
Squaraine-sensitized composite of a reduced graphene oxide/TiO2 photocatalyst: π–π stacking as a new method of dye anchoring†
K. L. Vincent
Joseph‡
a,
Jonghun
Lim‡
b,
A.
Anthonysamy
a,
Hyoung-il
Kim
b,
Wonyong
Choi
b and
Jin Kon
Kim
*a
aCreative Research Initiatives Centre for Smart Block Co-polymer Self Assembly, Department of Chemical Engineering, Pohang University of Science and Technology, Pohang, Kyungbuk 790-784, Republic of Korea. E-mail: jkkim@postech.ac.kr
bSchool of Environmental Science and Engineering Pohang University of Science and Technology (POSTECH), Pohang, Kyungbuk 790-784, Republic of Korea
First published on 29th October 2014
Abstract
We synthesized a near infra-red (NIR)-absorbing squaraine dye (VJ-S) showing strong absorption and emission maxima at 684 and 704 nm, respectively, with a high molar extinction coefficient (ε) of 1.277 × 105 M−1 cm−1 and a band gap of 1.77 eV. Its oxidation and reduction potentials were found to be 0.889 and −0.795 V, respectively, with HOMO and LUMO levels of −5.21 and −3.53 eV, respectively. We also prepared the self-assembled core/shell nanocomposite r-NGOT, where TiO2 is the core and reduced nano-sized graphene oxide (r-NGO) is the shell. When VJ-S was anchored on r-NGOT, it showed π–π stacking with r-NGO, which is confirmed by Fourier-transformed infrared spectroscopy, X-ray photoelectron spectroscopy, high-resolution transmission electron microscopy and electron energy loss spectroscopy. The optical absorption spectrum of the VJ-S/r-NGOT nanocomposite measured with diffuse reflectance UV/visible absorption spectroscopy covers the whole range of visible light wavelengths up to 800 nm. The photocatalytic activity of VJ-S/r-NGOT at visible light wavelengths (λ > 420 nm) is much higher than that of r-NGOT alone.
Introduction
Fossil fuels, as the traditional main energy source, have been over-exploited since the industrial revolution. The excessive consumption of fossil fuels leading to global warming has impelled researchers from all over the world to look for other renewable clean energy sources.1 Hydrogen, as a zero carbon emission fuel, is forecast to become a major source of energy in the future.2–4 Among the wide variety of hydrogen generation strategies, hydrogen production from light-induced water splitting in the presence of a photocatalyst has attracted enormous attention because it is cost effective and eco-friendly. Since Fujishima and Honda5 reported the photoelectrochemical splitting of water into H2 and O2 on a TiO2 semiconductor electrode, a large number of photocatalytic systems for light-driven hydrogen evolution have been developed.6–17 Among them, dye-sensitized photocatalysts have received remarkable attention because their optical and chemical properties can be understood and strategically tuned on the molecular level.7,8,13–17 However, two main limitations of TiO2, including a wide band gap (3.2 eV for anatase and 3.0 eV for rutile) and rapid charge recombination rate, significantly restrict the performance of pure TiO2.18,19 Numerous efforts have been made to overcome these shortcomings and to enhance photocatalytic H2 production activity, such as dye sensitization,20 metal and non-metal ion doping,21–25 combination with semiconductors26–29 and loading of co-catalysts.30,31Among these, graphene-based nanocomposites have been used as high performance photocatalysts.32–36 Typically, these composite photocatalysts were synthesized by preparing semiconductor powders and colloidal solutions of graphene oxide (GO) as precursors, followed by the reduction of GO to reduced graphene oxide (r-GO) via hydrothermal reduction, hydrazine reduction, or UV-irradiated reduction methods.37–42 Since some of the functional groups are still present in r-GO even after reduction, these could be attached to metal (or metal oxide) nanoparticles.43r-GO could be used as a good supporting site for the photocatalytic process because of good mechanical properties sufficient to stabilize the catalysis and wide two-dimensional planes for catalyst deposition.35,44–48 Many of the nanocomposites made of r-GO and metal oxides and sulfides, such as, r-GO/ZnO, r-GO/TiO2, r-GO/CdS and r-GO/ZnIn2S4, have been reported as photocatalyts.49–52
Recently, dye-sensitized r-GO nanocomposites with TiO2 have also received much attention. Min et al. first reported that Eosin Y (EY) and Rose Bengal (RB) dye-sensitized graphene/Pt nanocomposites showed better photocatalytic performance compared to EY or RB/TiO2/Pt photocatalysts.53 Chen et al. have reported that Ru(dcbpy)3/TiO2/r-GO/Pt (here, dcbpy is 4,4′-dicarboxy-2,2′-bipyridine) showed better photocatalytic activity compared to nanocomposites without r-GO (Ru(dcbpy)3/TiO2/Pt).54 Zhu et al. reported that the π–π stacked porphyrin (Porph) with r-GO nanocomposites (Porph/r-GO/TiO2/Pt) showed better performance in hydrogen production compared to the nanocomposites without r-GO (Porph/TiO2/Pt),55 suggesting that dye-sensitized nanocomposites with r-GO exhibit enhanced photocatalytic performance.
