
DNA nanotubes and helical nanotapes via self-assembly of ssDNA-amphiphiles†
Timothy R.
Pearce
a and
Efrosini
Kokkoli
*b
aDepartment of Biomedical Engineering, University of Minnesota, Minneapolis, MN 55455, USA
bDepartment of Chemical Engineering and Materials Science, University of Minnesota, Minneapolis, MN 55455, USA. E-mail: kokkoli@umn.edu
First published on 29th October 2014
Abstract
DNA nanotubes were created using molecular self-assembly of single-stranded DNA (ssDNA)-amphiphiles composed of a hydrophobic dialkyl tail and polycarbon spacer and a hydrophilic ssDNA headgroup. The nanotube structures were formed by bilayers of amphiphiles, with the hydrophobic components forming an inner layer that was shielded from the aqueous solvent by an outer layer of ssDNA. The nanotubes appeared to form via an assembly process that included transitions from twisted nanotapes to helical nanotapes to nanotubes. Amphiphiles that contained different ssDNA headgroups were created to explore the effect of the length and secondary structure of the ssDNA headgroup on the self-assembly behavior of the amphiphiles in the presence and absence of the polycarbon spacer. It was found that nanotubes could be formed using a variety of headgroup lengths and sequences. The ability to create nanotubes via ssDNA-amphiphile self-assembly offers an alternative to the other purely DNA-based approaches like DNA origami and DNA tile assembly for constructing these structures and may be useful for applications in drug delivery, biosensing, and electronics.
Introduction
The field of DNA nanotechnology has transformed DNA from a biological material that stores genetic information into a construction material that can be used to build 3-dimensional scaffolds, structures, and devices with nanoscale features.1,2 The ability to precisely control the organization of DNA relies on Watson–Crick base pairing, which acts as a molecular glue to hold strands of DNA together in a predictable manner. There are a variety of strategies that can be used to create DNA nanostructures, each that use a combination of different ssDNA sequences that when mixed together and subjected to specific annealing conditions (i.e., controlled cooling rates, specific ions, and pH) fold together to produce double stranded DNA segments that organize into highly uniform structures of the desired shape.3–5 The predictability of base pairing offers an opportunity to rationally select these ssDNA sequences, often with the aid of software, that can combine together to form tetrahedrons, cages, barrels, and tube structures while maintaining ssDNA overhangs that act as addressable locations and allow the structures to be further functionalized with drugs, dyes, and metals for use as therapeutics, diagnostics, electronics and photonics, and in molecular and cellular biophysical studies.2,5An alternative approach to form DNA nanostructures is to covalently link hydrophilic ssDNA sequences with hydrophobic tails (polymers or other hydrophobic moieties) to form amphiphilic molecules.6,7 The amphiphilic nature of the conjugates induces their spontaneous assembly when added to an aqueous environment, with the hydrophobic tails preferring to sequester themselves into a hydrophobic domain while the ssDNA sequences extend into the aqueous solution. With this structural arrangement the ssDNA is not required to base pair in order to create the nanostructure and remains available for base pairing with complimentary ssDNA sequences. Additionally, this approach to forming DNA nanostructures does not require base pairing prediction software and reduces the requirements for specific annealing conditions. However, this approach has not yet been used to create nanostructures with similar levels of complexity as those achieved by other approaches like DNA origami and DNA tile assembly.5 To date, the majority of structures created by ssDNA-amphiphile assembly have been spherical and cylindrical micelles.6,8
In pursuit of enhancing the level of structural complexity achievable through self-assembly of ssDNA-amphiphiles we recently tested how an additional building block, a spacer molecule used to link a ssDNA aptamer headgroup and hydrophobic lipid-like tail, could affect ssDNA-amphiphile assembly.9 It was found that globular micelles were formed when a 25 nucleotide aptamer was directly conjugated to a C16 dialkyl tail or conjugated to the tail via hydrophilic PEG4 or PEG8 spacers, but that flat and twisted nanotapes comprised of bilayers of amphiphiles were formed when hydrophobic C12 and C24 spacers were used.9 The nanotape morphology achieved by including a hydrophobic spacer in the design of the amphiphile was not predicted by the standard packing parameter analysis, leading to the hypothesis that polycarbon spacers, through attractive hydrophobic interactions, may force the aptamer headgroups into close proximity of each other, thus reducing the interfacial headgroup area and allowing the nanotapes to form.9 We have also recently shown that amphiphiles created with a 40 nucleotide ssDNA aptamer headgroup containing a large number of guanine nucleotides capable of forming intermolecular parallel G-quadruplexes with neighboring aptamer headgroups self-assembled into nanotapes in the absence of a polycarbon spacer.10 This finding suggested that the intermolecular interactions that produced the G-quadruplex structure may have reduced the effective headgroup area of the ssDNA in a manner analogous to the polycarbon spacer and encouraged the assembly of bilayer nanotapes.10
These previous findings suggested that variations in the ssDNA headgroups could influence the self-assembly behavior of ssDNA-amphiphiles. To investigate this possibility, ssDNA headgroups with random nucleotide sequences of variable length (10, 25, and 40 nucleotides) were conjugated to hydrophobic tails via the C12 spacer that was previously found to be important for forming twisted nanotape structures.9 ssDNA headgroups that lacked guanine nucleobases were selected to eliminate the possibility of G-quadruplex interactions within the ssDNA headgroups. Additional headgroups that contained guanine-rich sequences at the 5′ region of the headgroup were also created and directly conjugated to the hydrophobic tails to determine the possibility of using a guanine-rich sequence as a replacement for the C12 spacer. Finally, amphiphiles that contained both a guanine-rich headgroup and the C12 spacer were created to study the combined effect of these two variables.
It was found that amphiphiles containing the C12 spacers and the random guanine-free ssDNA headgroups of each length not only self-assembled into globular micelles and twisted nanotapes, as seen previously, but also helical nanotapes and nanotubes, nanostructures that have never before been created using ssDNA-amphiphiles. Amphiphiles created with these same headgroups but without the C12 spacer were unable to form the twisted or helical nanotapes or nanotubes, demonstrating the importance of the hydrocarbon spacer for forming these larger, more complex structures. Headgroups with oligo-guanine (G5) sequences designed to replace the C12 spacer and recapture the capability to form the nanotape and nanotube structures only succeeded in producing these larger structures when the headgroup was 40 nucleotides in length. It was also found that in the absence of the C12 spacer, 25 and 40 nucleotide headgroups that contained a (GGGT)3 sequence, created to form intermolecular G-quadruplex interactions, could produce the twisted nanotape structures but not the helical nanotape and nanotube structures. Finally, when the C12 spacer was combined with the G5-containing headgroups 25 and 40 nucleotides in length all of the nanostructures seen in the initial set of samples that contained the C12 spacer and guanine-free headgroups were again produced, while the amphiphiles with the G5-modified headgroups 10 nucleotides in length only produced short nanotubes.
