DOI:
10.1039/C5SC03104D
(Edge Article)
Chem. Sci., 2015,
6, 7169-7178
Metal-only Lewis pairs between group 10 metals and Tl(I) or Ag(I): insights into the electronic consequences of Z-type ligand binding†
Received
20th August 2015
, Accepted 17th September 2015
First published on 17th September 2015
Abstract
Complexes bearing electron rich transition metal centers, especially those displaying coordinative unsaturation, are well-suited to form reverse-dative σ-interactions with Lewis acids. Herein we demonstrate the generality of zerovalent, group 10 m-terphenyl isocyanide complexes to form reverse-dative σ-interactions to Tl(I) and Ag(I) centers. Structural and spectroscopic investigations of these metal-only Lewis pairs (MOLPs) has allowed insight into the electronic consequences of Lewis-acid ligation within the primary coordination sphere of a transition metal center. Treatment of the bis-isocyanide complex, Pt(CNArDipp2)2 (ArDipp2 = 2,6-(2,6-(i-Pr)2C6H3)2C6H3) with TlOTf (OTf = [O3SCF3]−) yields the Pt/Tl MOLP [TlPt(CNArDipp2)2]OTf (1). 1H NMR and IR spectroscopic studies on 1, and its Pd congener [TlPd(CNArDipp2)2]OTf (2), demonstrate that the M → Tl interaction is labile in solution. However, treatment of complexes 1 and 2 with Na[BArF4] (ArF = 3,5-(CF3)2C6H3) produces [TlPt(CNArDipp2)2]BArF4 (3) and [TlPd(CNArDipp2)2]BArF4 (4), in which Tl(I) binding is shown to be static by IR spectroscopy and, in the case of 3, 195Pt NMR spectroscopy as well. This result provides strong evidence that the M → Tl linkages can be attributed primarily to σ-donation from the group 10 metal to Tl, as loss of ionic stabilization of Tl by the triflate anion is compensated for by increasing the degree of M → Tl σ-donation. In addition, X-ray Absorption Near-Edge Spectroscopy (XANES) on the Pd/Tl and Ni/Tl MOLPs, [TlPd(CNArDipp2)2]OTf (2) and [TlNi(CNArMes2)3]OTf, respectively, is used to illustrate that the formation of a reverse-dative σ-interaction with Tl(I) does not alter the spectroscopic oxidation state of the group 10 metal. Also reported is the ability of M(CNArDipp2)2 (M = Pt, Pd) to form MOLPs with Ag(I), yielding the complexes [AgM(CNArDipp2)2]OTf (5, M = Pt; 6, M = Pd). As was determined for the Tl-containing MOLPs 1–4, it is shown that the spectroscopic oxidation states of the group 10 metal in 5 and 6 are essentially unchanged compared to the zerovalent precursors M(CNArDipp2)2. However, in the case of 5 and 6, the formation of a dative M → Ag σ-bonding interaction facilitates the binding of Lewis bases to the group 10 metal trans to Ag, illustrating the potential of acceptor fragments to open up new coordination sites on transition metal complexes without formal, two-electron oxidation.
Introduction
On account of their relatively electropositive nature and ability to act as formal acceptors toward Lewis bases, the transition metals in coordination complexes are traditionally viewed as Lewis acids. Classical “Werner-type” complexes utilize their empty nd, as well as (n + 1)s and (n + 1)p, orbitals to form dative bonds with electron-donor ligands. In the case of highly reduced and electron-rich complexes, the transition metal center may also be capable of exhibiting Lewis basic behavior.1 Although this phenomenon was initially invoked for the case of carbonyl metallates acting as Brønsted bases,2–4 it is now recognized as a central tenet of transition-metal bonding to π-acidic ligands5–7 as well as an essential component of many oxidative addition mechanisms.8–12 More recently, the extension of this concept to the binding of various main-group acceptor fragments (Z-type ligands)13 in a σ-fashion by electron-rich transition metals has been realized, and the study of such complexes continues to be of intense interest.14–27
In addition to these examples, a related topic concerning transition metal Lewis basicity is the ability to form dative interactions to another metal center. Judicious ligand design strategies that constrain an electron-rich metal center in close proximity to a coordinatively unsaturated metal fragment has proven to be a reliable approach for engendering metal–metal dative bonding.28–34 Furthermore, in certain instances, unsupported metal-only Lewis pairs (MOLPs), which do not rely on a ligand buttress, can be generated.2,35–39 The formation of such unsupported metal–metal interactions, while sometimes labile in solution, offers an interesting approach toward tuning the reactivity profiles of low-valent complexes, as the addition of metallic Lewis acids has been shown to enhance the rates of certain catalytic processes.40–43
While synthetic methods leading to MOLPs and their structural chemistry has advanced, a detailed understanding of how the presence of a metal–metal dative bond affects the electronic properties of the constituent fragments remains of significant interest. It is generally accepted that protonation of a transition metal complex is best viewed as involving a two-electron oxidation of the metal center to give a hydride ligand.44 As the electrons involved in the M–H bond have necessarily come from the metal, an increase of its valence by two units is required.45 In the case of other main group Lewis acids (e.g. boranes), the degree of charge transfer is often not as clear. As such, the adoption and assignment of formalisms to adequately describe the electronic structure of such adducts has been a point of debate in the community.46,47 Similar ambiguities in the electronic structures of MOLPs exist, although considerably less effort has been put toward uncovering satisfactory electronic descriptors for such compounds.39 Despite the fact that X-ray Absorption Near-Edge Spectroscopy (XANES) holds promise in this regard,30,34 its thus-far limited use in this capacity has not yet led to the development of general principles for properly describing the electronic structures of complexes containing metal–metal dative interactions.
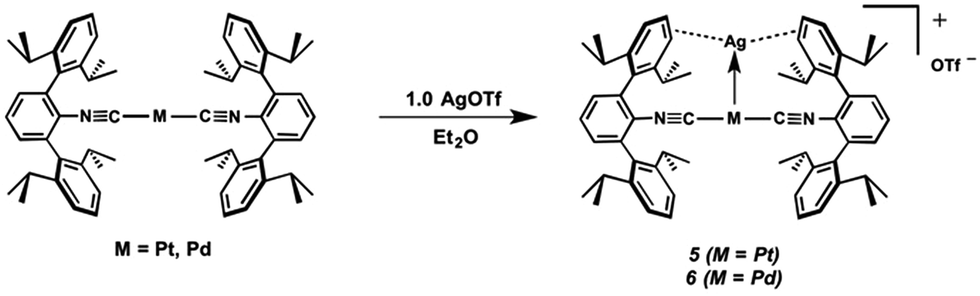
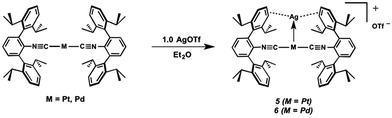
Work from our research group has demonstrated the utility of encumbering m-terphenyl isocyanides in stabilizing low-valent and coordinatively unsaturated complexes of late transition metals.27,48–55 Such electron-rich metal centers are prime candidates for acting as Lewis bases toward appropriate Lewis acidic substrates, a concept that has been demonstrated by the heterobimetallics [TlNi(η4-COD)(CNArMes2)2]X (X = OTf, BArF4),49 [TlNi(CNArMes2)3]OTf,49 and [TlPd(CNArDipp2)2]OTf (2),50 as well as by the recently-reported platinum (boryl)iminomethane complex Pt(κ2-N,B-Cy2BIM)(CNArDipp2).27 In addition, the response of the isocyanide ν(C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N) IR bands to the electron density on the Lewis-basic metal center renders them a convenient spectroscopic reporter on the degree of formal charge transfer upon binding a σ-acceptor fragment.25 In this work, we demonstrate the ability of the two-coordinate complexes M(CNArDipp2)2 (M = Pt, Pd)27,50 to form unsupported metal–metal linkages with Tl(I). Two Tl-containing MOLPs have also been examined by X-ray Absorption Near-Edge Spectroscopy (XANES), illustrating that the spectroscopic oxidation state of the group 10 metal is not affected by its interaction with Tl(I). We also show that the zero-valent platforms M(CNArDipp2)2 (M = Pt, Pd) can form metal-only Lewis pairs with Ag(I), yielding the heterobimetallic salts [AgM(CNArDipp2)2]OTf (5, M = Pt; 6, M = Pd). Spectroscopic and structural investigations provide insight into the nature of the M–Ag interactions in these compounds, and give strong evidence that formation of the M → Ag linkage results in only a marginal degree of metal-to-metal charge transfer. In the case of the Pt variant 5, further aggregation with additional AgOTf leads to dimeric {[Ag2Pt(CNArDipp2)2(η1-C6H6)]2(μ-OTf)2}(OTf)2 (7) containing triangulo-PtAg2 cores. It is shown that binding of one (compounds 5 and 6) and two (compound 7) equivalents of Ag(I) results in a sequential increase in the Lewis acidity of the group 10 metal center, thus illustrating how σ-acceptor fragments can be used to rationally tune the properties of electron-rich transition metal complexes.
N) IR bands to the electron density on the Lewis-basic metal center renders them a convenient spectroscopic reporter on the degree of formal charge transfer upon binding a σ-acceptor fragment.25 In this work, we demonstrate the ability of the two-coordinate complexes M(CNArDipp2)2 (M = Pt, Pd)27,50 to form unsupported metal–metal linkages with Tl(I). Two Tl-containing MOLPs have also been examined by X-ray Absorption Near-Edge Spectroscopy (XANES), illustrating that the spectroscopic oxidation state of the group 10 metal is not affected by its interaction with Tl(I). We also show that the zero-valent platforms M(CNArDipp2)2 (M = Pt, Pd) can form metal-only Lewis pairs with Ag(I), yielding the heterobimetallic salts [AgM(CNArDipp2)2]OTf (5, M = Pt; 6, M = Pd). Spectroscopic and structural investigations provide insight into the nature of the M–Ag interactions in these compounds, and give strong evidence that formation of the M → Ag linkage results in only a marginal degree of metal-to-metal charge transfer. In the case of the Pt variant 5, further aggregation with additional AgOTf leads to dimeric {[Ag2Pt(CNArDipp2)2(η1-C6H6)]2(μ-OTf)2}(OTf)2 (7) containing triangulo-PtAg2 cores. It is shown that binding of one (compounds 5 and 6) and two (compound 7) equivalents of Ag(I) results in a sequential increase in the Lewis acidity of the group 10 metal center, thus illustrating how σ-acceptor fragments can be used to rationally tune the properties of electron-rich transition metal complexes.
