
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDiscovery of low energy pathways to metal-mediated B![[double bond, length as m-dash]](https://www.rsc.org/images/entities/h2_char_e001.gif) N bond reduction guided by computation and experiment†
N bond reduction guided by computation and experiment†
Tyler J.
Carter
a,
Zachariah M.
Heiden
 *b and
Nathaniel K.
Szymczak
*a
*b and
Nathaniel K.
Szymczak
*a
aDepartment of Chemistry, University of Michigan, 930 N. University, Ann Arbor, MI 48109, USA. E-mail: nszym@umich.edu
bDepartment of Chemistry, Washington State University, PO Box 644630, Pullman, WA 99164, USA. E-mail: zachariah.heiden@wsu.edu
First published on 1st October 2015
Abstract
This manuscript describes a combination of DFT calculations and experiments to assess the reduction of borazines (B–N heterocycles) by η6-coordination to Cr(CO)3 or [Mn(CO)3]+ fragments. The energy requirements for borazine reduction are established as well as the extent to which coordination of borazine to a transition metal influences hydride affinity, basicity, and subsequent reduction steps at the coordinated borazine molecule. Borazine binding to M(CO)3 fragments decreases the thermodynamic hydricity by >30 kcal mol−1, allowing it to easily accept a hydride. These hydricity criteria were used to guide the selection of appropriate reagents for borazine dearomatization. Reduction was achieved with an H2-derived hydride source, and importantly, a pathway which proceeds through a single electron reduction and H-atom transfer reaction, mediated by anthraquinone was uncovered. The latter transformation was also carried out electrochemically, at relatively positive potentials by comparison to all prior reports, thus establishing an important proof of concept for any future electrochemical B![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N bond reduction.
N bond reduction.
1. Introduction
Due to a recognition of the finite availability of fossil fuels, considerable effort has been devoted to the development of new methods that generate, store, and use alternative energy vectors.1,2 Hydrogen is a particularly promising energy carrier; however, several key challenges must be overcome prior to implementation.3,4 In particular, reversible high-capacity hydrogen storage materials are required.5 Ammonia borane (AB; NH3BH3) and related C–B–N compounds show great promise due to their high gravimetric and volumetric hydrogen capacities as well as the relative ease with which they can be dehydrogenated.6–10 Dehydrogenation pathways of B–N compounds can vary depending on reaction conditions; however, thermal and catalytic dehydrogenation often affords cyclic oligomers, such as borazines and polyborazylene (PB), as spent fuel products.10–13Borazines (and other related B–N heterocycles) have been a topic of interest to inorganic chemists for decades.14,15 Early studies with this class of compounds probed electronic and reactivity parallels with arenes, since they are isostructural and isoelectronic with borazines.14–17 However, despite these similarities, borazines exhibit marked differences in their electronic structure and reactivity. Recently, borazines have been identified as a terminal dehydrogenation product from spent B–N hydrogen storage materials, and in this context, borazines pose a unique challenge to reversible H2 storage due to the thermodynamically strong B–N bonds.16–20 The highly exergonic nature of the dehydrogenation reaction of B–N compounds has made the development of low-energy methods (Scheme 1) for the net-hydrogenation of borazine, and PB, a topic of great interest to the hydrogen storage community. While a few notable examples of borazine/PB recycling to AB exist,21–26 all of these methods rely on energetically costly reagents which limit the net energy balance of the hydrogen storage system. Thus, a lower energy route to achieve reductive transformations of borazines would be beneficial. One strategy to lower the energetic barrier associated with reductive transformations of B–N heterocycles is through the coordination of borazine to a transition metal.27,28
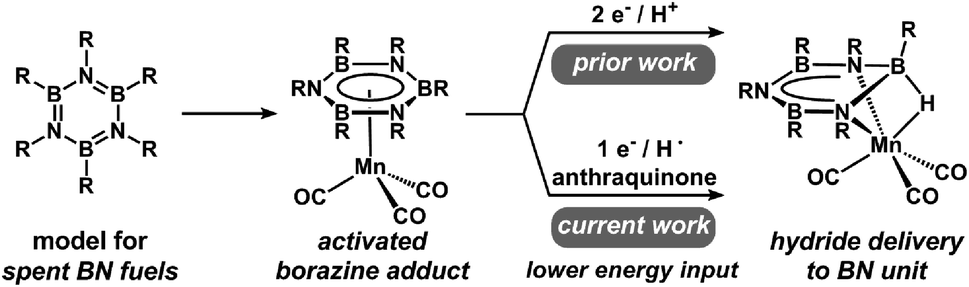 | ||
| Scheme 1 Reaction pathways for hydride delivery to a Mn–borazine complex. | ||
Recent work in our group demonstrated that η6-coordination of borazines to Cr(CO)3 or [Mn(CO)3]+ fragments facilitates the addition of H− or CH3− nucleophiles to afford dearomatized borazine complexes.27,28 Metal-mediated reduction of borazines was also achieved via successive delivery of two-electrons and an exogenous proton donor, which represents an important step toward a potential (electro)catalytic regeneration scheme. In order to further interrogate one- and two-electron pathways for BN bond reduction and to assess energetic costs, we sought to evaluate the thermodynamic requirements, as well as possible mechanistic pathways for these reactions. However, there are currently no data available that describe how the coordination of borazine to a transition metal fragment impacts reductive reactions.