Materials and methods
Materials
Toluene, chloroform, acetone, methanol, and triethylamine (TEA) were purchased from Fischer Chemical (Hanover Park, IL). ssDNA was purchased from Integrated DNA Technologies (Coralville, IA), cetyl trimethylammonium bromide from Acros Organics (Morris Plains, NJ), and hexafluroisopropanol (HFIP) from Oakwood Products Inc. (West Columbia, SC). Lacey Formvar/carbon 200 mesh copper grids were purchased from Ted Pella Inc. (Redding, CA). Atomic force microscopy contact mode rectangular Si cantilevers with an Al-coated backside (NSC36/Al BS) were acquired from MikroMasch (Lady's Island, SC). Ruby mica sheets, V2 quality, were purchased from S&J Trading Inc. (Glen Oaks, NY). All other chemicals and materials were purchased from Sigma-Aldrich (St Louis, MO).ssDNA-amphiphile synthesis
The ssDNA sequences with amino-C6 linkers attached to their 5′ ends were conjugated directly to the N-hydroxysuccinimide (NHS) activated (C16)2-Glu-C2 tails11 (NoSPR), or to the tails via a C12 spacer using a solution-phase synthesis as described previously9 to create ssDNA-amphiphiles. Unreacted ssDNA was separated from ssDNA-amphiphiles using reverse-phase high performance liquid chromatography (HPLC). HPLC information: Zorbax C8 300 Å SB column, 5–90% B over 25 min, buffer A: H2O + 10% methanol, 100 mM HFIP, 14.4 mM TEA, buffer B: methanol, 100 mM HFIP, 14.4 mM TEA. To confirm the success of the synthesis the molecular weights of the purified amphiphiles were identified by liquid chromatography-mass spectroscopy (LC-MS) (Zorbax C3 300 Å SB column, 50–80% B over 15 min, buffer A: H2O + 15 mM ammonium acetate, buffer B: acetonitrile). Mass spectroscopy data were acquired with an Agilent MSD ion trap.Cryogenic transmission electron microscopy (cryo-TEM)
4.5 μL of 500 μM amphiphile solutions were deposited onto lacey Formvar/carbon copper grids that had been treated with glow discharge for 60 s and vitrified in liquid ethane by Vitrobot (Vitrobot parameters: 4 s blot time, 0 offset, 3 s wait time, 3 s relax time, ambient humidity). The grids were kept under liquid nitrogen until they were transferred to a Tecnai G2 Spirit TWIN 20-120 kV/LaB6 TEM operated with an acceleration voltage of 120 keV. Images were captured using an Eagle 2k CCD camera.Fluorescent microscopy
Nile red was added to ssDNA-amphiphile solutions at a ratio of 3 μL Nile red solution (0.1 mg mL−1 in methanol) to 50 μL of 20 μM amphiphile solutions to stain the hydrophobic areas of the self-assembled structures. 3 μL of the amphiphile solutions were deposited onto clean glass slides and covered with clean glass coverslips. Fluorescent images were obtained using an EVOS FL microscope (Life Technologies, Grand Island, NY) with a Texas Red light cube (Ex: 585 nm, Em: 624 nm).Circular dichroism (CD)
500 μM solutions of ssDNA-amphiphiles were diluted to 20 μM with Milli-Q water or 20 mM KCl solution and transferred to a 0.1 cm path length cuvette. CD spectra from 320–200 nm were collected using a Jasco J-815 spectrapolarimeter using a read speed of 50 nm min−1 in 1 nm steps. 3 accumulations per amphiphile solution were recorded with the background spectrum from the water automatically subtracted. The accumulations were averaged and the raw ellipticity values were converted to molar ellipticity.Atomic force microscopy (AFM)
20 μL of 10 mM MgCl2 solution was added to a freshly cleaved mica surface for 10 s and then removed to enhance the adhesion of the ssDNA-amphiphiles to the mica substrate. 5 μL of a 500 nM solution of ssDNA-amphiphile was deposited onto the mica surface and left for 5 min to allow time for the amphiphiles to adhere to the surface. The surface was subsequently washed twice with 20 μL of Milli-Q water and allowed to dry in air prior to imaging. AFM imaging was performed with a Nanoscope V Multimode 8 SPM (Bruker, Santa Barbara, CA) in contact mode in air using rectangular Si cantilevers with a typical probe tip radius of 8 nm.Results
ssDNA-amphiphile synthesis
An initial ssDNA headgroup 40 nucleotides in length was created using only adenine (A), cytosine (C), and thymine (T) nucleobases selected at random. This guanine-free (NoG) 40 nucleotide headgroup was then used to create headgroups with 10 and 25 nucleotides that conserved the nucleotide order at the 5′ end of the headgroup (Fig. 1A). A second version of the 10 nucleotide sequence was created that conserved the 3′ end of the headgroup, which provided a headgroup with the same length but a different random nucleotide sequence. Nucleotides containing the guanine nucleobase were used to replace some nucleotides at the 5′ end of the headgroups, either as a single string of five guanines (G5) or as a repeat of (GGGT)3 (Fig. 1A) to produce headgroups that had potential to form intermolecular G-quadruplex interactions. The 5′ ends of the ssDNA headgroups were conjugated to dialkyl tails via C12 spacer molecules or directly to the tails without the use of a spacer (Fig. 1B). Successful conjugation was confirmed by LC-MS (Table S1†).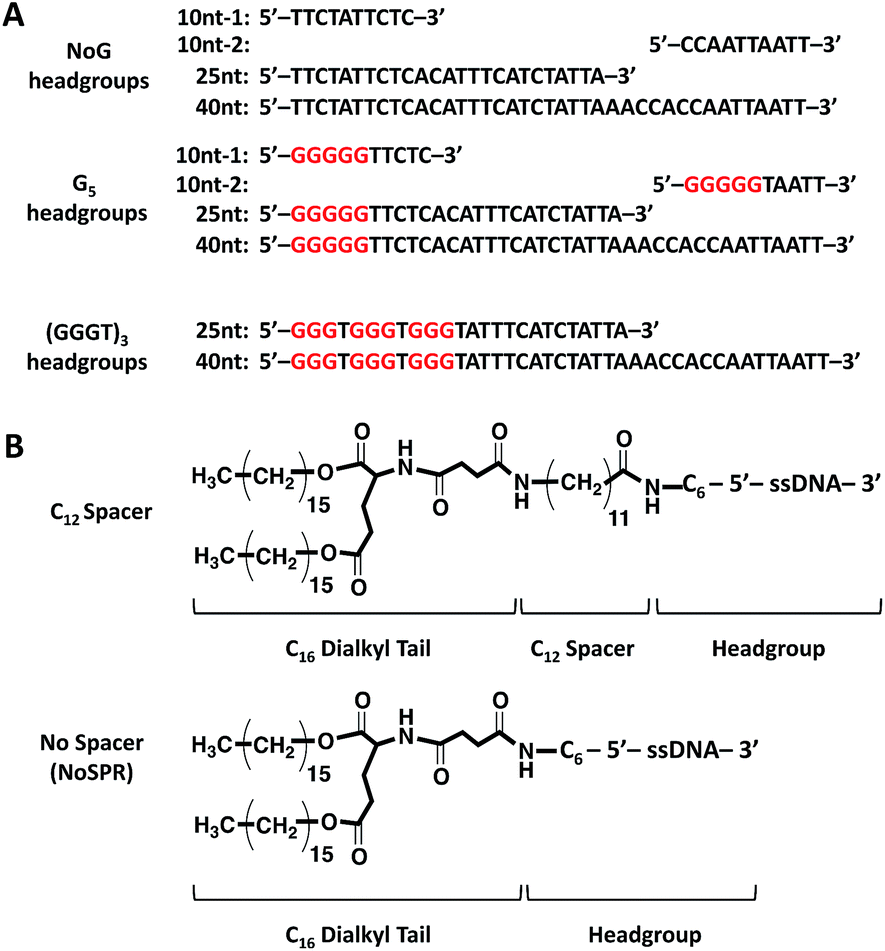 | ||
| Fig. 1 (A) Sequences of the 10 nucleotide (nt), 25 nucleotide, and 40 nucleotide guanine-free (NoG) and guanine-modified headgroups (having either a G5 or a (GGGT)3 sequence) used to create the ssDNA-amphiphiles. (B) Chemical structures of ssDNA-amphiphiles with a C16 dialkyl tail, a C12 spacer or without a spacer (NoSPR), and a ssDNA headgroup containing a C6 linker and having different sequences as shown in (A). | ||
Self-assembly of ssDNA-amphiphiles with NoG headgroups and with or without a C12 spacer
Amphiphiles with NoG headgroups attached to the hydrophobic tails via C12 spacers were dissolved in Milli-Q water to form 500 μM solutions and were immediately (within 30 min) deposited onto cryo-TEM grids, vitrified in liquid ethane, and imaged to visualize the morphology of the self-assembled structures formed by the amphiphiles. A variety of structures were present in each of the amphiphile solutions with either a 10, 25 or 40 nucleotide NoG headgroup and a C12 spacer: globular micelles, twisted nanotapes, helical nanotapes, and nanotubes (Fig. 2 and S1†). Of particular interest were the nanotube structures, which have never before been formed via self-assembly of ssDNA-amphiphiles. Analysis of an image of a nanotube created from amphiphiles with a 25 nucleotide NoG headgroup and a C12 spacer obtained at 0° and 45° stage-tilt (Fig. S2A and B†) showed that the diameter of the nanotube was unchanged when viewed from different angles, demonstrating its cylindrical shape. Line-scan analysis of the nanotube structure (Fig. S2C†) revealed a pattern of contrast consistent with that of a hollow tube, 34 nm in diameter with 10 nm thick walls, confirming the cylindrical structure observed in the sample is a nanotube. | ||