Results and discussion
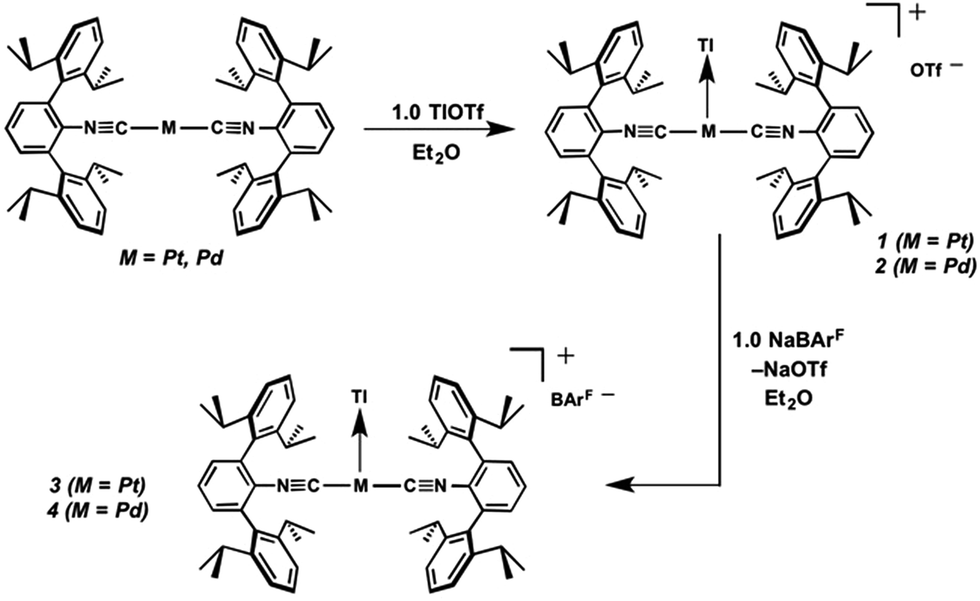
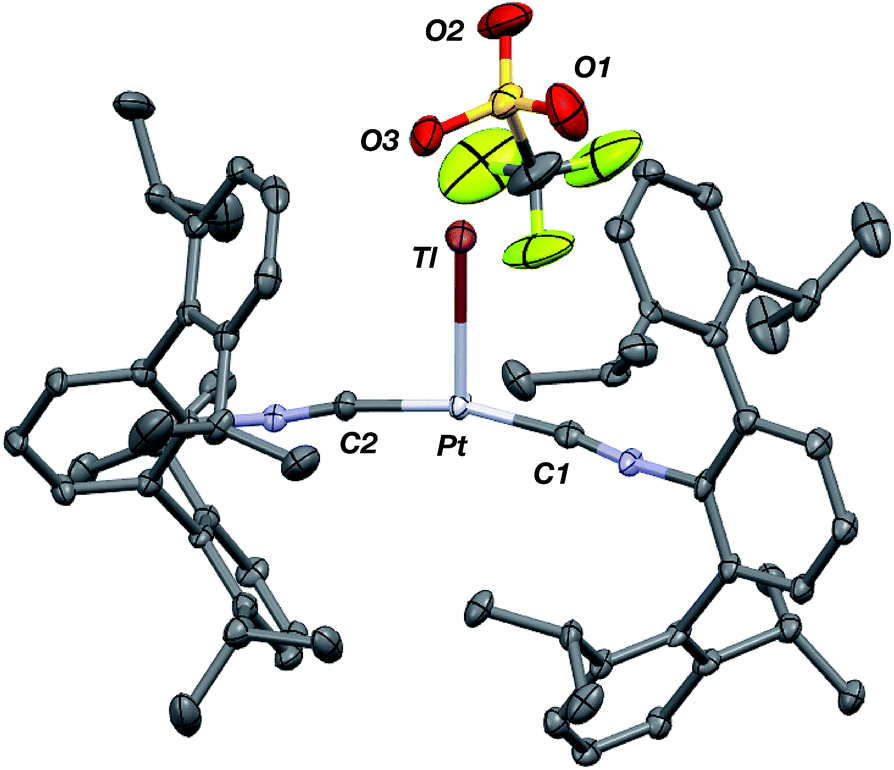
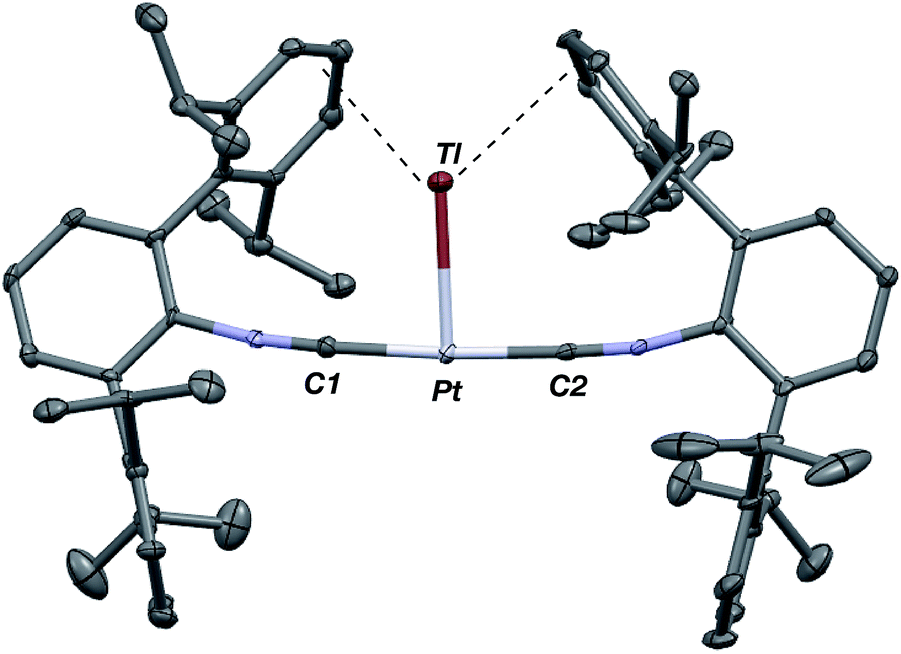
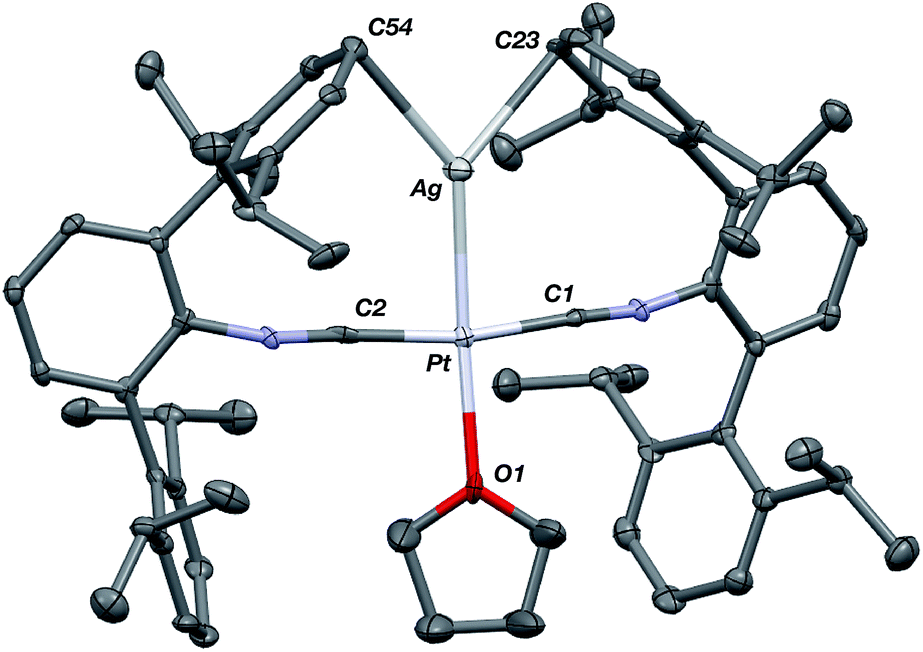
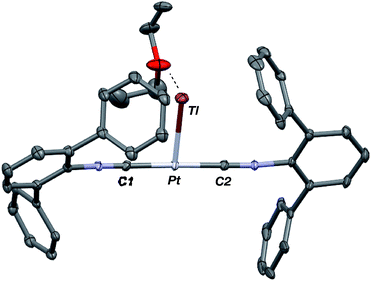
Similar to the zero-valent Pd congener, Pd(CNArDipp2)2,50 the addition of TlOTf to a solution of Pt(CNArDipp2)2 in Et2O yields the unsupported heterobimetallic compound [TlPt(CNArDipp2)2]OTf (1) as a yellow microcrystalline solid. Structural characterization of 1 (Scheme 1 and Fig. 1) reveals a T-shaped coordination geometry about Pt, while the Tl center makes long, but non-negligible contacts with the [OTf]− anion (d(Tl–O3) = 2.799(5) Å) and the Caryl atoms of the Dipp rings (shortest d(Tl–Caryl) = 3.355 Å). The presence of a Pt–Tl bonding interaction is apparent given their interatomic separation of 2.8617(3) Å. Importantly, this value is comparable to the most reasonable range for the sum of the covalent radii between Pt and Tl (2.67–2.84 Å),56 thereby suggesting that the solid-state structure of 1 does not simply arise from the co-crystallization of Pt(CNArDipp2)2 with TlOTf. While the role of closed-shell metallophilic interactions57 cannot be completely discounted, spectroscopic evidence indicates that this interaction is formed by a reverse-dative σ-bond, whereby Pt donates two electrons to an empty 6p orbital on Tl. Analysis of these solutions by FTIR spectroscopy shows a strong ν(CN) band at 2112 cm−1, which is shifted to higher energy relative to those of Pt(CNArDipp2)2 (2065, 2020 cm−1),27 consistent with a decrease in π-backbonding interactions to the isocyanides as a result of the formation of a Pt → Tl retrodative σ-bonding interaction. A similar blue-shift of this band for the palladium analogue [TlPd(CNArDipp2)2]OTf (2) with respect to Pd(CNArDipp2)2 was observed previously.50 Surprisingly, bonds between electron-rich, late transition metals (especially third-row metals) and Tl(I) have often been rationalized largely based on metallophilic interactions.58–61 However, the FTIR spectra of 1 and 2 compared with those of M(CNArDipp2)2 (M = Pt, Pd) provide strong experimental evidence that late-metal-Tl(I) bonds likely contain a substantial dative-bonding component in a manner analogous to that seen for complexes bearing main-group Z-type ligands.23–25
 |
| | Scheme 1 | |
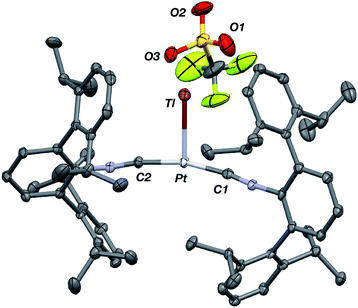 |
| | Fig. 1 Molecular structure of [TlPt(CNArDipp2)2]OTf (1). | |
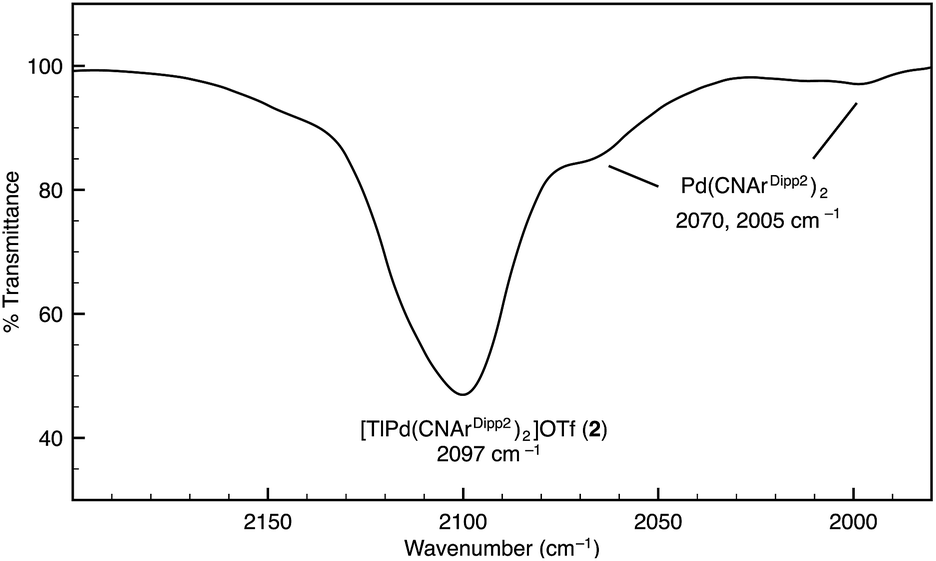
Although [TlPt(CNArDipp2)2]OTf (1) gives rise to a sharp set of 1H and 13C{1H} NMR resonances in benzene-d6, other spectroscopic data suggest that the metal–metal interaction is labile in solution. While the IR absorption bands of Pt(CNArDipp2)2 are not apparent in the IR spectrum of 1, it is important to note that ν(CN) bands corresponding to Pd(CNArDipp2)2 are readily observable as a minor component in the IR spectrum of [TlPd(CNArDipp2)2]OTf (2) in C6D6 solution, thereby suggesting the presence of an equilibrium between bound and unbound Tl(I) (Fig. 2). In addition, extended scanning failed to locate the 195Pt NMR62 resonance for the platinum analogue [TlPt(CNArDipp2)2]OTf (1). We suggest that this observation is indicative of lability in the Pt–Tl interaction on the NMR timescale, resulting in a broadening of this resonance that obviates its detection at room temperature.