In this study, we quantitatively describe the extent to which metal-mediated binding promotes the ionic hydrogenation of BN bonds. We assess these values by the direct comparison of pKa values and thermodynamic hydricities (ΔGH−) before and after coordination of the borazine to a transition-metal fragment. Additionally, the free energy values for the net-hydrogenation reactions have been used to guide the selection of mild reducing agents capable of promoting reduction. These insights provide direction for the targeted discovery of new pathways for the reduction of heterocyclic B–N bonds, which are relevant for the regeneration of spent AB fuels as well as other C–B–N heterocycles.6–8,25,29,30
2. Results and discussion
2.1. Free energy of hydrogen addition to H3B3N3H3 and Me3B3N3Me3
Alkyl substituted borazines have been used as model complexes to study the reactivity of borazine (and B–N spent fuels) due to their ease of synthesis, thermal stability, and low volatility. Despite these similarities, no direct comparison of the structural and thermodynamic properties of these two compounds has been reported. The parent borazine (H3B3N3H3) is volatile and readily cross links upon heating;12 properties which have prevented the isolation of H3B3N3H3 transition metal complexes. In contrast, substituted borazines are less volatile and thermally robust, thus, our initial studies focused on Cr(CO)3 (1) and [Mn(CO)3]+ (2) complexes of Me3B3N3Me3, which are readily isolable. We hypothesize that coordination of the B–N heterocycle to a transition metal can greatly influence its reduction chemistry. To evaluate this effect for M(CO)3 units, we computationally assessed differences in the thermodynamics of H2 addition reactions to borazines.Previous computational studies have explored the thermodynamics of B–N spent fuel hydrogenation18–20,31,32 and the nature of the M–borazine bond in theoretical H3B3N3H3 complexes.33,34 To our knowledge, a direct comparison of the free energy of H2 addition to the parent borazine vs. its alkyl substituted derivatives, as well as the extent of borazine activation and the effect on subsequent reduction following coordination, remains unexplored. To benchmark our computational model, the free energy of H2 addition was calculated for the parent and alkylated borazines using the B3LYP or M06-2X functionals with the 6-31G(d,p) basis set in the gas phase for geometry optimizations. More accurate energies were obtained through a single point energy calculation using the 6-311++G(d,p) basis set in Et2O (solvent selected for its compatibility with M–borazine complexes) using the polarized continuum solvation model.35,36
The B3LYP functional provided vibrational frequencies and redox potentials closest to the experimental values of the borazine metal complexes, vida infra, and was implemented in all subsequent computational analysis. The predicted free energy (Scheme 2, top) for the reduction of borazine (H3B3N3H3) to cyclotriborazane (H6B3N3H6) was compared with other computational studies and our calculated values slightly overestimate the free energy of borazine hydrogenation (53.5 kcal mol−1 for B3LYP and 45.0 for M06-2X in the gas phase) compared with the values predicted by Matus et al. (43.3 kcal mol−1) and Miranda and Ceder (approximately 48 kcal mol−1).19,20 The slight overestimation in the free energy for the reduction of borazine using the B3LYP functional is attributed to the limited ability of the B3LYP functional to describe dative interactions.19,37 However, our calculated enthalpy of H2 addition of 1.4 kcal mol−1 is in good agreement with the experimental value of 1 kcal mol−1 reported by Schellenberg and Wolf.38 Finally, we performed a NICS(0) calculation to evaluate aromaticity, and found a similar aromatic character between borazine and hexamethylborazine (see ESI†).39–42 This result further validates that hexamethylborazine is a suitable model for borazine.43
 | ||
| Scheme 2 Calculated free energy of hydrogenation for H3B3N3H3 and Me3B3N3Me3 in Et2O solutions using the B3LYP/6-311G++(d,p)//B3LYP/6-31G(d,p) level of theory. | ||
The only known transition metal complexes of borazines are those of alkyl substituted variants.27,28,44–49 Thus, determination of the thermodynamics of H2 addition for both the parent and alkylated borazines is essential to establish general relationships of alkyl borazine coordinated metal complexes,27,28 as model substrates for metal-mediated borazine reductions. In support, the addition of a single equivalent of H2 to form a 1,2-H2-borazine was predicted to have a nearly identical reaction free energy (ΔG = 31.9 vs. 28.8 kcal mol−1) for both the parent and alkylated borazines in Et2O solutions (Scheme 2).
Although the first BN bond reduction was found to be similar in energy for H3B3N3H3 and Me3B3N3Me3, the free energy for further reductions differed for each compound. For instance, while the reduction of H4B3N3H4 to cyclotriborazane (H6B3N3H6) is essentially thermo-neutral (ΔG = −1 kcal mol−1), the analogous H2 addition steps for Me3HB3N3HMe3 are substantially higher in energy (ΔG = 30 kcal mol−1). Given the similarity of free energies for the first addition of H2 to the borazine ring between H3B3N3H3 and Me3B3N3Me3, these data clearly demonstrate that Me3B3N3Me3 is a suitable model for mechanistic studies aimed at uncovering a route to reduce the thermodynamically most challenging BN bonds via metal-mediated ionic hydrogenations.
In addition to the reduction of free borazines, we evaluated the extent to which Me3B3N3Me3 is activated by η6-coordination to a Cr(CO)3 or [Mn(CO)3]+ fragment. Computational experiments replicated key experimental, structural, and spectroscopic metrics, which validated our computational parameters. With few exceptions, the calculated values matched our experimental νCO bands with an error of <20 cm−1 when the suggested correction factor (0.96)50 was applied (Table S-1†). For instance, the calculated CO stretching frequencies of 2069, 2002, and 2001 cm−1 agree well with the experimentally observed bands at 2072 and 1997 for compound 2. Additionally, the calculated redox potentials (Table S-2†) are likewise in good agreement with experimental data (when available). For instance, the calculated potential of −1.49 V (vs. Cp2Fe0/+) for the single electron reduction of 2 in Et2O agrees well with the experimental value of −1.38 V (vs. Cp2Fe0/+).
2.2. Effect of metal coordination on pKa and ΔGH− of Me3B3N3Me3
The C–C bonds of arenes are typically reduced by non-polar reagents (e.g. metal dihydrides and H2 with a variety of heterogeneous catalysts),51,52 though some exceptions have been reported.53–55 In contrast, the BN bonds of borazine require polar reagents for reduction, and thus, the two properties of borazines most relevant to ionic hydrogenation, pKa (Table S-4†) and hydricity (Scheme 3), were evaluated. Based on the absence of reactivity with hydride sources and many relatively strong acids,27,56 the free borazines are predicted to be inert to hydride addition and protonation. In contrast, coordination to a metal fragment induces dramatic changes in both of these values. The conjugate acid strength of the borazine ligand was increased (became a stronger acid) by 13 and 44 pKa units when coordinated in an η6-fashion to Cr(CO)3 and [Mn(CO)3]+ fragments, respectively. Furthermore, a dramatic decrease (>30 kcal mol−1) in the thermodynamic hydricity (became a poorer hydride donor) was observed for the corresponding hydrides [(Me3B3N3Me3)(μ-H)Cr(CO)3]Li (3; ΔGH− = 41 kcal mol−1) and (Me3B3N3Me3)(μ-H)Mn(CO)3 (4; ΔGH− = 69 kcal mol−1) when compared to [Me3B3HN3Me3]−. This is in agreement with our previous experimental results which showed that [Et3BH]− (ΔGH− = 23) was competent for hydride delivery to both metal complexes, but not to free Me3B3N3Me3.27,28 A NICS(0) calculation showed substantial reductions in the aromatic character upon hydride addition. Furthermore, when the ratio of the NICS(0) values between 1/3 and 2/4 was compared, a greater change in the aromaticity of the hexamethylborazine ring was noted for the former pair (see ESI, Table S-6†). This result suggests that hydride addition to Cr(CO)3 results in a greater decrease in the aromaticity than the corresponding [Mn(CO)3]+ unit.
