
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDesigning interchain and intrachain properties of conjugated polymers for latent optical information encoding†
Kyeongwoon
Chung
a,
Andrew
McAllister
b,
David
Bilby
c,
Bong-Gi
Kim
g,
Min Sang
Kwon
c,
Emmanouil
Kioupakis
c and
Jinsang
Kim
*acdef
aMacromolecular Science and Engineering, University of Michigan, Ann Arbor, MI 48109, USA. E-mail: jinsang@umich.edu
bDepartment of Applied Physics, University of Michigan, Ann Arbor, MI 48109, USA
cDepartment of Materials Science and Engineering, University of Michigan, Ann Arbor, MI 48109, USA
dDepartment of Biomedical Engineering, University of Michigan, Ann Arbor, MI 48109, USA
eDepartment of Chemistry, University of Michigan, Ann Arbor, MI 48109, USA
fDepartment of Chemical Engineering, University of Michigan, Ann Arbor, MI 48109, USA
gDepartment of Organic and Nano System Engineering, KonKuk University, Seoul 143-701, Republic of Korea
First published on 3rd September 2015
Abstract
Building molecular-design insights for controlling both the intrachain and the interchain properties of conjugated polymers (CPs) is essential to determine their characteristics and to optimize their performance in applications. However, most CP designs have focused on the conjugated main chain to control the intrachain properties, while the design of side chains is usually used to render CPs soluble, even though the side chains critically affect the interchain packing. Here, we present a straightforward and effective design strategy for modifying the optical and electrochemical properties of diketopyrrolopyrrole-based CPs by controlling both the intrachain and interchain properties in a single system. The synthesized polymers, P1, P2 and P3, show almost identical optical absorption spectra in solution, manifesting essentially the same intrachain properties of the three CPs having restricted effective conjugation along the main chain. However, the absorption spectra of CP films are gradually tuned by controlling the interchain packing through the side-chain design. Based on the tailored optical properties, we demonstrate the encoding of latent optical information utilizing the CPs as security inks on a silica substrate, which reveals and conceals hidden information upon the reversible aggregation/deaggregation of CPs.
Introduction
Conjugated polymers (CPs) are key materials for various optoelectronic applications, including chemical and bio sensors1–9 that detect from ions, biomolecules to dangerous chemical gases, and security inks10–12 for hidden information delivery or anti-counterfeiting. The optoelectronic properties of CPs are determined by both the intrachain (intrinsic) and the interchain (packing) properties of the polymer chains. While the intrachain properties are mainly determined by the chemical structure of the main chain, the interchain properties are driven by the polymer packing, which is influenced by various design factors. Therefore, the rational design of both the intrachain and the interchain properties of CPs is important for realizing CP applications with optimal characteristics.Various strategies for regulating the intrachain properties of CPs through the design of the conjugated main chain have been investigated, including atom substitution,13–15 addition of functional groups,16,17 and the modification of CP building blocks.18,19 These main chain design strategies also partly affect the interchain properties of CPs: for example, introducing planar building blocks and linkages, such as thiophene linked diketopyrrolopyrrole (DPP), promote close co-facial packing of CPs' main chain, which results in enhanced hole mobility in CP films.13
However, unlike the widely investigated main chain design of CPs, the effects of flexible side chains on the interpolymer assembly and packing are rather unexplored. Flexible side chains are often introduced mainly to provide solubility to the rigid-rod like CPs. For this purpose, long and branched side chains are usually adapted into CPs without a thorough consideration of their effects on the interchain packing. However, recent work has emphasized the role of the side chain and discusses it as an important design parameter to tune the properties of CPs. For instance, by investigating different side chains on the same conjugated main chain backbone, the side chain design has proven to be critical to the interchain properties.20–23 Our research group recently showed an increase in hole mobility of three orders of magnitude in aligned lyotropic liquid crystalline CPs by rationally designing the molecular architecture of side chains together with the main chain conformation and form factor, which rendered directed self-assembly and packing of CPs.24 Therefore, the optimization of CP characteristics requires a careful control for both intrachain and interchain properties: for this purpose, simultaneous design of conjugated main chain as well as flexible side chain is essential.
