
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Chiral ion-pair organocatalyst promotes highly enantioselective 3-exo iodo-cycloetherification of allyl alcohols†
Zhigao
Shen‡
ab,
Xixian
Pan‡
c,
Yisheng
Lai‡
b,
Jiadong
Hu
b,
Xiaolong
Wan
d,
Xiaoge
Li
d,
Hui
Zhang
e and
Weiqing
Xie
*a
aShaanxi Key Laboratory of Natural Products & Chemical Biology, College of Science, Northwest A&F University, 22 Xinong Road, Yangling 712100, China. E-mail: xiewqsioc@aliyun.com
bState Key Laboratory of Natural Medicines, Jiangsu Key Laboratory of Drug Discovery for Metabolic Diseases, Center of Drug Discovery, China Pharmaceutical University, 24 Tongjiaxiang, Nanjing 210009, China
cHubei Collaborative Innovation Center for Rare Metal Chemistry, Hubei Normal University, China
dShanghai Institute of Organic Chemistry, Chinese Academy of Sciences, China
eSchool of Science & Laboratory for Microstructures, Instrumental Analysis and Research Center, Shanghai University, China
First published on 27th August 2015
Abstract
By designing a novel chiral ion-pair organocatalyst composed of chiral phosphate and DABCO-derived quaternary ammonium, highly enantioselective 3-exo iodo-cycloetherification of allyl alcohols was achieved using NIS as a halogen source. Based on this reaction, one-pot asymmetric 3-exo iodo-cycloetherification/Wagner–Meerwein rearrangement of allyl alcohols en route to enantioenriched 2-iodomethyl-2-aryl cycloalkanones was subsequently developed. Due to the participation of adjacent iodine, the Wagner–Meerwein rearrangement of 2-iodomethyl-2-aryl epoxide proceeds with unusual retention of stereoconfiguration.
Halogenative functionalization of olefins is one of the most important transformations in organic synthesis, as it not only provides a versatile handle for further derivatization, but also delivers highly diastereoselective ring closure when the nucleophile and alkene are tethered together.1 Even though applications of halogenation reactions in total synthesis are well documented,2 catalytic enantioselective halogenation remains a significant challenge due to the rapid interexchange of the halonium complex between olefins, which leads to rapid racemization of the enantiopure halonium intermediate.3 Therefore, limited success has been achieved, despite enormous efforts being devoted to asymmetric halogenation reactions.4 Very recently, there has been impressive progress in this field after the landmark reports of Borhan,5a Tang,5b Fujioka,5c Jacobsen,5d and Yeung5e in 2010, taking advantage of organocatalysts to effect asymmetric halo-lactonization.5 Organocatalyzed enantioselective halocyclization of olefinic amines, alcohols and other substrates subsequently emerged.6–9 However, asymmetric halocyclization reactions are currently limited to the formation of four- to six-membered rings.5–9 The generation of enantioenriched, more strained three-membered rings via catalytic asymmetric halocyclization remains elusive. In this regard, although 3-exo halo-cycloetherification of allyl alcohols has long been known,10 reactive halogenating agents or harsh reaction conditions are needed to effect the energetically disfavored 3-exo halocyclization, which impedes the development of an asymmetric version of this reaction.
