
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAuICl-bound N-heterocyclic carbene ligands form MII4(LAuCl)6 integrally gilded cages†
William J.
Ramsay
,
Jonathan A.
Foster‡
 ,
Katharine L.
Moore
,
Tanya K.
Ronson
,
Raphaël J.
Mirgalet
,
David A.
Jefferson
and
Jonathan R.
Nitschke
*
,
Katharine L.
Moore
,
Tanya K.
Ronson
,
Raphaël J.
Mirgalet
,
David A.
Jefferson
and
Jonathan R.
Nitschke
*
Department of Chemistry, University of Cambridge, Lensfield Road, Cambridge CB2 1EW, UK. E-mail: jrn34@cam.ac.uk
First published on 24th September 2015
Abstract
The incorporation of an N-heterocyclic carbene (NHC) moiety into a self-assembled MII4L6 cage framework required the NHC first to be metallated with gold(I). Bimetallic cages could then be constructed using zinc(II) and cadmium(II) templates, showing weak luminescence. The cages were destroyed by the addition of further gold(I) in the form of AuI(2,4,6-trimethoxybenzonitrile)2SbF6, which caused the reversibly-formed cages to disassemble and controllably release the AuI-NHC subcomponent into solution. This release in turn induced the growth of gold nanoparticles. The rate of dianiline release could be tuned by capsule design or through the addition of chemical stimuli, with different release profiles giving rise to different nanoparticle morphologies.
Introduction
A variety of multitopic ligands have been designed to self-assemble with metal ions in order to construct three-dimensional metal–organic structures.1 Many of the novel applications of these supramolecular assemblies derive from the chemical functionalities of their incorporated ligands.2–13 As new rules are established to allow ligands incorporating more reactive groups to be built into supramolecular assemblies, new types of functionality can arise.14The shape and dimensions of discrete, self-assembled supramolecular structures are predominantly dictated by how ligands with a characteristic size and denticity come together in geometries defined by the coordination sphere of a given metal ion.15–28 The functionality29–36 of these assemblies can be shaped by incorporating ligands with active functional groups, which can also be modified in situ either pre-37–40 or post-synthetically.41–44 The response of a ligand to an external stimulus, such as light, can influence the guest binding affinity of the corresponding supramolecular structure.45 When a ligand contains a coordinated transition metal that is catalytically active, this active site can be orientated explicitly by the supramolecular framework, either protecting or exposing it;46 incorporating more than one metal center thus can allow for division of labor between structural and functional metal centers.
N-Heterocyclic carbenes (NHCs) are a useful class of electron-donating ligands that form strong metal–ligand bonds.47 Gold(I)-NHCs, in particular, have found applications in the fields of catalysis,48 pharmaceuticals,49 liquid crystals50 and optical devices.51,52 Despite the potential of incorporating metal-based NHC ligands into supramolecular constructs, opening new possibilities of control over their reactivity, few examples of self-assembled structures with this motif have been reported.53–56 We thus designed subcomponent A and its metallated carbene derivatives B–D (Scheme 1), to be capable of reversibly self-assembling with 2-formylpyridine and metal ions to form supramolecular structures, in order to probe the scope and limitations of the ability of this class of ligand to self-assemble into supramolecular structures. As AuI-NHC complexes can be reduced to form carbene-stabilized gold nanoparticles (Au NPs),57 we anticipated that incorporating this ligand motif into a reversibly formed supramolecular structure could allow for new means to be developed to control the nucleation and growth conditions of Au NPs.