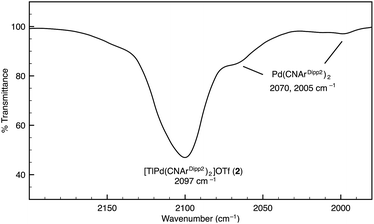 |
| | Fig. 2 FTIR spectrum (ν(CN) region) of [TlPd(CNArDipp2)2]OTf (2) showing the presence of Pd(CNArDipp2)2. Conditions: C6D6, 20 °C, KBr windows. | |
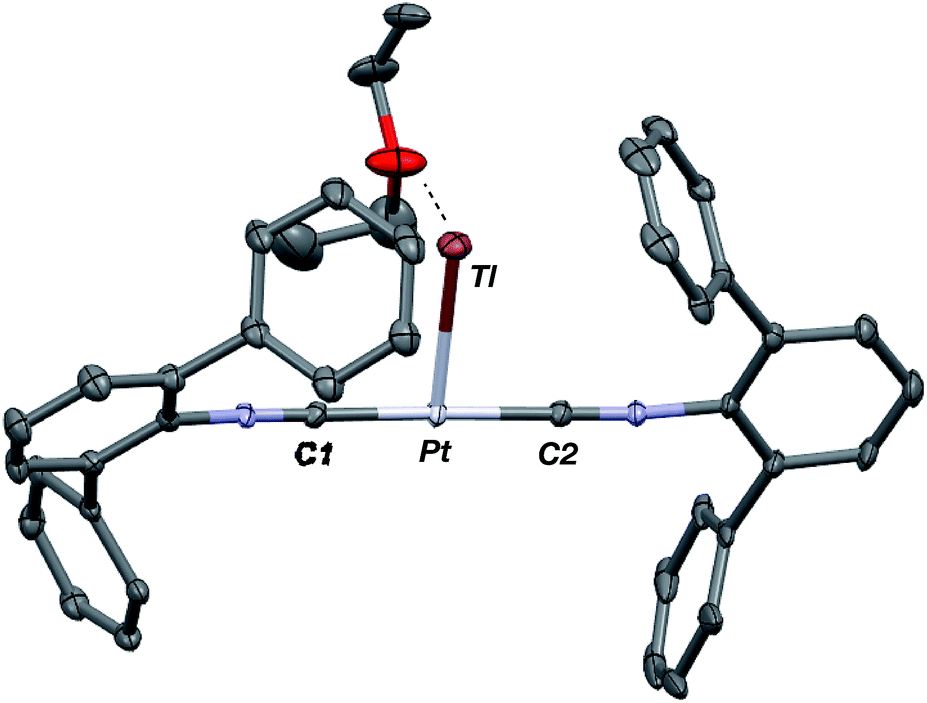
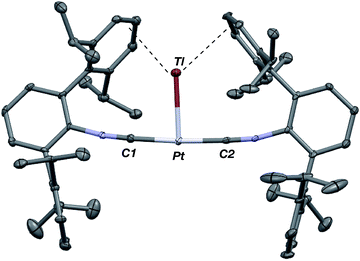
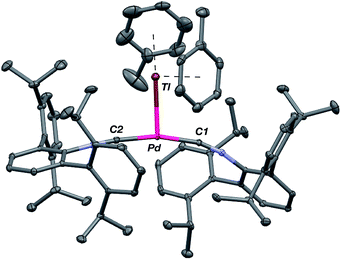
As the lability of unsupported M–Tl linkages has been observed to display a dependence on counteranion identity,58 we sought to explore the behavior of [TlM(CNArDipp2)2]+ (M = Pt, Pd) when accompanied by a traditionally non-coordinating anion. Addition of an Et2O solution of NaBArF4 (BArF4 = [B(3,5-(CF3)2C6H3)4]−) to 1 or 2 results in precipitation of NaOTf and smooth formation of [(Et2O)TlM(CNArDipp2)2]BArF4 (M = Pt (3(Et2O)), Pd (4(Et2O))) following crystallization from Et2O (Fig. 3). Structural determinations of 3(Et2O) and 4(Et2O) reveal discreet cation–anion pairs (two independent pairs per asymmetric unit). While no contact between the Tl center and the [BArF4]− anion is evident in the solid state, the Tl center is bound to a molecule of Et2O in both complexes (average d(Tl–O) = 2.760(3) Å (3) and 2.729(3) Å (4)). Furthermore, as noted for 1, long-range contacts (ca. 3.4 Å) between Tl and several Caryl atoms of the flanking Dipp rings are apparent. The Tl-bound ether molecules are easily liberated from crystalline samples upon prolonged exposure to vacuum (∼100 mTorr) as assayed by 1H NMR spectroscopy. Subsequent crystallization of 3 from toluene produces solvent-free [TlPt(CNArDipp2)2]BArF4 (3, Fig. 4), which contains Tl-(η2-arene) interactions with the flanking Dipp rings (d(Tl–C) = 3.188–3.616 Å).63 In the case of 4, cooling a saturated toluene solution to −35 °C yields [(η6-Tol)2TlPd(CNArDipp2)2]BArF4 (4(Tol)2, Fig. 5), which displays two toluene molecules bound in an η6-fashion to Tl (d(Tl–C) = 3.192–3.589 Å). Although the Tl–Carene distances in 3 and 4(Tol)2 are seemingly long, they are well in line with other structurally characterized examples of Tl-arene π-complexes.60,64–67
 |
| | Fig. 3 Molecular structure of [(Et2O)TlPt(CNArDipp2)2]BArF4 (3(Et2O)). Isopropyl groups and BArF4 counterion have been omitted for clarity. The Pd congener 4(Et2O) is isostructural. | |
 |
| | Fig. 4 Molecular structure of [TlPt(CNArDipp2)2]BArF4 (3). BArF4 counterion has been omitted for clarity. | |
 |
| | Fig. 5 Molecular structure of [(η6-Tol)2TlPd(CNArDipp2)2]BArF4 (4(Tol)2). BArF4 counterion has been omitted for clarity. | |
Examination of the solid-state and solution-phase behaviour of 1–4 reveals that replacement of the triflate anion with [BArF4]− has important ramifications for the lability of the M → Tl linkage. In both solvates of 3 and 4, the M–Tl distance is significantly contracted relative to 1 and 2 (Table 1), consistent with an increase in the degree of M → Tl donation. As the triflate anion likely stabilizes the Tl center through contact ion pairing, replacement of [OTf]− with neutral Et2O or arene donors appears to only partially compensate for the loss of this ionic association. Accordingly, in an attempt to recoup some of this stabilization, we contend that the degree of M → Tl σ-donation is increased. This notion is supported by the progression of the ν(CN) bands in 3 (2121 cm−1) and 4 (2116 cm−1) to higher energies relative to 1 and 2, as the increased withdrawal of electron density from the group 10 metal by Tl serves to attenuate backbonding interactions with the isocyanide ligands. Importantly, and in contrast to the triflate salt [TlPd(CNArDipp2)2]OTf (2), the solution FTIR spectra of 3 and 4 in benzene-d6 are devoid of ν(CN) features corresponding to M(CNArDipp2)2, signalling that Tl(I) dissociation in benzene can be significantly inhibited by the use of the weakly coordinating [BArF4]− anion. Interestingly, this replacement also allows for detection of the 195Pt NMR resonance of [TlPt(CNArDipp2)2]BArF4 (3), which appears as a doublet with well-resolved coupling to 205Tl (δ = −3802 ppm, 1JPt,Tl = 11.2 kHz).68 This resonance is shifted significantly downfield respective to that of Pt(CNArDipp2)2 (δ = −5993 ppm (s), C6D6), further suggestive of decreased electron density at the Pt center upon coordination of Tl(I).69,70 However, it is also important to note that dissolution of 1–4 in THF results in complete dissociation of the Tl(I) center and formation of M(CNArDipp2)2, according to FTIR spectroscopy. This result, which was similarly observed in the case of [TlNi(CNArDipp2)3]OTf,54 serves as a reminder of the weak dissociation energies inherent in most unsupported metal–metal dative bonds, as dissolution in solvents of moderate coordinating strength is sufficient to completely disrupt this interaction.
Table 1 Selected bond distances (X-ray structure) of Pt(CNArDipp2)2, Pd(CNArDipp2)2, and their Tl-containing MOLPs
| Complex |
d(M–Ciso) |
d(M–Tl) |
|
Data from ref. 27.
Data from ref. 50.