With recent development of high-end scanning and copying technologies, counterfeiting has become a serious problem due to easy accessibility as well as its high quality.10 In this trend, security inks for anti-counterfeiting or hidden information delivery have become a research field of great interest.10–12,25–28 Security inks provide invisible information in the initial stage which is not possible to read or photocopy; the hidden information such as color, pattern or letters are programed to be revealed under certain circumstances. Various functional organic molecules have been developed for security inks.25–28 In these molecular systems the hidden information was encoded based on fluorescence or color changes upon specific treatment (heat, chemical, or photo irradiation), and the concealed information was readable under irradiation of UV light. On the other hand, in case of CPs, polydiacetylenes that are prepared from unique molecular assembly of diacetylene monomers have mostly been investigated for security ink application, based on colorimetric change upon heat treatment.10–12 However, CPs with aromatic conjugated main chains have not been fully investigated for security ink application despite its great potential based on the readily tunable optimal properties through systematic molecular design.
In this work, we demonstrate the modification of the optical and electrochemical properties of diketopyrrolopyrrole (DPP)-based CPs by controlling both the interchain and intrachain properties with effective but simple polymer-design strategies. We synthesized three CPs having an alternating donor–acceptor structure (P1, P2 and P3 in Fig. 1) using a phenyl linker connecting the electron-withdrawing DPP unit and an electron-donating building block. Regardless of three different electron-donating building blocks, all three CPs have almost identical absorption in diluted solutions and deep highest occupied molecular orbitals (HOMOs). This is caused by the large dihedral angle of the phenyl linker, which ensures a restricted effective conjugation length. In contrast, the interchain packing of the three CPs in the aggregated-film state was gradually adjusted by changing the bulkiness and linking directions of the side chains (Fig. 1) to control the optical absorbance and HOMO levels. The color gradation of CPs in the aggregated state, in contrast to their identical color in solution, enables us to demonstrate the encoding of covert information utilizing CP solutions as security inks on a silica substrate, which immediately develop hidden information by water dipping.
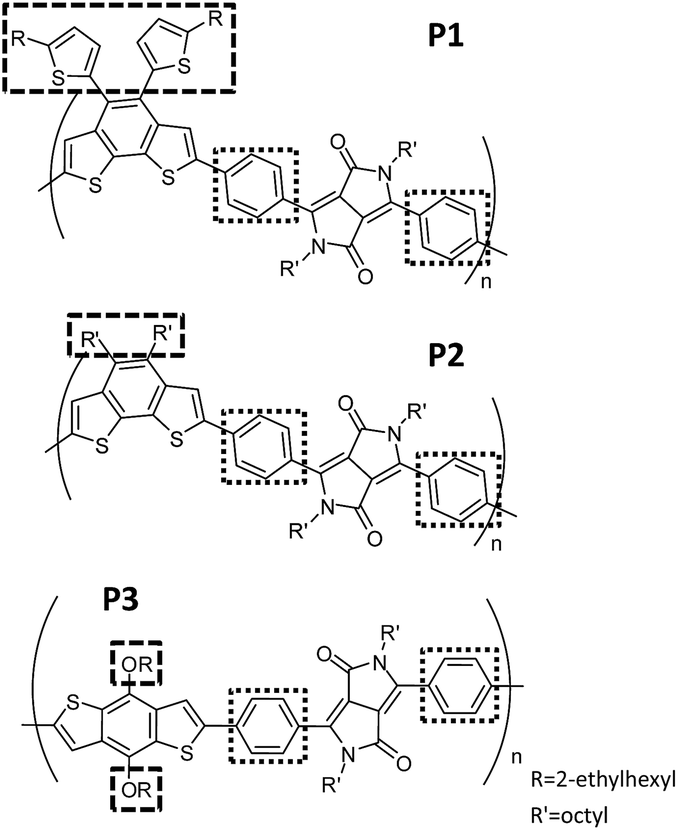 | ||
| Fig. 1 Chemical structures of the designed CPs: P1, P2 and P3. All three polymers have a phenyl linker between the electron-donating and electron-withdrawing building blocks to control the intrachain properties of CPs. Side chains with different bulkiness and direction are implemented to control the interchain interactions between CP chains. | ||
Results and discussion
The three designed CPs in Fig. 1 have distinct side chains, which differ in bulkiness and linking direction. Each CP also has a phenyl linker between the electron-donor and electron-acceptor building blocks. In comparison to CPs having the commonly used planar thienyl linkers for the DPP unit,18,29 the phenyl linker induces a large dihedral angle between building blocks, and hence the twisted conformation is expected to reduce the effective conjugation length. As a result, we anticipate largely blue-shifted absorption and deeper HOMO levels due to the enlarged band gap of the phenyl-linked CPs. This is similar to the cases of poly(p-phenylene) or polyfluorene, which exhibit a large band gap with bluish emission even in the high-molecular-weight regime because of the non-planar conformation of the phenyl linkage.30–32The optical properties of the three CPs are characterized by UV-vis spectrophotometry in diluted polymer solutions (10−5 M based on the repeating unit). Compared to the analogous CPs having the thienyl linker, whose optical band edge is around 800–950 nm,18,29 the optical band edge of the diluted solution of all three CPs is shifted to ca. 600 nm. This is a result of the modification of the intrachain properties by the phenyl linker incorporation (Fig. 2a and Table S1†). Interestingly, even though the three CPs are composed of different electron-donating building blocks (Fig. 1), the color of each CP solution is almost identical (Fig. 2c). This implies that the phenyl linker insertion effectively reduces the intramolecular charge transfer effect between the electron-donating and electron-withdrawing building blocks as well as the effective conjugation length. The identical intrachain properties, independent of the choice of electron-donating blocks, enables us to systematically investigate the effects of side chains on the interchain packing of CPs and, consequently, the alteration of their optoelectronic properties of P1, P2 and P3.