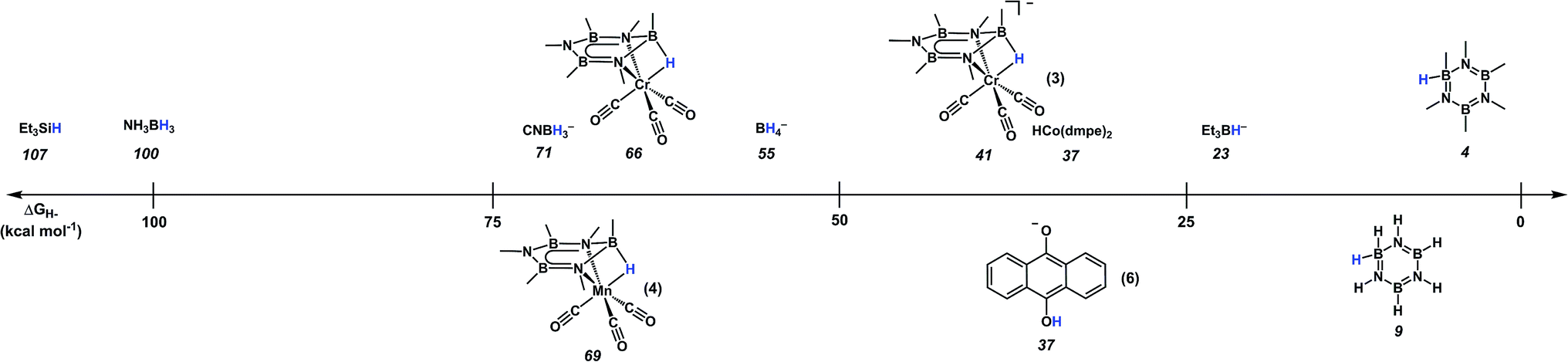 | ||
| Scheme 3 Computational thermodynamic hydricity values (ΔGH−) for borazines, M–borazine complexes, and several common hydride sources in Et2O solutions. | ||
Due to the high energy cost associated with hydride sources such as [HBEt3]−,57 these reagents have limited application in B–N spent fuel regeneration schemes. In contrast, metal hydrides derived from H2 have thermodynamic hydricity values that are tunable and can be of similar strength to many non-metal hydrides,58–61 and thus are attractive candidates for the development of low-energy regeneration pathways. Using the computed hydricity values to guide selection of a suitable hydride candidate, we evaluated the hydride transfer to 1 using HCo(dmpe)2, (ΔGH− = 37 kcal mol−1, dmpe = 1,2-bis(dimethylphosphino)ethane), which is an H2-derived hydride61 and much milder than LiEt3BH (ΔGH− = 23.2 kcal mol−1).27 Consistent with the calculated thermodynamic hydricities, when 1 was allowed to react with 1 eq. of HCo(dmpe)2 at −40 °C in 2-Me-THF, the hydride adduct (3) was obtained, as confirmed by NMR and IR spectroscopy. This finding is significant, as the delivery of H2-derived reducing equivalents to BN spent fuels has previously been a coveted goal for any regeneration cycle.25,61
2.3. Protonation reactions of dearomatized hydride adducts
Following hydride delivery, protonation reactions afford the partially saturated 1,2-H2-adduct, which completes BN bond reduction. Our previous experimental studies suggested that the products obtained from these reactions were highly dependent on the proton source, as well as the specific reaction conditions employed.27,28 In all cases, the products formed from the addition of acid were too unstable to be isolated and fully characterized, and further reactions with excess HX were used to trap the reactive intermediates by adding across the remaining BN bond(s). The substituted cyclotriborazanes (OAcF)2Me4B3N3Me3D3 and (OAcF)3Me3B3N3Me3D3 (OAcF = CF3COO−) were obtained following treatment of [(Me4B3N3Me3)Cr(CO)3]MgBr or Me3B3N3Me3 with excess trifluoroacetic acid (HOAcF). The former results from the trapping of the putative intermediate (Me4B3N3Me3H)Cr(CO)3 wherein a borazine BN bond has been reduced by the sequential addition of a Me− nucleophile and a proton.27
Given the challenges associated with studying such 1,2-H2-intermediates due to their limited stability, the feasibility of these reactions was explored computationally. For these studies, we evaluated both the relative basicity of the nitrogen atoms at the 2 and 4-positions, as well as at the hydride ligand (Scheme 4). Additionally, the free energy of hydrogen elimination from the 1,2-H2-adduct was calculated in order to assess its relative stability. Not surprisingly, H2 elimination is predicted to be favored by >20 kcal mol−1 from both metal complexes. This is consistent with our experimental data which suggests that the partially saturated borazine species are highly unstable. However, the basicity of both the hydride and the 2-position nitrogen atom are predicted to be within 5 pKa units for both complexes (ΔpKa = 4.3 for Mn and 4.1 for Cr). This is consistent with the experimental observations, vide supra, and suggests that such intermediates might be transiently accessible, and could be intercepted during a catalytic regeneration scheme.
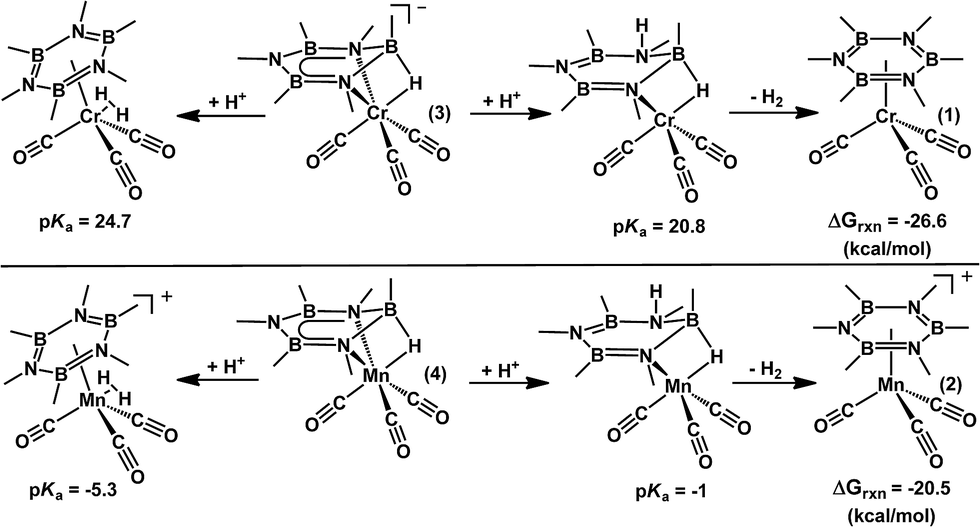 | ||
| Scheme 4 Calculated pKa values, and free energy of H2 loss, for borazine hydride adducts 3 and 4 in Et2O solution. | ||
2.4. Single electron transfer to 2 and subsequent reactivity
One of the main motivations for studying the coordination chemistry of borazines is to uncover low energy routes to generate new B–H bonds. The delivery of hydrides generated from electrons and hydrogen atoms (or protons) would be advantageous if reductions could be carried out at modest operating potentials. Complex 2 displays two reduction events at −1.19 and −1.70 V vs. FeCp2 (0.05 M [nBu4N][BAr′4]; BAr′4 = tetrakis-3,5-trifluoromethyl-phenyl borate) in 2-MeTHF, and at −1.38 and −1.88 V in 0.1 M nBu4NPF6/CH2Cl2, the first of which is quasi-reversible in both solvent systems. Prior work demonstrated that 2 eq. of Na/C10H8 and 1 eq. of HOAcF can be used to generate a hydride complex, (Me3B3N3Me3)(μ-H)Mn(CO)3 (4), from 2, through protonation of the reduced Mn center. As an alternative low-energy pathway, we targeted the 1 e− reduction of 2 as a potential intermediate en route to the hydride, 4.To explore single electron reductions of 2, we allowed 2 to react with a single equivalent of the mild reductant, Cp2Co (−1.30 V vs. Fc0/+).62 The reduction proceeded readily when 1 eq. of Cp2Co was added to a frozen 2-Me-THF solution of 2 (Fig. 1A). The resulting complex (5) was characterized by EPR spectroscopy (Fig. 1C), which revealed a rhombic spectrum (gx = 2.0238, gy = 2.0269, gz = 1.9803) with hyperfine coupling interactions to 55Mn (A55Mn = 169.7, 184.8, 223.8 mHz) and 14N (A14N = 107.8, 62.2, 119.1 mHz) nuclei. Further confirmation of single electron reduction was afforded by an Evans method analysis of a solution of 5 which revealed a μeff = 1.92, consistent with an s = 1/2 system. A solution IR spectrum of 5, revealed new νCO bands at 2011, 1944, and 1930 cm−1. The 61 cm−1 bathochromic shift of the A′ band is consistent with a Cs symmetric reduced Mn(CO)3 fragment. The lowered symmetry is likely the result of ring slippage from η6 to η4 upon reduction.63 Characterization of 5 by X-ray diffraction was precluded by its thermal instability.64 However, calculations provided insight into product structure. Importantly, a ΔνCO of 58 cm−1 was also predicted from theory, and the complex minimized to a structure featuring an η6 to η4 hapticity shift of the borazine unit. The calculated SOMO (Fig. 1B) is distributed between the Mn(CO)3 fragment and the nitrogen p-orbitals, which is consistent with the observed 14N hyperfine coupling.