| Fig. 2 Cryo-TEM images of ssDNA-amphiphiles forming (A) a twisted nanotape, (B) helical nanotapes and (C) nanotubes. All amphiphiles contained the C12 spacer and either the (A) 25nt NoG, (B) 10nt-2 NoG, or (C) 10nt-1 NoG headgroup. | ||
The cylindrical nanotube structures observed in the samples with headgroups containing 10 nucleotides had an overall average diameter of 30 ± 4 nm, while samples with the 25 and 40 nucleotide headgroups produced structures with average diameters of 32 ± 3 nm and 31 ± 1 nm, respectively. While the overall average diameters of the nanotubes produced by amphiphiles of different headgroup lengths were similar, the diameters of the nanotubes varied between different nanotubes in the same sample, and in some cases there was also variation along the length of a single nanotube. The lengths of the nanotubes formed by amphiphiles containing the 10, 25, and 40 nucleotide headgroups were variable, with each sample producing nanotubes 100s to 1000s of nm in length and no apparent difference in the typical length between amphiphiles with different headgroups. High aspect ratio structures with lengths greater than 10 μm were observed in fluorescent images of amphiphile samples (Fig. S3†), providing further evidence that nanotubes and nanotapes assemble under ambient conditions. However, the resolution and magnification of the fluorescent imaging was not sufficient to definitively determine if the structures observed in the fluorescent images were single structures or aggregates and the sizes observed may not accurately represent the lengths and widths of the individual nanostructures.
Twisted and helical nanotapes were also observed in all the samples, but in lower numbers than the nanotubes. The majority of the twisted nanotapes in each of the different amphiphile samples did not twist in a periodic manner and had widths ranging from 20 to 50 nm. However, in the few instances the twisted nanotapes were observed to twist in a periodic manner they had an average pitch length of 132 ± 6 nm and an average width of 24 ± 2 nm. The helical nanotapes observed in each of the different amphiphile samples displayed clear periodicity with an average pitch length of 129 ± 7 nm, similar to that observed in the twisted nanotape structures. However, the average width of the helical nanotapes was 38 ± 4 nm, substantially larger than that of the regularly twisted nanotapes. Also present in all of the samples were globular micelles, some of which were spherical and some were weakly ellipsoidal. Micelles formed by each of the amphiphile samples had diameters (or ellipsoid axes lengths) of 9–20 nm with no measurable difference in average size between the amphiphiles with different length headgroups.
The same NoG headgroups were also conjugated directly to hydrophobic tails without the use of the C12 spacer (NoSPR) and imaged with cryo-TEM. These amphiphiles also formed micelles but were not observed to form any of the larger, more complex, bilayer nanotape and nanotube structures (Table 1). The inability for amphiphiles with NoG headgroups and lacking the C12 spacer to form more complex bilayer structures was not surprising as it has been previously shown that amphiphiles with headgroups of similar lengths that lack G-quadruplex interactions only assemble into globular micelles.9,10,12
| Sample | Twisted nanotape | Helical nanotape | Nanotube |
|---|---|---|---|
| a Nanotubes were substantially shorter in this sample than in all others. b Structures were observed infrequently. | |||
| 10nt NoG C12 | Yes | Yes | Yes |
| 25nt NoG C12 | Yes | Yes | Yes |
| 40nt NoG C12 | Yes | Yes | Yes |
| 10nt G5 C12 | No | No | Yesa |
| 25nt G5 C12 | Yes | Yes | Yes |
| 40nt G5 C12 | Yes | Yes | Yes |
| 10nt NoG NoSPR | No | No | No |
| 25nt NoG NoSPR | No | No | No |
| 40nt NoG NoSPR | No | No | No |
| 10nt G5 NoSPR | No | No | No |
| 25nt G5 NoSPR | No | No | No |
| 40nt G5 NoSPR | Yesb | Yesb | Yesb |
| 25nt (GGGT)3 NoSPR | Yesb | No | No |
| 40nt (GGGT)3 NoSPR | Yesb | No | No |
Self-assembly of ssDNA-amphiphiles with guanine-modified headgroups and without a C12 spacer
To test if the presence of guanines positioned immediately adjacent to the site of conjugation to the hydrophobic tail could produce nanotape and nanotube structures in the absence of the C12 spacer a third set of amphiphiles was created that included the G5 modification in the 10, 25, and 40 nucleotide ssDNA headgroups, with the headgroups directly linked to the hydrophobic tails (as shown in Fig. 1). It was hypothesized that the inclusion of the five guanines would produce intermolecular parallel G-quadruplex interactions between the headgroups that would bring the headgroups together and minimize the headgroup area in a similar manner as the C12 spacer, thus allowing the nanotapes to form. These amphiphile samples were dissolved in Milli-Q water at 500 μM, vitrified and imaged with cryo-TEM to determine their self-assembly behavior. The only structures observed in the amphiphile samples with 10 (Fig. S4A†) and 25 nucleotide headgroups were spherical and weakly ellipsoidal micelles (Table 1) that were of similar sizes as observed in the amphiphile samples with the NoG headgroups. Micelles of similar shape and size were also the most prevalent structure observed in the amphiphile samples with the 40 nucleotide G5-modified headgroup, but twisted and helical nanotapes and nanotubes (Fig. S4B and C,†Table 1) that were similar to those produced by the NoG headgroups with the C12 spacer were also observed infrequently.CD was performed on the 40 nucleotide G5-modified amphiphiles to probe for the presence of G-quadruplex formations within the headgroups of these amphiphiles. Parallel G-quadruplex structures are tertiary DNA structures formed by the stacking of G-quartet structures, with each G-quartet formed by four guanine nucleotides arranged in a planar, square geometry held together by Hoogsteen hydrogen bonding. These unique structures are stabilized by small cations that fit between the G-quartets but can also be formed in pure water13 and produce a characteristic CD spectrum with a strong positive peak between 258–265 nm.14,15 With only five guanines a single headgroup could not form a G-quadruplex with itself but it could form an intermolecular parallel G-quadruplex by interacting with three adjacent headgroups.16 However, contrary to the hypothesis, the CD spectrum of the 40 nucleotide G5-modified amphiphiles had a maximum at 270 nm in water, characteristic of a stem-loop, and only 1 nm different than the free ssDNA sequence (maximum at 271 nm) suggesting that there were no significant G-quadruplex interactions occurring between the amphiphiles' headgroups following self-assembly (Fig. S5B and Table S2†). Both the 40 nucleotide G5-modified amphiphile and ssDNA sequence had a maximum at 269 nm upon addition of 20 mM KCl (Fig. S5C and Table S2†), that is outside the wavelength range typically attributed to a G-quadruplex (258–265 nm) or stem-loop (270–285 nm)17,18 secondary structure.