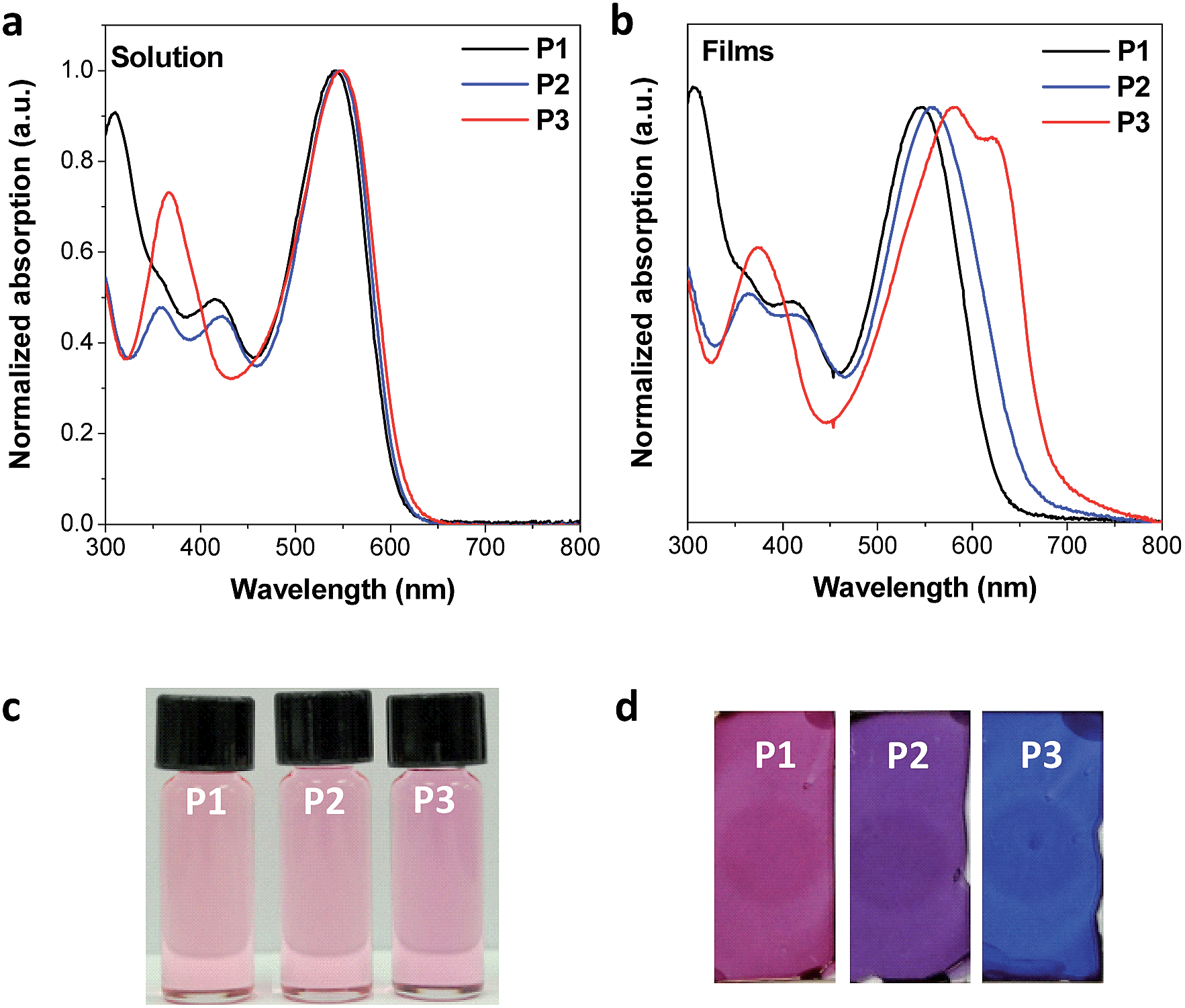 | ||
| Fig. 2 Optical properties of the synthesized CPs. (a) UV-vis absorption spectra of diluted CP solution samples in chloroform. The spectra for all three CPs are nearly identical, especially in λmax and optical band edge, because the CPs have essentially the same intrachain properties. (b) UV-vis absorption spectra of CP films, thermally annealed at 130 °C. The choice of side chains controls the interchain packing and modifies the optical properties among CPs. (c) Optical images of the CP solutions under room light. (d) Optical images of the CP films under room light. | ||
To further probe the role of the phenyl versus thienyl linkers on the structural and optical properties of the synthesized CPs, we performed structural-relaxation and electronic-structure calculations using Guassian09.33 The molecules were relaxed using density functional theory (DFT) with the B3LYP hybrid functional and the 6-31G** basis set. We started by investigating the dihedral angle of the monomer unit (Fig. 3). In the case of the thienyl linker, the dihedral angle between the electron-donating/withdrawing building blocks and the thienyl linker varies between 0.45° to 8.02°, which implies an almost planar chain conformation (Fig. 3b and Table S2†). On the other hand, the phenyl linker shows a much larger dihedral angle, between 20.42° to 23.65°, which is expected by the CP design (Fig. 3a and Table S2†). Using the optimized conformation of the monomeric units with phenyl or thienyl linker, we investigated the band gaps of the monomer, dimer, and trimer structures. It is revealed that the phenyl linked CPs do exhibit a larger band gap than the thienyl-linked CPs (Table 1). For the P1, P2 and P3 analogues, trimers bearing a phenyl linker have band gaps of 2.28, 2.27, 2.22 eV respectively, which is larger than those of thienyl-linked trimers (1.90, 