However, when r-GO was prepared from graphite (or graphene) by chemical oxidation followed by reduction through UV irradiation, the lateral sizes of the GO sheets widely varied from micrometer to nanometer size.56,57 The different sizes or shapes of graphene sheets induce two types of graphene edges, armchair and zigzag. The electronic structure of nanosized graphene (nanographene) is strongly dependent on the edge geometry: the nonbonding π-electron state (edge state) is present in the zigzag edge, while it is absent in the armchair edge.58,59 Therefore, many approaches have been tried to make size-controlled GO sheets with uniform size distribution.60–62 One of the groups of this paper prepared nanocomposites (r-NGOT) by hybridizing r-NGO (<50 nm) and TiO2 nanoparticles and achieved a self-assembled core/shell structure. This nanocomposite showed better photocatalytic activity compared with another composite made of larger-sized (μm scale) r-GO/TiO2.63
Since r-NGOT absorbs only UV wavelengths of solar radiation, the photocatalysis of r-NGOT under visible light is quite limited. To overcome this problem, in this study, we prepared a nanocomposite made of squaraine dye (VJ-S) sensitized r-NGOT (VJ-S/r-NGOT/Pt, as shown in Fig. 1) and investigated its photocatalytic activity and H2 evolution performance. Since VJ-S has a resonance-stabilized zwitterionic structure and contains an electron-deficient center resulting from a four-membered ring and two electron-donating groups in a donor–acceptor–donor (D–A–D) form, it is capable of π–π interaction with r-NGO and direct charge transfer to r-NGO. Also, since it shows intramolecular charge transfer (CT)-based strong absorption (ε > 105 M−1 cm−1), it is very suitable for photosensitization with enhanced photostability.64–66 When VJ-S was hybridized with only r-NGO without the TiO2 core, its photocatalytic activity is lowered due to aggregation of r-NGO. Because the VJ-S dye absorbs visible and near-IR wavelengths and gives a direct energy transfer to r-NGO, the photocatalytic production of hydrogen in VJ-S/r-NGOT/Pt is much higher than that of either VJ-S/TiO2/Pt or r-NGOT/Pt under visible light irradiation.
 | ||
| Fig. 1 Chemical structure of VJ-S and schematic diagram of VJ-S/r-NGOT/Pt. | ||
Experimental methods
All the solvents were distilled from appropriate drying agents prior to use. Commercially available reagents were used without further purification, unless otherwise stated.Synthesis of 4-(bromomethyl)-4′-methyl-2,2′-bipyridine (1)
This compound was prepared according to published procedures.67,68 The compounds, 4,4′-dimethyl-2,2′-bipyridine (5 g, 27.14 mmol), N-bromo-succinimide (5 g, 28.09 mmol), and azobis(isobutyronitrile) (0.16 g, 0.98 mmol) were added to benzene (150 mL), and heated at reflux under Ar for 3 h. The mixture was cooled to room temperature, filtered, washed with water, dried over anhydrous Na2SO4 and evaporated to near-dryness. The residue was redissolved in CH2Cl2 and purified by column chromatography (silica gel, CH2Cl2/acetone, 98![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2, v/v). The fractions containing the pure product were combined and evaporated to dryness to give 1.7 g (30% yield) of a white solid. 1H NMR (400 MHz, CDCl3): δ (ppm) = 8.65 (d, J = 6.8 Hz, 1H), 8.54 (d, J = 6.8 Hz, 1H), 8.41 (s, 1H), 8.24 (s, 1H), 7.34 (d, J = 9.2 Hz, 1H), 7.16 (d, J = 8 Hz, 1H), 4.48 (s, 1H), 2.45 (s, 3H) (see Fig. S1 in ESI†). Elemental analysis calculated for C12H11N2Br: C, 54.77; H, 4.21; N, 10.65%. Found: C, 54.72; H, 4.14; N, 10.65%.
:2, v/v). The fractions containing the pure product were combined and evaporated to dryness to give 1.7 g (30% yield) of a white solid. 1H NMR (400 MHz, CDCl3): δ (ppm) = 8.65 (d, J = 6.8 Hz, 1H), 8.54 (d, J = 6.8 Hz, 1H), 8.41 (s, 1H), 8.24 (s, 1H), 7.34 (d, J = 9.2 Hz, 1H), 7.16 (d, J = 8 Hz, 1H), 4.48 (s, 1H), 2.45 (s, 3H) (see Fig. S1 in ESI†). Elemental analysis calculated for C12H11N2Br: C, 54.77; H, 4.21; N, 10.65%. Found: C, 54.72; H, 4.14; N, 10.65%.
Synthesis of 4-(diethylphosphonomethyl)-4′-methyl-2,2′-bipyridine (2)68,69
A solution of 4-(bromomethyl)-4′-methyl-2,2′-bipyridine (2 g, 7.60 mmol), triethylphosphite (5 mL, 29 mmol) and a few drops of chloroform were heated at 110 °C for 10 h. The solution was then allowed to cool to room temperature, and triethylphosphite was removed under reduced pressure. The oily brown residue was re-dissolved in CH2Cl2 and purified by column chromatography (silica gel, acetone). The fractions containing the pure product were combined and evaporated to dryness to give a white solid in 85% yield. 1H NMR (400 MHz, CDCl3): δ (ppm) = 8.32 (d, J = 4.8 Hz, 1H), 8.24 (d, J = 4.8 Hz, 1H), 8.05 (s, 1H), 7.94 (s, 1H), 7.04 (d, J = 2.4 Hz, 1H), 6.84 (d, J = 4.4 Hz, 1H), 3.79 (m, 4H), 2.95 (d, J = 22 Hz, 2H), 2.14 (s, 3H), 0.98 (t, J = 14 Hz, 6H). 13C NMR (100 MHz, CDCl3): δ (ppm) = 155.9, 155, 148.6, 148.4, 147.4, 141.7, 141.6, 124.2, 122, 121.3, 61.8, 61.7, 33.7, 32.3, 20.6, 15.89, 15.8 (see Fig. S2a and S2b†). Elemental analysis calculated for C16H21N2O3P: C, 59.99; H, 6.61; N, 8.75%. Found: C, 59.97; H, 6.60; N, 8.75%.Synthesis of N-hexyl-pyrrole-2-carboxaldehyde (3)