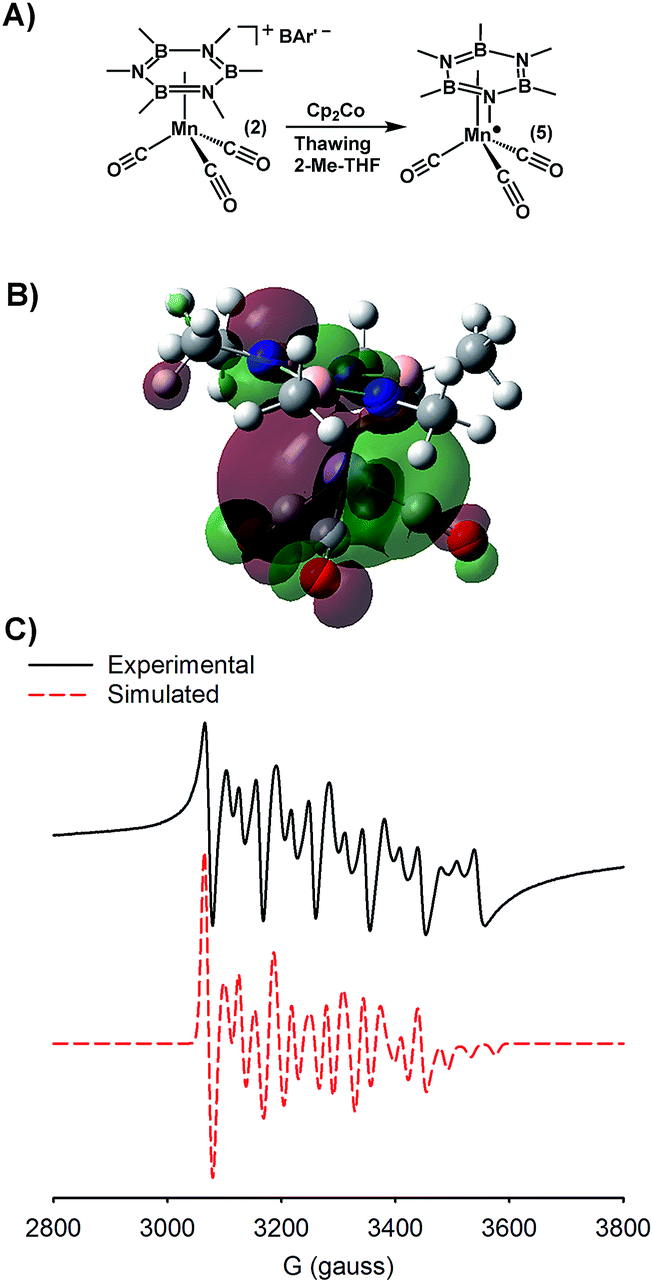 | ||
| Fig. 1 (A) Synthetic scheme for generation of reduced complex 5. (B) Calculated SOMO of 5. (C) Experimental (black) and simulated (red) X-band EPR spectra of 5. | ||
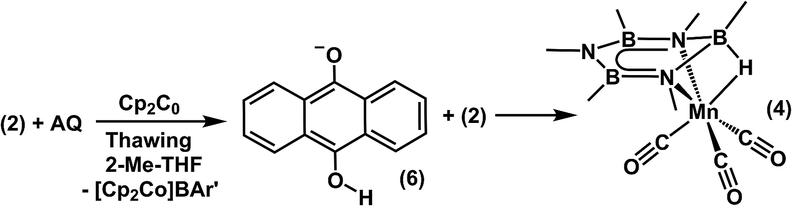
2.5. Anthraquinone mediated reduction of 2 to 4
Following the single electron reduction to afford 5, we assessed the viability of completing a hydrogen-atom transfer to the Mn–borazine unit. Quinones have been shown to act as redox mediators,65,66 and to abstract H-atoms from sacrificial sources (including solvents) when reduced under conditions that promote single electron transfer.67–69 These reactions afford reduced hydroquinone salts, that contain hydridic O–H bonds.70Based on our evaluation of hydride donor requirements for 2, we targeted a hydride reagent derived from a single equivalent of a reductant and an H-atom source. 9,10-anthraquinone (AQ) was selected because its corresponding hydroquinone salt (6) is sufficiently hydridic (37.2 kcal mol−1) to react with 2 to form 4. Consistent with our theoretical prediction of the hydricity of 4, when a mixture of AQ and 2 (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) was treated with Cp2Co (1 eq.) in thawing 2-Me-THF (Scheme 5), 4 was obtained in 90% yield. A control reaction showed no reaction occurred when combining Cp2Co with AQ in the absence of 2. This is consistent with the similar reduction potentials of Cp2Co and AQ (−1.3 vs. −1.2 V, vs. Fc0/+, respectively) in 2-Me-THF (0.05 M [nBu4N][BAr′4]). We note that the redox potential for the reduction of quinones is highly sensitive to hydrogen bonds and Lewis acid/base interactions with the quinone CO bond.71–74 Coordinated borazine likely acts as a Lewis acid in this regard, and consistent with a persistent interaction in solution, the anthraquinone wave is anodically shifted by ∼400 mV, (Fig. S12†) when 2 is also present in the solution. This redox tuning enables reduction of AQ to its corresponding semiquinone by Cp2Co. Following reduction, generation of the anthrahydroquinone salt, 6, likely results from H-atom transfer from the solvent to the semiquinone species, which can then transfer a hydride to 2.