One of the two crystallographically independent molecules in the X-ray structures of 3(Et2O) and 4(Et2O) contain two-site disorder of the Tl atom. We have used the major component of this disorder to calculate the average d(M–Tl).
|
| Pt(CNArDipp2)2a |
1.906(3) Å |
— |
| Pd(CNArDipp2)2b |
1.930(4) Å |
— |
|
1
|
1.918(5) Å |
2.8617(3) Å |
|
2
|
1.951(7) Å |
2.855(1) Å |
|
3(Et2O)c |
1.922(4) Å |
2.7710(4) Å |
|
4(Et2O)c |
1.958(4) Å |
2.7481(5) Å |
|
3
|
1.933(8) Å |
2.7659(5) Å |
|
4(Tol)2 |
1.965(3) Å |
2.7770(2) Å |
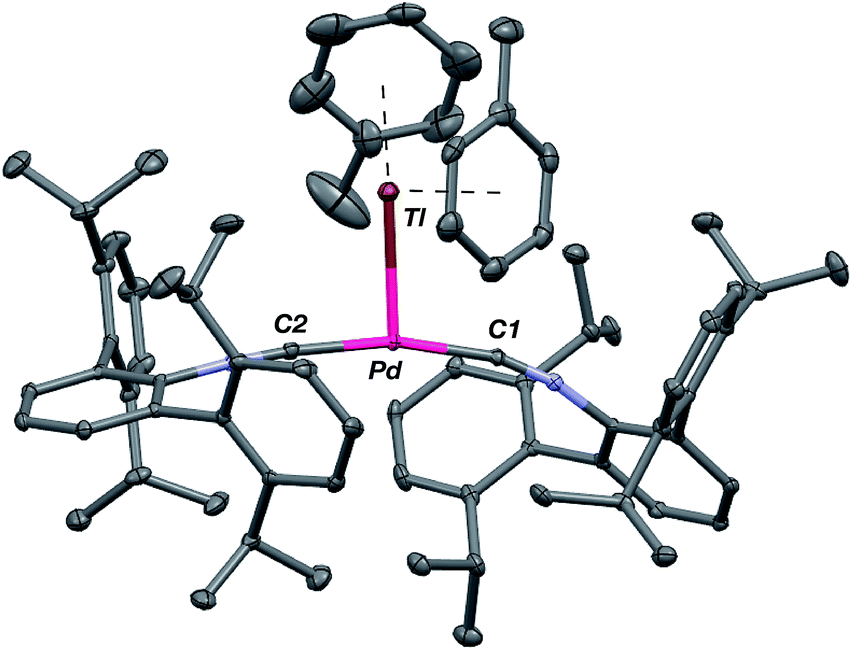
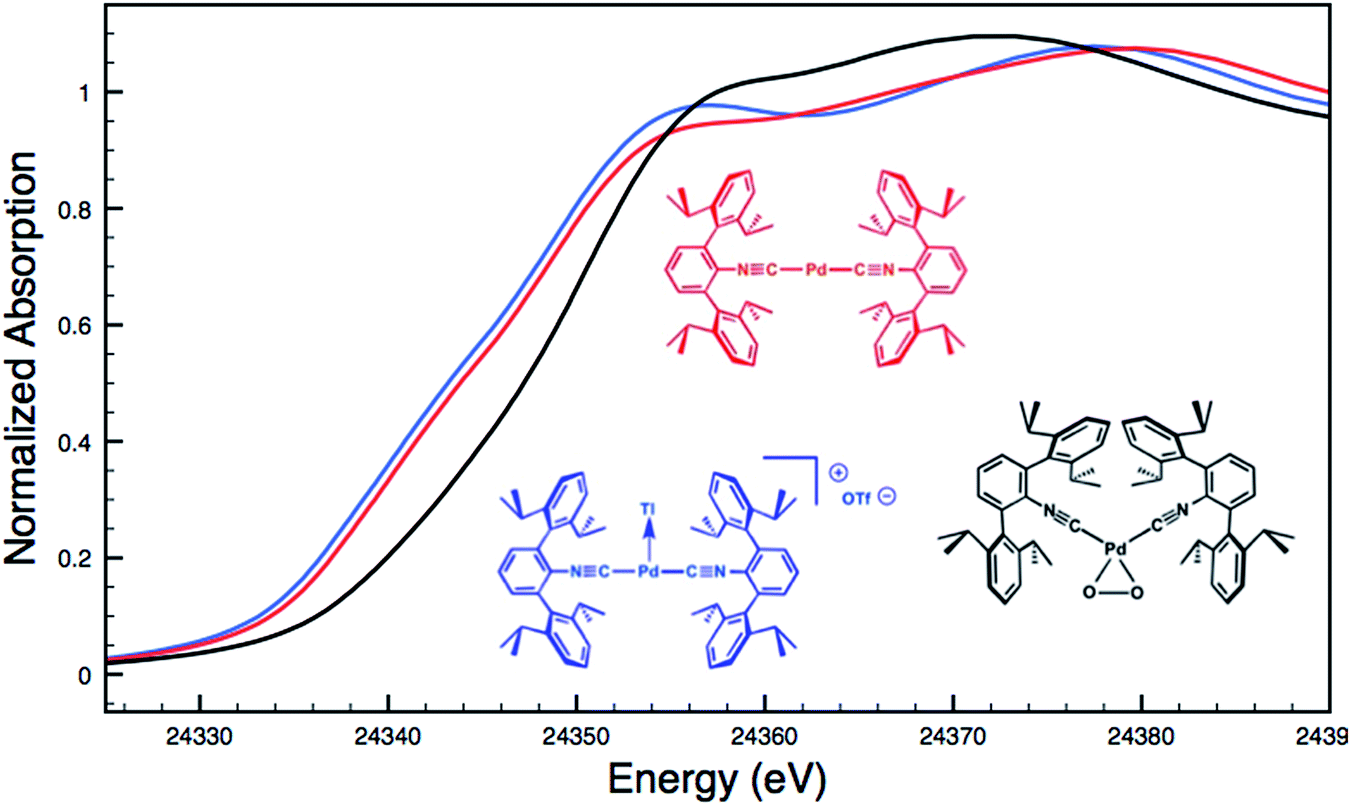
Although limited experimental techniques are capable of probing metal–metal dative interactions, X-ray Absorption Near-Edge Spectroscopy (XANES)71 has begun to find an important use in this regard.30,34 Importantly, its utility lies in its ability to decipher the spectroscopic oxidation states of the metals involved in a given bonding interaction. In order to assess the degree of charge transfer inherent in the formation of a reverse-dative σ-interaction to Tl(I), Pd K-edge XANES was carried out on the palladium–thallium adduct [TlPd(CNArDipp2)2]OTf (2, Fig. 6). While neither the Pd K-edge spectra of 2 nor that of Pd(CNArDipp2)2 display a discernable pre-edge feature, both exhibit nearly identical energies for the rising edge of the XANES region. In comparison, the rising edge energy of the Pd(II) peroxo complex50,72 Pd(η2-O2)(CNArDipp2)2 is shifted to higher energy by ca. 4.0 eV relative to that of Pd(CNArDipp2)2 and 2. Despite their differing geometries, the rising edge transition for each of these three Pd complexes should involve the promotion of a core 1s electron to a 5p orbital that is relatively unperturbed by ligand field effects. Accordingly, the marked shift of the rising edge to higher energy for Pd(η2-O2)(CNArDipp2)2 can be reasonably attributed to the presence of an oxidized Pd center relative to that found in Pd(CNArDipp2)2 or 2. However, the near-identical rising edge energies observed for Pd(CNArDipp2)2 and 2 strongly reflect that Tl(I) binding to an electron rich Pd center does not result in a formal oxidative event.
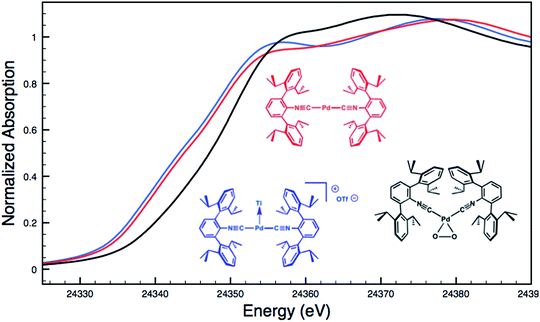 |
| | Fig. 6 Comparative Pd K-edge XANES spectra of Pd(CNArDipp2)2 (red), [TlPd(CNArDipp2)2]OTf (2, blue), and Pd(η2-O2)(CNArDipp2)2 (black). | |
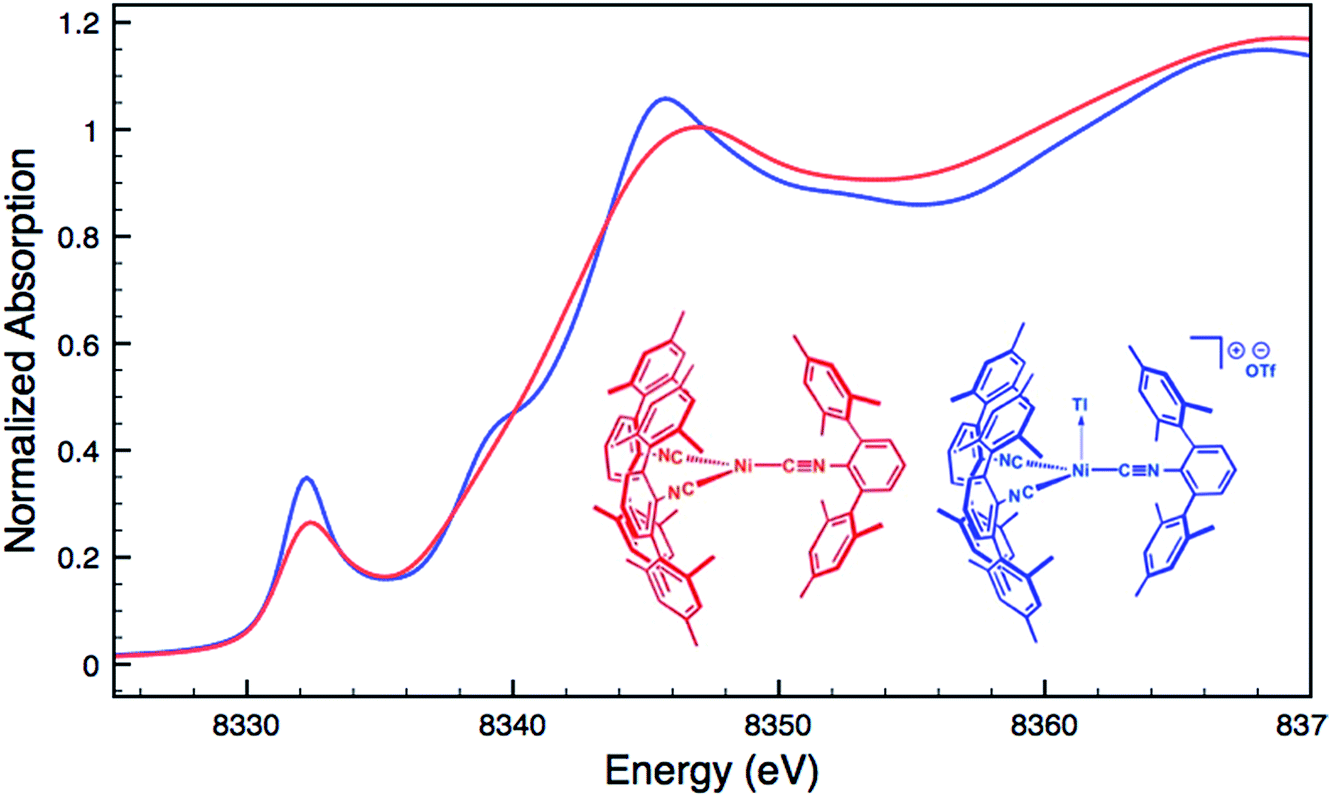
For an additional comparison, XANES measurements were carried out on the binary nickel tris-isocyanide complex Ni(CNArMes2)3 and its adduct with Tl(I), [TlNi(CNArMes2)3]OTf.49 Despite the unambiguous d10 configuration of Ni(CNArMes2)3, its Ni K-edge absorption spectrum (Fig. 7) displays a prominent pre-edge feature, which is likely the result of a 1s to isocyanide π* transition. The analogous absorption band for [TlNi(CNArMes2)3]OTf occurs at an identical energy, again signalling that the formation of a reverse-dative M → Tl σ-interaction does not result in significant formal charge transfer from the group 10 metal.
