
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThe approach to 4d/4f-polyphosphides†
Nicholas
Arleth
a,
Michael T.
Gamer
a,
Ralf
Köppe
a,
Nikolay A.
Pushkarevsky
bc,
Sergey N.
Konchenko
ab,
Martin
Fleischmann
c,
Michael
Bodensteiner
c,
Manfred
Scheer
c and
Peter W.
Roesky
*a
aInstitute of Inorganic Chemistry, Karlsruhe Institute of Technology, Engesserstrasse 15, 76131 Karlsruhe, Germany. E-mail: roesky@kit.edu
bNikolaev Institute of Inorganic Chemistry SB RAS, Novosibirsk State University, Prosp. Lavrentieva 3, Pirogova str. 2, 630090 Novosibirsk, Russia. E-mail: konch@niic.nsc.ru
cInstitute of Inorganic Chemistry, University of Regensburg, 93040 Regensburg, Germany. E-mail: Manfred.Scheer@chemie.uni-regensburg.de
First published on 29th September 2015
Abstract
The first 4d/4f polyphosphides were obtained by reaction of the divalent metallocenes [Cp*2Ln(thf)2] (Ln = Sm, Yb) with [{CpMo(CO)2}2(μ,η2:2-P2)] or [Cp*Mo(CO)2(η3-P3)]. Treatment of [Cp*2Ln(thf)2] (Ln = Sm, Yb) with [{CpMo(CO)2}2(μ,η2:2-P2)] gave the 16-membered bicyclic compounds [(Cp2*Ln)2P2(CpMo(CO)2)4] (Ln = Sm, Yb) as the major products. From the reaction involving samarocene, the cyclic P4 complex [(Cp*2Sm)2P4(CpMo(CO)2)2] and the cyclic P5 complex [(Cp*2Sm)3P5(CpMo(CO)2)3] were also obtained as minor products. In each reaction, the P2 unit is reduced and a rearrangement occurred. In dedicated cases, a P–P bond formation takes place, which results in a new aggregation of the central phosphorus scaffold. In the reactions of [Cp*2Ln(thf)2] (Ln = Sm, Yb) with [Cp*Mo(CO)2P3] a new P–P bond is formed by reductive dimerization and the 4d/4f hexaphosphides [(Cp*2Ln)2P6(Cp*Mo(CO)2)2] (Ln = Sm, Yb) were obtained.
Introduction
Phosphorus is one of the most common and well-established donor atoms in coordination chemistry, and it can form strong bonds with soft metals.1–3 Many transition metal phosphorus complexes are used as catalysts in various industrial processes.4 In contrast, rare-earth metal phosphorus complexes are far less common5 and only a few years ago the first molecular lanthanide polyphosphide complex, [(Cp*2Sm)4P8] (Cp* = η5-C5Me5) (A, Scheme 1),6 was reported. It was obtained by reaction of the divalent solvate-free samarocene [Cp*2Sm] with white phosphorus. The structure can be described as a realgar-shaped P84− ligand trapped in a cage of four samarocenes. Another example of direct activation of P4 to a ligand-stabilized P84− was reported by Diaconescu and coworkers. [{(NNfc)Sc}4P8] (NNfc = 1,1′-fc(NSitBuMe2)2, fc = ferrocenylene) was obtained by the reaction of P4 and 1 equiv. of the scandium naphthalene complex [{(NNfc)Sc}2(μ-C10H8)].7 Using this methodology, the P73− compounds [{(NNfc)Ln(THF)n}3P7] (Ln = Sc, Y, La, Lu; B, Scheme 1) were also reported.7,8 The first well-defined P3−-containing rare-earth metal compounds, in which six yttrium atoms coordinate to a μ6-P3− ligand, were synthesized by Chen and coworkers (C, Scheme 1).9 Recently, we reported the reactions of divalent samarium compounds with transition-metal-coordinated polyphosphides.10,11 In this context, the first P–P bond formation reactions between two [Cp*FeP5] molecules triggered by divalent lanthanide complexes to give [(Cp*Fe)2P10{Sm(η5-C5Me4R)2}2] (R = Me, nPr; D, Scheme 1) were reported. This intermolecular P–P bond formation arises from reductive dimerization.12,13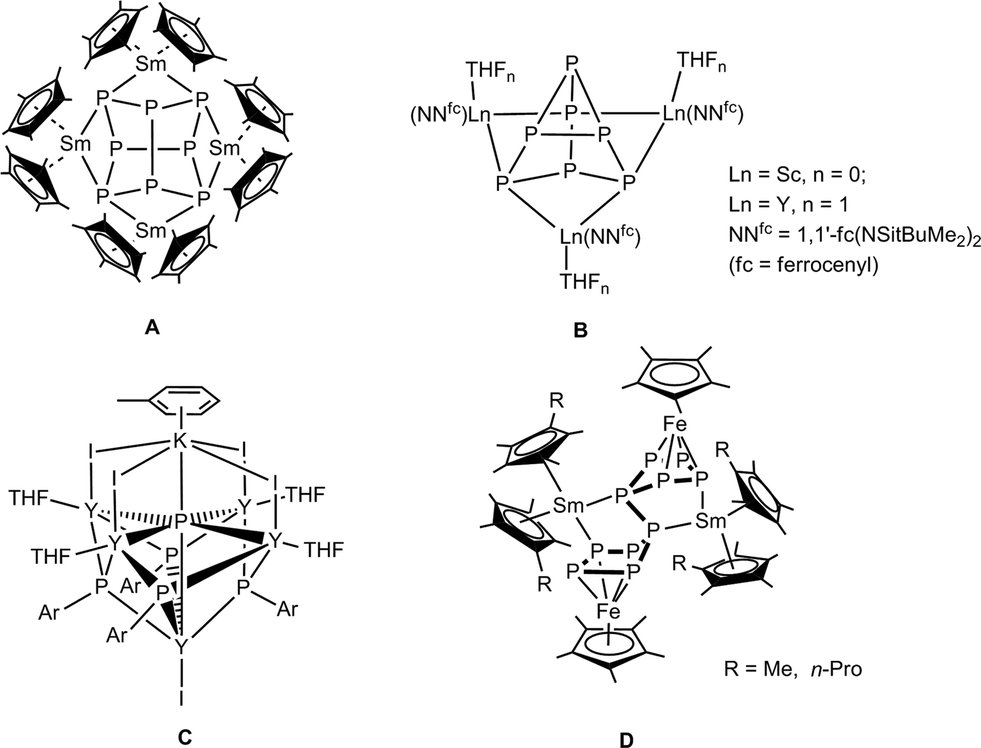 | ||
| Scheme 1 Known lanthanide polyphosphide complexes.6–9,13 | ||
Since the existing examples of lanthanide/polyphosphide complexes came from 3d transition metal complexes, the question arises, whether 4d metals will follow this reactivity mode or novel transformations will occur. Herein, we report the first 4d/4f polyphosphides containing unprecedented polyphosphorus moieties, which were formed by multiple intermolecular P–P bond formations and untypical metal fragment motions.