In order to enhance the probability that the ssDNA headgroups would form parallel G-quadruplexes and provide additional knowledge about the effect of G-quadruplex interactions on the self-assembly of ssDNA-amphiphiles, two additional headgroups were created from the random guanine-free 25 and 40 nucleotide headgroups. These headgroups had the first 12 nucleotides of the original sequences replaced with the sequence (GGGT)3, as shown in Fig. 1, which is capable of inducing intermolecular G-quadruplexes.10 The CD spectra in Milli-Q water of the 25 and 40 nucleotide (GGGT)3-modified ssDNA sequences measured prior to conjugation to the hydrophobic tails showed a maximum at 273 nm and 272 nm respectively, for each length, which can be attributed to the standard Watson–Crick base-pairing produced in stem-loop secondary structures that typically have a maximum between 270 and 285 nm.17,18 Addition of 20 mM KCl shifted the signal closer to that of a G-quadruplex sequence (258–265 nm), to 266 nm for the 25 nucleotide and 267 nm for the 40 nucleotide (GGGT)3-modified ssDNA sequences. Following conjugation to the hydrophobic tails and subsequent self-assembly in Milli-Q water the 25 nucleotide long sequence produced a CD spectrum characteristic of G-quadruplex secondary structure, whereas the 40 nucleotide sequence had a maximum at 267 nm, in between the wavelength range for the G-quadruplex and stem-loop structures. Addition of KCl had no effect on the location of the long wavelength maximum in the spectra of the amphiphiles. The CD spectra of the 40 and 25 nucleotide (GGGT)3-modified ssDNA sequences and amphiphiles in Milli-Q water and KCl are shown in Fig. S5–S6† and results are summarized in Table S2–S3† respectively. Cryo-TEM imaging of these two samples showed that both amphiphiles with the 25 and 40 nucleotide (GGGT)3-modified headgroups formed twisted nanotapes as well as micelles (Fig. S7†), although the nanotapes were observed very rarely and did not twist with a consistent periodicity. Thus, for the case of the 25 nucleotide headgroup, where the presence of the (GGGT)3 sequence was able to clearly induce the formation of G-quadruplexes between the headgroups of the amphiphiles, bilayer twisted nanotape structures were observed in the absence of the C12 spacer but helical nanotapes or nanotubes were not (Table 1).
Self-assembly of ssDNA-amphiphiles with G5-modified headgroups and a C12 spacer
As a final test of the influence of the guanine-modification of the headgroups, amphiphiles that contained both the G5-modified headgroups and the C12 spacers were created and their assembly compared to that of the amphiphiles with the C12 spacer and NoG headgroups. There were no apparent differences in the assembly behavior of amphiphiles with the C12 spacer containing the G5-modified (Fig S8†) and the NoG headgroups (Fig S1†) with 25 and 40 nucleotides, as each formed twisted and helical nanotapes and nanotubes. However, there was a dramatic difference in the nanotubes formed by the amphiphiles with headgroups containing only 10 nucleotides. Both amphiphile samples produced nanotubes with similar average diameters (NoG: 29.0 ± 3.6 nm; G5: 32.5 ± 1.3 nm), but amphiphiles with the NoG headgroup produced nanotubes that were microns in length while amphiphiles formed with the G5 headgroup produced nanotubes that were nearly two orders of magnitude shorter and varied between 60 and 350 nm (Fig. 3). Additional images of the short nanotubes produced by the amphiphiles with the G5-modified headgroup and C12 spacer are provided in Fig. S9.† These images contain end-on views of the short nanotubes, allowing the hollow morphology of these structures to be observed.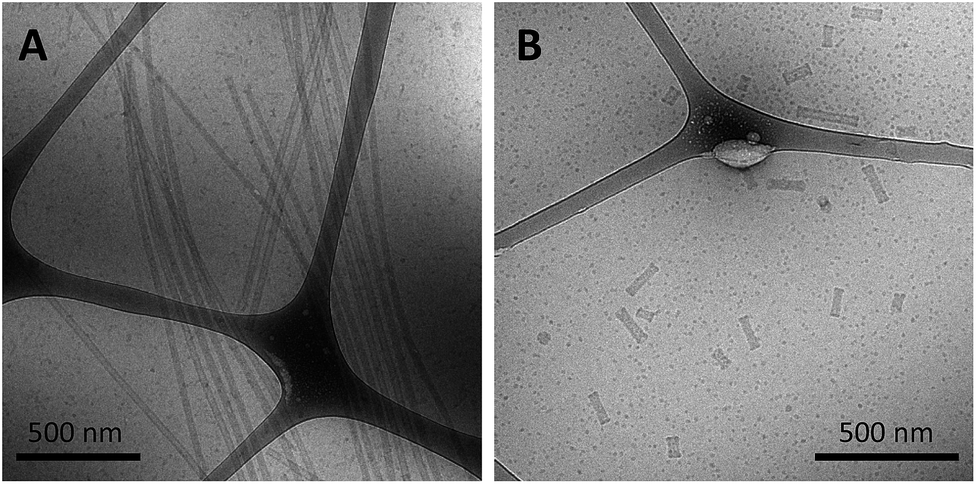 | ||
| Fig. 3 Cryo-TEM images of ssDNA nanotubes formed from the self-assembly of amphiphiles with a C12 spacer and (A) 10nt-1 NoG or (B) 10nt-1 G5 headgroups. | ||
AFM imaging of amphiphiles with the 25 nucleotide G5-modified headgroup and C12 spacer captured two sets of two nanotubes (Fig. S10†). The nanotubes were microns in length and each appeared to be around 65 nm in diameter based on the line-scan analysis of the friction image. The larger diameters and decreased heights of the nanotubes observed in the AFM images compared to cryo-TEM images is likely due to the flattening of the nanotubes during dehydration. It is possible that the parallel organization of the nanotubes was the result of the drying process that occurred during the sample preparation but it is also possible that the long nanotubes naturally align as observed in a number of cryo-TEM images including Fig. 2C and 3A.