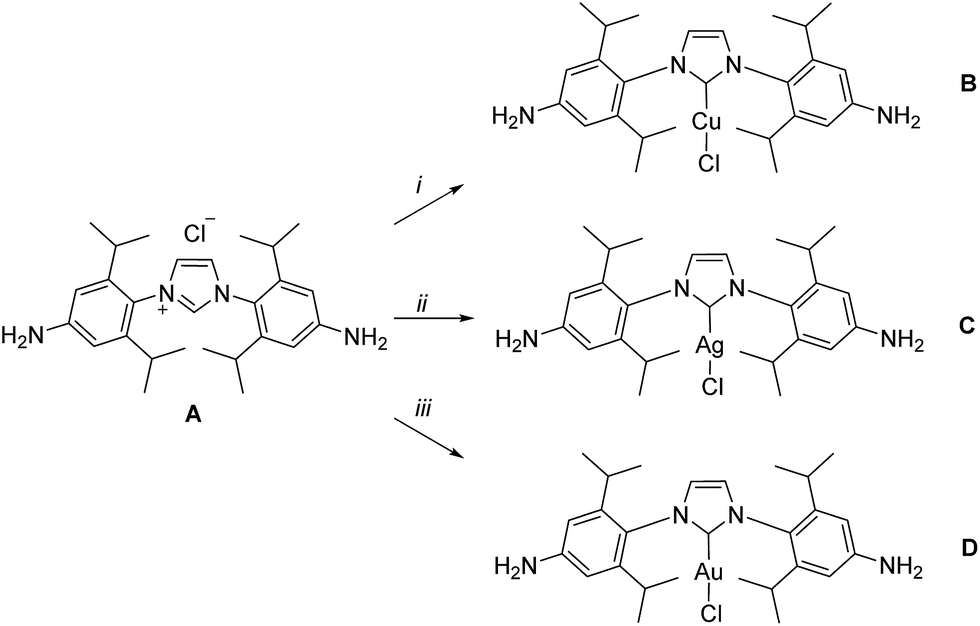 | ||
| Scheme 1 Syntheses of subcomponents A–D. Reaction conditions: (i) CuCl, Na2CO3, acetone, 60 °C, 16 h, 90%; (ii) Ag2O, DCM, 25 °C, 2 h, 90%; (iii) Au(tht)Cl, aq. Na2CO3, DCM, 25 °C, 0.5 h, 92%. | ||
Results and discussion
Subcomponent syntheses and cage preparation
The syntheses of NHC dianilines A–D were accomplished as shown in Scheme 1. Imidazolium A was prepared through reduction of a bis-azido imidazolium precursor (Scheme S1†), prepared as described in the ESI.† Taking advantage of the established ability of NHCs to coordinate to many metal ions,55 metallocarbenes B–D were prepared by metallating A following established, straightforward procedures.53,58,59These subcomponents were subjected to conditions that were anticipated to lead to the preparation of self-assembled MII4L6 structures.25 Each was treated in turn with 2-formylpyridine and the prospective metal templates ZnII, CdII, FeII and CoII. Although dianilines A–C did not yield discrete complexes with any of these templates, and D with FeII or CoII gave intractable mixtures of products, D (6 equiv.) was observed to generate metal–organic cages 1 and 2 following treatment with 2-formylpyridine (12 equiv.) and either zinc(II) or cadmium(II) di[bis(trifluoromethylsulfonyl)imide] (triflimide, NTf2−; 4 equiv.) respectively, as the uniquely observed products in solution (Fig. 1; ESI Sections 1.3 and 1.4†). Confirmation of the MII4L6 stoichiometry was provided by electrospray ionization mass spectrometry (ESI MS; Fig. S10a and S20b†), including high resolution MS (Fig. S10b and S20b†).
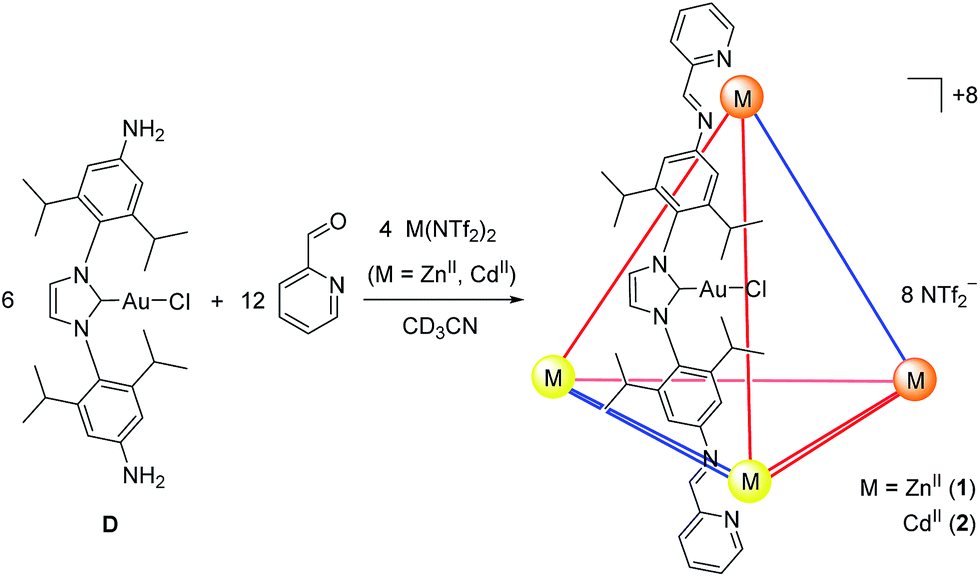 | ||
| Fig. 1 Dianiline D was the only dianiline observed to form ZnII4L6 (1) and CdII4L6 (2) cages through subcomponent self-assembly. These cages are inferred to have approximate S4 symmetry, consisting of two Δ (orange) and two Λ (yellow) metal vertices and two anti (blue) and four syn ligands (red). Only one (syn) ligand is shown for clarity. | ||
The observation that dianilines A–C did not lead to discrete cage formation suggested that a strong and inert metal-NHC bond was required in the ligand to construct a cage, which was provided by the gold(I) chloride moiety in D. Fluorescence spectroscopy for both 1 and 2 in dry acetonitrile (Fig. S11, S12, S21 and S22,† respectively) indicated weak luminescence, with bands that are attributed to both intra- and intermolecular AuI⋯AuI interactions, as observed in gold(I)-containing supramolecular architectures developed by Yam and co-workers.60 The removal of the gold-bound chloride from ligand D in cages 1 and 2 following the addition of chloride-binding metal cations, such as stoichiometric AgI or excess ZnII or CdII (Fig. S13, S14, S23 and S24†) caused disassembly, suggesting that coulombic repulsion between adjacent ligands destabilized the cage framework. Agents that engender the removal of chloride, therefore, provided a stimulus to trigger the response of cage destruction.