Results and discussion
Reactions of [Cp*2Ln(thf)2] (Ln = Sm, Yb),14–16 with [{CpMo(CO)2}2(μ,η2:2-P2)]17 at 60 °C in toluene for one week yielded a product mixture, which could be separated by fractional crystallization. The 16-membered bicyclic compounds [(Cp2*Ln)2P2(CpMo(CO)2)4] (Ln = Sm (1a), Yb (1b)) were the major products (Scheme 2). From the reaction with samarocene, the cyclic P4 complex [(Cp*2Sm)2P4(CpMo(CO)2)2] (2) and the cyclic P5 complex [(Cp*2Sm)3P5(CpMo(CO)2)3] (3) were obtained as minor products (Scheme 2).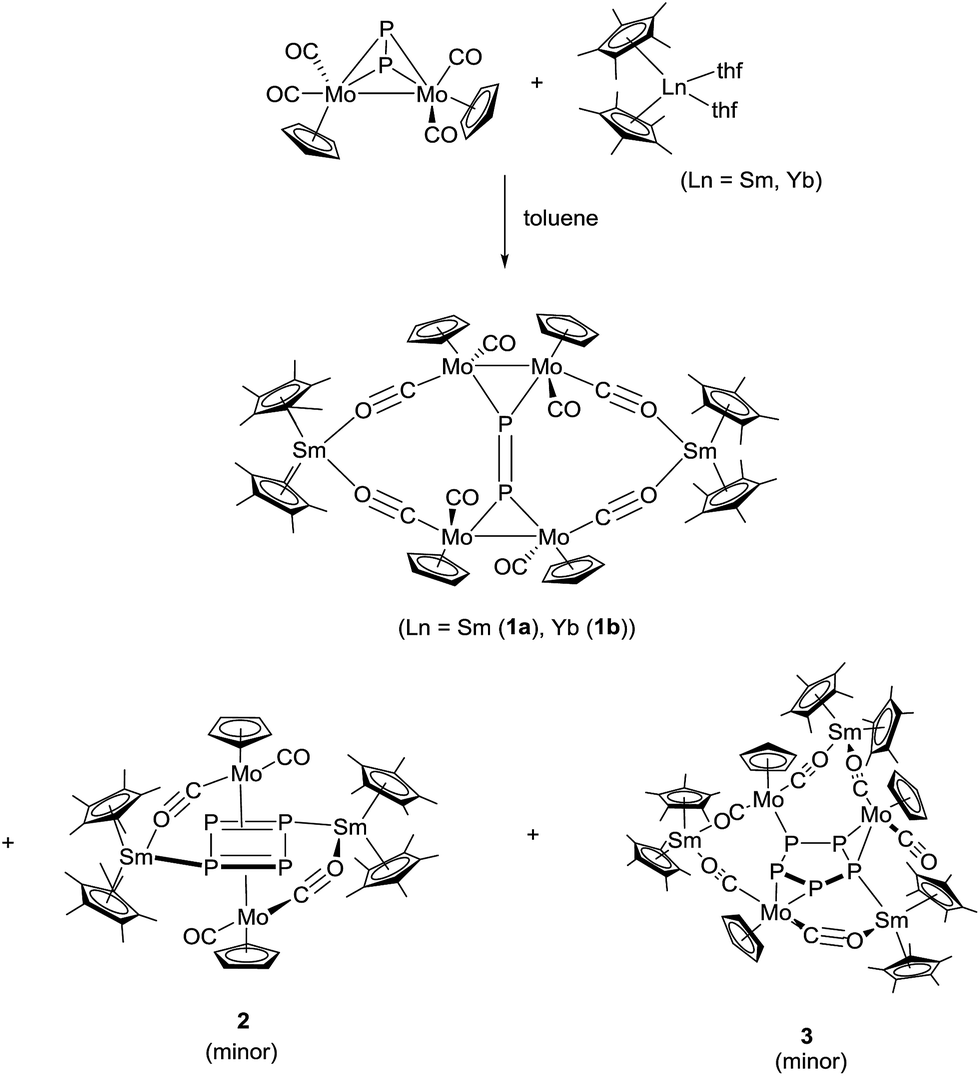 | ||
| Scheme 2 Synthesis of 1–3. | ||
Fractional crystallization of the mother liquor afforded 1a first, allowing for it to be fully characterized. Further concentration resulted in a mixture of 1a, 2 and 3, and some poorly soluble powder, which could not be further identified. The crystals of 2 and 3 could only be separated manually and, thus, a complete characterization was not possible. Nevertheless, the solid-state structures give insight into the reaction pathway.
In all cases, the lanthanide atoms in [Cp*2Ln(thf)2] are oxidized from oxidation state +2 to +3.18 The oxidation state of the samarium atom was clearly assigned by NIR spectroscopy (see below) and magnetic measurements. Concomitant with the oxidation of the lanthanide atom, the polyphosphide is reduced. Furthermore, upon reduction, in 2 and 3 the Mo–Mo bond is completely broken and {CpMo(CO)2} fragments without any metal-to-metal bond are formed. Each of these {CpMo(CO)2} units also coordinates to a polyphosphide for electronic saturation, resulting in 18 VE species.