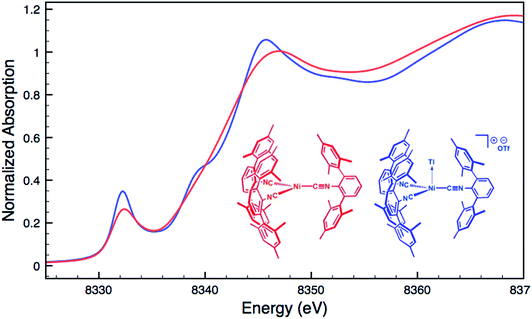 |
| | Fig. 7 Comparative Ni K-edge XANES spectra of Ni(CNArMes2)3 (red) and [TlNi(CNArMes2)3]OTf (blue). | |
The fact that neither Pd(CNArDipp2)2 nor Ni(CNArMes2)3 undergo significant charge transfer via the formation of a reverse-dative σ-interaction to Tl(I) suggests some important guidelines regarding the proper formalisms that should be used to describe such MOLPs. Although Tl(I) can exhibit Lewis basic properties under extraordinary conditions,73 the stabilization of its 6s2 “inert pair” due to relativistic effects74 should render it a very weak 2e− donor. As such, the electrons involved in a covalent interaction between an electron-rich transition metal and Tl(I) center will most plausibly be supplied by the former, meaning that the valence count of the transition metal must necessarily increase by two units.45 However, this interaction should not be described as effecting a two-unit increase in the formal oxidation state of the transition metal, as such an event would be readily apparent in the comparative XANES spectra of M(CNR)n and [TlM(CNR)n]+ complexes. This conclusion is further supported by the modest changes in the FTIR ν(CN) energies between the neutral parent compounds and their Tl(I) adducts (ca. 50 cm−1). For comparison, the Pd(II) and Ni(II) complexes trans-PdCl2(CNArDipp2)2 and trans-NiCl2(CNArMes2)2 display IR ν(CN) bands that are blue-shifted by ca. 200 cm−1 relative to Pd(CNArDipp2)2 and Ni(CNArMes2)3.49,75
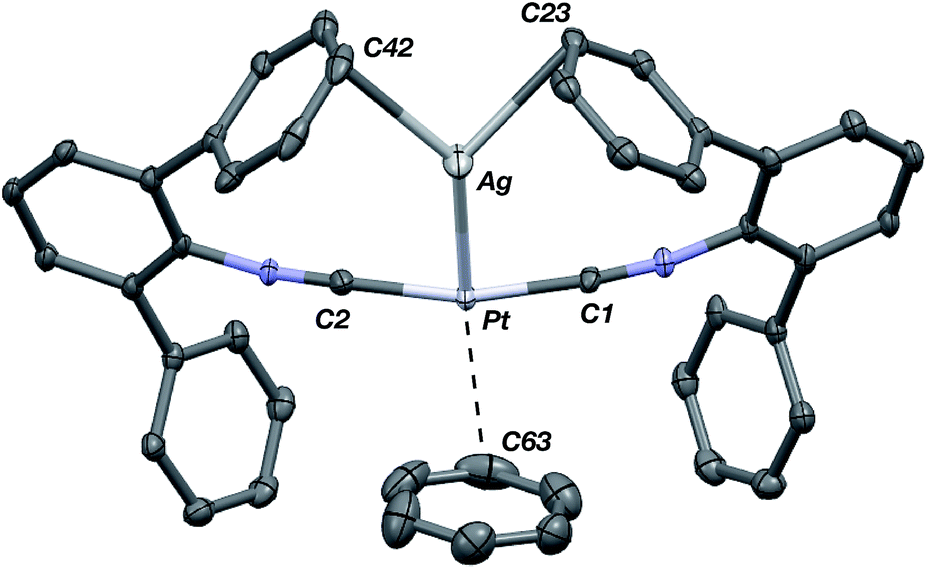
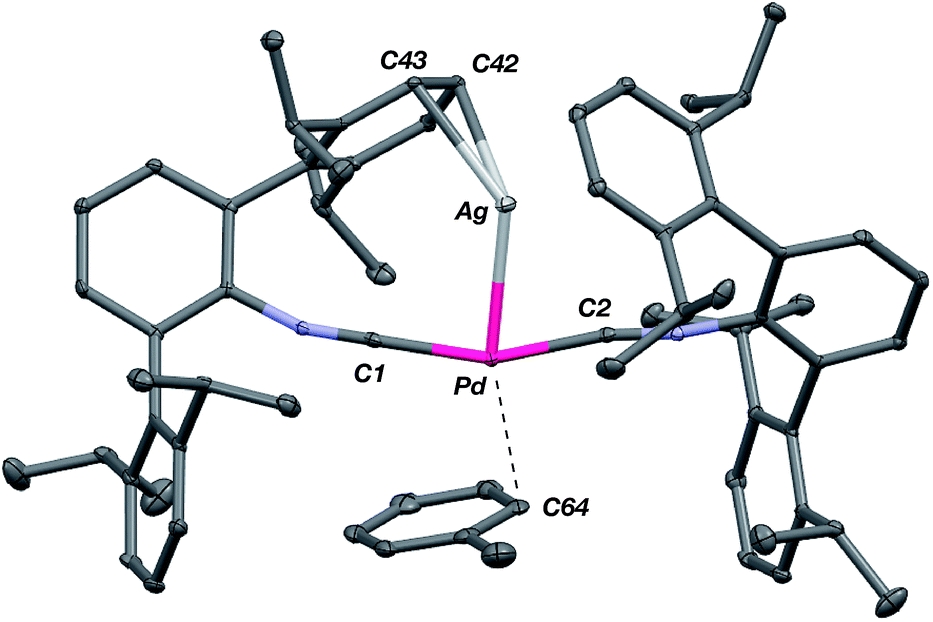
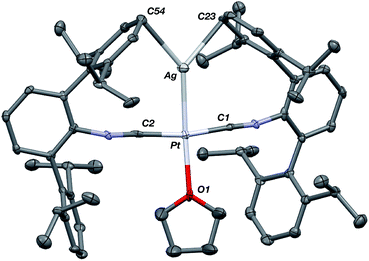
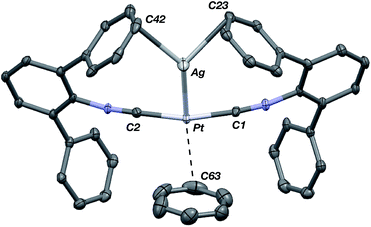
The abilities of M(CNArDipp2)2 (M = Pt, Pd) to act as the basic components of metal-only Lewis pairs can also be extended to Lewis acidic Ag(I) centers.76 Treatment of Pt(CNArDipp2)2 with AgOTf in Et2O results in precipitation of the heterobimetallic salt [AgPt(CNArDipp2)2]OTf (5) as a yellow powder. Attempts to synthesize the palladium analogue [AgPd(CNArDipp2)2]OTf (6) in the same fashion results in the formation of metallic mirrors and free CNArDipp2. However, performing the synthesis at reduced temperatures (ca. −100 °C) allows for 6 to be precipitated from solution as a pale yellow powder in modest yields (Scheme 2). Crystallization of 5 or 6 from THF/(TMS)2O at −35 °C yields trans-[AgM(CNArDipp2)2(THF)]OTf (5(THF), M = Pt; 6(THF), M = Pd), where a molecule of THF is bound to the group 10 metal trans to the coordinated Ag center (Fig. 8). The M–OTHF distances in 5(THF) (2.366(5) Å) and 6(THF) (2.326(7) Å) are long relative to Pd and Pt etherate complexes reported in the Cambridge Structural Database,77 thereby suggesting an attenuated interaction of THF with the group 10 metal center. Indeed, the 1H NMR spectra obtained from crystalline 5(THF) and 6(THF) in C6D6 show sharp peaks occurring at the expected chemical shift values for free THF.78 Further, prolonged exposure of crystalline samples to vacuum (∼100 mTorr) successfully liberates all THF as analyzed by 1H NMR spectroscopy. Subsequent recrystallization of these samples from Et2O/C6H6 (5) or n-hexane/toluene (6) yields 5(C6H6) and 6(C7H8) (Fig. 9 and 10), which display η1-Carene interactions between the group 10 metal and arene solvent in the position trans to Ag (for 5, d(Pt–C63 = 2.885(7) Å); for 6, d(Pd–C64) = 2.496(3)). These interactions are notable given the likely intermediacy of transient π-complexes79 in the C–H activation of arenes by electrophilic group 10 complexes.80,81
 |
| | Fig. 8 Molecular structure of [AgPt(CNArDipp2)2(THF)]OTf (5(THF)). The Pd congener 6(THF) is isostructural. Triflate counterion has been omitted for clarity. | |
 |
| | Fig. 9 Molecular structure of [AgPt(CNArDipp2)2(η1-C6H6)]OTf (5(C6H6)). Flanking isopropyl groups and the triflate counter ion have been omitted for clarity. | |
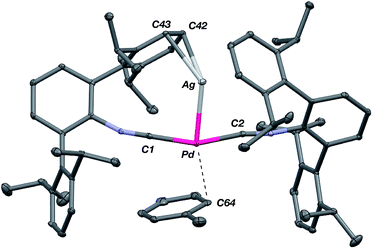 |
| | Fig. 10 Molecular structure of [AgPd(CNArDipp2)2(η1-C7H8)]OTf (6(C7H8)). Triflate counterion has been omitted for clarity. | |
 |
| | Scheme 2 | |
The M–Ag bond lengths in both solvates of 5 and 6 are among the shortest reported in the Cambridge Structural Database (see Table 2 and ESI S1†). As was seen for the M–Tl adducts above, the metal–metal interactions in 5 and 6 can be rationalized via M → Ag σ-donation, a notion that is supported by the increase in isocyanide ν(CN) stretching frequencies relative to M(CNArDipp2)2 (M = Pt, Pd) upon coordination of Ag(I) (ν(CN) = 2094 cm−1 (5); 2082 cm−1 (6)). An examination of the solid state structures of 5 and 6 also implicates a role of the flanking Dipp aryl rings in buttressing the M–Ag linkage through π-type interactions. Interestingly, these contacts are reflected in the room temperature 1H NMR spectra of 5 and 6 (measured in C6D6), for which the resonances corresponding to the Dipp aryl protons are broadened and shifted downfield by ca. 0.2 ppm relative to those typically observed for diamagnetic mononuclear complexes containing the CNArDipp2 ligand. It is also notable that the different solvates of both 5 and 6 display a square-planar coordination environment around the group 10 metal. While these geometries are certainly reminiscent of Pt(II) and Pd(II), it is critical to note that the progression of the IR ν(CN) stretching frequencies to higher energies upon binding of Ag(I) is quite modest and actually less than that seen for Tl(I). This observation serves to suggest that similar bonding descriptions laid out above for Tl-containing 1–4 can be extended to 5 and 6. While the use of electrons from the group 10 metal to form a covalent interaction with Ag requires an increase of two valence units,45 minimal charge transfer to Ag occurs. As such, these M/Ag MOLPs should not be described as containing formal M(II) centers (M = Pt, Pd).