:1) was treated with Cp2Co (1 eq.) in thawing 2-Me-THF (Scheme 5), 4 was obtained in 90% yield. A control reaction showed no reaction occurred when combining Cp2Co with AQ in the absence of 2. This is consistent with the similar reduction potentials of Cp2Co and AQ (−1.3 vs. −1.2 V, vs. Fc0/+, respectively) in 2-Me-THF (0.05 M [nBu4N][BAr′4]). We note that the redox potential for the reduction of quinones is highly sensitive to hydrogen bonds and Lewis acid/base interactions with the quinone CO bond.71–74 Coordinated borazine likely acts as a Lewis acid in this regard, and consistent with a persistent interaction in solution, the anthraquinone wave is anodically shifted by ∼400 mV, (Fig. S12†) when 2 is also present in the solution. This redox tuning enables reduction of AQ to its corresponding semiquinone by Cp2Co. Following reduction, generation of the anthrahydroquinone salt, 6, likely results from H-atom transfer from the solvent to the semiquinone species, which can then transfer a hydride to 2.
 | ||
| Scheme 5 Proposed mechanism of AQ mediated hydride reduction of 2 to 4. | ||
The anthrahydroquinone salt, 6, was independently prepared from the reaction of NaEt3BH with AQ, in order to verify its competence as a hydride donor to 2. Upon introduction of 6 to 2, complex 4 was afforded in 65% yield. No activity for reduction and/or hydride transfer was noted when 6 was replaced with p-benzoquinone, whose hydroquinone salt is a much weaker hydride donor (ΔGH− = 56.7 kcal mol−1).75 Furthermore, attempts to reduce 2 with p-benzoquinone and Cp2Co yielded none of the desired product under identical conditions. These results suggest that mixtures of 6 and 2 are thermodynamically matched to promote reduction and hydride addition.
2.6. Proposed mechanism of anthraquinone mediated hydride transfer
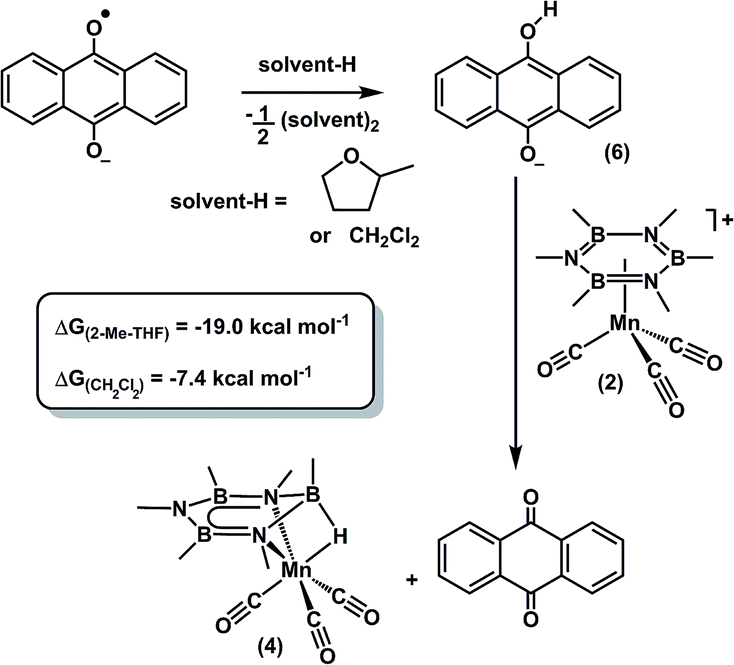
The anthrahydroquinone salt (6) that delivers a hydride to 2, vide supra, can be generated from the parent anthraquinone by either H-atom transfer (from solvent) or protonation pathways.66,67,70 To differentiate between these two limiting pathways, we evaluated whether an exogenous H-atom donor, such as solvent, participated in an H-atom transfer pathway to generate 6.66,67,70 The substitution of the 2-Me-THF solvent with C6F6, a solvent without hydrogen atoms, provided 4 in only trace amounts (<10%).76 Furthermore, the addition of 2 eq. of H2O to the reaction carried out in C6F6 also did not afford 4, which is inconsistent with adventitious water acting as the hydrogen donor. The reduction also proceeds readily (65% yield) when dichloromethane (CH2Cl2), a solvent with similar C–H BDFE values (86.4 kcal mol−1vs. 81.7 kcal mol−1 for 2-Me-THF), is substituted as the reaction solvent, suggesting that solvents with comparable BDFE values can be used as H-atom donors to drive the reaction.Calculation of the homolytic bond dissociation free energies (BDFE) for both 6 and 4, afforded values of 39.0 and 52.8 kcal mol−1 respectively, which are significantly lower than that of 2-Me-THF (81.7 kcal mol−1, Table S-4†). However, when the THF coupling product is included, the net reaction to reduce anthra-semiquinone to 6 is decreased to 12.5 kcal mol−1 (Scheme S-1†). Moreover, the reduction of 2 to 4, when using 6 as the hydride source, is exergonic by 31.5 kcal mol−1. The difference between these two reaction energies results in an overall free energy of −19 kcal mol−1 (Scheme 6). Furthermore, when identical calculations were carried out using CH2Cl2 as the H-atom donor (assuming formation of 1,1,2,2-tetrachloroethane as the radical coupling byproduct) the net reaction energy is −7.4 kcal mol−1. Thus, the products are sufficiently stable to drive the reaction forward and promote cleavage of the relatively strong solvent C–H bonds.
 | ||
| Scheme 6 Proposed net reaction, and calculated free energy, for formation of 2via AQ mediated H-atom transfer in 2-MeTHF and CH2Cl2. | ||
2.7. AQ mediated hydride reduction of 2 by applied potential
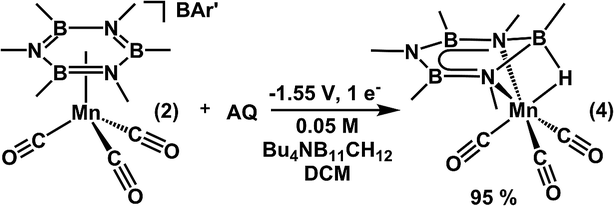
Electrochemical experiments were explored to further evaluate whether single or two electron redox pathways are operative to generate boron hydrides. The number of electron equivalents can be explicitly regulated during a controlled potential electrolysis, and thus can easily differentiate between single/two electron pathways. Furthermore, the operating potential can also be selected to coincide with a given redox event observed in the cyclic voltammogram.The selection of an appropriate solvent/electrolyte combination required careful consideration. For instance, when using [nBu4N]PF6 as the supporting electrolyte, fluorine transfer from the electrolyte was a competing side-reaction (40% yield) under the coulometry experimental conditions.77 Competitive fluorine transfer to borazine is perhaps not surprising, given the extremely high B–F bond strength (183 kcal mol−1).78 In an effort to circumvent F-transfer to the borazine, we targeted a fluorine-free solvent-electrolyte system for bulk electrolysis. Unfortunately, the high lability of the borazine unit further complicated solvent/electrolyte selection. For instance, borazine was displaced from the metal center with common electrolytes such as [nBu4N]ClO4 and [nBu4N]Cl, as well as in solvents such as tetrahydrofuran and acetonitrile. As an alternative, we turned to the [nBu4N]+ salt of mono-carborane (CB11H12−), as carborane electrolytes have been shown to be weakly coordinating anions that have high solubility and possess high electrochemical stability.79,80
Controlled potential electrolysis was conducted in a 0.05 M CH2Cl2 solution of [nBu4N][CB11H12] at −1.55 V (vs. Fc0/+). Gratifyingly, after the passage of 4.07 coulombs (0.049 mmol) to a 0.049 mmol solution of 2, 4 was obtained in 95% yield (Scheme 7) based on integration of the 11B NMR spectrum using (TMSO)3B as an internal standard (Fig. S6†). For further confirmation, the solution-cell IR spectrum was collected and 4 was the primary carbonyl containing species observed. Of particular note, the reaction yield of 95% after passing 1 eq. e− implies an operative single electron process, compared to a 2 e− process, where less than 50% yield would be expected. In agreement with our chemical reduction reactions, 4 was not observed when bulk electrolysis was conducted in the absence of 9,10-anthraquinone. Collectively, these results provide strong evidence that reduction of the borazine unit occurs through a single electron H-atom transfer pathway that is mediated by anthraquinone. The energy input required for this transformation is significantly lower than that required for a 2e−/H+ reduction sequence.