The reaction was performed under complete N2 atmosphere. Sodium hydride (1.51 g, 63.1 mmol) was added to DMF in a 500 mL RB flask and the suspension was degassed using N2. Pyrrole-2-carboxaldehyde (5 g, 52.6 mmol) dissolved in DMF (50 mL) was added slowly into the flask. After stirring for 30 min, 1-iodohexane (14.5 g, 68.35 mmol) dissolved in DMF (50 mL) was added slowly over a period of 30 minutes. The resulting solution was stirred at 25 °C overnight and then quenched with water. It was extracted with ethyl acetate (3 × 50 mL), the combined organic phases were washed with water and brine solution and dried over anhydrous Na2SO4. The solution was filtered, evaporated under vacuum and separated by column with hexane using silica gel. The eluted fractions containing pure product were combined and evaporated to give 3 in 80% yield as a colorless oil. 1H NMR (400 MHz, CDCl3): δ (ppm) = 9.53 (s, 1H), 6.92 (m, 2H), 6.21 (m, 2H), 4.59 (t, J = 14.4 Hz, 2H), 2.04 (t, J = 13.6 Hz, 2H), 1.17 (m, 3H). 13C NMR (100 MHz, CDCl3): δ (ppm) = 179.1, 131.4, 131.3, 124.7, 109.5, 49.1, 31.48, 31.45, 26.6, 14.08, 14.07 (see Fig. S3a and S3b†). Elemental analysis calculated for C11H17NO: C, 73.70; H, 9.56; N, 7.81%. Found: C, 73.71; H, 9.50; N, 7.79%.Synthesis of 4-(N-hexyl-pyrrole-2-ethenyl)-4′-methyl-2,2′-bipyridine (4)
Potassium-t-butoxide (0.632 g, 5.63 mmol) was added to a solution of 3 (1 g, 5.58 mmol) and 2 (1.61 g, 5 mmol) in THF (30 mL). The reaction mixture was stirred for 10 h at room temperature. After addition of water (10 mL), THF was removed under reduced pressure, the pH was adjusted to 7 and the aqueous residue was extracted with CH2Cl2 (3 × 25 mL). The combined organic layers were washed with water (50 mL), dried over Na2SO4, and filtered. After evaporation of the solvent, the precipitate was re-dissolved in CH2Cl2 and separated by column. The product was eluted with CH2Cl2/hexane (v/v = 1:1) to give 4 in 60% yield as a light brown oil. 1H NMR (400 MHz, CDCl3): δ (ppm) = 8.58 (m, 2H), 8.41 (d, J = 1.53 Hz, 1H), 8.25 (s, 1H), 7.30 (m, 2H), 7.16 (m, 1H), 6.90 (s, 1H), 6.74 (t, J = 5.43 Hz, 1H), 6.60 (t, J = 2.11 Hz, 1H), 6.20 (t, J = 8.56 Hz, 1H), 5.30 (s, 1H), 4.0 (t, J = 19.28, 2H), 2.45 (s, 3H), 1.80 (m, 3H), 1.30 (m, 4H), 0.88 (t, J = 15.52 Hz, 4H). 13C NMR (100 MHz, CDCl3): δ (ppm) = 156.5, 156.1, 149.4, 148.9, 148.1, 146.4, 130.3, 124.7, 123.9, 122.6, 122.1, 121.3, 120.3, 117, 108.8, 108.6, 53.5, 47.1, 31.6, 31.4, 26.5, 22.6, 21.2, 14 (see Fig. S4a and S4b†). Elemental analysis calculated for C23H27N3: C, 79.96; H, 7.88; N, 12.16%. Found: C, 79.93; H, 7.82; N, 12.16%.
Synthesis of squaraine dye (VJ-S)
4-(N-Hexyl-pyrrole-2-ethynyl)-4′-methyl-2,2′-bipyridine (4) (1 g, 2.89 mmol) and squaric acid (0.165 g, 1.44 mmol) were added into a 100 mL RB flask equipped with a Dean–Stark trap. A mixture of n-butanol (15 mL) and benzene (15 mL) was added and the reaction mixture was refluxed for 8 h without moisture. It was then cooled to room temperature and the precipitate was filtered and washed with hot ethyl acetate and methanol to give NMR-pure squaraine dye (VJ-S) in 35% yield as a pale green powder. 1H NMR (400 MHz, CDCl3): δ (ppm) = 8.70 (d, J = 4 Hz, 2H), 8.60 (d, J = 4 Hz, 2H), 8.50 (s, 2H), 8.27 (s, 2H), 7.91 (s, 2H), 7.39 (d, J = 1.2 Hz, 2H), 7.38 (s, 2H), 7.35 (s, 2H), 7.28 (d, J = 3.6 Hz, 2H), 7.19 (d, J = 4 Hz, 2H), 6.96 (d, J = 4 Hz, 2H), 4.88 (s, 4H), 2.46 (s, 6H), 1.81–1.74 (m, 8H), 1.41 (s, 4H), 1.32 (s, 10H), 0.87 (t, J = 10.8 Hz, 6H). 13C NMR (100 MHz, CDCl3): δ (ppm) = 206.9, 159.4, 157.1, 149.7, 149, 148.3, 144.2, 132.4, 131.1, 125, 124.2, 122, 120.6, 119, 118.5, 114.8, 46.9, 32.3, 31.5, 30.9, 26, 22.5, 21, 14, 8.1 (see Fig. S5a and S5b†). Elemental analysis calculated for C50H52N6O2: C, 78.09, H, 6.82, N, 10.93%. Found: C, 78.10; H, 6.80; N, 10.91%. HR MALDI-TOF mass calculated for C50H52N6O2: 769.00. Found: 769.4029 (see Fig. S5c†).Synthesis of NGO
NGO was prepared according to a previous paper.63 Graphite oxide was synthesized by the Hummers method from graphite powder (Aldrich, 20 μm). The obtained graphite oxide (0.05 g) was dissolved in concentrated H2SO4 solution (50 mL), and then the exact amount of KMnO4 (0.15 g for 300 wt% of graphite oxide) was slowly added to the above solution with vigorous stirring to produce NGO. Stirring was continued for 30 min at 35 °C and 2.5 h at 45 °C. This mixture was cooled in an ice bath, and H2O2 solution (100 mL = 95 mL of water + 5 mL of 30 wt% H2O2) was very slowly added and stirred for 1 h. The resulting solution was ultrasonicated for 30 min to obtain exfoliated NGO.Synthesis of NGOT
0.5 g of TiO2 (P25, Degussa) was dispersed in water with ultrasonication and then an exact amount of NGO (4 wt% to TiO2) was added. This mixture was vigorously stirred overnight and then filtered and washed several times with 1 M HCl aqueous solution containing 1% H2O2 and deionized water to remove impurities.Characterization