With the advent and booming of organocatalysis,11a–c ion-pairing of organocatalysts has emerged as a powerful strategy for designing new efficient organocatalysts.11d By cooperatively activating reactive partners, ion-pair catalysts have catalyzed enantioselective reactions that are otherwise difficult to achieve using other organocatalysts. In addition, the ion-pairing strategy also enables catalyst screening via combinational approaches, which greatly accelerates the catalyst screening process. Inspired by Toste's recent work8b–f and our work on enantioselective halogenation reactions using chiral anionic phase transfer catalysts,12 we postulated that an ion-pair catalyst could facilitate the enantioselective halogenation reaction by cooperative and synergistic activation of both reactants (Fig. 1), which has been responsible for the success of previous catalysts.5–9 To this end, chiral phosphate was judiciously chosen as counter anion for its fine-tunable chiral pocket as well as its Brønsted basicity to allow interaction with the substrate.8 Furthermore, DABCO-derived quaternary ammonium could serve as an excellent candidate for the cation moiety, since its tertiary amine moiety could act as a Lewis base to stabilize the halonium complex, an approach which has been utilized for the synthesis of well-known Selectfluor13 and other halogenating reagents.8d,9c,10b
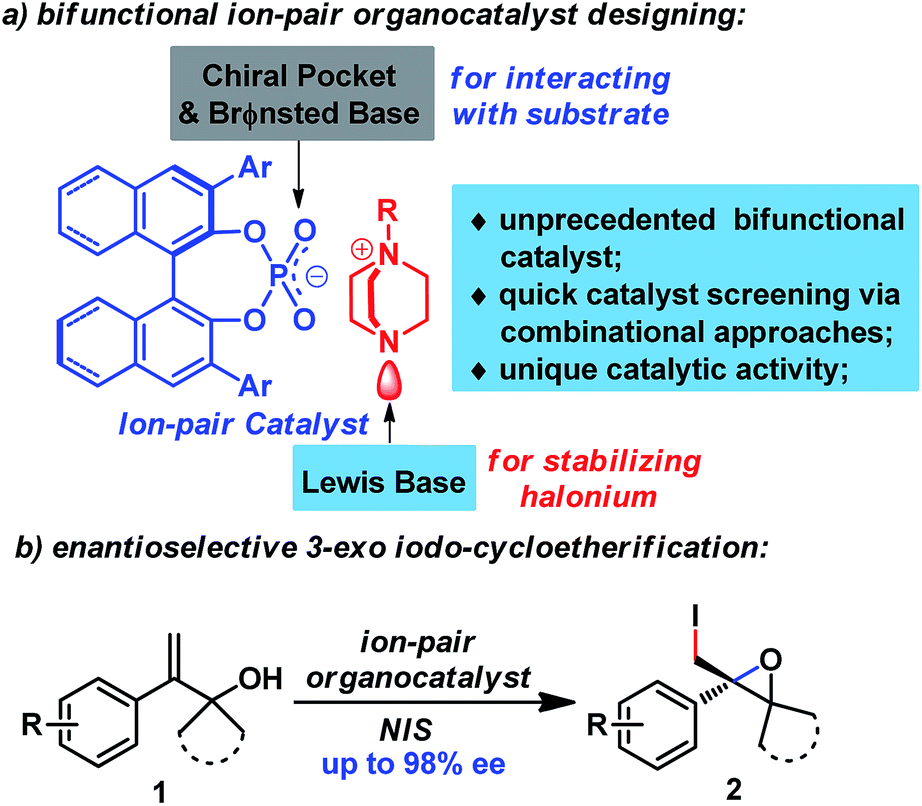 | ||
| Fig. 1 Ion-pair organocatalyst design for enantioselective 3-exo iodo-cycloetherification of allyl alcohols. | ||
Herein, we would like to report the success of implementation of the ion-paring strategy, leading to the discovery of a novel ion-pair organocatalyst. This unprecedented organocatalyst enables the first enantioselective 3-exo iodo-cycloetherification of allyl alcohols using commercially available NIS as a halogen source. Additionally, this protocol provides direct access to enantiopure 2-iodomethyl epoxides,14 which have previously been tedious to prepare from allyl alcohols, requiring an asymmetric Sharpless epoxidation/hydroxyl transformation procedure.15
To validate our hypothesis, enantioselective 3-exo-iodocyclization of allyl alcohol 1a was explored using an ion-pair organocatalyst generated in situ by combining silver phosphate with DABCO-derived quaternary ammonium salt for convenience of catalyst screening (Table 1). Initially, various ammonium salts were evaluated using 8H-R-TRIP-OAg L1 as a chiral counter-anion source. After extensive screening, A3 was determined to be a privileged scaffold, affording epoxide 2a with 77% ee in moderate yield (entries 1–3 and ESI†). In contrast, ammonium salt A2 derived from quinuclidine provided lower enantioselectivity, showing that the tertiary amine moiety of A1 played a pivotal role in the reaction (entries 1 and 2). Further structural modification of ammonium salt A3 revealed that A8 was the optimal cation fragment for the ion-pair organocatalyst, furnishing epoxide 2a with 92% ee (entries 3–9). As for the anion fragment, 8H-R-TRIP-OAg provided a better result than any other chiral silver phosphate evaluated (see ESI†). Importantly, both cationic and anionic fragments were indispensable for the reaction, as indicated by control experiments (entries 10–12). It should be pointed out that other organocatalysts (e.g. chiral phosphoric acid and quinine-derived catalysts) were also surveyed under identical reaction conditions but gave no desired cyclization product, with the starting material being fully recovered (Table S2, ESI†).