 | ||
| Scheme 7 Conversion of 2 to 4 by bulk electrolysis. | ||
3. Conclusions
Through a combination of computational and experimental studies, this study has established a low-energy pathway to reduce borazines, which are models for spent B–N fuels. Coordination of borazine to a M(CO)3 unit was found to dramatically change the requirements for hydride and proton transfer to the borazine unit. A decrease (>30 kcal mol−1) in the thermodynamic hydricity (complexes became more likely to accept a hydride) between free and coordinated borazines was imparted by coordination to Cr(CO)3 and [Mn(CO)3]+ fragments. This change in the hydride accepting ability accounted for the distinct hydride addition reactivity observed for free vs. metalated borazines. Hydricity values were also demonstrated to vary significantly with metal oxidation state. Importantly, borazine dearomatization via hydride addition was verified as the key first step in such a reduction scheme. Calculated hydricity requirements for borazine dearomatization were used to demonstrate hydride delivery from an H2-derived hydride source, and importantly, to uncover a unique hydride delivery mechanism which proceeds through a single electron reduction and H-atom transfer reaction, mediated by anthraquinone. The latter transformation was also carried out electrochemically, at relatively positive potentials by comparison to all prior reports, thus establishing an important proof of concept for any future electrochemical BN bond reduction sequences. These efforts may find utility in future regeneration schemes for spent AB fuels as well as other B–N heterocycles that are promising H2 storage molecules.
4. Experimental section
4.1. Computational details
All structures were fully optimized without symmetry constraints using either the M06-2X81 or the B3LYP82–85 functional as implemented in Gaussian 09,86 using the 6-31G** basis set.87,88 The ultrafine integration grid was employed in all calculations, which ensured the stability of the optimization procedure for the investigated molecules. Each stationary point was confirmed by a frequency calculation at the same level of theory to be a real local minimum on the potential energy surface without imaginary frequency. More accurate electronic energies were computed for the optimized geometries using the larger 6-311++G(d,p) basis set.89,90 All reported free energies are for Et2O solution at the standard state (T = 298 K, P = 1 atm, 1 mol L−1 concentration of all species in Et2O) as modelled by the polarized continuum model.91 The energy values given in the paper correspond to solvent-corrected Gibbs free energies that are based on B3LYP/6-311++G(d,p) electronic energies and all corrections calculated at the B3LYP/6-31G(d) level. All calculated pKa values are for Et2O solutions. Although no pKa scales have been experimentally determined in diethyl ether, we chose to employ [Et3NH]+ as the anchor of our pKa scale and assign it a pKa value of 12.5. A value of 12.5 was chosen because [Et3NH]+ has been previously employed as an anchor (pKa value of 12.5) for a structurally similar solvent, THF.35,36 Initial optimizations employed the M06-2X functional as the included dispersion corrections have been shown to provide better estimation of main-group systems.37,81,92 Although computations employing the M06-2X functional better modeled the thermochemistry of borazine and hexamethylborazine, all computations reported within made use of the B3LYP functional, which was found to model the manganese and chromium complexes better than the M06-2X functional. All computational vibrational frequencies reported within employed a scaling factor of 0.96.504.2. General experimental procedures
All experiments were conducted using standard Schlenk techniques or in a nitrogen filled glovebox. NMR spectra were recorded on Varian MR400, vnmrs 500, or vnmrs 700 MHz spectrometers. 1H spectra were referenced relative to the residual solvent resonance, and 11B, and 19F spectra were referenced indirectly based on the 1H spectrum.93 IR spectra were recorded on a Nicolet iS-10 spectrometer from Thermo Scientific in a KBr plate solution cell. X-band EPR spectra were recorded on a Bruker EMX ESR spectrometer equipped with a 4102-ST cavity. EPR simulations were conducted using the EasySpin software package for MATLAB.94 Mass spectra were collected by electron impact ionization on a VG 70-250-S magnetic sector, double-focusing mass spectrometer. The electron energy was set to 70 eV, and the source temperature was 240 °C. Nominal mass spectra were obtained scanning the magnet from m/z 1000 to m/z 35 at 8 seconds per decade, using an external mass calibration. Evan's method was conducted using a coaxial NMR tube insert from Wilmad glass with benzene or dichloromethane added to samples as the diamagnetic reference compound. 9,10-anthraquinone and p-benzoquinone were obtained from Sigma Aldrich and recrystallized from ethyl-acetate before use. Cobaltocene (Cp2Co) was purchased from Strem chemicals and used as received. HCo(dmpe)2 was prepared by a previously reported procedure.61 [nBu4N][CB11H12] was prepared by reacting the commercially available Cs[CB11H12] with [nBu4N]Cl in aqueous solution. The resulting precipitate was isolated by filtration and dried under vacuum at 100 °C for 48 h. (Me3B3N3Me3)Cr(CO)3 and [(Me3B3N3Me3)Mn(CO)3][BAr′4] were synthesized by previously reported routes.27,28 All solvents were passed through an S.G. Waters solvent purification system or dried and degassed by standard methods.95 All other chemicals were purchased from commercial vendors and used as received. All filtrations were conducted using a pipet blocked with Whatman glass fiber filter paper.Acknowledgements
The authors are grateful to Dr Michael Mock for a generous donation of the HCo(dmpe)2 used in this manuscript. This work was supported by the University of Michigan Department of Chemistry (NKS) and by startup funds from Washington State University (ZMH). NKS is an Alfred P. Sloan Research Fellow.Notes and references
- N. S. Lewis and D. G. Nocera, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 15729–15735 CrossRef CAS PubMed.
- M. Z. Jacobson, Energy Environ. Sci., 2009, 2, 148–173 Search PubMed.
- J. Graetz, Chem. Soc. Rev., 2009, 38, 73–82 RSC.
- Q.-L. Zhu and Q. Xu, Energy Environ. Sci., 2015, 8, 478–512 Search PubMed.
- http://www1.eere.energy.gov/vehiclesandfuels/pdfs/program/hstt_roadmap_june2013.pdf .
- Z. Huang and T. Autrey, Energy Environ. Sci., 2012, 5, 9257–9268 Search PubMed.