Compounds 1a,b are crystallographically similar but due to different amounts of lattice solvent not isostructural. They both crystallize in the triclinic space group P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) (Fig. 1). In 1a,b two phosphorus atoms form a central {P
(Fig. 1). In 1a,b two phosphorus atoms form a central {P![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) P}2− unit, which coordinates end-on to two {CpMo(CO)2}2 fragments. There is only one other example known of such a P2 unit four times coordinated by Cp*Re(CO)2 moieties.19 A crystallographic inversion center is observed in the center of the {PP}2− unit. The P–P bond distances of 2.029(4) Å (1a) and 2.007(4) Å (1b) are in the range of a PP double bond (e.g. 2.052(2) Å) in [(Cp′′′Co)2(μ,η2:2-P2)2] (Cp′′′ = 1,2,4-tBu3C5H2)20 and are slightly shorter than in the starting material [{CpMo(CO)2}2(μ,η2:2-P2)] (P–P 2.079(2) Å).17 The Mo–P distances in 1a (av. 2.366 Å) and 1b (av. 2.360 Å) are significantly shorter than in [{CpMo(CO)2}2(μ,η2:2-P2)] (2.552(1) Å and 2.463(1) Å). The Mo–Mo bonds of 3.1935(8) Å (1a) and 3.2073(9) Å (1b) are in the upper range of unsupported Mo–Mo bonds (2.88–3.32 Å).21
P}2− unit, which coordinates end-on to two {CpMo(CO)2}2 fragments. There is only one other example known of such a P2 unit four times coordinated by Cp*Re(CO)2 moieties.19 A crystallographic inversion center is observed in the center of the {PP}2− unit. The P–P bond distances of 2.029(4) Å (1a) and 2.007(4) Å (1b) are in the range of a PP double bond (e.g. 2.052(2) Å) in [(Cp′′′Co)2(μ,η2:2-P2)2] (Cp′′′ = 1,2,4-tBu3C5H2)20 and are slightly shorter than in the starting material [{CpMo(CO)2}2(μ,η2:2-P2)] (P–P 2.079(2) Å).17 The Mo–P distances in 1a (av. 2.366 Å) and 1b (av. 2.360 Å) are significantly shorter than in [{CpMo(CO)2}2(μ,η2:2-P2)] (2.552(1) Å and 2.463(1) Å). The Mo–Mo bonds of 3.1935(8) Å (1a) and 3.2073(9) Å (1b) are in the upper range of unsupported Mo–Mo bonds (2.88–3.32 Å).21
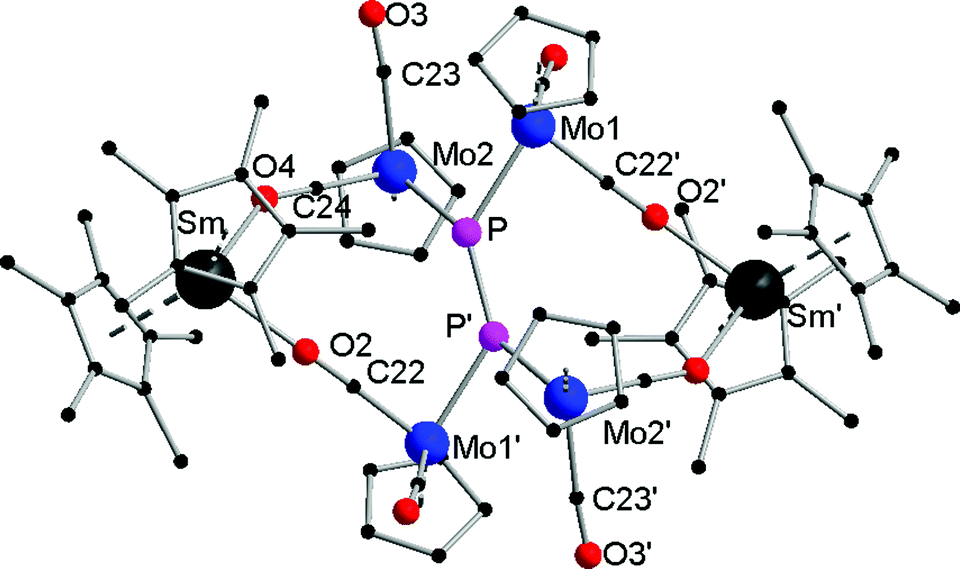 | ||
| Fig. 1 Solid-state structure of 1a (1b is similar). Hydrogen atoms are omitted for clarity. Selected distances [Å], angles [°]: 1a: Sm–O2 2.359(5), Sm–O4 2.389(5), Mo1–Mo2 3.1935(8), Mo1–P 2.367(2), Mo1–C21 1.976(8), Mo1–C22′ 1.881(8), Mo2–P 2.367(2), Mo2–C24 1.902(8), Mo2–C23 1.985(8), P–P′ 2.029(4); O2–Sm–O4 83.4(2), C21–Mo1–P 110.7(2), C22–Mo1–P′ 84.2(2), C21–Mo1–C22′ 79.7(3), C23–Mo2–C24 83.9(3), C24–Mo2–P 87.7(2), P–Mo2–Mo1 47.57(5), P′–P–Mo1 132.88(13), P′–P–Mo2 132.71(12), Mo2–P–Mo1 84.86(6). 1b: Yb–O2 2.231(5), Yb–O4 2.246(5), Mo1–Mo2 3.2073(9), Mo2–P 2.362(2), Mo1–C21 1.893(7), Mo1–C22 1.968(9), Mo2–P 2.362(2), Mo2–C23 1.974(8), Mo2–C24 1.863(7), P–P′ 2.007(4); O2–Yb–O4 83.5(2), P–Mo1–Mo2 47.25(5), C21–Mo1–P 111.0(2), C21–Mo1–C22 82.6(3), C22–Mo1–P 86.0(2), P–Mo2–Mo1 47.13(4), C23–Mo2–P 108.9(3), C23–Mo2–C24 84.7(3), C24–Mo2–P 85.6(2), Mo1–P–Mo2 85.61(7). | ||
The central {P2(CpMo(CO)2)4}2− subunit is end-capped by two {Cp*2Ln}+ units, each coordinating via two isocarbonyl bridges to the central core. Thus, a 16-membered bicyclic structure is formed, which consists of the {PP}2− unit, four Mo atoms, two Ln atoms, and four carbonyl ligands each bridging the Ln and Mo atoms. The Ln–O bond distances of the isocarbonyl bridges (Sm–O2 2.359(5) Å and Sm–O4 2.389(5) Å; Yb–O2 2.231(5) Å and Yb–O4 2.246(5) Å) are in the range of other isocarbonyl compounds, e.g. [(TpMe,Me)2Sm(μ-CO){CpMeMo(CO)2}] (2.335(4) Å; TpMe,Me = (2,4-dimethylpyrazolyl)3borate, CpMe = MeC5H4);22 and [(SmI2(thf)4)(μ-CO)(CpMo(CO)2)] (2.41(2) Å and 2.49(2) Å).20 The lanthanide atoms are coordinated to two isocarbonyl ligands and two Cp* rings, resulting in a distorted tetrahedral coordination geometry. As a result of the oxidation of the lanthanide atoms, the average Ln–C bond is shortened by about 0.2 Å in 1a (2.695 Å (Sm1–C(Cp*1)(av.)) and 2.684 Å (Sm1–C(Cp*2)(av.))) in comparison to [Cp*2Sm(thf)2] (av. 2.86(3) Å)14 and about 0.1 Å in 1b (2.590 Å (Yb1–C(Cp*1)(av.)) and 2.583 Å (Yb1–C(Cp*2) (av.))) in comparison to [Cp*2Yb(thf)] (av. 2.672 Å).23 Similar Ln–C bond distances as in 1a,b were observed in other trivalent complexes, e.g. in [Cp*2SmCl(thf)] (av. 2.71 Å and 2.72 Å).24