|
|
||||||
|---|---|---|---|---|---|---|
| Entry | Cat. (equiv.) | Additive (equiv.) | T (°C) | t (h) | Yieldb (%) | eec (%) |
| a CH2Cl2 (1 mL) was added to a mixture of silver salt L1 (0.01 mmol), ammonium salt A (0.012 mmol) and NIS (0.12 mmol), and the reaction mixture was cooled to 0 °C. Allyl alcohol 1a (0.1 mmol) in 0.5 mL CH2Cl2 was then added dropwise, and the reaction was quenched at the indicated time. b Isolated yield. c Determined by HPLC using a Chiralpak AD column. d CHCl3 as solvent. e EtOAc as solvent. ND = not detected. | ||||||
| 1 | L1 (0.1) | A1 (0.12) | 0 | 40 | 16 | 30 |
| 2 | L1 (0.1) | A2 (0.12) | 0 | 40 | 18 | 19 |
| 3 | L1 (0.1) | A3 (0.12) | 0 | 40 | 44 | 77 |
| 4 | L1 (0.1) | A4 (0.12) | 0 | 40 | 16 | 69 |
| 5 | L1 (0.1) | A5 (0.12) | 0 | 40 | 69 | 86 |
| 6 | L1 (0.1) | A6 (0.12) | 0 | 40 | 47 | 80 |
| 7 | L1 (0.1) | A7 (0.12) | 0 | 40 | 65 | 91 |
| 8 | L1 (0.1) | A8 (0.12) | 0 | 40 | 60 | 92 |
| 9 | L1 (0.1) | A9 (0.12) | 0 | 40 | 50 | 91 |
| 10 | — | — | 0 | 40 | ND | — |
| 11 | L1 (0.1) | — | 0 | 40 | ND | — |
| 12 | — | A8 (0.12) | 0 | 40 | ND | — |
| 13 | C1 (0.1) | — | 0 | 40 | 42 | 83 |
| 14 | C1 (0.1) | A8 (0.1) | 0 | 40 | 82 | 92 |
| 15 | C1 (0.1) | S![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) PPh3 (0.1) PPh3 (0.1) |
0 | 40 | 63 | 90 |
| 16d | C1 (0.1) | A8 (0.1) | 0 | 40 | 62 | 69 |
| 17e | C1 (0.1) | A8 (0.1) | 0 | 40 | 31 | 67 |
| 18 | C1 (0.1) | A8 (0.1) | −20 | 107 | 99 | 94 |

With the optimal anionic and cationic moiety of the catalyst identified, ion-pair organocatalyst C1 was synthesized directly from 8H-R-TRIP and ammonium A8 (see ESI†) and examined under otherwise identical reaction conditions. To our surprise, 2a was obtained with only moderate enantioselectivity (83% ee, entry 13). As a slight excess of A8 was used in the in situ procedure, we reasoned that A8 might be an effective promoter for this reaction. Indeed, comparable enantioselectivity (92% ee, entry 14) was obtained by adding a catalytic amount of A8 to the reaction. It is postulated that A8 might act as a Lewis base to stabilize the iodonium intermediate8d and facilitate the transfer of iodine from NIS to the DABCO moiety of the ion-pair organocatalyst, leading to an acceleration of the reaction rate and increased enantioselectivity. Employing SPPh3 (ref. 7c and e) as an additive also gave a comparable result, verifying the positive effect of a Lewis base as co-catalyst in this reaction (entry 15). With a suitable catalyst in hand, other reaction variations were subsequently evaluated. Other halogenating reagents such as NCS and NBS gave inferior results, leading to no reaction or a sharp drop in enantioselectivity (see ESI†). CH2Cl2 was determined to be the optimal solvent (entries 16, 17 and ESI†), and lowering the reaction temperature to −20 °C was beneficial for the reaction (entry 18).