CD was performed on each of the G5-modified ssDNA sequences and their amphiphiles with C12 spacers to determine the effect of the G5 sequence on the secondary structure of the ssDNA headgroup. CD spectra of all ssDNA sequences used in this study and their amphiphiles in Milli-Q water and KCl, as well as tables summarizing the CD data and the headgroup secondary structure assignment for all molecules are provided in the ESI (Fig. S5–S6 and S11 and Table S2–S4†). The CD spectra of the amphiphiles with the C12 spacer and G5-modified headgroups with 25 and 40 nucleotides had maxima at 268 and 270 nm respectively in water, which suggested that the headgroups of these amphiphiles formed either stem-loop structures or that the designation of the headgroup structure was unclear. For comparison, the CD spectra of the amphiphiles with a C12 spacer containing the NoG 25 and 40 nucleotide headgroups had maxima at 273 and 274 nm, indicative of a stem-loop structure. The spectra of the amphiphiles with the C12 spacer and the G5-modified 10 nucleotide headgroups (10nt-1 and 10nt-2) had maxima at 264 nm, characteristic of a parallel G-quadruplex structure, while the CD spectra of amphiphiles with a C12 spacer and the 10 nucleotide NoG headgroups were consistent with that of stem-loop structures (Fig. 4, S11 and Table S4†). This suggested that of the amphiphiles formed with the C12 spacer and a G5-modified headgroup only the amphiphiles with the shorter 10 nucleotide headgroups clearly produced G-quadruplex secondary structures. CD spectra of ssDNA and ssDNA-amphiphiles with guanine-modified headgroups were also collected in 20 mM KCl to test if the addition of the K+ cation would produce a substantial effect on the structure of the headgroups. Data show that the addition of KCl only produced minor changes in the CD spectra of the amphiphiles, suggesting that the presence of the G-quadruplex stabilizing K+ cation did not substantially influence the secondary structures adopted by headgroups of the amphiphiles towards the formation of G-quadruplexes.
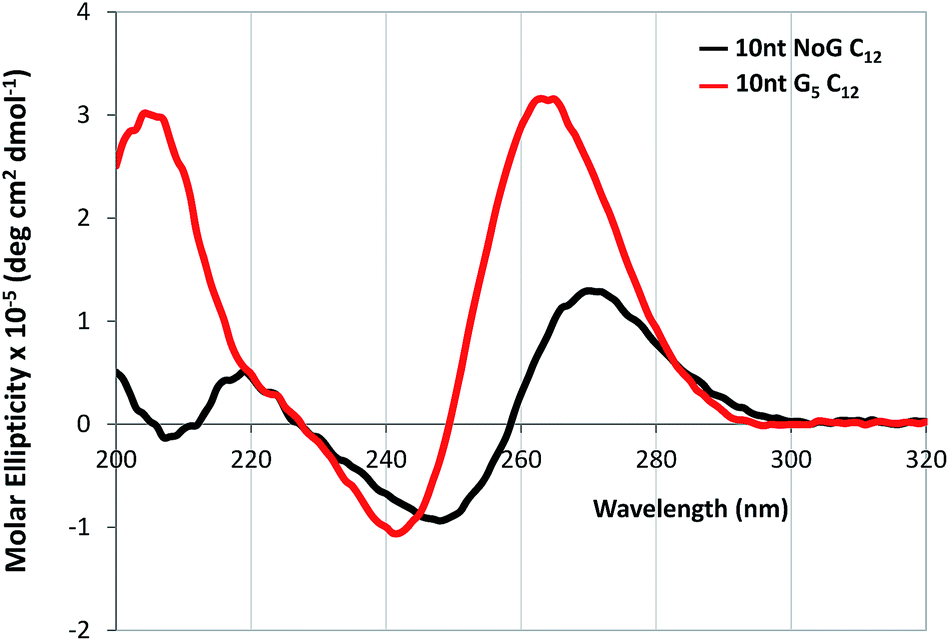 | ||
| Fig. 4 CD spectra in Milli-Q water of 20 μM ssDNA-amphiphiles with a C12 spacer and 10 nucleotide (10nt-1) NoG or G5-modified headgroups. | ||
Transitions between twisted nanotapes, helical nanotapes and nanotubes
Cryo-TEM images of the ssDNA-amphiphile nanostructures not only showed twisted nanotapes, helical nanotapes and nanotubes, but also captured the transition from twisted to helical nanotape as well as from helical nanotape to nanotube (Fig. 5 and S2†). These images provided direct evidence that the ssDNA-amphiphile nanostructures underwent transitions between these structures, likely in a similar manner as observed in other types of amphiphilic molecules as discussed in detail below. Analysis of cryo-TEM images that captured the transition from twisted nanotapes into helical nanotapes showed that the twisted nanotape segments had widths that were substantially smaller than the helical nanotape segments (24 ± 2 versus 38 ± 4 nm) but pitch lengths that were similar (132 ± 6 nm for the twisted nanotapes and 129 ± 7 nm for the helical nanotapes). | ||
| Fig. 5 Cryo-TEM images of ssDNA-amphiphiles formed by the 10nt-2 NoG headgroup and C12 spacer undergoing transitions from (A) twisted nanotapes to helical nanotapes and (B) a helical nanotape to a nanotube. | ||
To better understand the assembly mechanism of the ssDNA-amphiphiles a sample containing the 25 nucleotide G5-modified headgroup and C12 spacer was heated to 90 °C for 10 min to induce the structures to disassemble. Prior to thermal disruption this sample contained globular micelles, nanotapes and nanotubes (Fig. 6A). An aliquot of the sample was taken after 10 min of heating, while the solution was still at 90 °C, and was immediately vitrified and imaged to confirm the absence of any self-assembled structures following the heating regimen (Fig. 6B). The sample was cooled to room temperature and another aliquot vitrified upon reaching room temperature. The remaining sample was kept at room temperature for 3 weeks and aliquots of the sample were vitrified and imaged after 2 days, 9 days, and 21 days. Globular micelles were observed upon cooling to room temperature, and after 2 days of aging at room temperature short and thin nanostructures along with the globular micelles were observed to exist in the sample (Fig. 6C). These thin nanostructures had widths of ∼20 nm and their nanotape morphology was confirmed with stage tilting (Fig. S12†). After 9 days of aging, nanotapes that were longer, wider, and twisted (Fig. 6D) or helical (Fig. 6E) were observed. This suggested that the thin nanotapes broaden and begin twisting and transitioning to helical nanotapes over this timeframe. After 21 days, nanotubes were observed in the sample, suggesting the nanotubes reformed between 9 and 21 days after thermal disruption.