All reactions were monitored using pre-coated thin-layer chromatographic plates (0.20 mm) with fluorescent indicator UV254 (uvitec-LF-204.LS). 1H and 13C NMR spectra were recorded on a Bruker AMX 400 or AV 400 instrument. Mass spectra were obtained on a 4700 MALDI TOF/TOF™ analyzer (AB SCIEX, USA). Elemental analysis was carried out with a PerkinElmer 2400 CHN analyzer. Electronic absorption spectra were obtained on a Cary 50 Probe UV-visible-NIR spectrophotometer. The cyclic voltammograms were recorded on a CHI600A electrochemical analyzer. The transmission electron microscopy (TEM) and electron energy loss spectroscopy (EELS) mapping analyses were obtained using a JEM-2100 microscope with Cs-correction. X-ray photoelectron spectroscopy (XPS, Kratos, XSAM 800pci) with a Mg Kα source (1253.6 eV) was used to confirm the reduction of NGO and the presence of synthesized VJ-S. The binding energies of all peaks were referenced to the C 1s (284.6 eV) originating from the surface impurities. FT-IR spectra were recorded using thin pellets (referenced against a KBr pellet) on a Thermo Nicolet iS50 FT-IR instrument. Diffuse reflectance UV-visible spectra (DRUVS) of the powder samples were recorded using a spectrophotometer (Shimadzu UV-2600) with an integrating sphere attachment and BaSO4 was used as the reference.Photocatalytic activity
The photocatalytic hydrogen production was carried out in aqueous suspension ([cat.] = 0.5 g L−1, [VJ-S]0 = 50 μM, [EDTA]0 = 10 mM as an electron donor) with platinum under visible light (λ > 420 nm). The concentration of the VJ-S dye was fixed at 50 μM. When the dye concentration is larger (or smaller) than 50 μM, the H2 conversion efficiency becomes poor (see Fig. S6†). Platinum (0.05 wt%, as H2PtCl6) was photocatalytically deposited on NGO and NGOT in the presence of VJ-S in EDTA solution. The platinum deposition and the reduction of NGO were simultaneously achieved for r-NGO/Pt and r-NGOT/Pt under visible light irradiation (λ > 420 nm) over 45 min using a 300 W Xe arc lamp (Oriel). The filtered light was focused onto a 30 mL Pyrex reactor with a quartz window. The produced hydrogen was collected in the headspace of the closed reactor and analyzed by a gas chromatograph (GC, HP6890N) with a thermal conductivity detector and Ar as the carrier gas.Results and discussion
Synthesis of VJ-S
We synthesized the bipyridine functionalized squaraine dye VJ-S according to Scheme 1. Bromomethyl bipyridine (1) was synthesized by the bromination of dimethyl bipyridine using NBS. It was converted into its phosphate ester (2) by heating with triethylphosphite at 110 °C for 10 h. Pyrrole-2-carboxaldehyde was N-alkylated with 1-iodohexane using NaH as the base to give 3. The Wittig–Horner reaction of 2 and 3, using t-BuOK as the base, gave bipyridine functionalized pyrrole 4. The squaraine dye VJ-S was synthesized by refluxing 4 and squaric acid in n-BuOH/benzene with the exclusion of moisture. VJ-S was isolated as a pale green powder and was characterized using NMR, elemental analysis and MALDI-TOF mass spectra. | ||
| Scheme 1 Synthesis of VJ-S. | ||
Photophysical and electrochemical properties of VJ-S
The absorption and emission spectra of VJ-S measured in chloroform are shown in Fig. 2. It shows a strong absorption maximum at 684 nm with a high molar extinction coefficient (ε) of 1.277 × 105 M−1 cm−1 corresponding to the π–π* charge-transfer (CT) transition.65 When VJ-S is excited within the CT absorption band at room temperature, it exhibits a strong luminescence maximum at 704 nm. The optical band gap calculated from the intersection of the absorption and emission maxima is found to be 1.77 eV. | ||
| Fig. 2 Photophysical properties of VJ-S. | ||
Cyclic voltammetry of VJ-S was performed in a chloroform solution with 0.1 M tetrabutylammonium hexafluorophosphate as the supporting electrolyte using ferrocene as an internal standard (0.476 V vs. natural hydrogen electrode (NHE)) at a scan rate of 20 mV s−1 and the result is shown in Fig. 3. The oxidation and reduction potentials were 0.889 and −0.795 V vs. NHE, respectively. The HOMO and LUMO levels are found to be −5.21 and −3.53 eV, respectively (and thus, the band gap is 1.684 eV).
 | ||
| Fig. 3 Cyclic voltammogram of VJ-S. | ||
Characterization of VJ-S/r-NGOT
The chemical composition and microstructure of r-NGOT was investigated by high resolution transmission electron microscopy (HR-TEM) and electron energy loss spectroscopy (EELS) analysis for C and N atoms (Fig. 4). The TEM image clearly shows that the TiO2 surface is surrounded by thick graphene layers (Fig. 4a). The elemental mapping analysis exhibits the difference between r-NGOT without (Fig. 4b–d) and with (Fig. 4e–g) VJ-S. | ||
| Fig. 4 (a) HR-TEM image of r-NGOT, and EELS characterization for C and N atoms for r-NGOT without (b–d) and with (e–g) VJ-S. | ||
The X-ray photoelectron spectroscopy (XPS) results given in Fig. 5 also confirm that VJ-S is well-adsorbed on r-NGOT. Because the oxygen-containing groups (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O and C–O at 287.1 and 286.5 eV, respectively) on r-NGOT are photocatalytically reduced,63 the peak intensities of CO and C–O are decreased in both the r-NGOT and VJ-S/r-NGOT samples (Fig. 5a). On the other hand, the binding energy of the N 1s peak in the presence of VJ-S is clearly different from that without VJ-S. The peak at 400 eV appeared in VJ-S/r-NGOT, whereas it is not observed in r-NGOT (Fig. 5b).