In the IR spectra of 1a,b, the CO stretching frequencies of the terminal CO ligands are observed as strong bands at 1945 cm−1, 1905 cm−1 and 1871 cm−1 (1a), and 1906 cm−1 and 1873 cm−1 (1b) (Fig. S1 and S3†). Thus, they are in the range of [{CpMo(CO)2}2(μ,η2:2-P2)] (1965 cm−1, 1913 cm−1).17 As expected, the bridging CO groups show stretching frequencies at lower energy (1683 cm−1, 1636 cm−1 (1a) and 1687 cm−1, 1638 cm−1 (1b)), which is also in agreement with the literature, e.g. 1650 cm−1 in [(TpMe,Me)2Sm(μ-CO){CpMeMo(CO)2}].22
As a result of weak ligand field splitting, Sm3+ complexes exhibit a characteristic absorption pattern in the NIR spectrum even in the presence of strong visible absorption.25,26 The spectrum obtained for 1a shows spectral patterns that are comparable to Sm3+/POCl3/ZrCl4 (ref. 27) and Sm3+/SeOCl2/SnCl4.28 The observed bands at 9319 cm−1, 8084 cm−1, 7338 cm−1 and 6388 cm−1 for 1a are assigned to the transitions of states 6H5/2 → 6F9/2, 6F7/2, 6F5/2, and 6F1/2 respectively (Fig. S3†).27
The minor product 2 crystallizes in the triclinic space group P with one molecule and one equivalent of toluene in the unit cell (Fig. 2). It consists of a central planar P4 ring, which has a rectangular shape with two short (P1–P2 2.151(4) Å) and two long (P1–P2′ 2.265(3) Å) P–P bonds. Such a cyclo-tetraphosphabutadiene moiety was only recently reported in the Co(I) complex [(LDepCo)2(μ2:η4,η4-P4)] (LDep = CH[C(Me)N(2,6-Et2C6H3)]2) as a result of P4 activation.29 In contrast, here for the first time, it is formed by merging two P2 units together. A crystallographic inversion center is observed in the center of the P4 ring. The angles within the P4 ring (88.29(13)° and 91.71(13)°) are close to 90°. Two {CpMo(CO)2} fragments each coordinate side-on in an η2-mode to the shorter P–P bonds, forming a [P4(CpMo(CO)2)2]2− subunit. The angle between the P4 ring and the Mo–P1–P2 plane is 108.77(13)°. As observed in 1a, each {Cp*2Sm}+ unit coordinates via one isocarbonyl bridge to a {CpMo(CO)2} fragment. The Sm atom is tilted towards the ring with an O1′–Sm–P1 angle of 76.8(2)°. The Sm–P1 distance of 3.016(3) Å is in agreement with other samarium phosphide compounds such as [Cp*FeP5Sm(DIP2pyr)(THF)2] (2.847(2) Å to 3.0703(15) Å)11 and [(Cp*2Sm)4P8] (2.997(2) Å to 3.100(2) Å).6 The observed isocarbonyl bridge in 2 (Sm–O1 2.388(7) Å) is in the range of 1a.
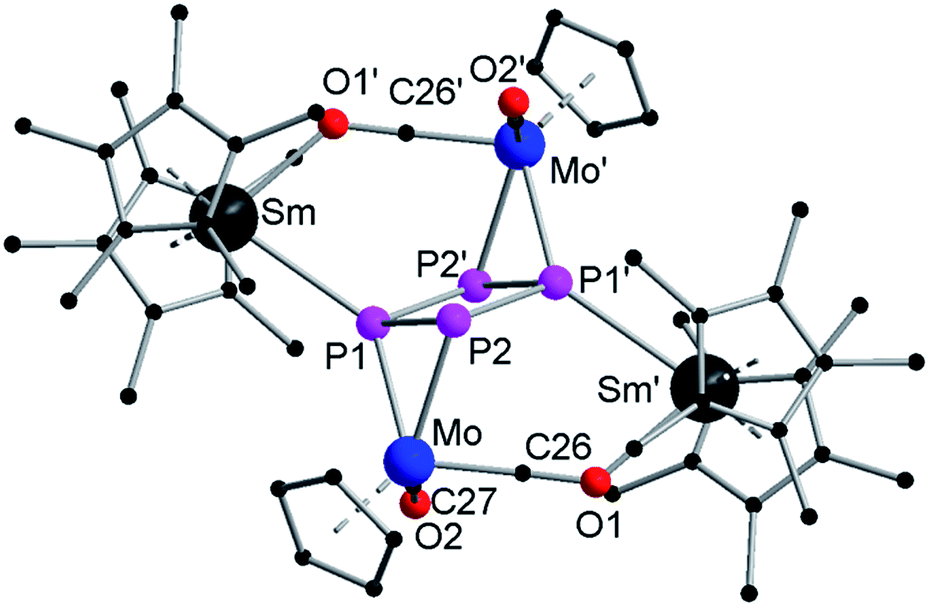 | ||
| Fig. 2 Solid-state structure of 2. Hydrogen atoms are omitted for clarity. Selected bond lengths [Å], angles [°]: Sm–P1 3.016(3), Sm–O1′ 2.388(7), Mo–P2 2.551(3), Mo–P1 2.525(3), Mo–C26 1.875(12), Mo–C27 1.968(11), P1–P2 2.151(4), P1–P2′ 2.265(3); O1′–Sm–P1 76.8(2), P1–Mo–P2 50.14(9), C26–Mo–C27 84.2(4), Mo–P1–Sm 137.26(10), P1–P2–Mo 64.31(10), P1–P2′–Mo′ 106.08(12), P1–P2–P1′ 88.29(13), P2–P1′–P2′ 91.71(13). | ||
Compound 3 crystallizes in the triclinic space group P (Fig. 3). As a result of strong disorder, bonding parameters are not discussed in detail. The disorder is caused by two superimposed molecules (Fig. S4 and S5†), which could be separately refined. The central part of 3 consists of a planar P5 ring. As observed in 2, two {CpMo(CO)2} fragments (Mo1, Mo2) coordinate side-on in an η2-mode to the phosphorus ring. Mo1 binds to P1 and P2, and Mo2 binds to P3 and P4. A third {CpMo(CO)2} fragment (Mo2′) binds to the remaining phosphorus atom (P5). The different coordination modes of Mo2 and Mo2′, and Sm1 and Sm1′, are a result of the disorder (see Fig. S4 and S5†). The two CO ligands of this unit each link Mo2′ to two {Cp*2Sm}+ units (Sm1′, Sm2) via isocarbonyl bridges. One additional isocarbonyl bridge from an adjacent {CpMo(CO)2} fragment is also observed for both {Cp*2Sm}+ units. Thus, Sm1′ and Sm2 are each coordinated to two Cp* rings and two isocarbonyl ligands. The samarium atom of the third {Cp*2Sm}+ unit (Sm1) is coordinated to only one isocarbonyl ligand (from Mo1) and one phosphorus atom (P3) from the planar P5 ring.