After establishing the optimal reaction conditions, the substrate scope of this reaction was examined (Scheme 1). Both electron-withdrawing groups (2aa–2af and 2ce-2cf) and electron-donating groups (2ag-2ah and 2ca–2ch) on the phenyl moiety were tolerated, affording the corresponding epoxides with good to excellent enantioselectivities (87% to 99% ee). Gem-substituents were crucial for the reaction, as 2f lacking gem-substituents was obtained in only 41% yield and 63% ee. Epoxides with cyclic gem-substituents were obtained with higher enantioselectivities (2c-2ch and ESI†) than those with acyclic gem-substituents (2a and 2b). A 2-alkyl substituted allyl alcohol was also smoothly converted to epoxide 2g, albeit with low enantioselectivity (37% ee). Furthermore, gram syntheses of epoxides 2a and 2c–2e were also smoothly realized by using 5 mol% C1 without affecting enantioselectivities, and the catalyst loading could even be reduced to 1 mol% affording comparable results (Scheme 1 and ESI†). The absolute configuration of epoxide 2 was determined to be R based on X-ray crystallographic analysis of epoxide 2ac,16 which was confirmed by vibrational circular dichroism (VCD) studies of epoxide 2c.17
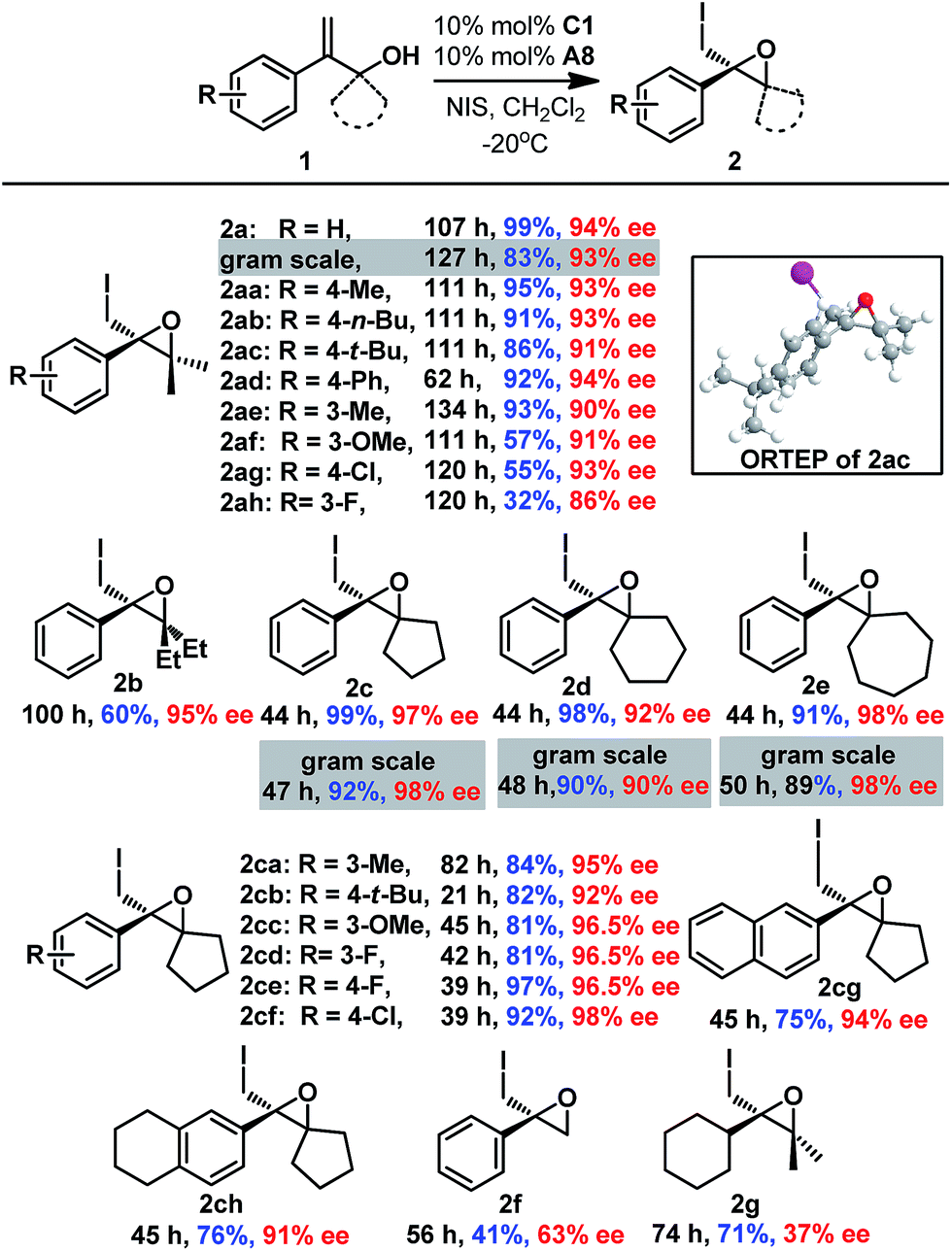 | ||
| Scheme 1 Substrate variation in the enantioselective 3-exo iodo-cycloetherification of allyl alcohols. | ||
Next, Wagner–Meerwein rearrangement18 of epoxide 2c was explored for the construction of 2-iodomethyl-2-aryl cyclohexanones with a chiral quaternary carbon center (Scheme 2). BF3·Et2O was determined to be the most efficient promoter (see ESI†), delivering cyclohexanone 3c in good yield with partial loss of enantioselectivity (93% ee vs. 97% ee for epoxide 2c). Surprisingly, the absolute configuration of 3c was established to be S by X-ray crystallographic analysis of hydrazone 4 derived from 3c,16 which indicated retention of stereoconfiguration in the Wagner–Meerwein rearrangement. This could be ascribed to the opening of the epoxide by the adjacent iodine to generate iodonium TS2, which then rearranged to ketone 3c with double inversion of configuration. Furthermore, derivatizations of 3c were also performed to demonstrate its synthetic utility. Substitution of the iodide with NaN3 provided azide ketone 5 smoothly, and the iodide could also be converted to an alcohol via formyloxylation/hydrolysis19 to give hydroxyl ketone 6 in satisfactory yield. It is noteworthy that no erosion of enantiopurity was detected in all these reactions.
