
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Catalytic arylsulfonyl radical-triggered 1,5-enyne-bicyclizations and hydrosulfonylation of α,β-conjugates†
Zhen-Zhen
Chen‡
a,
Shuai
Liu‡
a,
Wen-Juan
Hao
a,
Ge
Xu
a,
Shuo
Wu
a,
Jiao-Na
Miao
a,
Bo
Jiang
*a,
Shu-Liang
Wang
a,
Shu-Jiang
Tu
*a and
Guigen
Li
*bc
aJiangsu Key Laboratory of Green Synthetic Chemistry for Functional Materials, Jiangsu Normal University, Xuzhou, 211116, P. R. China. E-mail: jiangchem@jsnu.edu.cn; laotu@jsnu.edu.cn; Fax: +8651683500065; Tel: +8651683500065
bInstitute of Chemistry & BioMedical Sciences, Collaborative Innovation Center of Chemistry for Life Sciences, School of Chem. and Chem. Eng., Nanjing University, Nanjing 210093, P. R. China
cDepartment of Chemistry and Biochemistry, Texas Tech University, Lubbock, TX 79409-1061, USA. E-mail: guigen.li@ttu.edu
First published on 19th August 2015
Abstract
A catalytic bicyclization reaction of 1,5-enynes anchored by α,β-conjugates with arylsulfonyl radicals generated in situ from sulfonyl hydrazides has been established using TBAI (20 mol%) and Cu(OAc)2 (5 mol%) as co-catalysts under convenient conditions. In addition, the use of benzoyl peroxide (BPO) as the oxidant and pivalic acid (PivOH) as an additive was proven to be necessary for this reaction. The reactions occurred through 5-exo-dig/6-endo-trig bicyclizations and homolytic aromatic substitution (HAS) cascade mechanisms to give benzo[b]fluorens regioselectively. A similar catalytic process was developed for the synthesis of γ-ketosulfones. These reactions feature readily accessible starting materials and simple one-pot operation.
The search for efficient cyclization reactions, particularly for those in radical cascade processes, has been actively pursued in the past several decades because they are extremely useful for the total synthesis of numerous important targets.1,2 These reactions enable the rapid, reliable and straightforward creation of multicyclic ring systems using readily available starting materials with features such as unparalleled efficiencies, high functional tolerance and convenient conditions.2 Among these cyclization reactions, the majority of efforts have been devoted to conducting radical ene-cyclization cascades, in which terminal alkenes were utilized in most cases via either metal-free or transition-metal-mediated radical processes (Scheme 1, eqn (1)).3 However, the use of internal alkenes as radical acceptors has been highly challenging (Scheme 1, eqn (2))4 owing to their relatively low reactivity and larger steric hindrance as compared with their terminal counterparts.
 | ||
| Scheme 1 Two modes of radical ene-cyclizations. | ||
1,5-Enynes endowed with extra unsaturated moieties are remarkable building blocks, and have been widely used for direct and selective tandem cyclizations via synergistic additions across C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C and C
C and C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C bonds in a one-step operation.5 These cyclizations enhance both bond formation and annulation efficiencies with high levels of structural complexity and a reduced generation of waste. So far, two main methods for 1,5-enyne cyclizations have been developed through metal catalysis6 or electrophilic cyclization.7 However, the radical bicyclization of 1,5-enynes for generating multi-substituted polycycles has not been well documented. The literature survey revealed that sulfonyl radicals can be generated from sulfonyl hydrazides and utilized in situ for the radical sulfonylation of alkenes.8 Due to the importance of sulfonyl-containing compounds in photovoltaic materials, nonlinear optics and in general synthetic and medicinal areas,9 we envisioned that under suitable catalytic radical conditions, the in situ generated sulfonyl radicals would be able to be involved in cascade bond-forming events with the internal CC and CC bonds of 1,5-enyne conjugate systems, resulting in 5-exo-dig/6-endo-trig bicyclizations and homolytic aromatic substitutions (HASs) (Scheme 2). Herein, we would like to report the preliminary results of this endeavour (Scheme 2).