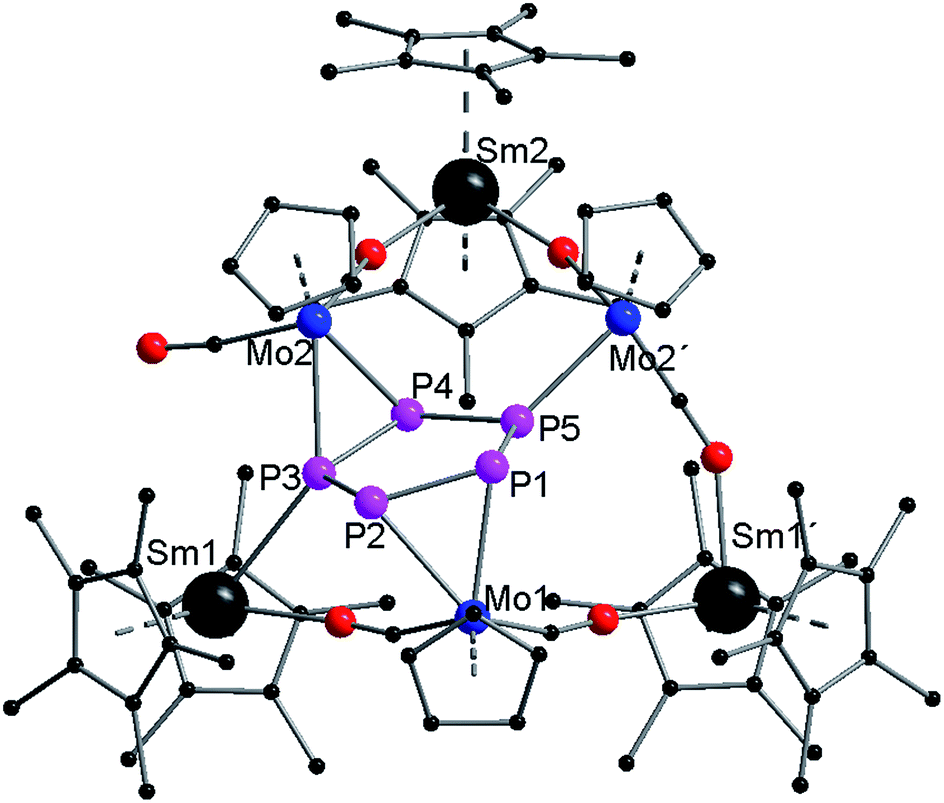 | ||
| Fig. 3 Solid-state structure of 3. Hydrogen atoms are omitted for clarity. | ||
The formation of 1–3 is in contrast to other reported coordination derivatives of [{CpMo(CO)2}2(μ,η2:2-P2)]. In the known derivatives, such as [CuBr(Cp2Mo2(CO)4P2)]n30 and [{(CpMo(CO)2P)2}4Ag2][Al{OC(CF3)3}4]2,31 the [CpMo(CO)2P]2 scaffold stayed intact. Even the addition of Lewis bases to the latter species did not cleave the Mo–P scaffold. Due to the reduction reported herein, the phosphorus atoms completely rearrange, forming P2, P4, and P5 units. Obviously, strong reducing agents are needed to cleave the [CpMo(CO)2P]2 scaffold since the reducing power of [Cp*2Eu(thf)2] is too weak to effect this reaction.
The nature of the bonding in 1a, 2 and 3 was investigated by quantum chemical RI-DFT calculations using the program system TURBOMOLE.32,33 The calculations were performed using the BP86 functional34–36 by using the R-IJ approximation.37 The basis sets for all atoms (except Sm) were of def-SV(P) quality.38,39 For Mo and Sm, a relativistic corrected effective core potential was used to simulate the 28 (Mo)40 and 51 inner electrons (ECP-51).41 Due to the expected ionic nature of the complex containing Sm in the formal trivalent oxidation state, the inclusion of the 5f-electrons into the ECP is allowed. The structural results obtained for 1a by crystallographic studies were confirmed by our theoretical calculations (1a (theory/exp.): d(P–P) 2.078/2.029, d(Mo–P) 2.406/2.367, d(Mo–Mo) 3.247/3.194 Å).
To gain insight into the nature of the bonding in 1a and 2, population analyses based on occupation numbers were performed (Roby–Davidson–Ahlrichs–Heinzmann population analysis).42,43 The results were compared with those of the starting material [{CpMo(CO)2}2(μ,η2:2-P2)] and the model compound (Na+)4(P2)2− possessing a PP double bond. Partial charges (Q) to interpret the ionicity of the bonds were obtained. Shared electron numbers (SEN) served as a measure for the covalent bond strength. The analysis confirmed the realization of an ionic complex 1a with regard to Sm (Q(Sm)) = +1.40); electron density was transferred from the π- and π*-type MOs of the P22− unit to Mo (Q(P) = 0.00, Q(Mo) = −0.54; (see MOs 6 b2u, 3 b1u and 6 b1g as well as 303 a, 312 a and 330 a in the MO diagrams given in the ESI†). This behavior leads to a reduced SEN as well as a larger distance of the P–P bond (SEN(P–P) = 1.38, r(P–P) = 207.8) compared to that of the typical PP double bond in (Na+)4(P2)2− (SEN(P–P) = 1.74, r(P–P) = 206.3). Comparison with the educt molecule [{CpMo(CO)2}2(μ,η2:2-P2)] (SEN(Mo–P) = 0.55, r(Mo–P) = 250.5) indicates a significant increase of the Mo–P bond strength in 1a.