NMR spectra for the products 1 and 2 (Fig. S5 and S15,† respectively) yielded three imine signals. In the case of 2, satellite signals associated with the imines were observed, attributed to J-coupling with the two spin-1/2 isotopes of cadmium.61 The three magnetically inequivalent ligand environments observed in both the 1H and 13C NMR spectra of the cages suggested that the achiral S4 (ΛΛΔΔ) diastereomer of a M4L6 framework was present in solution, but not the homochiral T (ΔΔΔΔ/ΛΛΛΛ) or heterochrial C3 (ΔΔΔΛ/ΛΛΛΔ) diastereomers.62,63 The steric bulk of the isopropyl groups is expected to hold the phenyl rings orthogonal to the central imidazolium ring; this coplanar orientation of the terminal phenylene rings has been shown to favor a syn arrangement of the ligands,64 making the S4-symmetric framework the lowest-energy conformation, and thus the unique product observed in solution. Diffusion ordered spectroscopy (DOSY) was consistent in each case with all signals belonging to a single species (Fig. S9 and S19†). The 19F NMR signal for NTf2− was unchanged from its free value in cages 1 and 2, consistent with the absence of anion encapsulation (Fig. S6 and S16†).
In order to visualize the cage geometry, an energy-minimized model of 2 in the S4 arrangement65 was constructed (Fig. 2b) based upon the X-ray crystal structure of D (Fig. 2a). The achiral framework is composed of two metal centers of the same handedness (Λ) and two metal centers of opposite handedness (Δ). Each pair of metal centers of the same stereochemistry is connected by ligands adopting anti conformations (where the –AuCl moieties are directed to the outside of the cage), and the other four ligands are syn (where the –AuCl moieties are directed inwards); we acknowledge that other conformations may be possible in solution. Our model suggests that the cages are sufficiently flexible to allow the gold(I) centers to approach each other, accounting for the weak luminescence associated with the presence of gold–gold interactions (further modelling of the intermolecular cage interactions in 2 is provided in Fig. S22†).
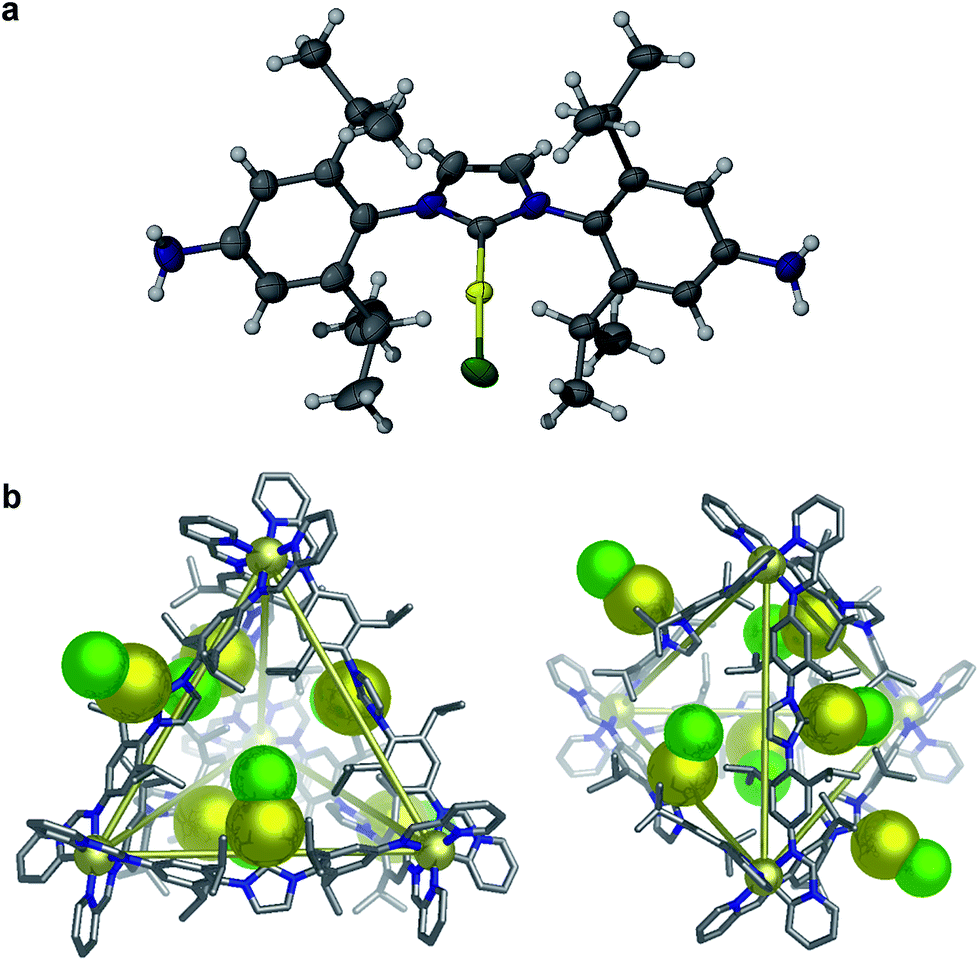 | ||
| Fig. 2 (a) ORTEP representation of the crystal structure of D, showing thermal ellipsoids at 40% probability level. Solvent molecules and disorder are omitted for clarity. (b) Two views of a model of the MM2 (ref. 66) energy-minimized structure of 2. The –AuCl units are shown in space-filling representation. Hydrogens and counterions are omitted for clarity (color scheme: carbon, grey; nitrogen, blue; cadmium, light yellow; gold, dark yellow; chloride, green). | ||
Nanoparticle synthesis