C bonds in a one-step operation.5 These cyclizations enhance both bond formation and annulation efficiencies with high levels of structural complexity and a reduced generation of waste. So far, two main methods for 1,5-enyne cyclizations have been developed through metal catalysis6 or electrophilic cyclization.7 However, the radical bicyclization of 1,5-enynes for generating multi-substituted polycycles has not been well documented. The literature survey revealed that sulfonyl radicals can be generated from sulfonyl hydrazides and utilized in situ for the radical sulfonylation of alkenes.8 Due to the importance of sulfonyl-containing compounds in photovoltaic materials, nonlinear optics and in general synthetic and medicinal areas,9 we envisioned that under suitable catalytic radical conditions, the in situ generated sulfonyl radicals would be able to be involved in cascade bond-forming events with the internal CC and CC bonds of 1,5-enyne conjugate systems, resulting in 5-exo-dig/6-endo-trig bicyclizations and homolytic aromatic substitutions (HASs) (Scheme 2). Herein, we would like to report the preliminary results of this endeavour (Scheme 2).
 | ||
| Scheme 2 Envisaged new reactivity of 1,5-enynes. | ||
At first, 3-(2-(phenylethynyl)phenyl)-1-(p-tolyl)prop-2-en-1-one 1a was selected as a benchmark substrate to investigate the additions by sulfonyl radicals. With 20 mol% tetrabutylammoniumiodide (TBAI) as the catalyst, the reaction of substrate 1a with tosylhydrazide 2a was performed in CH3CN in the presence of benzoperoxide (BPO) (4.0 equiv.) as an oxidant at 70 °C under air conditions, affording the expected benzo[b]fluorens 3a, albeit with a low yield of 18% (Table 1, entry 1). Other solvents, such as dichloromethane (DCM), 1,4-dioxane and toluene, were also examined, with CH3CN showing the best performance (entries 2–4). Raising the reaction temperature to 100 °C slightly ameliorates the yield of 3a (entry 5). A subsequent investigation of other catalysts was conducted in CH3CN. As illustrated in entries 6–8, different types of catalysts including I2, KI, and CuI were employed in the model reaction, and it turned out that I2 and KI hardly facilitate the reaction (entries 6 and 7), while CuI as a catalyst only led to a poor yield of 16%. Next, we turned our attention to evaluating different additives (entries 9–11). We found that the addition of PivOH (1.0 equiv.) delivered 3a in a 35% yield (entry 11). Notably, the reaction of 1a and 2a in the presence of 2.0 equiv. of PivOH gave 3a in a 71% yield using a co-catalyst of TBAI (20 mol%) and Cu(OAc)2 (5 mol%) with complete consumption of the starting material 1a (entry 15). Without PivOH, the yield of the expected product 3a decreased remarkably (entry 17). Further screening of other oxidants, such as TBHP (64% yield), DTBP (very poor yield) and H2O2 (no product) for this transformation showed that BPO was the best choice (See ESI†).
|
|
|||||
|---|---|---|---|---|---|
| Entry | Catalyst (mol%) | Additives (equiv.) | Solvent | T (°C) | Yieldb (%) |
| a Reaction conditions: 1,5-conjugated enyne (1a, 0.25 mmol), tosylhydrazide (2a, 0.50 mmol), BPO (1.0 mmol), solvent (2.5 mL), 12 h. b Isolated yields based on 1. | |||||
| 1 | TBAI (20) | — | MeCN | 70 | 18 |
| 2 | TBAI (20) | — | DCM | 70 | 10 |
| 3 | TBAI (20) | — | 1,4-Dioxane | 70 | Trace |
| 4 | TBAI (20) | — | Toluene | 70 | 0 |
| 5 | TBAI (20) | — | MeCN | 100 | 25 |
| 6 | I2 (15) | — | MeCN | 100 | Messy |
| 7 | KI (20) | — | MeCN | 100 | Messy |