The observed P–P distances in 2 were also confirmed by theoretical methods. It indicates that the nature of the bonding in the central P4 unit is comparable to that of a recently published Co(I) complex.29 Furthermore, some less significant similarities to the model compound D2h–P4 were seen (see ESI†). A similar situation is observed for the P–P bonds in 3. The distances are in agreement with the anionic building block in well-known P5− complexes.44,45 On the other hand the increase of the bond strength in the P5 unit is compared to a P–P single bond less pronounced than in the calculated model compounds Na2P4, NaP5, and Na3P52+ (distances and SEN given in the ESI†).
Due to the low symmetries and the sophisticated ligand sphere of metal complexes, complicated MO diagrams excluding a detailed bonding analysis were obtained for 2 and 3. Thus, it cannot be excluded that the P4 and P5 units are mainly stabilized by the coordinating metal complex fragments and the observed P–P interactions are less significant.
To gain more insight into the reduction chemistry of molybdenum phosphides, we further treated [Cp*2Ln(thf)2] (Ln = Sm, Yb) with [Cp*Mo(CO)2(η3-P3)],46 which features a P3 triangle in the coordination sphere. Reactions in toluene at 60 °C resulted in 4d/4f hexaphosphides [(Cp*2Ln)2P6(Cp*Mo(CO)2)2] (Ln = Sm (4), Yb (5)) (Scheme 3). The solid-state structures of both compounds were established by single crystal X-ray diffraction (Fig. 4). Compounds 4 and 5 are isostructural and crystallize in the triclinic space group P. The solid-state structures of 4 and 5 are disordered. The central P6 core shows four-fold disorder (Fig. S6 and S7†). Thus, bonding parameters are not discussed in detail. Fig. 4 shows only one of the disordered positions. The formation of 4 and 5 is easier to rationalize than the formation of 1–3. Due to the one-electron reduction of [Cp*Mo(CO)2(η3-P3)], one Mo–P bond is broken and a new P–P bond (P2–P2′ in 4) between two negatively charged 18 VE [Cp*Mo(CO)2P3]− units is formed. The P–P bond length of the newly formed bond is in the range of a P–P single bond (2.154(3) Å (4) and 2.216(6) Å (5)).47 A crystallographic inversion center is observed in the middle of this central P–P bond. The negative charge of the [Cp*Mo(CO)2P3]− units is a result of the one-electron reduction by the lanthanocenes. For charge balance, two {Cp*2Sm}+ units coordinate to the central {P6(Cp*Mo(CO)2)2}2– scaffold. The {Cp*Mo(CO)2} units in 4 and 5 bind side-on in an η2-mode to the central P6 core. As observed in 2, each {Cp*2Ln}+ cation coordinates to the molybdenum atom via one isocarbonyl bridge. The Ln–O distances (av. (including all disordered positions) 2.409 Å (4) and 2.311 Å (5)) are in the range of other isocarbonyl compounds, e.g. [(TpMe,Me)2Sm(μ-CO)(CpMo(CO)2)] (2.335(4) Å), [(Cp*2Sm)(μ-CO)2(Cp*Fe)]2 (2.348(4) Å), [(Cp*2Yb)(μ-CO)2Mn(CO)3]2 (2.271(2) Å) and [(Cp*2Yb(thf))(μ-CO)Co(CO)3] (2.258(2) Å).16,48 Furthermore, coordination of the lanthanide atoms to the P6 core is observed and Ln–P1 bonds of 2.963(2) Å (4) and 2.879(3) Å (5) are formed. The observed Yb–P distances correspond well with those of comparable compounds with Yb–PR3 interactions, e.g. [Li(thf)4][(Ph2PNPh)4Yb] (2.885(2)–3.031(2) Å).49 As already observed in 2, the lanthanide atoms are coordinated to two Cp* rings, one phosphorus, and one oxygen atom from the isocarbonyl bridge.
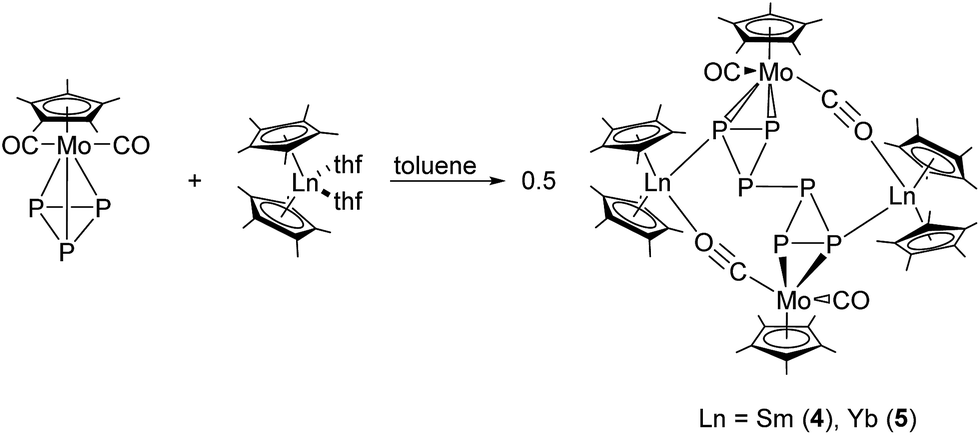 | ||
| Scheme 3 Synthesis of 4 and 5. | ||
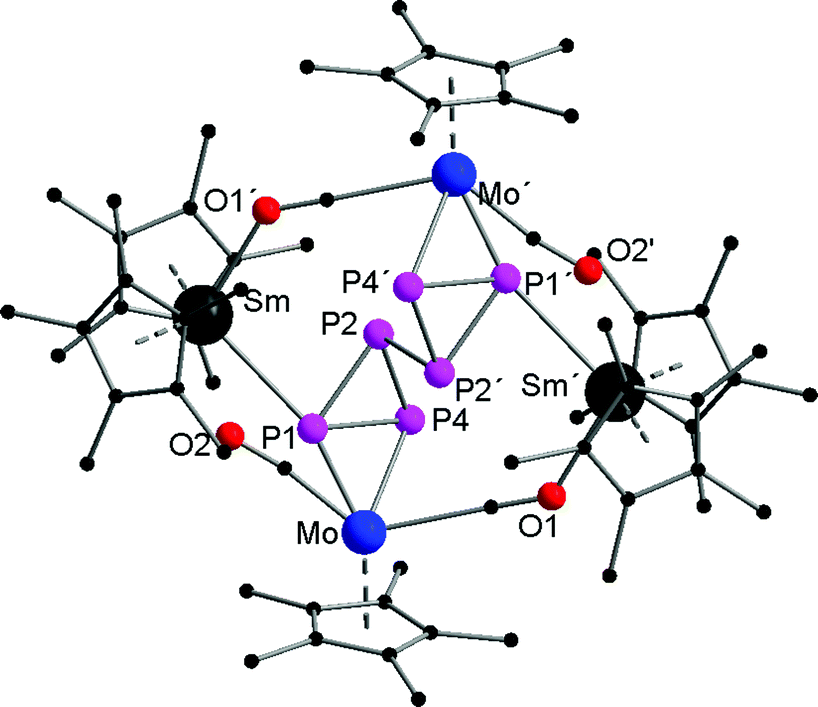 | ||
| Fig. 4 Solid-state structure of 4 (5 is isostructural). Hydrogen atoms are omitted for clarity. | ||
The IR spectra show intense bands for the CO stretching vibrations at 1917 cm−1 (4) and 1916 cm−1 (5), respectively, which can be assigned to the terminal non-bridging CO ligands. Absorption bands at 1701 cm−1 (4), and 1696 and 1669 cm−1 (5), are assigned to the isocarbonyl ligands (Fig. S9 and S11†). This assignment is in agreement with related compounds, e.g. [(Cp*2Yb)(μ-CO)2Mn(CO)3]2 and [(TpMe,Me)2Sm(μ-CO)(CpCr(CO)2)].16,22