Table 2 Selected bond distances (X-ray structure) of Ag-containing heterobimetallic complexes
| Complex |
d(M–Ciso) |
d(M–Ag) |
|
5(THF) |
1.926(8) Å |
2.6299(6) Å |
|
6(THF) |
1.941(9) Å |
2.6303(9) Å |
|
5(C6H6) |
1.916(4) Å |
2.6463(5) Å |
|
6(C7H8) |
1.950(3) Å |
2.6112(4) Å |
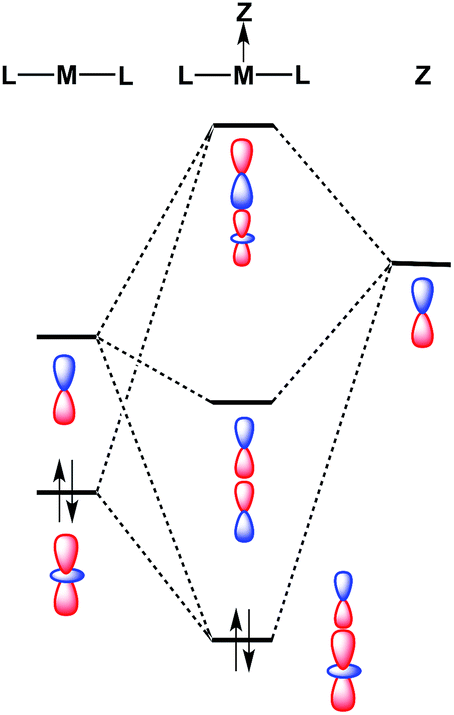
Despite the fact that formation of a M–Ag bonding interaction does not result in a formal oxidative event at Pt/Pd, it is remarkable that the Ag-containing heterobimetallics 5 and 6 will bind THF and arene molecules at the group 10 metal center in the solid state, whereas the zero-valent precursors M(CNArDipp2)2 (M = Pt, Pd) do not. Furthermore, Pt(CNArDipp2)2 and Pd(CNArDipp2)2 do not participate in addition reactions with stronger σ-donors (e.g. phosphines) to form species of the type ML(CNArDipp2)2, as the attempted syntheses of such compounds has led invariably to isocyanide dissociation and/or decomposition. While the ability of 5 and 6 to bind an additional Lewis base may be partly attributable to increased positive charge on the complexes, molecular orbital considerations provide a basis for enhanced Lewis acidity at the group 10 metal center of these MOLPs specifically. It has been suggested previously that coordination of a Z-type acceptor ligand to a square-planar d8 complex should result in enhanced affinity for Lewis bases at the open coordination site trans to the acceptor.82 Similarly, formation of a reverse-dative σ-interaction by M(CNArDipp2)2 (nominally from the ndz2 orbital) to an acceptor may have a stabilizing effect on the coaxial empty (n + 1)pz orbital of the group 10 metal (Fig. 11). While such stabilization may not be drastic, it is plausible that such effects could promote the binding of Lewis bases at a coordination site trans to the acceptor, resulting in square-planar [AgML2L′]+-type species. Similar behavior was observed by Peters in the trigonal-pyramidal Pt salt [(SiPPh3)Pt]BArF4,83 (SiPPh3 = (2-Ph2PC6H4)3Si) for which the crystal structure shows a molecule of toluene bound in the apical position trans to the silyl group. As silyl ligands can be viewed in certain systems as silylium Lewis acids,84 the binding of an arene molecule may be a result of Pt-to-Si σ-donation, thereby in effect enhancing the Lewis acidic nature of the Pt complex. In addition, similar phenomena have been observed by Gabbaï for a Hg(II) complex85 and by Berry for a bimetallic Mo2 system.86 In these examples, association of a Z-type fragment was shown to increase Lewis acidity at the coordination site trans to the acceptor ligand. However, to our knowledge, the MOLPs derived from M(CNArDipp2)2 (M = Pt, Pd) represent unique cases where Z-ligand-promoted Lewis acidity has been unambiguously observed for mononuclear transition metal complexes. Importantly, these observations highlight the ability of σ-acceptor ligands to open up a previously unavailable coordination site on a transition metal center without effecting a formal oxidative event. Furthermore, the observation that the Ag-containing complexes 5 and 6 bind solvent molecules at the group 10 metal center, while the Tl-containing complexes 1 and 2 exhibit binding at the Tl center, is likely attributable to the greater electronegativity of Ag relative to Tl.87 As stabilization of the empty pz orbital on the group 10 metal by a bound Lewis acid is expected to be marginal at best, Lewis acids possessing greater group electronegativity may be expected to more effectively stabilize this orbital and render it accessible to an exogenous Lewis base.
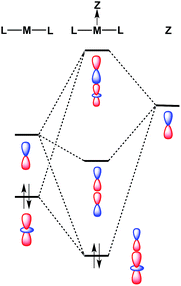 |
| | Fig. 11 Molecular orbital diagram for a transition metal (M) bound to a σ-acceptor fragment (Z), showing how the LUMO of the resulting adduct can be stabilized with respect to the acceptor-free complex. | |
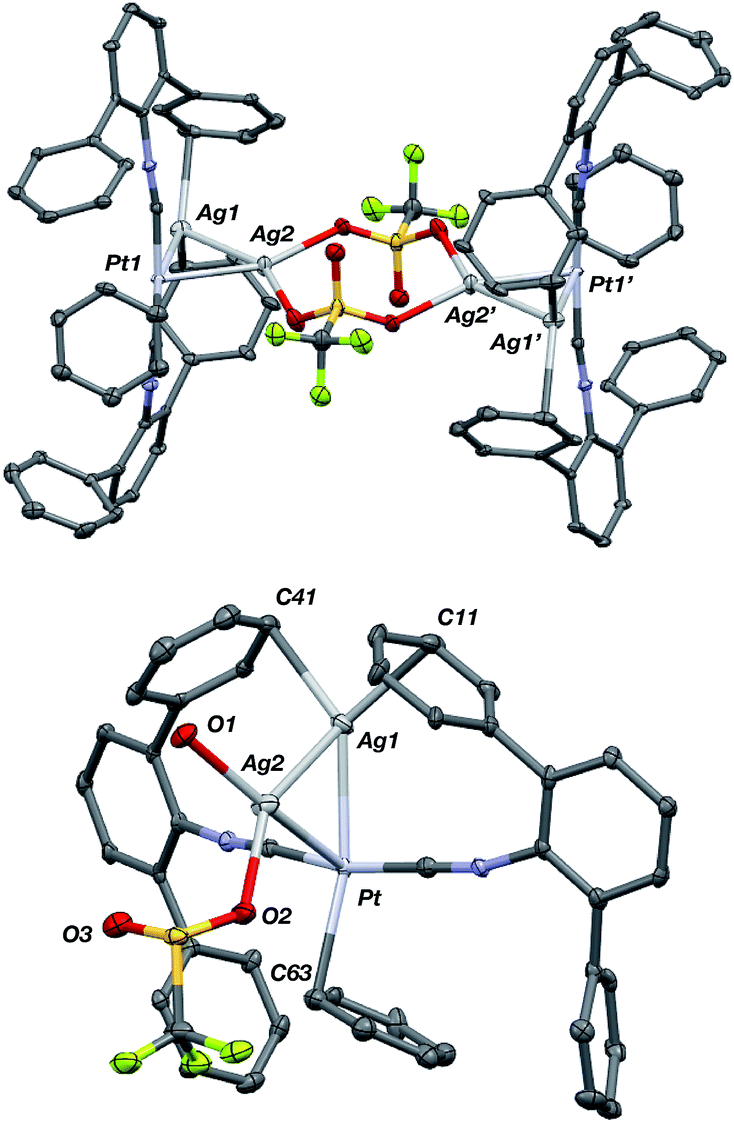
Although [AgPt(CNArDipp2)2]OTf (5) contains one acceptor fragment bound to platinum, its Pt–Ag unit can accommodate another equivalent of Ag(I). Stirring [AgPt(CNArDipp2)2]OTf (5) and equimolar AgOTf in THF followed by crystallization from benzene/THF (20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) yields {[Ag2Pt(CNArDipp2)2(η1-C6H6)]2(μ-OTf)2}(OTf)2 (7) as determined by X-ray diffraction. Attempts to synthesize a palladium analogue from [AgPd(CNArDipp2)2]OTf (6) resulted only in decomposition. The solid-state structure of 7 (Fig. 12) revealed a centro-symmetric dimer composed of triangulo-PtAg2 cores (average d(Pt–Ag) = 2.6843(6) Å; d(Ag–Ag) = 2.7684(8) Å) bridged by two triflate groups. Consistent with the coordination of an additional Lewis acid to the Pt–Ag unit in 5, the isocyanide stretching frequencies of 7 are shifted to higher energies (2132, 2169 cm−1, KBr pellet) compared to 5. In the solid state, the platinum centers in 7 also feature η1-C-bound benzene molecules trans to one of the silver atoms as seen in 5(C6H6). Interestingly however, the Pt–Cbenzene distance in 7 (d(Pt–C1) = 2.529(7) Å) is significantly contracted relative to that in 5(C6H6), a further indication of an increase in the Lewis acidity in the Pt center in 7 promoted by the presence of a second Ag center. It is also important to note that relative to complex 5, the second Ag atom in 7 (i.e. Ag(2), Fig. 12) can best be described as occupying the axial position of a nominally square-planar Pt center. As the binding of Lewis acids to the axial position square planar Pt(II) centers is known,88–92 complex 7 provides additional evidence that the presence of one Z-type ligand effectively results in the formation of a divalent Pt center capable of binding a second Z-type ligand. However, this electronic modulation occurs without formal oxidation of the Pt center.