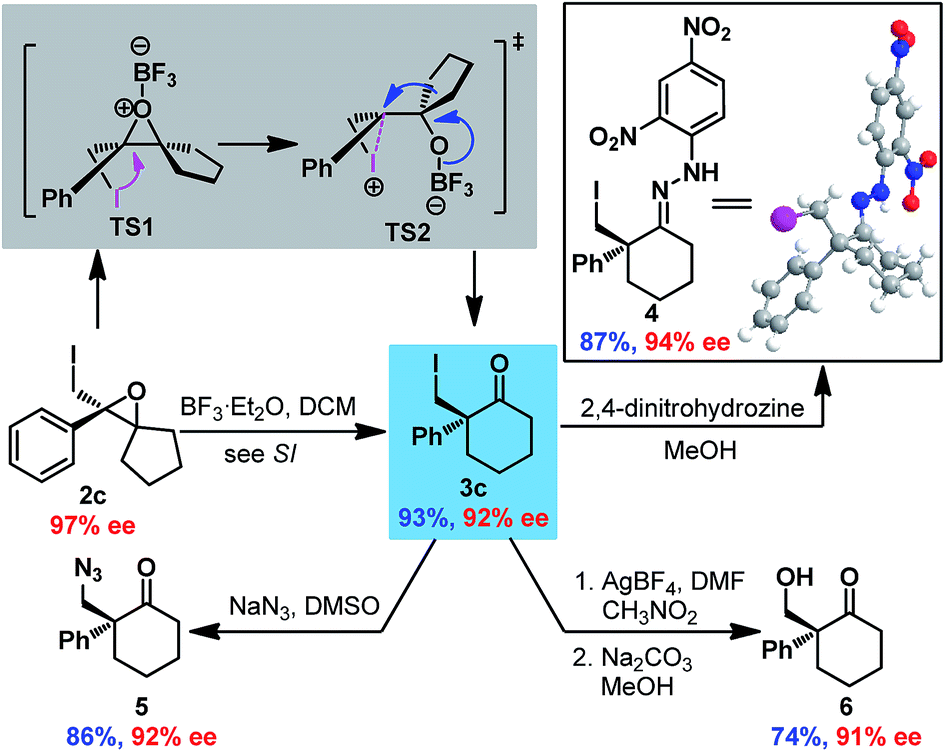 | ||
| Scheme 2 Transformations of spiro-epoxide 2c. | ||
To simplify the operation, one-pot asymmetric 3-exo iodo-cycloetherification/Wagner–Meerwein rearrangement was also developed (Scheme 3). Fortunately, when the iodo-cycloetherification reaction was completed, addition of BF3·OEt2 to the reaction mixture smoothly provided the desired cyclohexanone 3c without reducing enantioselectivity, even on a 2.7 mmol scale (92% ee). Different substituents on the phenyl group were found to be compatible with the one-pot process, affording the corresponding cyclohexanones 3c–3f in satisfactory enantiopurities. Furthermore, seven-membered cycloketone 3g could also be obtained via this one-pot cascade reaction with 91% ee (comparable with that of the corresponding epoxide 2d), providing a complementary route to previous protocols involving enantioselective halonium-induced semi-Pinacol rearrangement for the enantioselective construction of halogenated cycloheptanones.9a–e
 | ||
| Scheme 3 One-pot asymmetric 3-exo iodo-cycloetherification/Wagner–Meerwein rearrangement reaction. | ||
Conclusions
In conclusion, a novel ion-pair organocatalyst comprised of chiral phosphate and DABCO-derived quaternary ammonium was designed, which enabled the first asymmetric 3-exo iodo-cycloetherification of allyl alcohols using NIS as a halogenating reagent. By employing this novel catalyst, a variety of enantiopure 2-iodomethyl-2-aryl epoxides were successively prepared with good to excellent enantioselectivities, even on a gram scale. Subsequently, one-pot asymmetric 3-exo iodo-cycloetherification/Wagner–Meerwein rearrangement of 2-aryl-2-propen-3-ol was explored, which provided direct access to chiral 2-iodomethyl-2-aryl cycloalkanones with good enantioselectivities. Unusual retention of configuration owing to the assistance of the adjacent iodide was also observed in the Wagner–Meerwein rearrangement.Acknowledgements
We are grateful for financial support from the National Natural Science Foundation of China (grant No. 21372239, 21202187) and the Scientific Research Foundation of Northwest A&F University (grant No. Z111021501).Notes and references
- (a) F. A. Carey and R. J. Sundberg, Advanced Organic Chemistry, Part B, 5th edn, Plenum, New York, 2007, p. 298 Search PubMed; (b) R. C. Larock, Comprehensive Organic Transformations, 2nd edn, Wiley-VCH, New York, 1999, p. 638 Search PubMed.