To support the formation pathway of 4 and 5, the oxidation state of the samarium atom in 4 was determined by magnetic measurements (SQUID) and NIR spectroscopy. Low paramagnetic susceptibility χM (290 K) = 6238 × 10−6 cm3 mol−1 and magnetic moment μeff = 3.80 μB correspond to the two samarium(III) metal centers and two unpaired electrons from the Mo atoms in 4 (Fig. S9†). These data are comparable to the literature values for Sm3+ complexes.11
As described for 1, the oxidation state of the samarium atom in 4 was unambiguously assigned by NIR spectroscopy.25,26 The observed bands at 9224 cm−1, 7967 cm−1, 6702 cm−1 and 6395 cm−1 are assigned to the transitions of states 6H5/2 → 6F9/2, 6F7/2, 6F3/2 and 6F1/2 (Fig. S11†).27
Conclusions
In summary, we have synthesized and characterized the first 4d/4f polyphosphides. Reduction of molybdenum polyphosphides with the divalent lanthanocences [Cp*2Ln(thf)2] (Ln = Sm, Yb) resulted in the reduction of the molybdenum compounds in both cases. Depending on the nature of the molybdenum polyphoshide, different reaction pathways were followed. Reduction of the mononuclear complex [Cp*Mo(CO)2(η3-P3)] leads to a breaking of one Mo–P bond and the formation of a new P–P bond of a central P6 core in the {P6(Cp*Mo(CO)2)2}2– fragment.In contrast, the reduction pathway of [{CpMo(CO)2}2(μ,η2:2-P2)] is more diverse. This reduction always results in a rearrangement of the P2 unit. In the major product, a {PP}2− unit is observed, which is the result of a two electron reduction. In some cases also aggregation of the phosphorus scaffolds occurs, forming novel planar cyclo-tetraphosphabutadiene P4 and cyclo-P5 fragments by P–P bond formations in an unprecedented coordination environment. These polyphosphides are surrounded by {CpMo(CO)2} fragments.
In each compound, the lanthanocene cations {Cp*2Ln}+ are bound via isocarbonyl bridges, and in some cases also via Ln–P bonds, to the molybdenum polyphosphide core.
The present and earlier results contribute to our preliminary understanding of reduction reactions of transition metal polyphosphides by divalent lanthanide species. In each case a reduction and the rearrangement of the phosphorus scaffold takes place. This results predominantly in the aggregation of the polyphosphide units by the formation of novel P–P bonds.
Acknowledgements
This work was supported by the Deutsche Forschungsgemeinschaft (DFG) and the Russian Ministry of Education and Science. N.A.P. thanks the Russian Science Foundation for financial support (project No. 14-23-00013). We thank Dr Yanhua Lan for magnetic susceptibility measurements and Sibylle Schneider for X-ray single-crystal structure measurements.Notes and references
- J. H. Downing and M. B. Smith, in Comprehensive Coordination Chemistry II, ed. J. A. M. J. Meyer, Pergamon, Oxford, 2003, pp. 253–296 Search PubMed.
- K. B. Dillon, F. Mathey and J. F. Nixon, Phosphorus: The Carbon Copy, Wiley, Chichester, U.K., 1998 Search PubMed.
- H. Butenschön, Chem. Rev., 2000, 100, 1527–1564 CrossRef PubMed.
- M. Peruzzini and L. Gonsalvi, Phosphorus Compounds-Advanced Tools in Catalysis and Material Sciences, Springer, Netherlands, 2011 Search PubMed.
- T. Li, S. Kaercher and P. W. Roesky, Chem. Soc. Rev., 2014, 43, 42–57 RSC.
- S. N. Konchenko, N. A. Pushkarevsky, M. T. Gamer, R. Köppe, H. Schnöckel and P. W. Roesky, J. Am. Chem. Soc., 2009, 131, 5740–5741 CrossRef CAS PubMed.
- W. Huang and P. L. Diaconescu, Chem. Commun., 2012, 48, 2216–2218 RSC.
- W. Huang and P. L. Diaconescu, Eur. J. Inorg. Chem., 2013, 2013, 4090–4096 CrossRef CAS PubMed.
- Y. Lv, X. Xu, Y. Chen, X. Leng and M. V. Borzov, Angew. Chem., Int. Ed., 2011, 50, 11227–11229 CrossRef CAS PubMed.
- T. Li, M. T. Gamer, M. Scheer, S. N. Konchenko and P. W. Roesky, Chem. Commun., 2013, 49, 2183–2185 RSC.
- T. Li, J. Wiecko, N. A. Pushkarevsky, M. T. Gamer, R. Köppe, S. N. Konchenko, M. Scheer and P. W. Roesky, Angew. Chem., Int. Ed., 2011, 50, 9491–9495 CrossRef CAS PubMed.