- W. Luo, P. G. Campbell, L. N. Zakharov and S.-Y. Liu, J. Am. Chem. Soc., 2011, 133, 19326–19329 CrossRef CAS PubMed.
- N. C. Smythe and J. C. Gordon, Eur. J. Inorg. Chem., 2010, 2010, 509–521 CrossRef.
- A. Staubitz, A. P. M. Robertson and I. Manners, Chem. Rev., 2010, 110, 4079–4124 CrossRef CAS PubMed.
- F. H. Stephens, V. Pons and R. Tom Baker, Dalton Trans., 2007, 2613–2626 RSC.
- R. J. Keaton, J. M. Blacquiere and R. T. Baker, J. Am. Chem. Soc., 2007, 129, 1844–1845 CrossRef CAS PubMed.
- T. Wideman, P. J. Fazen, A. T. Lynch, K. Su, E. E. Remsen, L. G. Sneddon, T. Chen and R. T. Paine, in Inorganic Syntheses, John Wiley & Sons, Inc., 2007, pp. 232–242, DOI:10.1002/9780470132630.ch39.
- J.-M. Yan, X.-B. Zhang, S. Han, H. Shioyama and Q. Xu, Angew. Chem., Int. Ed., 2008, 47, 2287–2289 CrossRef CAS PubMed.
- E. K. Mellon and J. J. Lagowski, Advances in inorganic chemistry and radiochemistry, Academic Press., New York, 1959 Search PubMed.
- E. L. Muetterties, Boron hydride chemistry, Academic Press, New York, 1975 Search PubMed.
- R. Boese, A. H. Maulitz and P. Stellberg, Chem. Ber., 1994, 127, 1887–1889 CrossRef CAS.
- R. Islas, E. Chamorro, J. Robles, T. Heine, J. Santos and G. Merino, Struct. Chem., 2007, 18, 833–839 CrossRef CAS.
- A. S. Lisovenko and A. Y. Timoshkin, Inorg. Chem., 2010, 49, 10357–10369 CrossRef CAS PubMed.
- M. H. Matus, K. D. Anderson, D. M. Camaioni, S. T. Autrey and D. A. Dixon, J. Phys. Chem. A, 2007, 111, 4411–4421 CrossRef CAS PubMed.
- C. R. Miranda and G. Ceder, J. Chem. Phys., 2007, 126, 184703 CrossRef PubMed.
- P. V. Ramachandran and P. D. Gagare, Inorg. Chem., 2007, 46, 7810–7817 CrossRef CAS PubMed.
- B. L. Davis, D. A. Dixon, E. B. Garner, J. C. Gordon, M. H. Matus, B. Scott and F. H. Stephens, Angew. Chem., Int. Ed., 2009, 48, 6812–6816 CrossRef CAS PubMed.
- A. D. Sutton, A. K. Burrell, D. A. Dixon, E. B. Garner, J. C. Gordon, T. Nakagawa, K. C. Ott, J. P. Robinson and M. Vasiliu, Science, 2011, 331, 1426–1429 CrossRef CAS PubMed.
- C. Reller and F. O. R. L. Mertens, Angew. Chem., Int. Ed., 2012, 51, 11731–11735 CrossRef CAS PubMed.
- O. T. Summerscales and J. C. Gordon, Dalton Trans., 2013, 42, 10075–10084 RSC.
- E. M. Leitao and I. Manners, Eur. J. Inorg. Chem., 2015, 13, 2199–2205 CrossRef.
- T. J. Carter, J. W. Kampf and N. K. Szymczak, Angew. Chem., Int. Ed., 2012, 51, 13168–13172 CrossRef CAS PubMed.
- T. J. Carter, J. Y. Wang and N. K. Szymczak, Organometallics, 2014, 33, 1540–1543 CrossRef CAS.
- P. G. Campbell, L. N. Zakharov, D. J. Grant, D. A. Dixon and S. Y. Liu, J. Am. Chem. Soc., 2010, 132, 3289–3291 CrossRef CAS PubMed.
- Y. Tan and X. Yu, RSC Adv., 2013, 3, 23879–23894 RSC.
- W. R. Nutt and M. L. McKee, Inorg. Chem., 2007, 46, 7633–7645 CrossRef CAS PubMed.
- S. Bhunya, P. M. Zimmerman and A. Paul, ACS Catal., 2015, 5, 3478–3493 CrossRef CAS.
- H. S. Kang, J. Phys. Chem. A, 2005, 109, 1458–1467 CrossRef CAS PubMed.
- A. J. Bridgeman, Polyhedron, 1998, 17, 2279–2288 CrossRef CAS.
- K. Abdur-Rashid, T. P. Fong, B. Greaves, D. G. Gusev, J. G. Hinman, S. E. Landau, A. J. Lough and R. H. Morris, J. Am. Chem. Soc., 2000, 122, 9155–9171 CrossRef CAS.
- T. Rodima, I. Kaljurand, A. Pihl, V. Maemets, I. Leito and I. Koppel, J. Org. Chem., 2002, 67, 1873–1881 CrossRef CAS PubMed.
- L. Goerigk and S. Grimme, Phys. Chem. Chem. Phys., 2011, 13, 6670–6688 RSC.
- R. Schellenberg, J. Kriehme and G. Wolf, Thermochim. Acta, 2007, 457, 103–108 CrossRef CAS.
- Z. Chen, C. S. Wannere, C. Corminboeuf, R. Puchta and P. V. R. Schleyer, Chem. Rev., 2005, 105, 3842–3888 CrossRef CAS PubMed.
- P. V. R. Schleyer, B. Kiran, D. V. Simion and T. S. Sorensen, J. Am. Chem. Soc., 2000, 122, 510–513 CrossRef CAS.
- C. Corminboeuf, T. Heine and J. Weber, Phys. Chem. Chem. Phys., 2003, 5, 246–251 RSC.
- A. Velian and C. C. Cummins, Science, 2015, 348, 1001–1004 CrossRef CAS PubMed.
- N. D. Charistos, A. G. Papadopoulos and M. P. Sigalas, J. Phys. Chem. A, 2014, 118, 1113–1122 CrossRef CAS PubMed.
- Werner and co-workers have characterized a Cr(CO)3 complex of H3B3N3Me3 through ring metathesis, however, no structural data or reactivity studies for this complex have been reported.
- K. Deckelmann and H. Werner, Helv. Chim. Acta, 1970, 53, 139–141 CrossRef.
- K. Deckelmann and H. Werner, Helv. Chim. Acta, 1971, 54, 2189–2193 CrossRef CAS.
- J. J. Lagowski, Coord. Chem. Rev., 1977, 22, 185–194 CrossRef CAS.
- R. Prinz and H. Werner, Angew. Chem., Int. Ed., 1967, 6, 91–92 CrossRef CAS.
- H. Werner, R. Prinz and E. Deckelmann, Chem. Ber., 1969, 102, 95–103 CrossRef CAS.
- M. P. Andersson and P. Uvdal, J. Phys. Chem. A, 2005, 109, 2937–2941 CrossRef CAS PubMed.
- P. J. Dyson, Dalton Trans., 2003, 2964–2974 RSC.