1.94, 1.84 eV). Furthermore, the band gap decreases faster in thienyl-linker analogues as the chain length increases from the monomer to the trimer. This difference in band gaps supports the idea that the effective conjugation is more restricted in twisted phenyl linker analogues compared to planar thienyl linker analogues. In addition, the phenyl linker insertion shifts the calculated HOMO levels of the CPs deeper (Table S3†). The deep HOMO level is highly plausible to be advantageous when CPs are utilized in energy device application in terms of open circuit voltage (Voc).
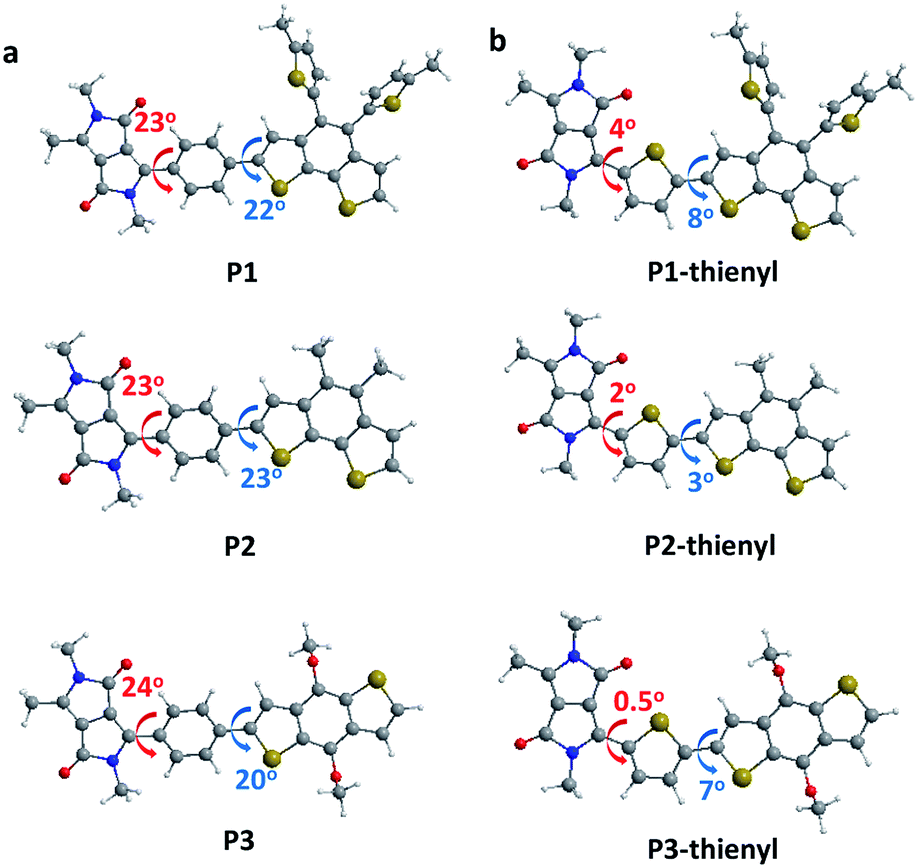 | ||
| Fig. 3 Calculated dihedral angles of the monomeric units of P1 (top), P2 (middle), P3 (bottom) with (a) a phenyl linker and (b) a thienyl linker. | ||
| Calculated band gap [eV] | |||
|---|---|---|---|
| Monomer | Dimer | Trimer | |
| P1 | 2.77 | 2.37 | 2.28 |
| P1-thienyl | 2.53 | 2.03 | 1.90 |
| P2 | 2.77 | 2.36 | 2.27 |
| P2-thienyl | 2.52 | 2.02 | 1.94 |
| P3 | 2.76 | 2.34 | 2.22 |
| P3-thienyl | 2.51 | 1.96 | 1.84 |
Even though the three CPs show almost identical optical properties in dilute solutions, their solid-state optical properties differ significantly (Fig. 2b and d). The variation of the solid-state properties is originated from the molecular design, as each CPs has a different side chain with distinct bulkiness and orientation, which affects the interchain packing. P1 has two branched 2-ethylhexyl side chains connected through a bulky thienyl ring to the CP main chain. Our calculations show that the thienyl rings of the side chains are not in the same plane as the electron-donating benzo[2,1-b:3,4-b′]dithiophene that they are connected to, but have instead adopted a largely twisted conformation (ca. 69°) to minimize steric hindrance (P1 in Fig. 3a). Because of the branched bulky 2-ethylhexyl chains together with the twisted thienyl