| 8 | CuI (20) | — | MeCN | 100 | 16 |
| 9 | TBAI (20) | HOAc (1.0) | MeCN | 100 | 28 |
| 10 | TBAI (20) | L-Proline (1.0) | MeCN | 100 | 33 |
| 11 | TBAI (20) | PivOH (1.0) | MeCN | 100 | 35 |
| 12 | TBAI (20)/CuI (5) | PivOH (1.0) | MeCN | 100 | 49 |
| 13 | TBAI (30)/Cu(OAc)2 (5) | PivOH (1.0) | MeCN | 100 | 53 |
| 14 | TBAI (20)/Cu(OAc)2 (5) | PivOH (1.0) | MeCN | 100 | 61 |
| 15 | TBAI (20)/Cu(OAc)2 (5) | PivOH (2.0) | MeCN | 100 | 71 |
| 16 | TBAI (20)/Cu(OAc)2 (10) | PivOH (2.0) | MeCN | 100 | 63 |
| 17 | TBAI (20)/Cu(OAc)2 (5) | — | MeCN | 100 | 33 |

With the optimized reaction conditions in hand, we examined the substrate scope of the sulfonyl hydrazides 2 by treating them with 1,5-enynes 1a (Scheme 3). As anticipated, the substituents on the phenyl ring of the arylsulfonyl hydrazides 2 did not hamper the catalytic process, but affected the reaction efficiency. Reactions of methyl- or bromo, t-butyl-substituted arylsulfonyl hydrazide 2 with 1a afforded the desired products in moderate to good yields. Additionally, benzenesulfonohydrazide exhibited a higher reactivity, allowing 1,5-enyne-bicyclization cascades toward the formation of benzo[b]fluorens 3c in an 84% yield. 1,5-Enynes bearing an electron-donating or electron-withdrawing group (methoxy and chloro) at the para position of the aromatic ring (Ar1) directly bound to the CC bond gave the corresponding sulfonated products 3e and f in 55% and 68% yields, respectively. Alternatively, a naphthalen-1-yl substituent linked to the CC bond was also well-tolerated, affording the product 3g in a 60% chemical yield. Similarly, either electron-donating (methyl) or electron-withdrawing (bromo) groups (R1) at the para position of the phenyl ring tethered to the enone unit were well-suited for these radical 1,5-enyne-bicyclizations (3a–3k). 1,5-Enynes 1 carrying electron-neutral groups were also smoothly converted into the corresponding sulfonated benzo[b]fluorens 3l–3n in 39–67% yields. Notably, 2-naphthalenylethanone-derived 1,5-enynes furnished the unprecedented pentacyclic indeno[2,1-b]phenanthren-7-ol 3n in a 67% chemical yield though sulfonyl radicals triggered the 1,5-enyne-bicyclization. Unfortunately, a bulky ortho-Br substituent and benzylsulfonyl hydrazide did not work at all (3o and 3p). Besides the NMR and HR-MS spectroscopic analysis for benzo[b]fluorens 3, the X-ray diffraction for product 3f has been performed as shown in Fig. 1.
 | ||
| Scheme 3 Substrate scope of the hydrosulfonylation reaction. Reaction conditions: 1,5-conjugated enyne (1, 0.25 mmol), sulfonyl hydrazide (2, 0.50 mmol), TBAI (0.05 mmol), Cu(OAc)2 (0.0125 mmol), PivOH (0.50 mmol), BPO (1.0 mmol), CH3CN (2.5 mL), 100 °C, 12 h. Isolated yields based on 1. | ||
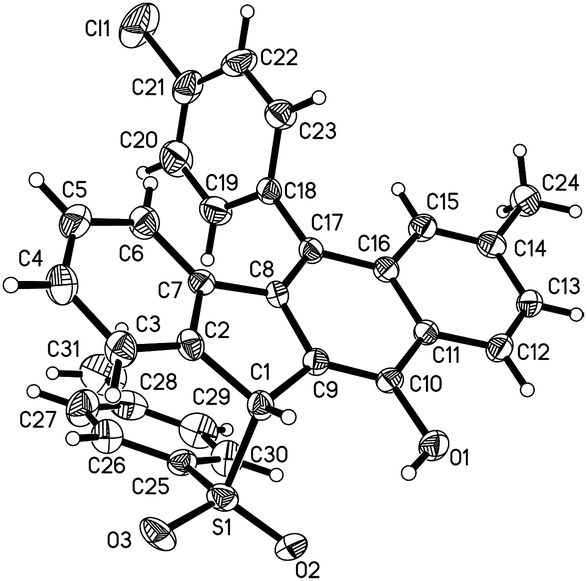 | ||
| Fig. 1 The ORTEP drawing of 3f. | ||
In view of our success with the synthesis of functional benzo[b]fluorens 3, we reasoned that in the absence of alkyne moieties, chalcones 4 would be able to accept sulfonyl radicals via typical 1,4-additions, which would expand their utility for the synthesis of γ-ketosulfones. We thus explored this possibility through a one-pot reaction of (4-chlorophenyl)-3-phenylprop-2-en-1-one (4a) with 2a under the conditions described above. The expected γ-ketosulfone 5a was obtained but with a lower yield (15%) initially. After careful optimizations were performed, we found that although Cu(OAc)2 and PivOH did promote this catalytic process, the use of co-oxidants of BPO (2.0 equiv.) and TBHP (1.0 equiv., 70% in water) in 20 mol% of TBAI proved to be suitable for the current hydrosulfonylation, furnishing product 5a in a 77% yield. Subsequently, we further studied the reaction scope by reacting arylsulfonyl hydrazides 2 with various chalcones 4 under these conditions (Scheme 4). It turned out that the presence of various substituents, including methoxyl, methyl, chloro and bromo groups, on the aryl rings of the chalcones all worked well, giving access to a wide range of γ-ketosulfones 5a–5o with yields ranging from 50% to 90%. Alternatively, arylsulfonyl hydrazides 2 carrying either electronically neutral or rich groups can be successfully engaged in this catalysis. Unfortunately, aliphatic sulfonyl hydrazide (5p) was proven not to be an adaptable substrate for this reaction, which may be ascribed to the relative instability of the sulfonyl radicals generated in situ from aliphatic sulfonyl hydrazides. Joining previously reported work,10 this catalytic radical addition provided a new protocol for the formation of γ-ketosulfones, which are important building blocks in organic synthesis.
 | ||
| Scheme 4 Substrate scope of the synthesis of γ-ketosulfones. Reaction conditions: chalcone (4, 0.25 mmol), sulfonyl hydrazide (2, 0.50 mmol), TBAI (0.05 mmol), BPO (0.50 mmol), TBHP (0.25 mmol, 70% in water), CH3CN (2.5 mL), 100 °C, 6 h. Isolated yields based on 4. | ||
To understand the mechanism, several control experiments were conducted. The treatment of 1,5-enyne 1h with tosylhydrazide 2a in the presence of radical scavenger TEMPO (4.0 equiv.) under standard conditions gave complex mixtures without the observation of the desired product 3l, confirming the existence of a radical mechanism (Scheme 5, eqn (1)). In the absence of BPO, the reaction did not show the desired product (eqn (2)). To further confirm the sulfonylation sequence, subjecting 1,5-enyne 1h to the standard conditions in the absence of 2a failed to generate any desired benzo[b]fluoren product 6 (eqn (3)). These control experiments suggest that BPO is essential for the catalytic cycles and the in situ generated sulfonyl radical triggers a 5-exo-dig/6-endo-trig bicyclization cascade.
 | ||
| Scheme 5 Control reactions. Reaction conditions: 1,5-conjugated enyne (1h, 0.25 mmol), tosylhydrazide (2a, 0.50 mmol), TBAI (0.05 mmol), Cu(OAc)2 (0.0125 mmol), PivOH (0.50 mmol), BPO (1.0 mmol), CH3CN (2.5 mL), 100 °C, 12 h. | ||
On the basis of the above observations and those reported in literature,8,11 a mechanism is proposed and shown in Scheme 6. The first step is to form the sulfonyl radical A from the sulfonyl hydrazide using the benzoyloxy radical generated in situ from the I− anion-assisted decomposition of BPO. The intermolecular addition of the resulting sulfonyl radical A and the 1,5-conjugated enyne 1 followed by a 5-exo-dig cyclization gives intermediate B, in which the homolysis of carbon–copper(III) affords vinyl radical C. Intermediate C is converted into aryl radical Dvia a 6-endo-trig cyclization. Intermediate D undergoes SET (single electron transfer) oxidation and subsequent deprotonation to provide intermediate F. The tautomerization of F leads to the formation of benzo[b]fluorens 3. Although the generation of sulfonyl radicals triggered by various oxidants has been achieved well,8 the bicyclizations12 towards fused carbocycles via sulfonyl radical initiated bifunctionalization of enynes is very rare in organic chemistry as mentioned earlier.