O and C–O at 287.1 and 286.5 eV, respectively) on r-NGOT are photocatalytically reduced,63 the peak intensities of CO and C–O are decreased in both the r-NGOT and VJ-S/r-NGOT samples (Fig. 5a). On the other hand, the binding energy of the N 1s peak in the presence of VJ-S is clearly different from that without VJ-S. The peak at 400 eV appeared in VJ-S/r-NGOT, whereas it is not observed in r-NGOT (Fig. 5b).
 | ||
| Fig. 5 XPS spectra of r-NGOT and VJ-S/r-NGOT. | ||
It is known that when graphene is not reduced, FT-IR spectra should show two distinguished peaks at 1728 and 1620 cm−1 (CO and CC).53 But, as shown in Fig. S7,† only the peak corresponding to CC in r-NGOT and VJ-S/r-NGOT is observed, indicating the reduction of graphene, which is consistent with the XPS data. The difference between r-NGOT with and without VJ-S is also confirmed in the FT-IR spectra. The C–H peak located at 2926 cm−1 appeared for VJ-S/r-NGOT, whereas it is absent for r-NGOT. From the results obtained by HR-TEM, EELS mapping, XPS, and FT-IR spectra, we conclude that VJ-S is well-adsorbed on r-NGOT through π–π stacking.
The DRUV spectra of r-NGOT and VJ-S/r-NGOT are shown in Fig. 6. r-NGOT exhibits absorptions below 380 nm (namely, in the UV-region). Once VJ-S was adsorbed on r-NGOT through π–π interaction with r-NGO, the absorption of VJ-S/r-NGOT covers all visible light wavelengths up to 800 nm. The red-shift in the DRUV spectrum of the VJ-S/r-NGOT nanocomposite compared to the absorption spectrum of VJ-S (Fig. 2) is due to the π–π-stacking of VJ-S with r-NGOT.70–73
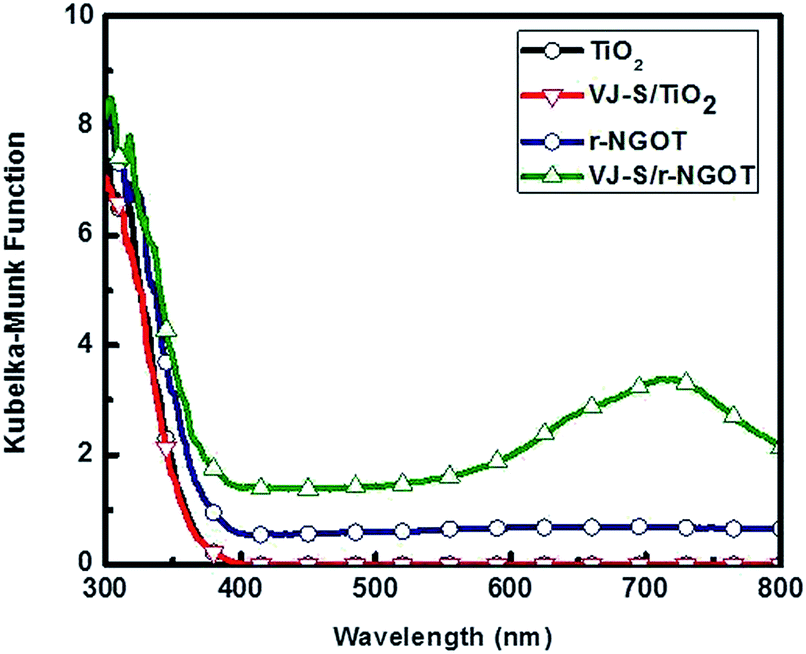 | ||
| Fig. 6 Diffuse reflectance UV-Visible spectra of TiO2, VJ-S/TiO2, r-NGOT and VJ-S/r-NGOT. | ||
Photocatalytic activity of VJ-S/r-NGOT
The visible light photocatalytic activity of VJ-S/r-NGOT was evaluated for the production of hydrogen with EDTA (ethylenediaminetetraacetic acid) as an electron donor. As shown in Fig. 7, since VJ-S cannot be anchored onto the TiO2 surface without r-NGO, the photoinduced electron transfer from the dye to TiO2 is not allowed under visible light irradiation, resulting in a negligible amount of hydrogen evolution. The photocatalytic activity of the VJ-S/r-NGO nanocomposite was slightly enhanced compared to VJ-S/TiO2. This result indicates that squaraine dye is adsorbed onto the r-NGO surface through π–π interaction, which facilitates the interfacial electron transfer. The photo-generated electron is transferred from squaraine dye to graphene and hydrogen is subsequently evolved on the Pt-loaded graphene surface under visible light. However, the photocatalytic activity of VJ-S/r-NGO was still lower because of self-aggregation of r-NGO through π–π stacking.74 However, when r-NGO is deposited to form shells on a TiO2 core, its aggregation is greatly reduced. Therefore, VJ-S/r-NGOT exhibits the highest photocatalytic activity under visible light.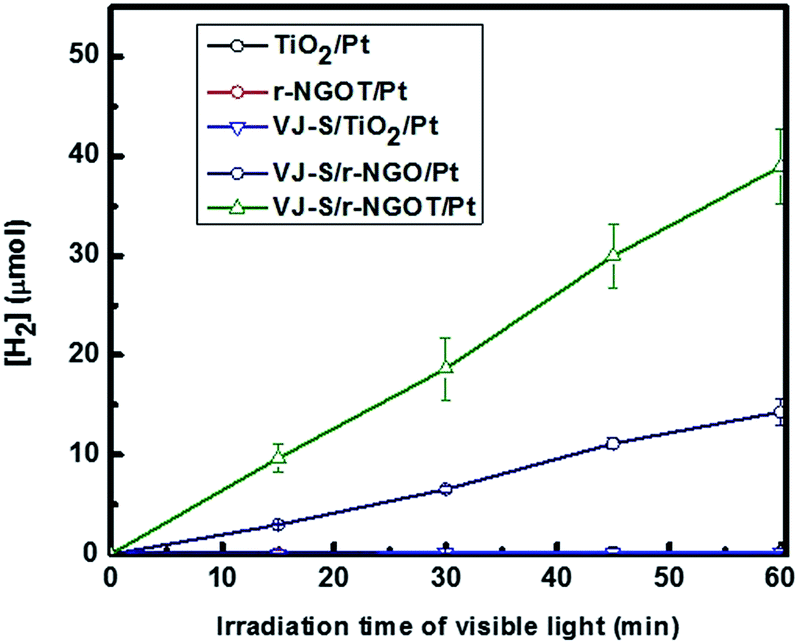 | ||
| Fig. 7 Photocatalytic activity under visible light wavelengths (λ > 420 nm) for VJ-S/r-NGOT/Pt, VJ-S/TiO2/Pt, VJ-S/r-NGO and r-NGOT/Pt. | ||
The apparent photonic efficiency (APE) of hydrogen production is defined as: APE (%) = (2Phydrogen/Iin) × 100, where Phydrogen (mol h−1) represents the rate of hydrogen production. The incident light intensity measured using an optical power meter (1918-R, Newport) was 215 mW cm−2 with a cutoff filter (λ > 420 nm). Thus, the APE of VJ-S/r-NGOT/Pt for H2 production was 0.91%.