- For selected recent examples, see: (a) D. K. Bedke, G. M. Shibuya, A. Pereira, W. H. Gerwick, T. H. Haines and C. D. Vanderwal, J. Am. Chem. Soc., 2009, 131, 7570 CrossRef CAS PubMed; (b) S. A. Snyder, Z.-Y. Tang and R. J. Gupta, J. Am. Chem. Soc., 2009, 131, 5744 CrossRef CAS PubMed; (c) C. Nilewski, R. W. Geisser and E. M. Carreira, Nature, 2009, 457, 573 CrossRef CAS PubMed.
- (a) R. S. Brown, R. W. Nagorski, A. J. Bennet, R. E. D. McClung, G. H. M. Aarts, M. Klobukowski, R. McDonald and B. D. Santarsiero, J. Am. Chem. Soc., 1994, 116, 2448 CrossRef CAS; (b) A. A. Neverov and R. S. Brown, J. Org. Chem., 1996, 61, 962 CrossRef CAS; (c) R. S. Brown, Acc. Chem. Res., 1997, 30, 131 CrossRef CAS; (d) S. E. Denmark, M. T. Burk and A. J. Hoover, J. Am. Chem. Soc., 2010, 132, 1232 CrossRef CAS PubMed.
- For selected examples, see: (a) I. Sakurada, S. Yamasaki, R. Gottlich, T. Iida, M. Kanai and M. Shibasaki, J. Am. Chem. Soc., 2000, 122, 1245 CrossRef CAS; (b) A. K. El-Qisairi, H. A. Qaseer, G. Katsigras, P. Lorenzi, U. Trivedi, T. S. Tracz, A. Hartman, J. A. Miller and P. M. Henry, Org. Lett., 2003, 5, 439 CrossRef CAS PubMed; (c) S. H. Kang, S. B. Lee and C. M. Park, J. Am. Chem. Soc., 2003, 125, 15748 CrossRef CAS PubMed; (d) A. Sakakura, A. Ukai and K. Ishihara, Nature, 2007, 445, 900 CrossRef CAS.
- For enantioselective halo-lactonization reactions, see: (a) D. C. Whitehead, R. Yousefi, A. Jaganathan and B. Borhan, J. Am. Chem. Soc., 2010, 132, 3298 CrossRef CAS PubMed; (b) W. Zhang, S. Zheng, N. Liu, J. B. Werness, I. A. Guzei and W. Tang, J. Am. Chem. Soc., 2010, 132, 3664 CrossRef CAS PubMed; (c) K. Murai, T. Matsushita, A. Nakamura, S. Fukushima, M. Shimura and H. Fujioka, Angew. Chem., Int. Ed., 2010, 49, 9174 CrossRef CAS PubMed; (d) G. E. Veitch and E. N. Jacobsen, Angew. Chem., Int. Ed., 2010, 49, 7332 CrossRef CAS PubMed; (e) L. Zhou, C. K. Tan, X. Jiang, F. Chen and Y.-Y. Yeung, J. Am. Chem. Soc., 2010, 132, 15474 CrossRef CAS PubMed; (f) M. C. Dobish and J. N. Johnston, J. Am. Chem. Soc., 2012, 134, 6068 CrossRef CAS PubMed; (g) X. Jiang, C. K. Tan, L. Zhou and Y.-Y. Yueng, Angew. Chem., Int. Ed., 2012, 51, 7771 CrossRef CAS PubMed; (h) D. H. Paull, C. Fang, J. R. Donald, A. D. Pansick and S. F. Martin, J. Am. Chem. Soc., 2012, 134, 11128 CrossRef CAS PubMed; (i) K. Ikeuchi, S. Ido, S. Yoshimura, T. Asakawa, M. Inai, Y. Hamashima and T. Kan, Org. Lett., 2012, 14, 6016 CrossRef CAS PubMed; (j) M. Wilking, C. Mück-Lichtenfeld, C. G. Daniliuc and U. Hennecke, J. Am. Chem. Soc., 2013, 135, 8133 CrossRef CAS PubMed.