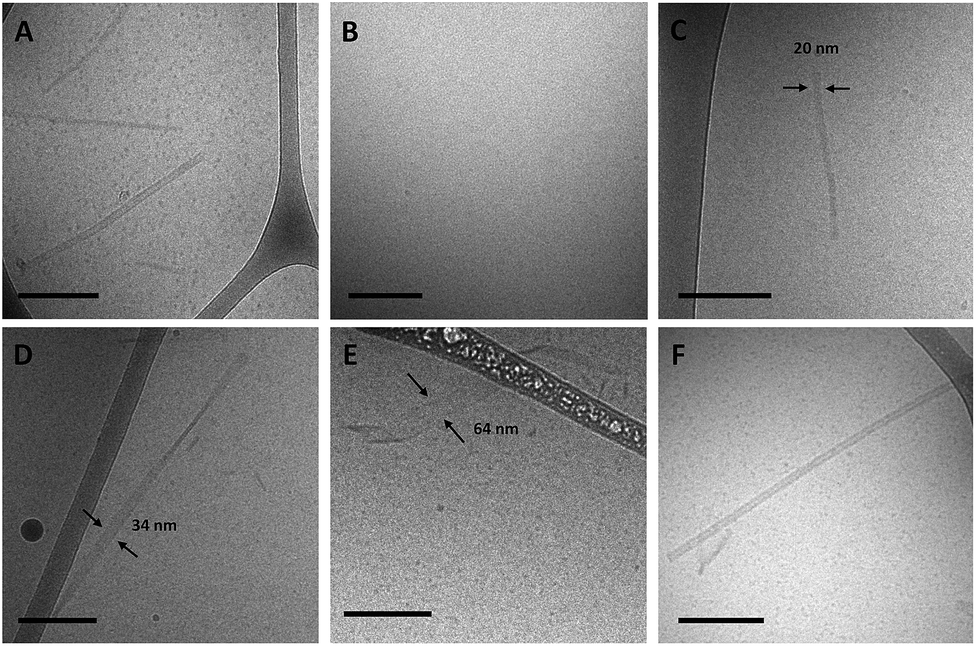 | ||
| Fig. 6 Cryo-TEM images of 25 nucleotide G5-modified amphiphiles containing the C12 spacer at various times in the thermal disruption timeline. (A) Prior to heating. (B) From a solution heated at 90 °C for 10 min. (C) After 2 days at room temperature. (D) and (E) After 9 days at room temperature. (F) After 21 days at room temperature. All scale bars are 200 nm. | ||
Discussion
In this work three building blocks were used to create ssDNA-amphiphiles: a hydrophobic tail, a hydrophilic ssDNA headgroup, and a spacer molecule that links the tail and the headgroup. Our previous work identified the hydrophobic force produced by the dialkyl tails as a major driving force for the assembly of a ssDNA aptamer–amphiphile and that the inclusion of a hydrophobic spacer is important for the assembly of the ssDNA-amphiphiles into flat or twisted nanotapes.9 This work explored the influence of the headgroup length and sequence on the self-assembly behavior of ssDNA-amphiphiles created with the same dialkyl C16 tail and C12 spacer to expand our understanding of the role of the ssDNA headgroup in the self-assembly of ssDNA-amphiphiles. Our current data demonstrated that ssDNA-amphiphiles with C12 spacers and NoG headgroups of 10, 25, or 40 nucleotides not only produced the twisted nanotapes previously seen, but also helical nanotapes and nanotubes. Each of these structures is formed from bilayers of amphiphiles with the hydrophobic tails organized into an interior core and the ssDNA headgroups forming an exterior shell (Fig. 7 and S13†).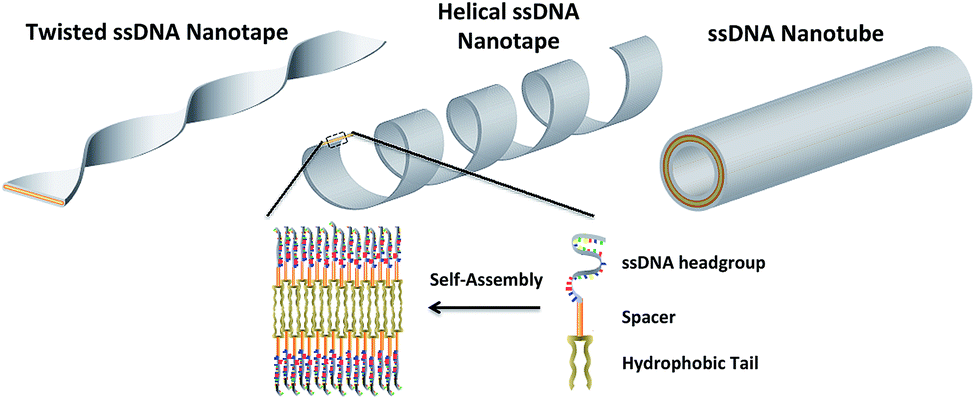 | ||
| Fig. 7 An artistic rendering of the self-assembly of ssDNA-amphiphiles into an ordered bilayer structure and the twisted and helical nanotapes and nanotubes that they form. The amphiphile contains three building blocks: a hydrophobic tail, a spacer, and a hydrophilic headgroup (the secondary structure of the headgroup is not shown). | ||
Similar nanotape and nanotube structures were observed in solutions of different amphiphilic molecules including glycolipids, peptide-amphiphiles, and bolaamphiphiles.19–21 In each case the nanotape and nanotube structures were created from bilayers of amphiphiles, with the hydrophobic moieties sequestered into an inner layer and surrounded with the hydrophilic headgroups to form the exterior of the nanostructure. The chirality of the individual amphiphile requires that the amphiphiles organize with their neighboring molecules at non-zero angles, generating a preferred orientation of each amphiphile tail and headgroup within the self-assembled bilayer, which induces twisting.22 The ssDNA-amphiphiles we have created are rich in chirality, with chiral centers in the hydrophobic tails as well as the nucleotides of the ssDNA headgroups. As such, it is likely that the chirality of the individual ssDNA-amphiphile is responsible for producing the twisting that was observed in the ssDNA-amphiphile nanotapes.
The ability for self-assembled structures to transition from a twisted nanotape morphology to a helical nanotape morphology has been captured and described in a number of publications.20,22–26 For example, a peptide–amphiphile that contained three phenylalanine residues that were capable of intermolecular π–π stacking was observed to form short twisted bilayer nanotapes 30 s after dissolution in water.20 These short structures grew into long twisted nanotapes within ten min, coexisted with helical tapes after two weeks and transitioned entirely to helical tapes after four weeks. Similarly, single amino acid amphiphiles dissolved in water were found to form twisted nanotapes after 24 h, a mixture of twisted and helical nanotapes after one week, which were almost entirely helical after four weeks, and finally transitioned into nanotubes between one and four months.23
These and other reports propose that the transition from a twisted to helical nanotape morphology requires a change in membrane curvature from Gaussian (saddle-like) to cylindrical, an event that is often attributed to a rearrangement of the individual amphiphiles into a molecular organization that is more ordered or crystalline.20,21,27,28 The forces that are often identified as causing the order or crystallinity are hydrogen-bonding and π–π stacking between individual amphiphiles although electrostatic and hydrophobic forces are also likely important.20,23 The C12 spacer has previously been found to play an important role in producing the bilayer nanotapes, possibly by forcing the aptamer headgroups into close proximity of each other, thus reducing their interfacial headgroup area, which allows the nanotapes to form.9,10 The C12 spacer may also be helping to ensure that the amphiphiles can organize into crystalline or well-ordered bilayers by extending the large ssDNA headgroups away from the interface and relieving some of the electrostatic or steric constraints that could impede close and ordered packing of the amphiphiles. This may be especially important in the case of the NoG headgroups that do not appear to form significant interactions with each other.