Fig. 8 compares the wavelength-dependent photocatalytic hydrogen production in the presence and absence of VJ-S on r-NGOT. r-NGOT without the dye exhibited negligible photocatalytic activity for all visible light wavelengths despite the broad absorption background of r-NGOT at these wavelengths (see Fig. 6). Thus, r-NGO alone has no sensitizing effect. On the other hand, the photocatalytic activity of the dye-loaded r-NGOT is significant throughout the visible wavelengths, although it gradually decreased from 420 nm to 650 nm. This wavelength-dependent activity is not consistent with the main absorption spectral band of VJ-S/r-NGOT (Fig. 6), which shows an absorption maximum around 700 nm. That is, the main absorption band of VJ-S is not responsible for the sensitized production of hydrogen and therefore, the visible light sensitization mechanism in this case is different from that of the conventional dye-sensitized system. A plausible explanation for this phenomenon is that the visible light sensitization is mediated by ligand-to-metal charge-transfer (LMCT).75 Instead of the conventional dye-sensitization, that an electron in the excited LUMO state of the dye is injected to the conduction band (CB) of TiO2, an electron in the ground state (HOMO) of the dye can be directly transferred to the CB of TiO2 without involving the excited LUMO state by absorbing visible light photons. The presence of r-NGO between VJ-S and the TiO2 surface should facilitate the LMCT-type electron transfer through a strong π–π interaction.
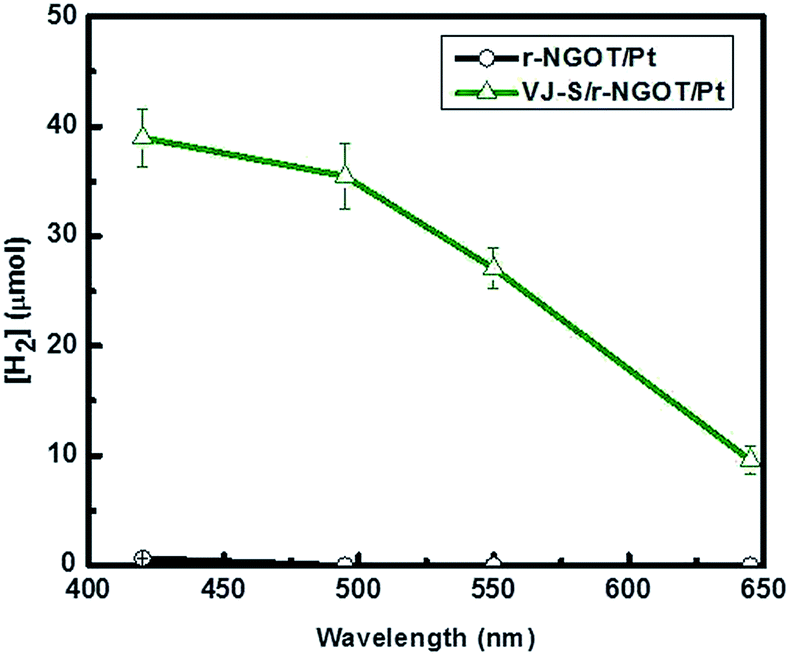 | ||
| Fig. 8 Visible light photocatalytic generation of hydrogen for r-NGOT/Pt and VJ-S/r-NGOT/Pt over 60 min irradiation as a function of wavelength. The wavelength in the x-axis refers to the cut-off wavelength (λc) of the filter and the wavelengths above λc are utilized for hydrogen generation. | ||
Finally, we carried out the stability test of the VJ-S/r-NGOT/Pt nanocomposite. We found that H2 production first increases with irradiation time up to 60 min, then it remains almost the same (less than ∼1% loss) until 120 min of light irradiation (see Fig. S8†).
Conclusion
We have designed and synthesized a NIR-absorbing squaraine dye (VJ-S) that has π-stacking ability with r-NGOT. We observed that the VJ-S was well-adsorbed on r-NGOT through π–π interaction between r-NGO and VJ-S. The photo-generated electrons in the VJ-S dye were transferred through r-NGO to platinum and hydrogen was evolved. Because VJ-S dye absorbs visible and near IR wavelengths and gives a direct energy transfer to r-NGO, the photocatalytic production of hydrogen for VJ-S/r-NGOT/Pt is much higher than that for either VJ-S/TiO2/Pt or r-NGOT/Pt under visible light irradiation.Acknowledgements
This work was supported by the National Creative Research Initiative Program supported by the National Research Foundation of Korea (2013R1A3A2042196).References
- E. Chornet and S. Czernik, Nature, 2002, 418, 928–929 CrossRef CAS PubMed.
- D. Pimentel, M. Herz, M. Glickstein, M. Zimmerman, R. Allen, K. Becker, J. Evans, B. Hussain, R. Sarsfeld and A. Grosfeld, Bioscience, 2002, 52, 1111–1120 CrossRef.
- J. M. Thornton and D. Raftery, ACS Appl. Mater. Interfaces, 2012, 4, 2426–2431 CAS.
- J. Zhang, Y. Wang, J. Zhang, Z. Lin, F. Huang and J. Yu, ACS Appl. Mater. Interfaces, 2013, 5, 1031–1037 CAS.
- A. Fujishima and K. Honda, Nature, 1972, 238, 37–38 CrossRef CAS.
- X. Chen, S. Shen, L. Guo and S. S. Mao, Chem. Rev., 2010, 110, 6503–6570 CrossRef CAS PubMed.
- J. L. Dempsey, B. S. Brunschwig, J. R. Winkler and H. B. Gray, Acc. Chem. Res., 2009, 42, 1995–2004 CrossRef CAS PubMed.