:1) yields {[Ag2Pt(CNArDipp2)2(η1-C6H6)]2(μ-OTf)2}(OTf)2 (7) as determined by X-ray diffraction. Attempts to synthesize a palladium analogue from [AgPd(CNArDipp2)2]OTf (6) resulted only in decomposition. The solid-state structure of 7 (Fig. 12) revealed a centro-symmetric dimer composed of triangulo-PtAg2 cores (average d(Pt–Ag) = 2.6843(6) Å; d(Ag–Ag) = 2.7684(8) Å) bridged by two triflate groups. Consistent with the coordination of an additional Lewis acid to the Pt–Ag unit in 5, the isocyanide stretching frequencies of 7 are shifted to higher energies (2132, 2169 cm−1, KBr pellet) compared to 5. In the solid state, the platinum centers in 7 also feature η1-C-bound benzene molecules trans to one of the silver atoms as seen in 5(C6H6). Interestingly however, the Pt–Cbenzene distance in 7 (d(Pt–C1) = 2.529(7) Å) is significantly contracted relative to that in 5(C6H6), a further indication of an increase in the Lewis acidity in the Pt center in 7 promoted by the presence of a second Ag center. It is also important to note that relative to complex 5, the second Ag atom in 7 (i.e. Ag(2), Fig. 12) can best be described as occupying the axial position of a nominally square-planar Pt center. As the binding of Lewis acids to the axial position square planar Pt(II) centers is known,88–92 complex 7 provides additional evidence that the presence of one Z-type ligand effectively results in the formation of a divalent Pt center capable of binding a second Z-type ligand. However, this electronic modulation occurs without formal oxidation of the Pt center.
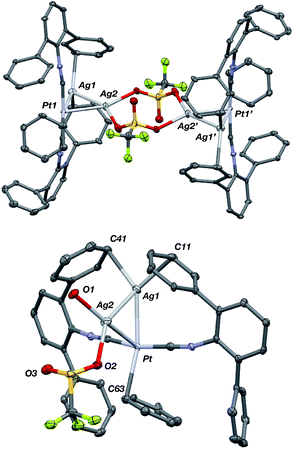 |
| | Fig. 12 Molecular structure of the cationic portion of {[Ag2Pt(CNArDipp2)2(η1-C6H6)]2(μ-OTf)2}(OTf)2 (7). Top: Full view of the centro-symmetric dication. Bottom: Truncated view of one triangulo-Ag2Pt core. In both views, isopropyl groups and non-coordinating triflate anions have been removed for clarity. | |
Conclusions
Zero-valent binary m-terphenyl isocyanide complexes of Pt and Pd are excellent candidates for acting as the Lewis basic component of metal-only Lewis pairs (MOLPs). In addition to stabilizing low oxidation states, the steric encumbrance of the m-terphenyl isocyanide ligand promotes coordinative unsaturation, yielding electron-rich metal centers that can accommodate an exogenous Lewis acid in the primary coordination sphere. Furthermore, the IR ν(CN) resonances provide a convenient handle to assess the degree of group 10 metal σ-donation in these complexes. In this work, it has been demonstrated that Pt(CNArDipp2)2 and Pd(CNArDipp2)2 can form discreet and unsupported adducts with Tl(I). Although these bonding interactions are not particularly robust, use of the weakly coordinating anion BArF4− diminishes the lability of the M–Tl bond in non-coordinating solvents. Analysis of two M → Tl adducts by XANES spectroscopy provides compelling evidence that any degree of formal charge transfer inherent in these metal–metal interactions is minimal, and that therefore no formal oxidative event takes place upon binding of Tl(I). The ability of Pt(CNArDipp2)2 and Pd(CNArDipp2)2 to form Lewis pairs with Ag(I) has also been demonstrated, with FTIR spectroscopy and X-ray crystallography suggesting, again, that no significant charge transfer to Ag occurs in the adducts. Despite this fact, the binding of Ag(I) activates the group 10 metal toward the ligation of Lewis bases trans to the Ag acceptor, thus highlighting how σ-acceptor ligands can be utilized to tune the reactivity profiles of electron-rich transition metal complexes. The Pt/Ag MOLP [AgPt(CNArDipp2)2]OTf (5) can also accommodate an additional equivalent of AgOTf to form dimeric 7, which further increases the Lewis acidity of the Pt center. These results indicate that the presence of a reverse-dative σ-interaction can activate the coordination site trans to it for binding of Lewis bases despite the high trans influence exhibited by Z-type ligands.24,25 Such modulation can be thought of as a novel type of cooperative effect between a Lewis acid and Lewis base, whereby the former alters the reactivity profile of the electron rich metal without directly participating in the reaction with an incoming substrate. A more detailed understanding of the possibilities afforded by such cooperative effects is currently being pursued in our laboratory.
Acknowledgements
We are grateful to the U.S. National Science Foundation for support of this work (CHE-0954710, CHE-1464978 and a Graduate Research Fellowship to B. R. B.). J. S. F is a Camille Dreyfus Teacher-Scholar (2012–2017). S. D. acknowledges the Max Planck Foundation for funding. Portions of the data in this manuscript were obtained at SSRL. SSRL is supported by the U.S. Department of Energy, Office of Science, Office of Basic Energy Sciences under Contract No. DE-AC02-76SF00515. The SSRL Structural Molecular Biology Program is supported by the DOE Office of Biological and Environmental Research, and by the National Institutes of Health, National Institute of General Medical Sciences (including P41GM103393).
Notes and references
- D. F. Shriver, Acc. Chem. Res., 1970, 3, 231–238 CrossRef CAS.
-
W. Hieber, in Advances in Organometallic Chemistry, ed. F. G. A. Stone and R. West, Academic Press, 1970, vol. 8, pp. 1–28 Search PubMed.
- W. Hieber and F. Leutert, Naturwissenschaften, 1931, 19, 360–361 CrossRef CAS.
- W. Hieber, Angew. Chem., 1936, 49, 463–464 CrossRef CAS.
- D. M. P. Mingos, J. Organomet. Chem., 2001, 635, 1–8 CrossRef CAS.
- J. Chatt and L. A. Duncanson, J. Chem. Soc., 1953, 2939 RSC.
-
D. M. P. Mingos, in Advances in Organometallic Chemistry, ed. F. G. A. Stone and R. West, Academic Press, 1977, vol. 15, pp. 1–51 Search PubMed.
- L. Vaska, Acc. Chem. Res., 1968, 1, 335–344 CrossRef CAS.
- L. M. Rendina and R. J. Puddephatt, Chem. Rev., 1997, 97, 1735–1754 CrossRef CAS PubMed.
- J. K. Stille and K. S. Y. Lau, Acc. Chem. Res., 1977, 10, 434–442 CrossRef CAS.
- C. E. Johnson and R. Eisenberg, J. Am. Chem. Soc., 1985, 107, 3148–3160 CrossRef CAS.
-
J. P. Collman and W. R. Roper, in Advances in Organometallic Chemistry, ed. F. G. A. Stone and R. West, Academic Press, 1969, vol. 7, pp. 53–94 Search PubMed.
- M. L. H. Green, J. Organomet. Chem., 1995, 500, 127–148 CrossRef CAS.
- J. M. Burlitch, M. E. Leonowicz, R. B. Petersen and R. E. Hughes, Inorg. Chem., 1979, 18, 1097–1105 CrossRef CAS.
- S. J. la Placa and J. A. Ibers, Inorg. Chem., 1966, 5, 405–410 CrossRef CAS.
- K. W. Muir and J. A. Ibers, Inorg. Chem., 1969, 8, 1921–1928 CrossRef CAS.
- R. A. Fischer and J. Weiss, Angew. Chem., Int. Ed., 1999, 38, 2830–2850 CrossRef PubMed.
- A. F. Hill, G. R. Owen, A. J. P. White and D. J. Williams, Angew. Chem., Int. Ed., 1999, 38, 2759–2761 CrossRef CAS PubMed.
- J. S. Figueroa, J. G. Melnick and G. Parkin, Inorg. Chem., 2006, 45, 7056–7058 CrossRef CAS PubMed.
- K. Pang, J. M. Tanski and G. Parkin, Chem. Commun., 2008, 1008 RSC.
- M.-E. Moret and J. C. Peters, J. Am. Chem. Soc., 2011, 133, 18118–18121 CrossRef CAS PubMed.
- W. H. Harman and J. C. Peters, J. Am. Chem. Soc., 2012, 134, 5080–5082 CrossRef CAS PubMed.
- H. Braunschweig, R. D. Dewhurst and A. Schneider, Chem. Rev., 2010, 110, 3924–3957 CrossRef CAS PubMed.
- H. Braunschweig and R. D. Dewhurst, Dalton Trans., 2010, 40, 549 RSC.
- A. Amgoune and D. Bourissou, Chem. Commun., 2011, 47, 859 RSC.
- M. Devillard, G. Bouhadir and D. Bourissou, Angew. Chem., Int. Ed., 2015, 54, 730–732 CrossRef CAS PubMed.
- B. R. Barnett, C. E. Moore, A. L. Rheingold and J. S. Figueroa, J. Am. Chem. Soc., 2014, 136, 10262–10265 CrossRef CAS PubMed.
- P. Gualco, T.-P. Lin, M. Sircoglou, M. Mercy, S. Ladeira, G. Bouhadir, L. M. Pérez, A. Amgoune, L. Maron, F. P. Gabbaï and D. Bourissou, Angew. Chem., Int. Ed., 2009, 48, 9892–9895 CrossRef CAS PubMed.
- T.-P. Lin, C. R. Wade, L. M. Pérez and F. P. Gabbaï, Angew. Chem.,
Int. Ed., 2010, 49, 6357–6360 CrossRef CAS PubMed.
- C. R. Wade, T.-P. Lin, R. C. Nelson, E. A. Mader, J. T. Miller and F. P. Gabbaï, J. Am. Chem. Soc., 2011, 133, 8948–8955 CrossRef CAS PubMed.
- C. R. Wade and F. P. Gabbaï, Angew. Chem., Int. Ed., 2011, 50, 7369–7372 CrossRef CAS PubMed.
- T.-P. Lin, I.-S. Ke and F. P. Gabbaï, Angew. Chem., Int. Ed., 2012, 51, 4985–4988 CrossRef CAS PubMed.
- J. S. Jones, C. R. Wade and F. P. Gabbaï, Angew. Chem., Int. Ed., 2014, 53, 8876–8879 CrossRef CAS PubMed.