- M. V. Butovskiy, G. Balázs, M. Bodensteiner, E. V. Peresypkina, A. V. Virovets, J. Sutter and M. Scheer, Angew. Chem., Int. Ed., 2013, 52, 2972–2976 CrossRef CAS PubMed.
- T. Li, N. Arleth, M. T. Gamer, R. Köppe, T. Augenstein, F. Dielmann, M. Scheer, S. N. Konchenko and P. W. Roesky, Inorg. Chem., 2013, 52, 14231–14236 CrossRef CAS PubMed.
- W. J. Evans, I. Bloom, W. E. Hunter and J. L. Atwood, J. Am. Chem. Soc., 1981, 103, 6507–6508 CrossRef CAS.
- W. J. Evans, T. A. Ulibarri, H. Schumann and S. Nickel, in Inorg. Synth., John Wiley & Sons, 2007, pp. 155–157 Search PubMed.
- J. M. Boncella and R. A. Andersen, Inorg. Chem., 1984, 23, 432–437 CrossRef CAS.
- O. J. Scherer, H. Sitzmann and G. Wolmershäuser, J. Organomet. Chem., 1984, 268, C9–C12 CrossRef CAS.
- C. E. Kefalidis, S. Essafi, L. Perrin and L. Maron, Inorg. Chem., 2014, 53, 3427–3433 CrossRef CAS PubMed.
- O. J. Scherer, M. Ehses and G. Wolmershäuser, Angew. Chem., Int. Ed., 1998, 37, 507–510 CrossRef CAS.
- G. Lin and W.-T. Wong, J. Organomet. Chem., 1996, 522, 271–275 CrossRef CAS.
- G. Parkin, Struct. Bonding, 2010, 136, 123 CrossRef.
- A. C. Hillier, S. Y. Liu, A. Sella, O. Zekria and M. R. J. Elsegood, J. Organomet. Chem., 1997, 528, 209–215 CrossRef CAS.
- T. D. Tilley, R. A. Andersen, B. Spencer, H. Ruben, A. Zalkin and D. H. Templeton, Inorg. Chem., 1980, 19, 2999–3003 CrossRef CAS.
- S. N. Konchenko, T. Sanden, N. A. Pushkarevsky, R. Köppe and P. W. Roesky, Chem.–Eur. J., 2010, 16, 14278–14280 CrossRef CAS PubMed.
- C. M. Lukehart, Fundamental Transition Metal Organometallic Chemistry, Brooks Cole, Belmont, 1988 Search PubMed.
- W. J. Evans, J. W. Grate, K. R. Levan, I. Bloom, T. T. Peterson, R. J. Doedens, H. Zhang and J. L. Atwood, Inorg. Chem., 1986, 25, 3614–3619 CrossRef CAS.
- A. C. Hillier, A. Sella and M. R. J. Elsegood, J. Chem. Soc., Dalton Trans., 1998, 3871–3874 RSC.
- A. Recknagel, A. Steiner, S. Brooker, D. Stalke and F. T. Edelmann, Chem. Ber., 1991, 124, 1373–1375 CrossRef CAS PubMed.
- S. Yao, N. Lindenmaier, Y. Xiong, S. Inoue, T. Szilvási, M. Adelhardt, J. Sutter, K. Meyer and M. Driess, Angew. Chem., Int. Ed., 2015, 54, 1250–1254 CrossRef CAS PubMed.
- J. Bai, E. Leiner and M. Scheer, Angew. Chem., Int. Ed., 2002, 41, 783–786 CrossRef CAS.
- M. Scheer, L. J. Gregoriades, M. Zabel, J. Bai, I. Krossing, G. Brunklaus and H. Eckert, Chem.–Eur. J., 2008, 14, 282–295 CrossRef CAS PubMed.
- O. Treutler and R. Ahlrichs, J. Chem. Phys., 1995, 102, 346–354 CrossRef CAS PubMed.
- R. Ahlrichs, M. Bär, M. Häser, H. Horn and C. Kölmel, Chem. Phys. Lett., 1989, 162, 165–169 CrossRef CAS.
- A. D. Becke, Phys. Rev. A, 1988, 38, 3098–3100 CrossRef CAS.
- J. P. Perdew, Phys. Rev. B: Condens. Matter Mater. Phys., 1986, 33, 8822–8824 CrossRef.
- J. P. Perdew, Phys. Rev. B: Condens. Matter Mater. Phys., 1986, 34, 7406 CrossRef.
- M. Sierka, A. Hogekamp and R. Ahlrichs, J. Chem. Phys., 2003, 118, 9136–9148 CrossRef CAS PubMed.
- F. Weigend, Phys. Chem. Chem. Phys., 2006, 8, 1057–1065 RSC.
- F. Weigend and R. Ahlrichs, Phys. Chem. Chem. Phys., 2005, 7, 3297–3305 RSC.
- D. Andrae, U. Häußermann, M. Dolg, H. Stoll and H. Preuß, Theor. Chim. Acta, 1990, 77, 123–141 CrossRef CAS.
- M. Dolg, H. Stoll, A. Savin and H. Preuss, Theor. Chim. Acta, 1989, 75, 173–194 CrossRef CAS.
- R. Ahlrichs and C. Ehrhardt, Chem. Unserer Zeit, 1985, 19, 120–124 CrossRef CAS PubMed.
- R. Heinzmann and R. Ahlrichs, Theor. Chim. Acta, 1976, 42, 33–45 CrossRef CAS.
- O. J. Scherer and T. Brück, Angew. Chem., Int. Ed., 1987, 26, 59 Search PubMed.
- M. Detzel, T. Mohr, O. J. Scherer and G. Wolmershäuser, Angew. Chem., Int. Ed., 1994, 33, 1110–1112 CrossRef PubMed.
- O. J. Scherer, H. Sitzmann and G. Wolmershäuser, Angew. Chem., 1985, 97, 358–359 CrossRef CAS PubMed.
- M. Häser and O. Treutler, J. Chem. Phys., 1995, 102, 3703–3711 CrossRef PubMed.
- T. D. Tilley and R. A. Andersen, J. Chem. Soc., Chem. Commun., 1981, 985–986 RSC.
- T. G. Wetzel, S. Dehnen and P. W. Roesky, Angew. Chem., Int. Ed., 1999, 38, 1086–1088 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1402049-1402054. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5sc02252e |
| This journal is © The Royal Society of Chemistry 2015 |