 | ||
| Scheme 6 Proposed mechanism for forming products 3. | ||
Conclusions
In summary, we have discovered new 1,5-enyne-bicyclization and hydrosulfonylation reactions of α,β-conjugates under convenient co-catalytic conditions. The addition of in situ generated sulfonyl radicals across the activated double bond is able to trigger a cascade 5-exo-dig/6-endo-trig bicyclization and HAS sequence, delivering tetracyclic sulfonylated benzo[b]fluorens in a successive C–S and C–C bond-forming process. Using chalcones as replacements for 1,5-conjugated enynes, this reaction enables the hydrosulfonylation of alkenes to form γ-ketosulfones with good to excellent yields. These two methods allow easy access to important functional sulfones for potential applications in organic and medicinal chemistry.Acknowledgements
We are grateful for financial support from the NSFC (No. 21232004, 21272095, 21472071 and 21332005), PAPD of Jiangsu Higher Education Institutions, the Qing Lan Project (12QLG006), Robert A. Welch Foundation (D-1361, USA) and NIH (R33DA031860, USA), the Outstanding Youth Fund of Jiangsu Normal University (YQ2015003), and Natural Science Foundation of Jiangsu Normal University (14XLR005).Notes and references
- For selected reviews, see: (a) M. Malacria, Chem. Rev., 1996, 96, 289 CrossRef CAS PubMed; (b) D. P. Curran, Aldrichimica Acta, 2000, 33, 104 CAS; (c) S. Z. Zard, Radical Reactions in Organic Synthesis, Oxford University Press, Oxford, UK, 2003 Search PubMed; (d) H. Togo, Advanced Free Radical Reactions for Organic Synthesis, Elsevier, Amsterdam, 2004 Search PubMed; (e) B. B. Snider, Oxidative Free-Radical Cyclizations and Additions with Mono and β-Dicarbonyl Compounds, in Handbook of C-H Transformations: Applications in Organic Synthesis, ed. G. Dyker, Wiley-VCH, Weinheim, 2005, vol. 2, pp. 371–377 Search PubMed; (f) M. Albert, L. Fensterbank, E. Lacote and M. Malacria, Topics in Current Chemistry, ed. A. Gansäuer, Springer, Berlin, 2006, vol. 264, p. 1 Search PubMed; (g) P. Renaud and M. P. Sibi, Radicals in Organic Synthesis, Wiley-VCH, Weinheim, 2008 Search PubMed; (h) E. Godineau and Y. Landais, Chem.–Eur. J., 2009, 15, 3044 CrossRef CAS PubMed; (i) S. Hata and M. P. Sibi, Science of Synthesis-Stereoselective Reactions of C–C double bonds, ed. J. G. de Vries, Georg Thieme Verlag, Stuttgart, 2011, p. 873 Search PubMed.
- For selected examples, see: (a) W. Kong, N. Fuentes, A. Garcia-Dominguez, E. Merino and C. Nevado, Angew. Chem., Int. Ed., 2015, 54, 2487 CrossRef CAS PubMed; (b) N. Fuentes, W. Kong, L. Fernandez-Sanchez, E. Merino and C. Nevado, J. Am. Chem. Soc., 2015, 137, 964 CrossRef CAS PubMed; (c) W. Kong, E. Merino and C. Nevado, Angew. Chem., Int. Ed., 2014, 53, 5078 CAS; (d) W. Kong, M. Casimiro, N. Fuentes, E. Merino and C. Nevado, Angew. Chem., Int. Ed., 2013, 52, 13086 CrossRef CAS PubMed; (e) B. Jiang, B.-M. Feng, S.-L. Wang, S.-J. Tu and G. Li, Chem. - Eur. J., 2012, 18, 9823 CrossRef CAS PubMed; (f) B. Jiang, M.-S. Yi, S. Feng, S.-J. Tu, S. Pindi, M. Patrick and G. Li, Chem. Commun., 2012, 48, 808 RSC.
- For selected examples, see: (a) W.-T. Wei, M.-B. Zhou, J.-H. Fan, W. Liu, R.-J. Song, Y. Liu, M. Hu, P. Xie and J.-H. Li, Angew. Chem., Int. Ed., 2013, 52, 3638 CrossRef CAS PubMed; (b) M.-B. Zhou, C.-Y. Wang, R.-J. Song, Y. Liu, W.-T. Wei and J.-H. Li, Chem. Commun., 2013, 49, 10817 RSC; (c) Y. Meng, L.-N. Guo, H. Wang and X.-H. Duan, Chem. Commun., 2013, 49, 7540 RSC; (d) H. Wang, L.-N. Guo and X.-H. Duan, Chem. Commun., 2013, 49, 10370 RSC; (e) Z. Li, Y. Zhang, L. Zhang and Z.-Q. Liu, Org. Lett., 2014, 16, 382 CrossRef CAS PubMed.