Amines have been shown to reduce cationic gold species to Au0 and to cap the gold nanoparticles (Au NPs) formed following growth.67–70 Dianiline D thus possesses two useful characteristics in this context: it can reduce a gold source to Au0 (which can be stabilized by the oxidized aniline) and the NHC coordinated AuI can also participate in the formation of Au NPs, stabilized by the available carbene. The reduction of a soluble AuI source in acetonitrile (Au(tmbn)2SbF6; tmbn = 2,4,6-trimethoxybenzonitrile) for Au NP growth was thus investigated in two cases: with dianiline D alone, and when D was incorporated into cages 1 and 2.Following the addition of Au(tmbn)2SbF6 (6 equiv.) to a solution of 1 in dry acetonitrile (ESI Section 3.1†), the surface plasmon resonance (SPR) band in the UV-Vis spectrum, attributable to Au NPs,71 was observed to grow in following a sigmoidal time course with steps corresponding to nucleation, growth, and saturation (Fig. 3a); consistent growth features were observed in a second run (Fig. S26†). When cadmium-containing cage 2 was mixed with Au(tmbn)2SbF6 (6 equiv.) (ESI Section 3.2†), the SPR band in the UV-Vis spectra began to grow in intensity approximately 22 min after mixing (Fig. 3a), a delay 10 min longer than was observed in the case of 1 (Fig. S36†). In both experiments, NP growth was observed only after part of the cage had first disassembled (Fig. S27, S28, S37 and S39†). When cage 1 decomposed to provide the reducing agent, TEM (Fig. S29†) and AFM (Fig. S30†) images displayed aggregates (250 nm in diameter) of approximately 5 nm Au NPs, together with Au NPs approximately 50 nm in diameter (Fig. 3b), following 90 min of NP growth. In contrast, when cage 2 disassembled {111}-faceted triangular prisms approximately 300 nm in edge length were observed (Fig. 3b), in addition to aggregates of 5 nm Au NPs (Fig. S40 and S41†). Particle size distribution analysis of AFM measurements confirmed the presence of the larger triangular species alongside numerous small NP (Fig. S41d–h†); the triangular prisms constituted approximately 10% of the species counted, but approximately 52% of the total gold.
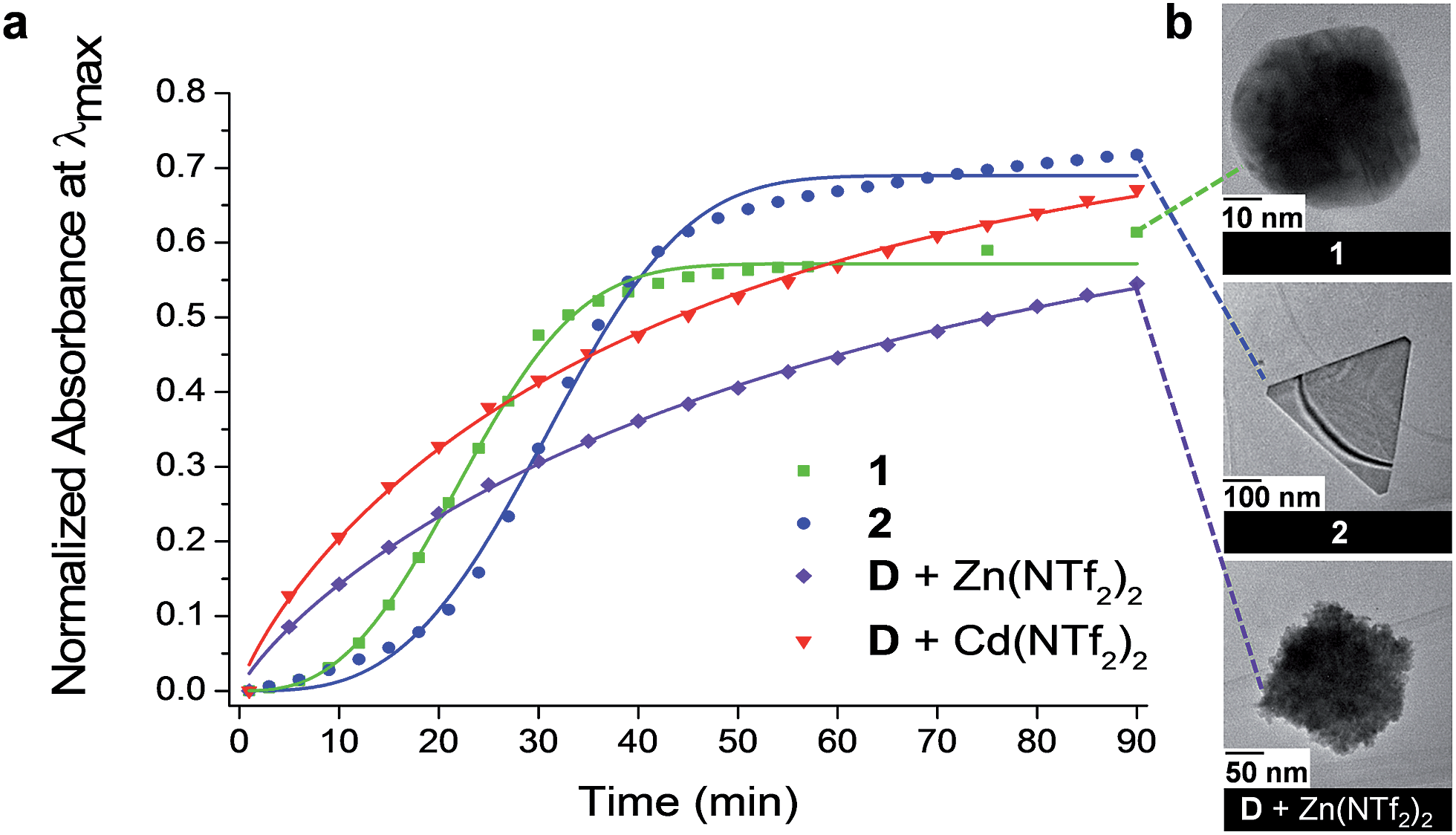 | ||
| Fig. 3 (a) The evolution of the SPR UV-Vis band was monitored during Au NP formation when the reducing agent employed was provided by cages 1 and 2 and with D and either Zn(NTf2)2 or Cd(NTf2)2; the curves were fitted using the Avrami equation72,73 (ESI Section 3.6†). (b) Differences in Au NP morphology observed in the TEM images for growth conditions shown in (a). | ||
We also observed that when reagent grade acetonitrile (0.01% H2O) was used in place of the anhydrous solvent (<0.001% H2O) during the synthesis of Au NPs with cage 2,74 the SPR band in the UV-Vis spectra appeared more rapidly (Fig. S42†). We infer that the higher water content of the solvent increased the rate of imine hydrolysis in 2, resulting in a shortening of the nucleation stage. Instead of the triangular prisms observed with anhydrous solvent, TEM images revealed hexagonal plates (Fig. S42†) and aggregates of 5 nm particles. This difference in Au NP morphology may be attributed to the variation in the rate of cage breakdown.