rings, the main chains of P1 are anticipated to have minimal co-facial π–π stacking. On the other hand, P2 has two linear octyl side chains connected on the same side of the benzo[2,1-b:3,4-b′]dithiophene unit but without the thienyl linkage, which is expected to have a mediocre propensity for interchain packing. The n-octyl side chains of P2 might stretch out to the opposite out-of-plane directions of the main chain plane due to steric hindrance as shown in Fig. S1.† In case of P3, the two 2-ethylhexyloxy side chains are not bulky enough to prevent P3 from aggregating.24 Moreover, they are attached on the opposite sides of the benzo[1,2-b:4,5-b′]dithiophene donor unit such that the side chains do not have steric repulsion either. As a result, P3 is expected to have strong π–π stacking between the main chains.
Indeed, the colors and the UV-vis absorption spectra of the thermally annealed CP films show distinct gradation despite the similar absorption spectra in solution (Fig. 2). The absorption λmax of the P3 film is red-shifted by 36 nm compared to that of the P3 solution and a new aggregation band appears at 620 nm (Fig. 2b and Table S1†). The red-shifted new absorption band in P3 film may also be ascribed to an extended effective conjugation length caused by planarization of the conjugated main chain in the aggregated film state. In contrast, P1 essentially shows almost identical absorption in the solid film and in solution due to weak interchain interactions, while the absorption λmax of the P2 film is red-shifted by 9 nm compared to the P2 solution (Fig. 2b and Table S1†). The grazing incidence X-ray diffraction (GIXRD) analysis also revealed the same trend in the interpolymer packing: P3 shows a distinct peak at about 5° corresponding to the developed interchain packing, while P1 shows amorphous packing due to the out-of-plane bulky side chains (Fig. S2†).