- For reviews, see: (a) A. Castellanos and S. P. Fletcher, Chem.–Eur. J., 2011, 17, 5766 CrossRef CAS PubMed; (b) C. K. Tan, L. Zhou and Y.-Y. Yeung, Synlett, 2011, 1335 CAS; (c) U. Hennecke, Chem.–Asian J., 2012, 7, 456 CrossRef CAS PubMed; (d) S. E. Denmark, W. E. Kuester and M. T. Burk, Angew. Chem., Int. Ed., 2012, 51, 10938 CrossRef CAS PubMed; (e) C. K. Tan and Y.-Y. Yeung, Chem. Commun., 2013, 49, 7985 RSC; (f) K. Brak and E. N. Jacobsen, Angew. Chem., Int. Ed., 2013, 52, 534 CrossRef CAS; (g) J. Chen and L. Zhou, Synthesis, 2014, 46, 586 CrossRef.
- For halo-cycloetherification reactions, see: (a) U. Hennecke, C. H. Müller and R. Frölich, Org. Lett., 2011, 13, 860 CrossRef CAS; (b) D. Huang, H. Wang, F. Xue, H. Guan, L. Li, X. Peng and Y. Shi, Org. Lett., 2011, 13, 6350 CrossRef CAS; (c) S. E. Denmark and M. T. Burk, Org. Lett., 2012, 14, 256 CrossRef CAS PubMed; (d) X. H. Zeng, C. X. Miao, S. F. Wang, C. G. Xia and W. Sun, Chem. Commun., 2013, 49, 2418 RSC; (e) S. E. Denmark and M. T. Burk, Chirality, 2014, 26, 344 CrossRef CAS PubMed; (f) D. W. Tay, G. Y. C. Leung and Y.-Y. Yeung, Angew. Chem., Int. Ed., 2014, 53, 5161 CAS; (g) Z. Ke, C. K. Tan, F. Chen and Y.-Y. Yeung, J. Am. Chem. Soc., 2014, 136, 5627 CrossRef CAS PubMed.
- For chiral phosphate directed reactions, see: (a) R. J. Phipps, G. L. Hamilton and F. D. Toste, Nat. Chem., 2012, 4, 603 CrossRef CAS; (b) M. Mahlau and B. List, Angew. Chem., Int. Ed., 2013, 52, 518 CrossRef CAS PubMed. For selected halogenation reactions using chiral phosphoric acid as a phase transfer catalyst, see: (c) V. Rauniyar, A. D. Lackner, G. L. Hamilton and F. D. Toste, Science, 2011, 334, 1681 CrossRef CAS PubMed; (d) Y.-M. Wang, J. Wu, C. Hoong, V. Rauniyar and F. D. Toste, J. Am. Chem. Soc., 2012, 134, 12928 CrossRef CAS PubMed; (e) T. Honjo, R. J. Phipps, V. Rauniyar and F. D. Toste, Angew. Chem., Int. Ed., 2012, 51, 9684 CrossRef CAS PubMed; (f) H. P. Shunatona, N. Früh, Y.-M. Wang, V. Rauniyar and F. D. Toste, Angew. Chem., Int. Ed., 2013, 52, 7724 CrossRef CAS.