- L. Foppa and J. Dupont, Chem. Soc. Rev., 2015, 44, 1886–1897 RSC.
- M. P. Boone and D. W. Stephan, J. Am. Chem. Soc., 2013, 135, 8508–8511 CrossRef CAS PubMed.
- M. P. Boone and D. W. Stephan, Chem.– Eur. J., 2014, 20, 3240 CrossRef CAS.
- T. Mahdi, Z. M. Heiden, S. Grimme and D. W. Stephan, J. Am. Chem. Soc., 2012, 134, 4088–4091 CrossRef CAS PubMed.
- Strong acids with nucleophillic anions (e.g. HCl) react readily with borazines through concerted addition. In our hands, Brookhart's acid (HBAr′4) and HCl:AlCl3 did not produce a reaction with borazine suggesting that protonated ether (pKa = -9.7) is not sufficiently acidic to protonate borazine. To our knowledge, the only example of non-concerted borazine protonation was reported by Noth and co-workers, who used the superacid HBr:AlBr3 in non-polar solvent. See: B. Gemünd, B. Günther and H. Nöth, ARKIVOC, 2008, 136 Search PubMed.
- Ullmann's Fine Chemicals, ed. B. Elvers, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, Germany, 2014, vol. 3 Search PubMed.
- Z. M. Heiden and A. P. Lathem, Organometallics, 2015, 34, 1818–1827 CrossRef CAS.
- C. J. Curtis, A. Miedaner, W. W. Ellis and D. L. DuBois, J. Am. Chem. Soc., 2002, 124, 1918–1925 CrossRef CAS PubMed.
- A. J. Price, R. Ciancanelli, B. C. Noll, C. J. Curtis, D. L. DuBois and M. R. DuBois, Organometallics, 2002, 21, 4833–4839 CrossRef CAS.
- M. T. Mock, R. G. Potter, M. J. O'Hagan, D. M. Camaioni, W. G. Dougherty, W. S. Kassel and D. L. DuBois, Inorg. Chem., 2011, 50, 11914–11928 CrossRef CAS PubMed.
- N. G. Connelly and W. E. Geiger, Chem. Rev., 1996, 96, 877–910 CrossRef CAS PubMed.
- Attempts to verify the geometry of this species by paramagnetic 1H NMR were unsuccessful as no signals were observed for complex 5.
- Slow decomposition was noted even when solutions of 5 were held at −78 °C.
- X. Guo, H. Zipse and H. Mayr, J. Am. Chem. Soc., 2014, 136, 13863–13873 CrossRef CAS PubMed.
- N. Song, C. J. Gagliardi, R. A. Binstead, M.-T. Zhang, H. Thorp and T. J. Meyer, J. Am. Chem. Soc., 2012, 134, 18538–18541 CrossRef CAS PubMed.
- X. Ci, R. Silveira da Silva, D. Nicodem and D. G. Whitten, J. Am. Chem. Soc., 1989, 111, 1337–1343 CrossRef CAS.
- C. Rüchardt, M. Gerst and J. Ebenhoch, Angew. Chem., Int. Ed., 1997, 36, 1406–1430 CrossRef.
- F. Wurche, W. Sicking, R. Sustmann, F.-G. Klärner and C. Rüchardt, Chem.– Eur. J., 2004, 10, 2707–2721 CrossRef CAS PubMed.
- X.-Q. Zhu and C.-H. Wang, J. Org. Chem., 2010, 75, 5037–5047 CrossRef CAS PubMed.
- J. Yuasa, T. Suenobu and S. Fukuzumi, Chem. Phys. Chem., 2006, 7, 942–954 CrossRef CAS PubMed.
- G. T. Cheek, ECS Trans., 2013, 50, 79–85 CrossRef.
- J. Q. Chambers, in The Quinonoid Compounds (1988), John Wiley & Sons, Inc., 2010, pp. 719–757, DOI:10.1002/9780470772119.ch12.
- R. S. Kim and T. D. Chung, Bull. Korean Chem. Soc., 2014, 35, 3143–3155 CrossRef CAS.
- We computationally investigated the possibility of dative bonding between AQ and 4. The resulting complex of 4 and AQ is predicted to have a hydide donor abilitiy of 37 kcal mol−1. See ESI† for additional details.
- The C–H bonds of borazine are also possible H-atom donors, which can account for the 10% yield observed in C6F6.
- See ESI† for additional details.
- J. A. Dean and N. A. Lange, Lange's handbook of chemistry, McGraw-Hill, 1992 Search PubMed.
- T. J. Carter, R. Mohtadi, T. S. Arthur, F. Mizuno, R. Zhang, S. Shirai and J. W. Kampf, Angew. Chem., Int. Ed., 2014, 53, 3173–3177 CrossRef CAS PubMed.
- J. W. Johnson and A. H. Thompson, J. Electrochem. Soc., 1981, 128, 932–933 CrossRef CAS.
- Y. Zhao and D. G. Truhlar, J. Chem. Theory Comput., 2008, 4, 1849–1868 CrossRef CAS PubMed.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS.
- C. Lee, W. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785–789 CrossRef CAS.
- S. H. Vosko, L. Wilk and M. Nusair, Can. J. Phys., 1980, 58, 1200–1211 CrossRef CAS.
- P. J. Stephens, F. J. Devlin, C. F. Chabalowski and M. J. Frisch, J. Phys. Chem., 1994, 98, 11623–11627 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr., J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, T. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Revision D.01, Gaussian, Inc., Wallingford, CT, 2013 Search PubMed.
- J. D. Dill and J. A. Pople, J. Chem. Phys., 1975, 62, 2921–2923 CrossRef CAS.
- W. J. Hehre, R. Ditchfield and J. A. Pople, J. Chem. Phys., 1972, 56, 2257–2261 CrossRef CAS.
- R. Krishnan, J. S. Binkley, R. Seeger and J. A. Pople, J. Chem. Phys., 1980, 72, 650–654 CrossRef CAS.
- M. M. Francl, W. J. Pietro, W. J. Hehre, J. S. Binkley, M. S. Gordon, D. J. DeFrees and J. A. Pople, J. Chem. Phys., 1982, 77, 3654–3665 CrossRef CAS.
- M. Cossi, N. Rega, G. Scalmani and V. Barone, J. Comput. Chem., 2003, 24, 669–681 CrossRef CAS PubMed.
- Y. Zhao and D. G. Truhlar, Theor. Chem. Acc., 2008, 120, 215–241 Search PubMed.
- R. K. Harris, E. D. Becker, S. M. C. D. Menezes, R. Goodfellow and P. Granger, Pure Appl. Chem., 2001, 73, 1795–1818 CrossRef CAS.
- S. Stoll and A. Schweiger, J. Magn. Reson., 2006, 178, 42 CrossRef CAS PubMed.
- W. L. F. Armarego and C. L. L. Chai, Knovel and ScienceDirect, Purification of Laboratory Chemicals, Elsevier/Butterworth-Heinemann, Amsterdam, Boston, 2009 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Spectral and electrochemical data and computational details. See DOI: 10.1039/c5sc02348c |
| This journal is © The Royal Society of Chemistry 2015 |