We further demonstrate the encoding of covert optical information on a silica substrate using the three CPs as security inks by exploiting the difference among their interpolymer packing. A highly polar silica gel is likely to interact strongly with the CPs in the dry-state due to polar–polar interactions, which can prevent the aggregation of CPs. When dilute solutions of the CPs (ca. 5 × 10−4 M based on the repeating unit) were painted on polar silica gel substrates, the CPs retained identical solution-like colors even after evaporation of the chloroform solvent (Fig. 4a). However, because water molecules are highly polar and favorable for hydrogen bonding, they interact strongly with the silica substrates and break the CP-substrate interaction. The different aggregation tendencies of the CPs caused by the side chains were manifested by a vivid color difference among the CP films when the substrate was dipped into water: P1 is red, P2 is purple, and P3 is bluish purple (Fig. 4a). By using P1 and P3 as inks, we demonstrated the encoding of latent patterns and messages. An array of 5 × 5 dots of P1 and P3 inks with identical colors were patterned on a silica substrate (Fig. 4b). When the dot array was immersed into water, the latent arrow image was developed immediately (Movie S1†). By increasing the array size to 11 × 7, we could demonstrate the covert letter “M” (Fig. 4b and Movie S2†). Moreover, this phenomenon is completely reversible: the patterns developed by water are completely erased by chloroform dipping or under chloroform vapor treatment (Fig. 4a and Movie S2†).
 | ||
| Fig. 4 Encoding latent optical information on a silica substrate using the CPs as a security ink. (a) Reversible color change of CP on a silica substrate upon water and chloroform treatment. The color change is particularly prominent for P2 and P3. (b) The latent pattern appears upon water-induced aggregation. Before dipping in water, the colors of P1 and P3 on a silica substrate are undistinguishable (left panels). Only P3 clearly changes its color from red to bluish purple upon aggregation by water treatment, revealing the covert arrow pattern or the hidden letter “M” (right panels). | ||
The design strategies to control the intrachain and interchain properties of CPs also essentially link to the electrochemical properties of the CP films. Each CP shares the same phenyl linker so as to has a deeper HOMO than the conventional analogous CPs with thienyl linkers. Cyclic voltammetry (CV) measurements were conducted to measure the energy levels of the three CPs. CP films for the measurements were spin cast onto ITO substrates, followed by thermal annealing. The energy levels of regioregular poly(3-hexylthiophene) (P3HT) and [6,6]-phenyl-C61-butyric acid methyl ester (PCBM) are presented together with the CPs for comparison (Fig. 5b). The three CPs indeed have deep HOMO energy levels (−5.34 to −5.69 eV) (Fig. 5), which are deeper by about 0.3 eV than the HOMO levels of the analogous CPs with thienyl linkers reported in the literature.18,34 Furthermore, the interpolymer packing propensity difference of the three CPs also affects the electrochemical properties of CP films. Similar to the variation of the optical properties, the different interchain properties of the CPs result in a gradual change of the HOMO levels: the restricted interchain interaction via the side chain design results in deeper HOMO levels. As a result, P1 with an amorphous nature has the deepest HOMO level (−5.69 eV), while P3 with a relatively high interpolymer packing propensity exhibits the shallowest HOMO level (−5.34 eV) among the three CPs. A similar trend of the HOMO levels were reported in recent publications, emphasizing the role of side chains in designing the electrochemical properties.20,21,35