Notably, control experiments involving equimolar quantities of D, Zn(NTf2)2 or Cd(NTf2)2 and Au(tmbn)2SbF6 (ESI Sections 3.1.1 and 3.2.1†) resulted in a SPR band in the UV-Vis spectra immediately following mixing (Fig. 3a) and significantly different NP morphologies. When equimolar quantities of D and Zn(NTf2)2 were mixed with Au(tmbn)2SbF6, only aggregates were observed in the TEM images (Fig. 3b); nanoparticles greater than 5 nm in diameter were not observed in contrast to the case when 1 provided reductant D through its hydrolysis. In addition, none of the anisotropic Au NP morphologies grown starting from 2 were observed in the control experiment involving equimolar quantities of D, Cd(NTf2)2 and Au(tmbn)2SbF6. Further control experiments in which only A or D were used in the reduction of AuI (ESI Sections 3.3 and 3.4,† respectively) revealed similarly rapid rates of formation and small nanoparticle morphologies in the presence of either CdII or ZnII. Energy-dispersive X-ray spectroscopy (EDS) confirmed the presence of gold (with small quantities of nitrogen) across all experiments (Fig. S29, S32, S40 and S44†), with a higher organic content observed in the aggregates of smaller Au NPs compared to the larger anisotropic species; spectral features consistent with Zn or Cd were not observed, suggesting that these metals were not incorporated into the NPs. The aggregation of the Au NPs and the observation of organic material in the EDS spectra suggested that the oxidized bis-monodentate ligand D may have acted to bridge between Au NPs, as has been observed with similar ligands.75
The kinetics of Au NP formation were assessed with the Avrami theoretical model for crystallization and growth72,73,76 (ESI Section 3.6†). In the Avrami equation, the overall kinetics are described by the apparent rate parameter kapp which depends principally upon the nucleation rate – a slower kapp is attributed to a lengthier nucleation stage.77 The kapp of Au NP growth when 1 provided the reducing agent was 1.8 × 10−4 (the units are min−n for all rate constants), with an Avrami exponent (n) of 2.59; in a control experiment using D and Zn(NTf2)2 as reductant, the rate constant was 1.70 × 10−2 with an exponent of 0.97. Similarly, Au NP growth where the reducing agent was provided by 2 displayed a rate constant of 1.5 × 10−6 with an exponent of 3.4, and the control experiment using D and Cd(NTf2)2 gave a kapp of 3.8 × 10−2 with an exponent of 0.90 (Table S1†). As the Avrami exponent n78–82 has been inferred to reflect the nucleation mechanism and directionality of growth, the constant nucleation rate (n ≈ 1) observed when D served as the reductant with different metal salts is consistent with the observation of spherical Au NPs. Similarly, the multi-dimensional growth (n > 1) observed when 1 or 2 were used as reductants is consistent with a heterogeneous nucleation mechanism being responsible for the anisotropic features.72 The rate constants and Avrami exponents observed when 1 and 2 were used as reductants suggest that the supramolecular structures impeded Au0 nucleation, in contrast to the faster nucleation observed when free D was present in solution.
Mechanistically, we infer that the addition of AuI to a solution of 1 or 2 first resulted in the abstraction of chloride, as noted above in the cases of other metal salts. This destabilizes the cage and results in its disassembly and release of free subcomponent D. We attribute the difference in rate of dianiline release between 1 and 2 to the differences in thermodynamic stabilities between these two structures. The shorter nucleation stage provided by the breakdown of 1 in comparison to the longer nucleation stage provided by 2 is consistent with the labile ZnII centers that have faster ligand exchange kinetics;83 it is this delay during the nucleation stage of Au NP growth that ultimately determines the final NP size and shape. The slow rate of D release from 1 and 2 thus brought about a longer nucleation stage and slower rate of AuI reduction, as required to produce thermodynamically-favored shapes with low-index facets, such as the {111}-faceted triangular prisms.84 Several other factors, including the presence of small amounts of different salts (such as ZnCl2 or CdCl2 from cage decomposition, or the metal triflate salts) could also influence the final particle shape and kinetics of nanoparticle growth,84 but the control experiments involving D and ZnII or CdII were not consistent with this mechanism. The different growth rates and morphologies of the observed Au NPs were thus inferred to result from the incorporation of an AuI-NHC dianiline ligand into the walls of metal–organic capsule as the diimine.