- (a) S.-L. Zhou, L.-N. Guo, S. Wang and X.-H. Duan, Chem. Commun., 2014, 50, 3589 RSC; (b) W.-P. Mai, J.-T. Wang, L.-R. Yang, J.-W. Yuan, Y.-M. Xiao, P. Mao and L.-B. Qu, Org. Lett., 2014, 16, 204 CrossRef CAS PubMed; (c) W.-P. Mai, G.-C. Sun, J.-T. Wang, G. Song, P. Mao, L.-R. Yang, J.-W. Yuan, Y.-M. Xiao and L.-B. Qu, J. Org. Chem., 2014, 79, 8094 CrossRef CAS PubMed.
- L. Zhang, J. Sun and S. A. Kozmin, Adv. Synth. Catal., 2006, 348, 2271 CrossRef CAS PubMed.
- For selected examples, see: (a) K. Speck, K. Karaghiosoff and T. Magauer, Org. Lett., 2015, 17, 1982 CrossRef CAS PubMed; (b) C. Blaszykowski, Y. Harrak, M.-H. Goncalves, J.-M. Cloarec, A.-L. Dhimane, L. Fensterbank and M. Malacria, Org. Lett., 2004, 6, 3771 CrossRef CAS PubMed; (c) C.-H. Chen, Y.-C. Tsai and R.-S. Liu, Angew. Chem., Int. Ed., 2013, 52, 4599 CrossRef CAS PubMed; (d) E. Tudela, J. Gonzalez, R. Vicente, J. Santamaria, M. A. Rodriguez and A. Ballesteros, Angew. Chem., Int. Ed., 2014, 53, 12097 CrossRef CAS PubMed; (e) K. Fukamizu, Y. Miyake and Y. Nishibayashi, Angew. Chem., Int. Ed., 2009, 48, 2534 CrossRef CAS PubMed; (f) J. Zhang, D. Wu, X. Chen, Y. Liu and Z. Xu, J. Org. Chem., 2014, 79, 4799 CrossRef CAS PubMed; (g) H. Zheng, R. J. Felix and M. R. Gagne, Org. Lett., 2014, 16, 2272 CrossRef CAS PubMed.
- (a) F. Huber and S. F. Kirsch, J. Org. Chem., 2013, 78, 2780 CrossRef CAS PubMed; (b) A. Pradal, A. Nasr, P. Y. Toullec and V. Michelet, Org. Lett., 2010, 12, 5222 CrossRef CAS PubMed; (c) B. Crone, S. F. Kirsch and K.-D. Umland, Angew. Chem., Int. Ed., 2010, 49, 4661 CrossRef CAS PubMed.
- For selected sulfonyl radicals generated in situ from sulfonyl hydrazides, see: (a) X. Li, X. Xu and C. Zhou, Chem. Commun., 2012, 48, 12240 RSC; (b) J. Zhang, Y. Shao, H. Wang, Q. Luo, J. Chen, D. Xu and X. Wan, Org. Lett., 2014, 16, 3312 CrossRef CAS PubMed; (c) X. Li, Y. Xu, W. Wu, C. Jiang, C. Qi and H. Jiang, Chem.–Eur. J, 2014, 20, 7911 CrossRef CAS PubMed; (d) S. Tang, Y. Wu, W. Liao, R. Bai, C. Liu and A. Lei, Chem. Commun., 2014, 50, 4496 RSC; (e) W. Wei, C. Liu, D. Yang, J. Wen, J. You, Y. Suo and H. Wang, Chem. Commun., 2013, 49, 10239 RSC; (f) T. Taniguchi, Y. Sugiura, H. Zaimoku and H. Ishibashi, Angew. Chem., Int. Ed., 2010, 49, 10154 CrossRef CAS PubMed; (g) G. Rong, J. Mao, H. Yan, Y. Zheng and G. Zhang, J. Org. Chem., 2015, 80, 4697 CrossRef CAS PubMed.