- A. J. Esswein and D. G. Nocera, Chem. Rev., 2007, 107, 4022–4047 CrossRef CAS PubMed.
- M. Kitano and M. Hara, J. Mater. Chem., 2010, 20, 627–641 RSC.
- A. Kudo and Y. Miseki, Chem. Soc. Rev., 2009, 38, 253–278 RSC.
- K. Maeda, J. Photochem. Photobiol., C, 2011, 12, 237–268 CrossRef CAS PubMed.
- F. E. Osterloh, Chem. Mater., 2007, 20, 35–54 CrossRef.
- M. Wang, Y. Na, M. Gorlov and L. Sun, Dalton Trans., 2009, 6458–6467 RSC.
- M. Zhu, M. Han, Y. Du, P. Yang and X. Wang, Dyes Pigm., 2010, 86, 81–86 CrossRef CAS PubMed.
- M. Zhu, Z. Li, Y. Du, Z. Mou and P. Yang, ChemCatChem, 2012, 4, 112–117 CrossRef CAS.
- M. Zhu, Y. Lu, Y. Du, J. Li, X. Wang and P. Yang, Int. J. Hydrogen Energy, 2011, 36, 4298–4304 CrossRef CAS PubMed.
- M. Zhu, Y. Dong, Y. Du, Z. Mou, J. Liu, P. Yang and X. Wang, Chem.–Eur. J., 2012, 18, 4367–4374 CrossRef CAS PubMed.
- X. Chen and S. S. Mao, Chem. Rev., 2007, 107, 2891–2959 CrossRef CAS PubMed.
- C. Liu, X. Han, S. Xie, Q. Kuang, X. Wang, M. Jin, Z. Xie and L. Zheng, Chem.–Asian J., 2013, 8, 282–289 CrossRef CAS PubMed.
- E. Reisner, D. J. Powell, C. Cavazza, J. C. Fontecilla-Camps and F. A. Armstrong, J. Am. Chem. Soc., 2009, 131, 18457–18466 CrossRef CAS PubMed.
- X. Chen, Y. B. Lou, A. C. Samia, C. Burda and J. L. Gole, Adv. Funct. Mater., 2005, 15, 41–49 CrossRef CAS.
- P. Ji, M. Takeuchi, T.-M. Cuong, J. Zhang, M. Matsuoka and M. Anpo, Res. Chem. Intermed., 2010, 36, 327–347 CrossRef CAS PubMed.
- D. Y. Leung, X. Fu, C. Wang, M. Ni, M. K. Leung, X. Wang and X. Fu, ChemSusChem, 2010, 3, 681–694 CrossRef CAS PubMed.
- G. Liu, C. Sun, X. Yan, L. Cheng, Z. Chen, X. Wang, L. Wang, S. C. Smith, G. Q. M. Lu and H.-M. Cheng, J. Mater. Chem., 2009, 19, 2822–2829 RSC.
- G. Zhang, X. Ding, Y. Hu, B. Huang, X. Zhang, X. Qin, J. Zhou and J. Xie, J. Phys. Chem. C, 2008, 112, 17994–17997 CAS.
- D. R. Baker and P. V. Kamat, Adv. Funct. Mater., 2009, 19, 805–811 CrossRef CAS.
- R. Brahimi, Y. Bessekhouad, A. Bouguelia and M. Trari, Catal. Today, 2007, 122, 62–65 CrossRef CAS PubMed.
- H. Tada, T. Mitsui, T. Kiyonaga, T. Akita and K. Tanaka, Nat. Mater., 2006, 5, 782–786 CrossRef CAS PubMed.
- H. Yang, L. Guo, W. Yan and H. Liu, J. Power Sources, 2006, 159, 1305–1309 CrossRef CAS PubMed.
- Y. Mizukoshi, K. Sato, T. J. Konno and N. Masahashi, Appl. Catal., B, 2010, 94, 248–253 CrossRef CAS PubMed.
- G. Wu, T. Chen, G. Zhou, X. Zong and C. Li, Sci. China, Ser. B: Chem., 2008, 51, 97–100 CrossRef CAS PubMed.
- X. Cao, G. Tian, Y. Chen, J. Zhou, W. Zhou, C. Tian and H. Fu, J. Mater. Chem. A, 2014, 2, 4366–4374 CAS.
- Y. Wang, J. Yu, W. Xiao and Q. Li, J. Mater. Chem. A, 2014, 2, 3847–3855 CAS.
- Q. Xiang and J. Yu, J. Phys. Chem. Lett., 2013, 4, 753–759 CrossRef CAS.
- Q. Xiang, J. Yu and M. Jaroniec, Chem. Soc. Rev., 2012, 41, 782–796 RSC.
- H. Yu, Y. Zhao, C. Zhou, L. Shang, Y. Peng, Y. Cao, L.-Z. Wu, C.-H. Tung and T. Zhang, J. Mater. Chem. A, 2014, 2, 3344–3351 CAS.
- V. Krishna, D. Yanes, W. Imaram, A. Angerhofer, B. Koopman and B. Moudgil, Appl. Catal., B, 2008, 79, 376–381 CrossRef CAS PubMed.
- D. K. Lee, I.-S. Cho, S. Lee, S.-T. Bae, J. H. Noh, D. W. Kim and K. S. Hong, Mater. Chem. Phys., 2010, 119, 106–111 CrossRef CAS PubMed.
- Y. Park, S.-H. Kang and W. Choi, Phys. Chem. Chem. Phys., 2011, 13, 9425–9431 RSC.
- H. Zhang, X. Lv, Y. Li, Y. Wang and J. Li, ACS Nano, 2009, 4, 380–386 CrossRef PubMed.
- W. Fan, Q. Lai, Q. Zhang and Y. Wang, J. Phys. Chem. C, 2011, 115, 10694–10701 CAS.
- G. Williams, B. Seger and P. V. Kamat, ACS Nano, 2008, 2, 1487–1491 CrossRef CAS PubMed.
- R. Mohan, K. Krishnamoorthy and S.-J. Kim, Solid State Commun., 2012, 152, 375–380 CrossRef CAS PubMed.