Conclusions
We have reported the synthesis of NHC-containing dianilines A–D, and demonstrated that subcomponent D alone was able to form supramolecular cages with zinc(II) and cadmium(II), thanks to the protection afforded to the reactive carbene center by the AuICl moiety; we anticipate that similarly inert metal-bound NHC complexes, such as Pt(II)-NHC,85 could also be used to this effect. The removal of this gilding resulted in cage decomposition, which could be productively used in order to build up Au NPs of controllably variable morphologies. This supramolecular approach to modulating the rate of release of reducing agent affords a means of indirectly programming the morphology of Au NPs through reversible protection of the reducing amines within dynamic imine linkages. Our method circumvents the need to directly control the rate of reductant release, as is currently required in order to control Au NP morphology.84 The reagent release profile is governed by the thermodynamic stability of the supramolecular structure and can be purposely altered by adding chemical stimuli that disrupt cage stability. Although this method of control is prohibitively expensive, the controllable nonlinearity of response that we observed may be of interest in the context of signal transduction in complex chemical networks.86Acknowledgements
This work was supported by the Marie Curie Academic-Industrial Initial Training Network on Dynamic Molecular Nanostructures (DYNAMOL) and the Engineering and Physical Sciences Research Council (EPSRC).Notes and references
- T. R. Cook and P. J. Stang, Chem. Rev., 2015, 115, 7001–7045 CrossRef CAS PubMed
.
- J. M. Rivera, T. Martín and J. Rebek, Science, 1998, 279, 1021–1023 CrossRef CAS
- T. Liu, Y. Liu, W. Xuan and Y. Cui, Angew. Chem., Int. Ed., 2010, 49, 4121–4124 CrossRef CAS PubMed
- P. D. Frischmann, S. H. M. Mehr, B. O. Patrick, F. Lelj and M. J. MacLachlan, Inorg. Chem., 2012, 51, 3443–3453 CrossRef CAS PubMed
- S. Mirtschin, A. Slabon-Turski, R. Scopelliti, A. H. Velders and K. Severin, J. Am. Chem. Soc., 2010, 132, 14004–14005 CrossRef CAS PubMed
- M. M. J. Smulders, S. Zarra and J. R. Nitschke, J. Am. Chem. Soc., 2013, 135, 7039–7046 CrossRef CAS PubMed
- J. S. Mugridge, G. Szigethy, R. G. Bergman and K. N. Raymond, J. Am. Chem. Soc., 2010, 132, 16256–16264 CrossRef CAS PubMed
- R. Custelcean, P. V. Bonnesen, N. C. Duncan, X. Zhang, L. A. Watson, G. van Berkel, W. B. Parson and B. P. Hay, J. Am. Chem. Soc., 2012, 134, 8525–8534 CrossRef CAS PubMed
- S. K. Samanta and M. Schmittel, Org. Biomol. Chem., 2013, 11, 3108–3115 CAS
- R. Frantz, C. S. Grange, N. K. Al-Rasbi, M. D. Ward and J. Lacour, Chem. Commun., 2007, 1459–1461 RSC
- S. Mecozzi and J. J. Rebek, Chem.–Eur. J., 1998, 4, 1016–1022 CrossRef CAS
- G. Zhang, O. Presly, F. White, I. M. Oppel and M. Mastalerz, Angew. Chem., Int. Ed., 2014, 53, 5126–5130 CAS
- M. Han, J. Hey, W. Kawamura, D. Stalke, M. Shionoya and G. H. Clever, Inorg. Chem., 2012, 51, 9574–9576 CrossRef CAS PubMed
- K. Liu, Y. Kang, Z. Wang and X. Zhang, Adv. Mater., 2013, 25, 5530–5548 CrossRef CAS PubMed
- B. Olenyuk, J. A. Whiteford, A. Fechtenkotter and P. J. Stang, Nature, 1999, 398, 796–799 CrossRef CAS PubMed
- B. F. Abrahams, S. J. Egan and R. Robson, J. Am. Chem. Soc., 1999, 121, 3535–3536 CrossRef CAS
- M. Eddaoudi, J. Kim, J. B. Wachter, H. K. Chae, M. O'Keeffe and O. M. Yaghi, J. Am. Chem. Soc., 2001, 123, 4368–4369 CrossRef CAS
- H. Arora, J. Cano, F. Lloret and R. Mukherjee, Dalton Trans., 2011, 40, 10055–10062 RSC
- R. W. Saalfrank, A. Stark, K. Peters and H. G. von Schnering, Angew. Chem., Int. Ed. Engl., 1988, 27, 851–853 CrossRef PubMed
- N. Takeda, K. Umemoto, K. Yamaguchi and M. Fujita, Nature, 1999, 398, 794–796 CrossRef CAS