Hydrogen bonding can occur between guanine nucleobases and produce the G-quartet structures that can stack into G-quadruplexes, which led us to test whether guanine-rich headgroups that can form parallel G-quadruplexes could be used in place of the C12 spacer to produce nanotapes and nanotubes. Amphiphiles with the (GGGT)3-modified headgroups of 25 and 40 nucleotides in length and without the C12 spacer (NoSPR) were found to assemble into twisted nanotapes but did not appear to progress into helical nanotapes or nanotubes while amphiphiles with a NoG sequence and without a spacer formed only micelles. This result is in agreement with our previous study that found that amphiphiles with a different 40 nucleotide headgroup containing the (GGGT)3 sequence and directly conjugated to the hydrophobic tail (NoSPR) also formed twisted nanotapes but were not observed to form helical nanotapes or nanotubes.10 Additionally, we have also observed previously that amphiphiles formed with C12 hydrophobic spacers and a Muc-1 aptamer headgroup (25 nucleotides long) that adopted parallel G-quadruplex secondary structure only assembled into globular micelles and twisted nanotapes.9 Thus, the findings of this and our previous studies suggest that long headgroups (with 25 or 40 nucleotides) with additional hydrogen bonding interactions present in G-quadruplex structures may encourage the formation of the bilayer nanotape structures in the absence of a hydrophobic C12 spacer, but may not allow for the change in membrane curvature required for twisted nanotapes to transition into helical nanotapes and nanotubes.
The literature offers insight into the transition from twisted to helical nanotapes and from helical nanotapes to nanotubes. Recent theoretical and experimental work shows that the width of the nanotape is a critical parameter in determining the morphology of the nanotape.23,24,29 Specifically, as the bilayer grows in width it becomes energetically favorable for the bilayer to transition from Gaussian to cylindrical curvature, thus producing the transition from a twisted to helical morphology. Theoretical studies also pointed out that shape selection in self-assembled chiral molecules may involve a geometrical frustration, and thus a competition between bending and stretching.29,30 The transition from twisted to helical ribbons (or nanotapes) to nanotubes has been described by two competing theories: a “closing-pitch model” and a “growing width model”.31 The closing-pitch model assumes that a helical nanotape maintains its width while the pitch shortens until the edges of the nanotape meet to form a nanotube, while the growing width model assumes the pitch remains constant and the nanotape widens until a closed nanotube is formed. An alternate possibility is that some of the twisted and helical nanotapes are at equilibrium and never transition into nanotubes as observed previously in other amphiphilic systems.32
Analysis of cryo-TEM images that captured the transition from twisted nanotapes into helical nanotapes, like those shown in Fig. 5, showed that the twisted nanotape segments had widths that were substantially smaller than the helical nanotape segments. This suggests that the transition from twisted to helical nanotape occurs as the width of the nanotape increases and that the “growing width” model rather than the “shortening pitch” model better describes the mechanism of transitioning from twisted to helical nanotapes as well as nanotube formation. However, based on the presence of twisted and helical nanotapes in an amphiphile solution aged for 6 months (Fig. S14†), it is possible that not all nanotapes will progress into nanotubes. This can be either because there are no more micelles to contribute to the growth mechanism (as seen in Fig. S14†) or because some nanostructures may get locked into a twisted or helical nanotape morphology, an outcome that has recently been theorized24 and has been observed experimentally.32
Further support for the “growing width” mechanism is provided by the observed progression from thin nanotapes to nanotubes following thermal disruption of the nanostructures. The heating process was found to cause all self-assembled structures to disassemble, allowing the reassembly process to be monitored over time. Immediately after cooling to room temperature only globular micelles were observed. After 2 days of aging, globular micelles and thin nanotapes were present. After 9 days, wider, longer nanotapes that were twisted, as well as much wider helical nanotapes were observed. And finally, after 21 days, nanotubes were observed (images were not collected between 9 and 21 days). The reassembly progression seen over time in the cryo-TEM images of Fig. 6, along with the images shown in Fig. 5, suggest that nanotapes transition into nanotubes due to the increasing width of the nanotapes.
Interestingly, the timescale of nanotape and nanotube assembly appeared to be substantially slower following the thermally induced disassembly than after the sample was initially synthesized, purified and dissolved in water. The heat treatment did not appear to cause appreciable degradation of the amphiphiles, as LC-MS of an amphiphile sample following the thermal disruption procedure showed that over 98% of the amphiphiles still had the expected molecular weight. Following thermal disruption and cooling to room temperature, samples were imaged at four time points: immediately after cooling, and at 2, 9 and 21 days after cooling. The twisted nanotapes were observed after 9 days of aging and the nanotubes after 21 days, a timescale similar to that observed in other amphiphilic systems.20,23,33 However, nanotapes and nanotubes were observed within 30 min after the amphiphiles were dissolved in Milli-Q water following their synthesis and purification. At this time the cause of the dramatic difference in assembly dynamics between the ssDNA-amphiphiles and other amhiphilic molecules remains unclear. One possible cause though for the apparent rapid assembly of the ssDNA nanotubes following synthesis and purification of the ssDNA-amphiphiles is that the amphiphiles may began to assemble during the purification steps used to separate the ssDNA-amphiphiles from the unreacted ssDNA, hydrophobic tails and other reaction inputs. During this purification process the newly formed ssDNA-amphiphiles, as well as other amphiphilic molecules, are exposed to an aqueous environment that contains salts, elevated temperatures, and mixtures of aqueous and organic solvents (including methanol and ethanol). These factors have all been implicated in the formation of tubular structures by self-assembling amphiphiles,34–37 and may play a role in accelerating the assembly of the ssDNA-amphiphiles more than other amphiphiles.
Conclusions
This work demonstrates that ssDNA-amphiphiles containing a random nucleic acid headgroup can adopt a variety of self-assembled structures including twisted and helical bilayer nanotapes and nanotubes, structures that are substantially more complex than spherical and cylindrical micelles observed by others in the literature. The ability to create DNA nanotubes from ssDNA-amphiphiles is particularly exciting, as nanotubes have been utilized for targeted drug delivery of small molecules and siRNA, as templates for nanowires and as tracks for molecular motors. For many of these applications there is no need for the complex designs made possible by other DNA nanotechnology approaches that rely entirely on DNA base pairing. ssDNA-amphiphile assembly into nanotubes occurs rapidly via the association of the hydrophobic tails and does not require stringent annealing conditions as demonstrated by the nanotube formation min after amphiphile dissolution in water. Furthermore, DNA nanotubes were formed using a single ssDNA sequence, that varied in length and sequence, and the addition of a guanine-rich sequence in the headgroup was found to be capable of modifying the assembly, all of which demonstrate the versatility of the amphiphile-based self-assembly strategy for forming DNA nanostructures.Acknowledgements
This work was supported by the CAREER award NSF/CBET-0846274. Cryo-TEM and AFM imaging were carried out in the Characterization Facility, University of Minnesota, which receives partial support from NSF through the MRSEC program.Notes and references
- N. C. Seeman, Annu. Rev. Biochem., 2010, 79, 65–87 CrossRef CAS PubMed.