- For halonium induced semi-pinacol rearrangements, see: (a) Z.-M. Chen, Q.-W. Zhang, Z.-H. Chen, H. Li, Y.-Q. Tu, F.-M. Zhang and J.-M. Tian, J. Am. Chem. Soc., 2011, 133, 8818 CrossRef CAS PubMed; (b) C. H. Muller, M. Wilking, A. Ruhlmann, B. Wibbeling and U. Hennecke, Synlett, 2011, 2043 Search PubMed; (c) F. Romanov-Michailidis, L. Guénée and A. Alexakis, Org. Lett., 2013, 15, 5890 CrossRef CAS; (d) F. Romanov-Michailidis, L. Guénée and A. Alexakis, Angew. Chem., Int. Ed., 2013, 52, 9266 CrossRef CAS PubMed; (e) Q. Yin and S.-L. You, Org. Lett., 2014, 16, 1810 CrossRef CAS PubMed. For selected other reactions, see: (f) K. C. Nicolaou, N. L. Simmons, Y. Ying, P. M. Heretsch and J. S. Chen, J. Am. Chem. Soc., 2011, 133, 8134 CrossRef CAS PubMed; (g) F. Chen, C. K. Tan and Y.-Y. Yeung, J. Am. Chem. Soc., 2013, 135, 1232 CrossRef CAS PubMed; (h) K. Mori, Y. Ichikawa, M. Kobayashi, Y. Shibata, M. Yamanaka and T. Akiyama, J. Am. Chem. Soc., 2013, 135, 3964 CrossRef CAS PubMed; (i) Y. Zhao, X. Jiang and Y.-Y. Yeung, Angew. Chem., Int. Ed., 2013, 52, 8597 CrossRef CAS PubMed; (j) D. Huang, X. Liu, L. Li, Y. Cai, W. Liu and Y. Shi, J. Am. Chem. Soc., 2013, 135, 8101 CrossRef CAS PubMed; (k) C. S. Brindle, C. S. Yeung and E. N. Jacobsen, Chem. Sci., 2013, 4, 2100 RSC.
- For selected examples, see: (a) F. Straus and R. Kuhnel, Chem. Ber., 1933, 66, 1834 CrossRef PubMed; (b) R. D. Evans, J. W. Magee and J. H. Schauble, Synthesis, 1988, 862 CrossRef CAS; (c) P. Galatsis and S. D. Millan, Tetrahedron Lett., 1991, 32, 7493 CrossRef CAS; (d) I. Rawal, Tetrahedron Lett., 1992, 33, 4687 CrossRef.
- For selected reviews, see: (a) New Frontiers in Asymmetric Catalysis, ed. K. Mikami and M. Lautens, Wiley-Interscience, Hoboken, NJ, 2007, vol. 3 Search PubMed; (b) Enantioselective Organocatalysis, ed. P. I. Dalko, Wiley-VCH, Weinheim, 2007 Search PubMed; (c) K. L. Jensen, G. Dickmeiss, H. Jiang, L. Albrecht and K. A. Jørgensen, Acc. Chem. Res., 2012, 45, 248 CrossRef CAS; (d) J.-F. Brière, S. Oudeyer, V. Dallab and V. Levacher, Chem. Soc. Rev., 2012, 41, 1696 RSC.
- (a) W. Xie, G. Jiang, H. Liu, J. Hu, X. Pan, H. Zhang, X. Wan, Y. Lai and D. Ma, Angew. Chem., Int. Ed., 2013, 52, 12924 CrossRef CAS PubMed; (b) H. Liu, G. Jiang, X. Pan, X. Wan, Y. Lai, D. Ma and W. Xie, Org. Lett., 2014, 16, 1908 CrossRef CAS PubMed.
- G. S. Lal, J. Org. Chem., 1993, 58, 2791–2796 CrossRef CAS.
- For a recent review on organocatalyzed epoxidation, see: R. L. Davis, J. Stiller, T. Naicker, H. Jiang and K. A. Jögensen, Angew. Chem., Int. Ed., 2014, 53, 7406 CrossRef CAS.
- For selected examples, see: (a) T. Ichige, Y. Okano, N. Kanoh and M. Nakata, J. Am. Chem. Soc., 2007, 129, 9862 CrossRef CAS; (b) A. R. van Dyke and T. F. Jamison, Angew. Chem., Int. Ed., 2009, 48, 4430 CrossRef CAS PubMed; (c) Q. Yang, J. T. Njardarson, C. Draghici and F. Li, Angew. Chem., Int. Ed., 2013, 52, 8648 CrossRef CAS PubMed.
- CCDC 1023013 and 1028455 contain the supplementary crystallographic data for compounds 2ac and 4, respectively. For a rationale of the observed stereoselectivity, see the ESI.†.
- See ESI† for detailed experimental data..
- For selected examples, see: (a) Y. Kita, K. Higuchi, Y. Yoshida, K. Iio, S. Kitagaki, K. Ueda, S. Akai and H. Fujioka, J. Am. Chem. Soc., 2001, 123, 3214 CrossRef CAS PubMed; (b) Y. M. Shen, B. Wang and Y. Shi, Angew. Chem., Int. Ed., 2006, 45, 1429 CrossRef CAS.
- A. Abad, C. Agulló, A. C. Cuñat and I. Navarro, Synthesis, 2005, 3355 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental procedures and characterization for all new compounds. CCDC 1023013 and 1028455. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5sc02485d |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2015 |