- P. Mal, B. Breiner, K. Rissanen and J. R. Nitschke, Science, 2009, 324, 1697–1699 CrossRef CAS PubMed
- H. T. Chifotides, I. D. Giles and K. R. Dunbar, J. Am. Chem. Soc., 2013, 135, 3039–3055 CrossRef CAS PubMed
- M.-K. Chung, K. Severin, S. J. Lee, M. L. Waters and M. R. Gagne, Chem. Sci., 2011, 2, 744–747 RSC
- T. Weilandt, U. Kiehne, J. Bunzen, G. Schnakenburg and A. Lützen, Chem.–Eur. J., 2010, 16, 2418–2426 CrossRef CAS PubMed
- T. K. Ronson, S. Zarra, S. P. Black and J. R. Nitschke, Chem. Commun., 2013, 49, 2476–2490 RSC
- J. Dömer, J. C. Slootweg, F. Hupka, K. Lammertsma and F. E. Hahn, Angew. Chem., 2010, 122, 6575–6578 CrossRef PubMed
- X.-P. Zhou, Y. Wu and D. Li, J. Am. Chem. Soc., 2013, 135, 16062–16065 CrossRef CAS PubMed
- S. Pasquale, S. Sattin, E. C. Escudero-Adán, M. Martínez-Belmonte and J. de Mendoza, Nat. Commun., 2012, 3, 785 CrossRef PubMed
- L. Isaacs, Acc. Chem. Res., 2014, 47, 2052–2062 CrossRef CAS PubMed
- P. Ballester, Chem. Soc. Rev., 2010, 39, 3810–3830 RSC
- H.-J. Schneider and A. K. Yatsimirsky, Chem. Soc. Rev., 2008, 37, 263–277 RSC
- M. I. Sánchez, J. Mosquera, M. E. Vázquez and J. L. Mascareñas, Angew. Chem., Int. Ed., 2014, 53, 9917–9921 CrossRef PubMed
- S. J. Edwards, H. Valkenier, N. Busschaert, P. A. Gale and A. P. Davis, Angew. Chem., Int. Ed., 2015, 54, 4592–4596 CrossRef CAS PubMed
- K. Yazaki, Y. Sei, M. Akita and M. Yoshizawa, Nat. Commun., 2014, 5, 5179 CrossRef CAS PubMed
- J. Kang, J. Santamaría, G. Hilmersson and J. Rebek, J. Am. Chem. Soc., 1998, 120, 7389–7390 CrossRef CAS
- D. J. Cram, M. E. Tanner and R. Thomas, Angew. Chem., Int. Ed. Engl., 1991, 30, 1024–1027 CrossRef PubMed
- L. M. Hancock, L. C. Gilday, S. Carvalho, P. J. Costa, V. Félix, C. J. Serpell, N. L. Kilah and P. D. Beer, Chem.–Eur. J., 2010, 16, 13082–13094 CrossRef CAS PubMed
- G. Barin, M. Frasconi, S. M. Dyar, J. Iehl, O. Buyukcakir, A. A. Sarjeant, R. Carmieli, A. Coskun, M. R. Wasielewski and J. F. Stoddart, J. Am. Chem. Soc., 2013, 135, 2466–2469 CrossRef CAS PubMed
- I. Pochorovski, M.-O. Ebert, J.-P. Gisselbrecht, C. Boudon, W. B. Schweizer and F. Diederich, J. Am. Chem. Soc., 2012, 134, 14702–14705 CrossRef CAS PubMed
- Y. Li and A. H. Flood, Angew. Chem., Int. Ed., 2008, 47, 2649–2652 CrossRef CAS PubMed
- K. K. Tanabe and S. M. Cohen, Chem. Soc. Rev., 2011, 40, 498–519 RSC
- P. L. Golas and K. Matyjaszewski, Chem. Soc. Rev., 2010, 39, 1338–1354 RSC
- D. Konkolewicz, A. Gray-Weale and S. Perrier, J. Am. Chem. Soc., 2009, 131, 18075–18077 CrossRef CAS PubMed
- D. A. Roberts, A. M. Castilla, T. K. Ronson and J. R. Nitschke, J. Am. Chem. Soc., 2014, 136, 8201–8204 CrossRef CAS PubMed
- M. Han, R. Michel, B. He, Y.-S. Chen, D. Stalke, M. John and G. H. Clever, Angew. Chem., Int. Ed., 2013, 52, 1319–1323 CrossRef CAS PubMed
- H. J. Yoon, J. Kuwabara, J.-H. Kim and C. A. Mirkin, Science, 2010, 330, 66–69 CrossRef CAS PubMed
- M. N. Hopkinson, C. Richter, M. Schedler and F. Glorius, Nature, 2014, 510, 485–496 CrossRef CAS PubMed
- D. Gatineau, J.-P. Goddard, V. Mouriès-Mansuy and L. Fensterbank, Isr. J. Chem., 2013, 53, 892–900 CrossRef CAS PubMed
- E. R. T. Tiekink, Crit. Rev. Oncol. Hematol., 2002, 42, 225–248 CrossRef
- P. Espinet, Gold Bull., 1999, 32, 127–134 CrossRef CAS
- R. Visbal, I. Ospino, J. M. López-de-Luzuriaga, A. Laguna and M. C. Gimeno, J. Am. Chem. Soc., 2013, 135, 4712–4715 CrossRef CAS PubMed
-
D. M. Roundhill and J. P. Fackler, Optoelectronic Properties of Inorganic Compounds, Springer, USA, 2013 Search PubMed
- C. E. Willans, K. M. Anderson, P. C. Junk, L. J. Barbour and J. W. Steed, Chem. Commun., 2007, 3634–3636 RSC
- T. Fahlbusch, M. Frank, G. Maas and J. Schatz, Organometallics, 2009, 28, 6183–6193 CrossRef CAS
- I. J. B. Lin and C. S. Vasam, Can. J. Chem., 2005, 83, 812–825 CrossRef CAS
- C. Mejuto, G. Guisado-Barrios, D. Gusev and E. Peris, Chem. Commun., 2015, 51, 13914–13917 RSC
