. Miller and S. J. Danishefsky, J. Am. Chem. Soc., 2004, 126, 736 CrossRef CAS PubMed; (c) V. Y. Dudkin, J. S. Miller, A. S. Dudkina, C. Antczak, D. A. Scheinberg and S. J. Danishefsky, J. Am. Chem. Soc., 2008, 130, 13598 CrossRef CAS PubMed.
- C. Unverzagt, G. Gundel, S. Eller, R. Schuberth, J. Seifert, H. Weiss, M. Niemietz, M. Pischl and C. Raps, Bioorg. Chem., 2009, 15, 12292 CAS.
- (a) D. Crich, W. Li and H. Li, J. Am. Chem. Soc., 2004, 126, 15081 CrossRef CAS PubMed; (b) L. Yan and D. Kahne, J. Am. Chem. Soc., 1996, 118, 9239 CrossRef CAS.
- Y. Liu, Y. M. Chan, J. Wu, C. Chen, A. Benesi, J. Hu, Y. Wang and G. Chen, ChemBioChem, 2011, 12, 685 CrossRef CAS PubMed.
- T. Bamhaoud, S. Sanchez and J. Prandi, Chem. Commun., 2000, 659 RSC.
- H. Paulsen, M. Heume and H. Nürnberger, Carbohydr. Res., 1990, 200, 127 CrossRef CAS.
- Y. Liu, J. Marshall, Q. Li, N. Edwards and G. Chen, Bioorg. Med. Chem. Lett., 2013, 23, 2328 CrossRef CAS PubMed.
- G. Sugiarto, K. Lau, Y. Li, Z. Khedri, H. Yu, D. T. Le and X. Chen, Mol. Biosyst., 2011, 7, 3021 RSC.
- H. Yu, S. Huang, H. Chokhawala, M. Sun, H. Zheng and X. Chen, Angew. Chem., Int. Ed., 2006, 45, 3938 CrossRef CAS PubMed.
- S. W. Lin, T. M. Yuan, J. R. Li and C. H. Lin, Biochemistry, 2006, 45, 8108 CrossRef CAS PubMed.
- X. Chen and A. Varki, ACS Chem. Biol., 2010, 5, 163 CrossRef CAS PubMed.
- A. Varki, Am. J. Phys. Anthropol., 2001,(33), 54 CrossRef CAS PubMed.
- H. Yu, H. Chokhawala, R. Karpel, H. Yu, B. Wu, J. Zhang, Y. Zhang, Q. Jia and X. Chen, J. Am. Chem. Soc., 2005, 127, 17618 CrossRef CAS PubMed.
- H. Shimizu, K. Ochiai, K. Ikenaka, K. Mikoshiba and S. Hase, J. Biochem., 1993, 114, 334 CAS.
- (a) L. Li, Y. Liu, Y. Wan, Y. Li, X. Chen, W. Zhao and P. G. Wang, Org. Lett., 2013, 15, 5528 CrossRef CAS PubMed; (b) L. Muinelo-Romay, C. Vazquez-Martin, S. Villar-Portela, E. Cuevas, E. Gil-Martin and A. Fernandez-Briera, Int. J. Cancer, 2008, 123, 641 CrossRef CAS PubMed; (c) A. Kuno, Y. Ikehara, Y. Tanaka, T. Angata, S. Unno, M. Sogabe, H. Ozaki, K. Ito, J. Hirabayashi, M. Mizokami and H. Narimatsu, Clin. Chem., 2011, 57, 48 CrossRef CAS PubMed.
- (a) K. Cheng, R. Chen, D. Seebun, M. Ye, D. Figeys and H. Zou, J. Proteomics, 2014, 110, 145 CrossRef CAS PubMed; (b) D. Kolarich, P. H. Jensen, F. Altmann and N. H. Packer, Nat. Protoc., 2012, 7, 1285 CrossRef CAS PubMed.
- S. Serna, S. Yan, M. Martin-Lomas, I. B. Wilson and N. C. Reichardt, J. Am. Chem. Soc., 2011, 133, 16495 CrossRef CAS PubMed.
- Y. Liu and G. Chen, J. Org. Chem., 2011, 76, 8682 CrossRef CAS PubMed.
- W. Wang, T. Hu, P. A. Frantom, T. Zheng, B. Gerwe, D. S. Del Amo, S. Garret, R. D. Seidel III and P. Wu, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 16096 CrossRef CAS PubMed.
- V. Y. Dudkin, M. Orlova, X. Geng, M. Mandal, W. C. Olson and S. J. Danishefsky, J. Am. Chem. Soc., 2004, 126, 9560 CrossRef CAS PubMed.
- C. Unverzagt, Carbohydr. Res., 1997, 305, 423 CrossRef CAS.
- (a) Z. Tang, R. S. Varghese, S. Bekesova, C. A. Loffredo, M. A. Hamid, Z. Kyselova, Y. Mechref, M. V. Novotny, R. Goldman and H. W. Ressom, J. Proteome Res., 2010, 9, 104 CrossRef CAS PubMed; (b) Z. Kyselova, Y. Mechref, M. M. Al Bataineh, L. E. Dobrolecki, R. J. Hickey, J. Vinson, C. J. Sweeney and M. V. Novotny, J. Proteome Res., 2007, 6, 1822 CrossRef CAS PubMed; (c) P. Babu, S. J. North, J. Jang-Lee, S. Chalabi, K. Mackerness, S. R. Stowell, R. D. Cummings, S. Rankin, A. Dell and S. M. Haslam, Glycoconjugate J., 2009, 26, 975 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Materials, detailed experimental protocols, supporting schemes and figures, synthetic methods, analytical data including HPLC profiles, mass and NMR spectra of synthesized N-glycans. See DOI: 10.1039/c5sc02025e |
| ‡ Contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2015 |