- (a) N. Simpkins, in Sulfones in Organic Synthesis, ed. J. E. Baldwin and P. D. Magnus, Pergamon Press, Oxford, 1993 Search PubMed; (b) E. Block, Reaction of Organosulfur Compounds, Academic Press, New York, 1978 Search PubMed; (c) P. D. Magnus, Tetrahedron, 1977, 33, 2019 CrossRef CAS; (d) E. N. Prilezhaeva, Russ. Chem. Rev., 2000, 69, 367 CrossRef CAS; (e) A. Costa, C. Najera and J. M. Sansano, J. Org. Chem., 2002, 67, 5216 CrossRef CAS PubMed; (f) M. S. Vedula, A. B. Pulipaka, C. Venna, V. K. Chintakunta, S. Jinnapally, V. A. Kattuboina, R. K. Vallakati, V. Basetti, V. Akella, S. Rajgopal, A. K. Reka, S. K. Teepireddy, P. K. Mamnoor, R. Rajagopalan, G. Bulusu, A. Khandelwal, V. V. Upreti and S. R. Mamidi, Eur. J. Med. Chem., 2003, 38, 811 CrossRef CAS; (g) S. F. Barbuceanu, G. L. Almajan, I. Saramet, C. Draghici, A. I. Tarcomnicu and G. Bancescu, Eur. J. Med. Chem., 2009, 44, 4752 CrossRef CAS PubMed; (h) T. M. Williams, T. M. Ciccarone, S. C. MacTough, C. S. Rooney, S. K. Balani, J. H. Condra, E. A. Emini, M. E. Goldman, W. J. Greenlee, L. R. Kauffman, J. A. O'Brien, V. V. Sardana, W. A. Schleif, A. D. Theoharides and P. S. Anderson, J. Med. Chem., 1993, 36, 1291 CrossRef CAS.
- (a) N. K. Konduru, S. Dey, M. Sajid, M. Owais and N. Ahmed, Eur. J. Med. Chem., 2013, 59, 23 CrossRef CAS PubMed; (b) Y. Chen, Y. Lam and Y.-H. Lai, Org. Lett., 2003, 5, 1067 CrossRef CAS PubMed; (c) B. Sreedhar, M. A. Reddy and P. S. Reddy, Synlett, 2008, 1949 CAS; (d) H. Gilman and L. F. Cason, J. Am. Chem. Soc., 1950, 72, 3469 CrossRef CAS; (e) M. Fernandez, U. Uria, L. Orbe, J. L. Vicario, E. Reyes and L. Carrillo, J. Org. Chem., 2014, 79, 441 CrossRef CAS PubMed; (f) T. A. Kovalchuk, N. M. Kuzmenok and A. M. Zvonok, Chem. Heterocycl. Compd., 2005, 41, 1237 CrossRef CAS.
- (a) S. Mondal, B. Gold, R. K. Mohamed, H. Phan and I. V. Alabugin, J. Org. Chem., 2014, 79, 7491 CrossRef CAS PubMed; (b) S. Mondal, R. K. Mohamed, M. Manoharan, H. Phan and I. V. Alabugin, Org. Lett., 2013, 15, 5650 CrossRef CAS PubMed; (c) G.-B. Deng, Z.-Q. Wang, J.-D. Xia, P.-C. Qian, R.-J. Song, M. Hu, L.-B. Gong and J.-H. Li, Angew. Chem., Int. Ed., 2013, 52, 1535 CrossRef CAS PubMed; (d) Y. Liu, J.-L. Zhang, M.-B. Zhou, R.-J. Song and J.-H. Li, Chem. Commun., 2014, 50, 14412 RSC; (e) Y. Liu, J.-L. Zhang, R.-J. Song, P.-C. Qian and J.-H. Li, Angew. Chem., Int. Ed., 2014, 53, 9017 CrossRef CAS PubMed.
- (a) O. Miyata, A. Nishiguchi, I. Ninomiya, T. Naito, K. Aoe and K. Okamura, Tetrahedron Lett., 1996, 37, 229 CrossRef CAS; (b) O. Miyata, Y. Ozawa, I. Ninomiya and T. Naito, Tetrahedron, 2000, 56, 6199 CrossRef CAS; (c) O. Miyata, Y. Ozawa, I. Ninomiya and T. Naito, Synlett, 1997, 275 CrossRef CAS; (d) T. Naito, Y. Honda, O. Miyata and I. Ninomiya, J. Chem. Soc., Perkin Trans. 1, 1995, 19 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 1406678 (3f). For ESI and crystallographic data in CIF or other electronic formats see DOI: 10.1039/c5sc02343b |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2015 |