- A. V. Pinheiro, D. Han, W. M. Shih and H. Yan, Nat. Nanotechnol., 2011, 6, 763–772 CrossRef CAS PubMed.
- T. Tørring, N. V. Voigt, J. Nangreave, H. Yan and K. V. Gothelf, Chem. Soc. Rev., 2011, 40, 5636–5646 RSC.
- C. Lin, Y. Liu, S. Rinker and H. Yan, ChemPhysChem, 2006, 7, 1641–1647 CrossRef CAS PubMed.
- F. A. Aldaye, A. L. Palmer and H. F. Sleiman, Science, 2008, 321, 1795–1799 CrossRef CAS PubMed.
- M. Kwak and A. Herrmann, Chem. Soc. Rev., 2011, 40, 5745–5755 RSC.
- A. Patwa, A. Gissot, I. Bestel and P. Barthélémy, Chem. Soc. Rev., 2011, 40, 5844–5854 RSC.
- M.-P. Chien, A. M. Rush, M. P. Thompson and N. C. Gianneschi, Angew. Chem., Int. Ed., 2010, 49, 5076–5080 CrossRef CAS PubMed.
- T. R. Pearce, B. Waybrant and E. Kokkoli, Chem. Commun., 2014, 50, 210–212 RSC.
- B. Waybrant, T. R. Pearce and E. Kokkoli, Langmuir, 2014, 30, 7465–7474 CrossRef CAS PubMed.
- A. Mardilovich and E. Kokkoli, Biomacromolecules, 2004, 5, 950–957 CrossRef CAS PubMed.
- H. Liu, Z. Zhu, H. Kang, Y. Wu, K. Sefan and W. Tan, Chem.–Eur. J., 2010, 16, 3791–3797 CrossRef CAS PubMed.
- E. W. Choi, L. V. Nayak and P. J. Bates, Nucleic Acids Res., 2010, 38, 1623–1635 CrossRef CAS PubMed.
- J. Kypr, I. Kenjovska, D. Renciuk and M. Vorlickova, Nucleic Acids Res., 2009, 37, 1713–1725 CrossRef CAS PubMed.
- D. M. Gray, J.-D. Wen, C. W. Gray, R. Repges, C. Repges, G. Raabe and J. Fleischhauer, Chirality, 2008, 20, 431–440 CrossRef CAS PubMed.
- A. Rajendran, M. Endo, K. Hidaka, P. L. T. Tran, J.-L. Mergny and H. Sugiyama, Nucleic Acids Res., 2013, 41, 8738–8747 CrossRef CAS PubMed.
- C.-H. Leung, D. S.-H. Chan, B. Y.-W. Man, C.-J. Wang, W. Lam, Y.-C. Cheng, W.-F. Fong, W.-L. W. Hsiao and D.-L. Ma, Anal. Chem., 2011, 83, 463–466 CrossRef CAS PubMed.
- E. K. Efthimiadou, Y. Sanakis, M. Katsarou, C. P. Raptopoulou, A. Karaliota, N. Katsaros and G. Psomas, J. Inorg. Biochem., 2006, 100, 1378–1388 CrossRef CAS PubMed.
- A. S. Cuvier, J. Berton, C. V. Stevens, G. C. Fadda, F. Babonneau, I. N. A. Van Bogaert, W. Soetaert, G. Pehau-Arnaudet and N. Baccile, Soft Matter, 2014, 10, 3950–3959 RSC.
- E. T. Pashuck and S. I. Stupp, J. Am. Chem. Soc., 2010, 132, 8819–8821 CrossRef CAS PubMed.
- H. Shao, J. Seifert, N. C. Romano, M. Gao, J. J. Helmus, C. P. Jaroniec, D. A. Modarelli and J. R. Parquette, Angew. Chem., Int. Ed., 2010, 49, 7688–7691 CrossRef CAS PubMed.
- A. Sorrenti, O. Illa and R. M. Ortuño, Chem. Soc. Rev., 2013, 42, 8200–8219 RSC.
- L. Ziserman, H.-Y. Lee, S. R. Raghavan, A. Mor and D. Danino, J. Am. Chem. Soc., 2011, 133, 2511–2517 CrossRef CAS PubMed.
- L. Ziserman, A. Mor, D. Harries and D. Danino, Phys. Rev. Lett., 2011, 106, 238105 CrossRef.
- Z. Chen, C. Majidi, D. J. Srolovitz and M. Haataja, Appl. Phys. Lett., 2011, 98, 011906 CrossRef PubMed.
- A. Perino, M. Schmutz, S. Meunier, P. J. Mesini and A. Wagner, Langmuir, 2011, 27, 12149–12155 CrossRef CAS PubMed.
- J. V. Selinger, M. S. Spector and J. M. Schnur, J. Phys. Chem. B, 2001, 105, 7157–7169 CrossRef CAS.
- M. S. Spector, A. Singh, P. B. Messersmith and J. M. Schnur, Nano Lett., 2001, 1, 375–378 CrossRef CAS.
- S. Armon, H. Aharoni, M. Moshe and E. Sharon, Soft Matter, 2014, 10, 2733–2740 RSC.
- R. Ghafouri and R. Bruinsma, Phys. Rev. Lett., 2005, 94, 138101 CrossRef.
- T. Shimizu, M. Masuda and H. Minamikawa, Chem. Rev., 2005, 105, 1401–1443 CrossRef CAS PubMed.
- R. Oda, I. Huc, M. Schmutz, S. J. Candau and F. C. MacKintosh, Nature, 1999, 399, 566–569 CrossRef CAS PubMed.
- J. Adamcik, V. Castelletto, S. Bolisetty, I. W. Hamley and R. Mezzenga, Angew. Chem., Int. Ed., 2011, 50, 5495–5498 CrossRef CAS PubMed.
- V. Castelletto, I. W. Hamley, C. Cenker and U. Olsson, J. Phys. Chem. B, 2010, 114, 8002–8008 CrossRef CAS PubMed.
- V. Castelletto, I. W. Hamley, P. J. Harris, U. Olsson and N. Spencer, J. Phys. Chem. B, 2009, 113, 9978–9987 CrossRef CAS PubMed.
- B. N. Thomas, C. R. Safinya, R. J. Plano and N. A. Clark, Science, 1995, 267, 1635–1638 CrossRef CAS PubMed.
- T. G. Barclay, K. Constantopoulos, W. Zhang, M. Fujiki and J. G. Matisons, Langmuir, 2012, 28, 14172–14179 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: LC-MS data, CD data, fluorescent images, AFM images, cryo-TEM images, tables. See DOI: 10.1039/c4sm01332h |
| This journal is © The Royal Society of Chemistry 2015 |