- A. Ferrari, J. Meyer, V. Scardaci, C. Casiraghi, M. Lazzeri, F. Mauri, S. Piscanec, D. Jiang, K. Novoselov and S. Roth, Phys. Rev. Lett., 2006, 97, 187401 CrossRef CAS.
- Y. Yang, L. Ren, C. Zhang, S. Huang and T. Liu, ACS Appl. Mater. Interfaces, 2011, 3, 2779–2785 CAS.
- A. K. Agegnehu, C.-J. Pan, J. Rick, J.-F. Lee, W.-N. Su and B.-J. Hwang, J. Mater. Chem., 2012, 22, 13849–13854 RSC.
- G. Liao, S. Chen, X. Quan, H. Yu and H. Zhao, J. Mater. Chem., 2012, 22, 2721–2726 RSC.
- H. Seema, K. C. Kemp, V. Chandra and K. S. Kim, Nanotechnology, 2012, 23, 355705 CrossRef PubMed.
- X. Bai, L. Wang, R. Zong, Y. Lv, Y. Sun and Y. Zhu, Langmuir, 2013, 29, 3097–3105 CrossRef CAS PubMed.
- Y. T. Liang, B. K. Vijayan, K. A. Gray and M. C. Hersam, Nano Lett., 2011, 11, 2865–2870 CrossRef CAS PubMed.
- X. Liu, L. Pan, T. Lv, G. Zhu, Z. Sun and C. Sun, Chem. Commun., 2011, 47, 11984–11986 RSC.
- J. Zhou, G. Tian, Y. Chen, X. Meng, Y. Shi, X. Cao, K. Pan and H. Fu, Chem. Commun., 2013, 49, 2237–2239 RSC.
- S. Min and G. Lu, Int. J. Hydrogen Energy, 2012, 37, 10564–10574 CrossRef CAS PubMed.
- Y. Chen, Z. Mou, S. Yin, H. Huang, P. Yang, X. Wang and Y. Du, Mater. Lett., 2013, 107, 31–34 CrossRef CAS PubMed.
- M. Zhu, Z. Li, B. Xiao, Y. Lu, Y. Du, P. Yang and X. Wang, ACS Appl. Mater. Interfaces, 2013, 5, 1732–1740 CAS.
- M. J. Allen, V. C. Tung and R. B. Kaner, Chem. Rev., 2009, 110, 132–145 CrossRef PubMed.
- O. C. Compton and S. T. Nguyen, Small, 2010, 6, 711–723 CrossRef CAS PubMed.
- S. Fujii and T. Enoki, J. Am. Chem. Soc., 2010, 132, 10034–10041 CrossRef CAS PubMed.
- L. Jiao, L. Zhang, X. Wang, G. Diankov and H. Dai, Nature, 2009, 458, 877–880 CrossRef CAS PubMed.
- Y. Cui, Q. Fu, H. Zhang and X. Bao, Chem. Commun., 2011, 47, 1470–1472 RSC.
- Y. Li, Y. Hu, Y. Zhao, G. Shi, L. Deng, Y. Hou and L. Qu, Adv. Mater., 2011, 23, 776–780 CrossRef CAS PubMed.
- Q. Zheng, W. H. Ip, X. Lin, N. Yousefi, K. K. Yeung, Z. Li and J.-K. Kim, ACS Nano, 2011, 5, 6039–6051 CrossRef CAS PubMed.
- H.-i. Kim, G.-h. Moon, D. Monllor-Satoca, Y. Park and W. Choi, J. Phys. Chem. C, 2011, 116, 1535–1543 Search PubMed.
- S. Das, K. G. Thomas and M. V. George, Mol. Supramol. Photochem., 1997, 1, 467–518 CAS.
- J.-H. Yum, P. Walter, S. Huber, D. Rentsch, T. Geiger, F. Nüesch, F. De Angelis, M. Grätzel and M. K. Nazeeruddin, J. Am. Chem. Soc., 2007, 129, 10320–10321 CrossRef CAS PubMed.
- Y. Shi, R. Hill, J. H. Yum, A. Dualeh, S. Barlow, M. Grätzel, S. R. Marder and M. K. Nazeeruddin, Angew. Chem., 2011, 123, 6749–6751 CrossRef.
- S. Gould, G. F. Strouse, T. J. Meyer and B. P. Sullivan, Inorg. Chem., 1991, 30, 2942–2949 CrossRef CAS.
- S.-R. Jang, C. Lee, H. Choi, J. J. Ko, J. Lee, R. Vittal and K.-J. Kim, Chem. Mater., 2006, 18, 5604–5608 CrossRef CAS.
- O. Maury, L. Viau, K. Sénéchal, B. Corre, J. P. Guégan, T. Renouard, I. Ledoux, J. Zyss and H. Le Bozec, Chem.–Eur. J., 2004, 10, 4454–4466 CrossRef CAS PubMed.
- D. M. Cropek, A. Metz, A. M. Müller, H. B. Gray, T. Horne, D. C. Horton, O. Poluektov, D. M. Tiede, R. T. Weber and W. L. Jarrett, Dalton Trans., 2012, 41, 13060–13073 RSC.
- A. S. Jalilov, S. Patwardhan, A. Singh, T. Simeon, A. A. Sarjeant, G. C. Schatz and F. D. Lewis, J. Phys. Chem. B, 2013, 118, 125–133 CrossRef PubMed.
- T. Nakano, Polym. J., 2010, 42, 103–123 CrossRef CAS.
- J.-H. Yang, Y. Gao, W. Zhang, P. Tang, J. Tan, A.-H. Lu and D. Ma, J. Phys. Chem. C, 2013, 117, 3785–3788 CAS.
- N. H. Le, H. Seema, K. C. Kemp, N. Ahmed, J. N. Tiwari, S. Park and K. S. Kim, J. Mater. Chem. A, 2013, 1, 12900–12908 CAS.
- G. Zhang, G. Kim and W. Choi, Energy Environ. Sci., 2014, 7, 954–966 CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: NMR, MALDI-TOF and FT-IR spectra, optimized concentration of VJ-S dye and effect of dye amount on H2 production. See DOI: 10.1039/c4ta04313h |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2015 |