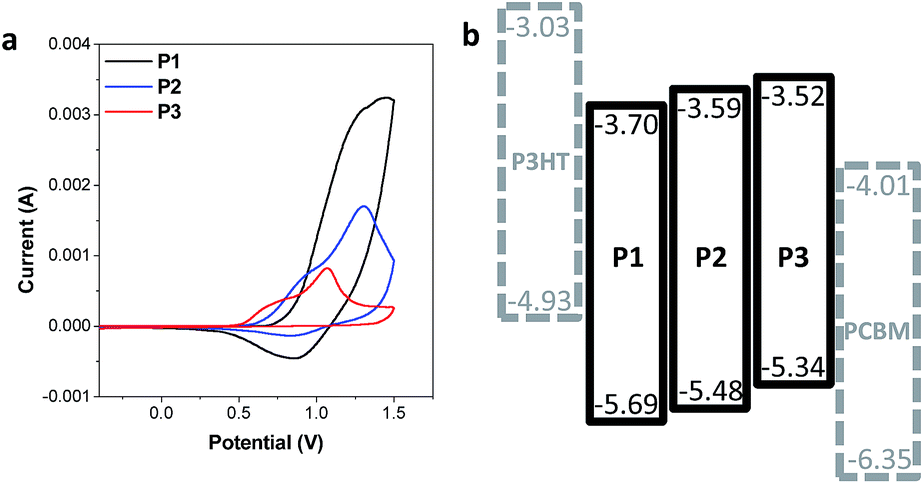 | ||
| Fig. 5 Electrochemical properties of P1, P2, and P3. (a) CV measurement of CPs. (b) The energy level diagram of the synthesized CPs, P3HT, and PCBM. | ||
Conclusions
In summary, we present an effective but simple design strategy for controlling the optical and electrochemical properties of CPs. Controlling the dihedral angle along the CP main chain with a phenyl linker combined with side chains of varying bulkiness and orientation can modulate both the intrachain and interchain properties of CPs. Regardless of the difference in electron-donating building blocks, the synthesized CPs, P1, P2 and P3, exhibit almost identical absorption with deep HOMO energy levels, due to a restricted effective conjugation length resulting from the large dihedral angle of the phenyl linker. The side chain design decisively affects the interpolymer packing of the CPs and renders gradual shifts in optical properties and HOMO energy levels in the aggregated state. We further demonstrate the encoding of latent optical information on a silica substrate by exploiting the interpolymer packing difference of the three CPs. Hidden patterns recorded in the dry state are immediately revealed upon water treatment in a completely reversible manner.Acknowledgements
The authors thank Dr D. Lee for assistance on GIXRD analysis. The experimental work was supported as part of the Center for Solar and Thermal Energy Conversion, and Energy Frontier Research Center funded by the U.S. Department of Energy (DoE), Office of Science, Basic Energy Sciences (BES) (DE-SC0000957). The computational work was supported by the National Science Foundation CAREER award through Grant No. DMR-1254314. K. C. was also partly supported by Rackham Summer Award and the Converging Research Center Program funded by the Ministry of Science, ICT and Future Planning (NFR-2014M3C1A8048791). A. M. acknowledges support from the National Science Foundation Graduate Research Fellowship Program through Grant No. DGE 1256260. This research used resources of both the National Energy Research Scientific Computing Center, a DOE Office of Science User Facility supported by the Office of Science of the U.S. Department of Energy under Contract No. DE-AC02-05CH11231 and of Advanced Research Computing at the University of Michigan, Ann Arbor.Notes and references
- J. Kim, D. T. McQuade, S. K. McHugh and T. M. Swager, Angew. Chem., 2000, 112, 4026–4030 CrossRef.
- J. Lee, H. Jun and J. Kim, Adv. Mater., 2009, 21, 3674–3677 CrossRef CAS PubMed.
- H.-A. Ho, M. Boissinot, M. G. Bergeron, G. Corbeil, K. Doré, D. Boudreau and M. Leclerc, Angew. Chem., Int. Ed., 2002, 41, 1548–1551 CrossRef CAS.
- K. Lee, J.-M. Rouillard, B.-G. Kim, E. Gulari and J. Kim, Adv. Funct. Mater., 2009, 19, 3317–3325 CrossRef CAS PubMed.
- K. Lee, L. K. Povlich and J. Kim, Analyst, 2010, 135, 2179–2189 RSC.
- G. Jang, J. Kim, D. Kim and T. S. Lee, Polym. Chem., 2015, 6, 714–720 RSC.
- S. J. Toal and W. C. Trogler, J. Mater. Chem., 2006, 16, 2871–2883 RSC.
- L. O. Péres and J. Gruber, Mater. Sci. Eng., C, 2007, 27, 67–69 CrossRef PubMed.
- J. Lee, S. Seo and J. Kim, Adv. Funct. Mater., 2012, 22, 1632–1638 CrossRef CAS PubMed.
- B. Yoon, J. Lee, I. S. Park, S. Jeon, J. Lee and J.-M. Kim, J. Mater. Chem. C, 2013, 1, 2388–2403 RSC.
- B. Yoon, H. Shin, O. Yarimaga, D.-Y. Ham, J. Kim, I. S. Park and J.-M. Kim, J. Mater. Chem., 2012, 22, 8680–8686 RSC.
- B. Yoon, D.-Y. Ham, O. Yarimaga, H. An, C. W. Lee and J.-M. Kim, Adv. Mater., 2011, 23, 5492–5497 CrossRef CAS PubMed.
- J. S. Ha, K. H. Kim and D. H. Choi, J. Am. Chem. Soc., 2011, 133, 10364–10367 CrossRef CAS PubMed.