- J. Vignolle and T. D. Tilley, Chem. Commun., 2009, 7230–7232 RSC
- A. Hospital, C. Gibard, C. Gaulier, L. Nauton, V. Thery, M. El-Ghozzi, D. Avignant, F. Cisnetti and A. Gautier, Dalton Trans., 2012, 41, 6803–6812 RSC
- O. Santoro, A. Collado, A. M. Z. Slawin, S. P. Nolan and C. S. J. Cazin, Chem. Commun., 2013, 49, 10483–10485 RSC
- X.-F. Jiang, F. K.-W. Hau, Q.-F. Sun, S.-Y. Yu and V. W.-W. Yam, J. Am. Chem. Soc., 2014, 136, 10921–10929 CrossRef CAS PubMed
- W. Meng, T. K. Ronson and J. R. Nitschke, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 10531–10535 CrossRef CAS PubMed
- A. M. Castilla, W. J. Ramsay and J. R. Nitschke, Acc. Chem. Res., 2014, 47, 2063–2073 CrossRef CAS PubMed
- D. L. Caulder, C. Brückner, R. E. Powers, S. König, T. N. Parac, J. A. Leary and K. N. Raymond, J. Am. Chem. Soc., 2001, 123, 8923–8938 CrossRef CAS PubMed
- W. Meng, J. K. Clegg, J. D. Thoburn and J. R. Nitschke, J. Am. Chem. Soc., 2011, 133, 13652–13660 CrossRef CAS PubMed
- T. Beissel, R. E. Powers, T. N. Parac and K. N. Raymond, J. Am. Chem. Soc., 1999, 121, 4200–4206 CrossRef CAS
-
CAChe Workspace, WorkSystem Pro Version 7.5.0.85
Search PubMed
- M. Aslam, L. Fu, M. Su, K. Vijayamohanan and V. P. Dravid, J. Mater. Chem., 2004, 14, 1795–1797 RSC
- S. Gomez, K. Philippot, V. Colliere, B. Chaudret, F. Senocq and P. Lecante, Chem. Commun., 2000, 1945–1946 RSC
- X. Lu, M. S. Yavuz, H.-Y. Tuan, B. A. Korgel and Y. Xia, J. Am. Chem. Soc., 2008, 130, 8900–8901 CrossRef CAS PubMed
- C. Subramaniam, R. Tom and T. Pradeep, J. Nanopart. Res., 2005, 7, 209–217 CrossRef CAS
- W. Haiss, N. T. K. Thanh, J. Aveyard and D. G. Fernig, Anal. Chem., 2007, 79, 4215–4221 CrossRef CAS PubMed
- Y. Zhou, W. Lin, F. Yang, W. Fang, J. Huang and Q. Li, Chem. Phys., 2014, 441, 23–29 CrossRef CAS PubMed
- M. Avrami, J. Chem. Phys., 1940, 8, 212–224 CrossRef CAS PubMed
- HPLC grade acetonitrile was purchased from Fisher Scientific with a 0.008% water content (max. 0.01%). Anhydrous acetonitrile (99.8%) was purchased from Sigma-Aldrich with a <0.001% water content.
- M. Orbach, M. Lahav, P. Milko, S. G. Wolf and M. E. van der Boom, Angew. Chem., Int. Ed., 2012, 51, 7142–7145 CrossRef CAS PubMed
- P. N. Njoki, J. Luo, M. M. Kamundi, S. Lim and C.-J. Zhong, Langmuir, 2010, 26, 13622–13629 CrossRef CAS PubMed
- M. L. Di Lorenzo and C. Silvestre, Prog. Polym. Sci., 1999, 24, 917–950 CrossRef CAS
- A. T. W. Kempen, F. Sommer and E. J. Mittemeijer, J. Mater. Sci., 2002, 37, 1321–1332 CrossRef CAS
- T. A. Baker, O. L. A. Monti and D. J. Nesbitt, J. Phys. Chem. C, 2011, 115, 9861–9870 CAS
- S. Ranganathan and M. von Heimendahl, J. Mater. Sci., 1981, 16, 2401–2404 CrossRef CAS
- J. P. Gaviría, L. G. Navarro and A. E. Bohé, J. Phys. Chem. A, 2012, 116, 2062–2070 CrossRef PubMed
- G. Oyama, Y. Yamada, R.-I. Natsui, S.-I. Nishimura and A. Yamada, J. Phys. Chem. C, 2012, 116, 7306–7311 CAS
- I. A. Riddell, Y. R. Hristova, J. K. Clegg, C. S. Wood, B. Breiner and J. R. Nitschke, J. Am. Chem. Soc., 2013, 135, 2723–2733 CrossRef CAS PubMed
- M. L. Personick and C. A. Mirkin, J. Am. Chem. Soc., 2013, 135, 18238–18247 CrossRef CAS PubMed
- E. A. Baquero, J. C. Flores, J. Perles, P. Gómez-Sal and E. de Jesús, Organometallics, 2014, 33, 5470–5482 CrossRef CAS
- D. Ray, J. T. Foy, R. P. Hughes and I. Aprahamian, Nat. Chem., 2012, 4, 757–762 CrossRef CAS PubMed
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental procedures, synthesis and characterization data for subcomponents and cages, and optical, microscopy, ESI-MS and NMR data. CCDC 1061936. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5sc03065j |
| ‡ Current address: Department of Chemistry, University of Sheffield, Sheffield, S3 7HF, UK. |
| This journal is © The Royal Society of Chemistry 2015 |