- J. P. Krogman, J. R. Gallagher, G. Zhang, A. S. Hock, J. T. Miller and C. M. Thomas, Dalton Trans., 2014, 43, 13852–13857 RSC.
- G. M. Sheldrick and R. N. F. Simpson, J. Chem. Soc. A, 1968, 1005 RSC.
- W. Clegg and P. J. Wheatley, J. Chem. Soc. A, 1971, 3572 RSC.
- B. A. Sosinsky, R. G. Shong, B. J. Fitzgerald, N. Norem and C. O'Rourke, Inorg. Chem., 1983, 22, 3124–3129 CrossRef CAS.
- T. Tanase, Y. Yamamoto and R. J. Puddephatt, Organometallics, 1996, 15, 1502–1504 CrossRef CAS.
- J. Bauer, H. Braunschweig and R. D. Dewhurst, Chem. Rev., 2012, 112, 4329–4346 CrossRef CAS PubMed.
- R. Grigg, V. Loganathan, V. Santhakumar and A. Teasdale, Tetrahedron Lett., 1991, 32, 687–690 CrossRef CAS.
- R. Grigg, P. Kennewell and A. J. Teasdale, Tetrahedron Lett., 1992, 33, 7789–7792 CrossRef CAS.
- R. Grigg and V. Sridharan, Tetrahedron Lett., 1993, 34, 7471–7474 CrossRef CAS.
- C. C. Roberts, D. M. Matias, M. J. Goldfogel and S. J. Meek, J. Am. Chem. Soc., 2015, 137, 6488–6491 CrossRef CAS PubMed.
- R. G. Pearson, Chem. Rev., 1985, 85, 41–49 CrossRef CAS.
- G. Parkin, J. Chem. Educ., 2006, 83, 791 CrossRef CAS.
- G. Parkin, Organometallics, 2006, 25, 4744–4747 CrossRef CAS.
- A. F. Hill, Organometallics, 2006, 25, 4741–4743 CrossRef CAS.
- B. J. Fox, Q. Y. Sun, A. G. DiPasquale, A. R. Fox, A. L. Rheingold and J. S. Figueroa, Inorg. Chem., 2008, 47, 9010–9020 CrossRef CAS PubMed.
- B. J. Fox, M. D. Millard, A. G. DiPasquale, A. L. Rheingold and J. S. Figueroa, Angew. Chem., Int. Ed., 2009, 48, 3473–3477 CrossRef CAS PubMed.
- L. A. Labios, M. D. Millard, A. L. Rheingold and J. S. Figueroa, J. Am. Chem. Soc., 2009, 131, 11318–11319 CrossRef CAS PubMed.
- G. W. Margulieux, N. Weidemann, D. C. Lacy, C. E. Moore, A. L. Rheingold and J. S. Figueroa, J. Am. Chem. Soc., 2010, 132, 5033–5035 CrossRef CAS PubMed.
- A. E. Carpenter, G. W. Margulieux, M. D. Millard, C. E. Moore, N. Weidemann, A. L. Rheingold and J. S. Figueroa, Angew. Chem., Int. Ed., 2012, 51, 9412–9416 CrossRef CAS PubMed.
- A. E. Carpenter, I. Wen, C. E. Moore, A. L. Rheingold and J. S. Figueroa, Chem.–Eur. J., 2013, 10452–10457 CrossRef CAS PubMed.
- B. M. Emerich, C. E. Moore, B. J. Fox, A. L. Rheingold and J. S. Figueroa, Organometallics, 2011, 30, 2598–2608 CrossRef CAS.
- C. C. Mokhtarzadeh, G. W. Margulieux, A. E. Carpenter, N. Weidemann, C. E. Moore, A. L. Rheingold and J. S. Figueroa, Inorg. Chem., 2015, 54, 5579–5587 CrossRef CAS PubMed.
- Given the uncertainty in values for the covalent radii of transition metals and heavy main-group elements, we present a range for the sum of the covalent radii of Pt and Tl based on several commonly used tabulations (in Å): rcov(Pt) 1.36 + rcov(Tl) 1.45 = 2.81,
(a) B. Cordero, V. Gómez, A. E. Platero-Prats, M. Revés, J. Echeverría, E. Cremades, F. Barragán and S. Alvarez, Dalton Trans., 2008, 2832 RSC. rcov(Pt) 1.30 + rcov(Tl) 1.44 = 2.74,
(b)
L. Pauling, The Nature of the Chemical Bond, Cornell University Press, Cornell, NY, 3rd edn, 1960, ch. 11, p. 403 Search PubMed. rcov(Pt) 1.23 + rcov(Tl) 1.44 = 2.67,
(c) P. Pyykkö and M. Atsumi, Chem.–Eur. J., 2009, 15, 186 CrossRef PubMed.
- P. Pyykkö, Chem. Rev., 1997, 97, 597–636 CrossRef.
- V. J. Catalano, B. L. Bennett, S. Muratidis and B. C. Noll, J. Am. Chem. Soc., 2001, 123, 173–174 CrossRef CAS PubMed.
- S. Wang, J. P. Fackler, C. King and J. C. Wang, J. Am. Chem. Soc., 1988, 110, 3308–3310 CrossRef CAS.
- E. J. Fernández, A. Laguna, J. M. A. Lopez-de-Luzuriaga, M. Elena Olmos and J. Perez, Chem. Commun., 2003, 1760 RSC.
- S. Jamali, M. M. Ashtiani, Z. Jamshidi, E. Lalinde, M. T. Moreno, H. Samouei, E. Escudero-Adán and J. Benet-Buchholz, Inorg. Chem., 2013, 52, 10729–10731 CrossRef CAS PubMed.
- B. M. Still, P. G. A. Kumar, J. R. Aldrich-Wright and W. S. Price, Chem. Soc. Rev., 2007, 36, 665–686 RSC.
- For each arene ring seen to be interacting with Tl in 3 (two per molecule, two independent molecules per asymmetric unit), there are two Tl–C bond lengths that are appreciably shorter than the others for that given arene ring. As such, we believe this interaction is best characterized as having η2 hapticity.
- E. J. Fernández, A. Laguna, J. M. López-de-Luzuriaga, M. Monge, M. Montiel and M. E. Olmos, Inorg. Chem., 2007, 46, 2953–2955 CrossRef PubMed.
- S. H. Strauss, M. D. Noirot and O. P. Anderson, Inorg. Chem., 1986, 25, 3850–3851 CrossRef CAS.
- M. D. Noirot, O. P. Anderson and S. H. Strauss, Inorg. Chem., 1987, 26, 2216–2223 CrossRef CAS.
- Y. Sarazin, N. Kaltsoyannis, J. A. Wright and M. Bochmann, Organometallics, 2007, 26, 1811–1815 CrossRef CAS.
- While naturally-occuring thallium also consists of 29.5% 203Tl (I = 1/2), complexes containing Pt–Tl bonds typically only resolve coupling to 205Tl in their 195Pt NMR spectra. For example, see ref. 58.
- S. Fuertes, C. H. Woodall, P. R. Raithby and V. Sicilia, Organometallics, 2012, 31, 4228–4240 CrossRef CAS.
- U. Belío, S. Fuertes and A. Martín, Dalton Trans., 2014, 43, 10828–10843 RSC.
- F. de Groot, Chem. Rev., 2001, 101, 1779–1808 CrossRef CAS PubMed.
- N. C. Tomson, L. A. Labios, T. Weyhermüller, J. S. Figueroa and K. Wieghardt, Inorg. Chem., 2011, 50, 5763–5776 CrossRef CAS PubMed.
- R. J. Wright, A. D. Phillips, S. Hino and P. P. Power, J. Am. Chem. Soc., 2005, 127, 4794–4799 CrossRef CAS PubMed.
- P. Pyykkö, Chem. Rev., 1988, 88, 563–594 CrossRef.
- A. E. Carpenter, A. J. McNeece, B. R. Barnett, A. L. Estrada, C. C. Mokhtarzadeh, C. E. Moore, A. L. Rheingold, C. L. Perrin and J. S. Figueroa, J. Am. Chem. Soc., 2014, 136, 15481–15484 CrossRef CAS PubMed.
- For other examples of metal-only Lewis pairs with Ag as the Lewis acidic component, see ref. 39 and references cited therein.
- See the ESI, section S2.†.
- G. R. Fulmer, A. J. M. Miller, N. H. Sherden, H. E. Gottlieb, A. Nudelman, B. M. Stoltz, J. E. Bercaw and K. I. Goldberg, Organometallics, 2010, 29, 2176–2179 CrossRef CAS.
- S. M. Hubig, S. V. Lindeman and J. K. Kochi, Coord. Chem. Rev., 2000, 200–202, 831–873 CrossRef CAS.
- S. S. Stahl, J. A. Labinger and J. E. Bercaw, J. Am. Chem. Soc., 1996, 118, 5961–5976 CrossRef CAS.
- D. D. Wick and K. I. Goldberg, J. Am. Chem. Soc., 1997, 119, 10235–10236 CrossRef CAS.
- G. Aullón and S. Alvarez, Inorg. Chem., 1996, 35, 3137–3144 CrossRef.
- C. Tsay, N. P. Mankad and J. C. Peters, J. Am. Chem. Soc., 2010, 132, 13975–13977 CrossRef CAS PubMed.
- V. K. Dioumaev and J. F. Harrod, Organometallics, 1996, 15, 3859–3867 CrossRef CAS.
- T.-P. Lin, R. C. Nelson, T. Wu, J. T. Miller and F. Gabbai, Chem. Sci., 2012, 3, 1128–1136 RSC.
- B. S. Dolinar and J. F. Berry, Inorg. Chem., 2013, 52, 4658–4667 CrossRef CAS PubMed.
- C. H. Suresh and N. Koga, J. Am. Chem. Soc., 2002, 124, 1790–1797 CrossRef CAS PubMed.
- M.-E. Moret and P. Chan, J. Am. Chem. Soc., 2009, 131, 5675–5690 CrossRef CAS PubMed.
- T. Yamaguchi, F. Yamazaki and T. Ito, J. Am. Chem. Soc., 2001, 123, 743–744 CrossRef CAS PubMed.
- R. Uson, J. Fornies, M. Tomas, I. Ara, J. M. Casas and A. Martin, J. Chem. Soc., Dalton Trans., 1991, 2253–2264 RSC.
- T. Yamaguchi, F. Yamazaki and T. Ito, J. Am. Chem. Soc., 1999, 121, 7405–7406 CrossRef CAS.
- L. R. Falvello, J. Fornies, A. Martin, R. Navarro, V. Sicilia and P. Villarroya, Inorg. Chem., 1997, 36, 6166–6171 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Synthetic and spectroscopic details, crystallographic structure determinations and CSD search results. CCDC 1414413 – 1414422. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5sc03104d |
|
| This journal is © The Royal Society of Chemistry 2015 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a