- A. J. Kronemeijer, E. Gili, M. Shahid, J. Rivnay, A. Salleo, M. Heeney and H. Sirringhaus, Adv. Mater., 2012, 24, 1558–1565 CrossRef CAS PubMed.
- C. M. Amb, S. Chen, K. R. Graham, J. Subbiah, C. E. Small, F. So and J. R. Reynolds, J. Am. Chem. Soc., 2011, 133, 10062–10065 CrossRef CAS PubMed.
- L. Huo, S. Zhang, X. Guo, F. Xu, Y. Li and J. Hou, Angew. Chem., Int. Ed. Engl., 2011, 50, 9697–9702 CrossRef CAS PubMed.
- J. You, L. Dou, K. Yoshimura, T. Kato, K. Ohya, T. Moriarty, K. Emery, C.-C. Chen, J. Gao, G. Li and Y. Yang, Nat. Commun., 2013, 4, 1446 CrossRef PubMed.
- L. Huo, J. Hou, H.-Y. Chen, S. Zhang, Y. Jiang, T. L. Chen and Y. Yang, Macromolecules, 2009, 42, 6564–6571 CrossRef CAS.
- M. Yuan, A. H. Rice and C. K. Luscombe, J. Polym. Sci., Part A: Polym. Chem., 2011, 49, 701–711 CrossRef CAS PubMed.
- J. Mei and Z. Bao, Chem. Mater., 2014, 26, 604–615 CrossRef CAS.
- L. Yang, H. Zhou and W. You, J. Phys. Chem. C, 2010, 114, 16793–16800 CAS.
- J. M. Szarko, J. Guo, Y. Liang, B. Lee, B. S. Rolczynski, J. Strzalka, T. Xu, S. Loser, T. J. Marks, L. Yu and L. X. Chen, Adv. Mater., 2010, 22, 5468–5472 CrossRef CAS PubMed.
- K. Chung, M. S. Kwon, B. M. Leung, A. G. Wong-Foy, M. S. Kim, J. Kim, S. Takayama, J. Gierschner, A. J. Matzger and J. Kim, ACS Cent. Sci., 2015, 1, 94–102 CrossRef CAS.
- B.-G. Kim, E. J. Jeong, J. W. Chung, S. Seo, B. Koo and J. Kim, Nat. Mater., 2013, 12, 659–664 CrossRef CAS PubMed.
- D. R. Whang, Y. You, W.-S. Chae, J. Heo, S. Kim and S. Y. Park, Langmuir, 2012, 28, 15433–15437 CrossRef CAS PubMed.
- X. Zhu, R. Liu, Y. Li, H. Huang, Q. Wang, D. Wang, X. Zhu, S. Liu and H. Zhu, Chem. Commun., 2014, 50, 12951–12954 RSC.
- A. Kishimura, T. Yamashita, K. Yamaguchi and T. Aida, Nat. Mater., 2005, 4, 546–549 CrossRef CAS PubMed.
- S. Kobatake, H. Imagawa, H. Nakatani and S. Nakashima, New J. Chem., 2009, 33, 1362–1367 RSC.
- L. Dou, J. You, J. Yang, C.-C. Chen, Y. He, S. Murase, T. Moriarty, K. Emery, G. Li and Y. Yang, Nat. Photonics, 2012, 6, 180–185 CrossRef CAS PubMed.
- M. Remmers, B. Müller, K. Martin, H.-J. Räder and W. Köhler, Macromolecules, 1999, 32, 1073–1079 CrossRef CAS.
- D. Neher, Macromol. Rapid Commun., 2001, 22, 1365–1385 CrossRef CAS.
- V. Cimrová, M. Remmers, D. Neher and G. Wegner, Adv. Mater., 1996, 8, 146–149 CrossRef PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Ö. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Revision C.01, Gaussian, Inc., Wallingford CT, 2009 Search PubMed.
- C. Kanimozhi, P. Balraju, G. D. Sharma and S. Patil, J. Phys. Chem. B, 2010, 114, 3095–3103 CrossRef CAS PubMed.
- C. Piliego, T. W. Holcombe, J. D. Douglas, C. H. Woo, P. M. Beaujuge and J. M. J. Fréchet, J. Am. Chem. Soc., 2010, 132, 7595–7597 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Details on methods, materials and synthesis are available. See DOI: 10.1039/c5sc02403j |
| This journal is © The Royal Society of Chemistry 2015 |