
Fabrication and characterisation of spin coated oxidised PMMA to provide a robust surface for on-chip assays†
M.
Rowinska
a,
S. M.
Kelleher
*a,
F.
Soberon
a,
A. J.
Ricco
a and
S.
Daniels
ab
aBiomedical Diagnostics Institute, Dublin City University, Glasnevin, Dublin 9, Ireland. E-mail: susan.kelleher@dcu.ie; Fax: +353 (0)1 700 6558; Tel: +353 (0)1 700 6337
bNational Centre for Plasma Research and Technology, Dublin City University, Glasnevin, Dublin 9, Ireland
First published on 29th October 2014
Abstract
A method to prepare a highly stable carboxylic acid functional surface on various substrates for use in bioassays is reported. A thin layer of a poly(methylmethacrylate) (PMMA) is achieved by spin coating dissolved PMMA onto a variety of underlying substrates at various thicknesses in a range of c. 5–27 nm. Varying the PMMA concentration, time and spinning speed controls the thickness of the spin coated layer. The root-mean-squared roughness values of the spin coated PMMA are less than 1.5 nm, resulting in smooth and uniform layer. Substrate functionalisation is carried out by either ultraviolet/ozone (UV/O3) or oxygen plasma oxidation. Both techniques result in initially stable, highly functional films as demonstrated by the covalent attachment of amino-modified oligonucleotides, however longevity studies comparing the stability of films attached following oxidative activation show better stability for UV/O3 activated substrates when compared to oxygen plasma activated substrates. PMMA films activated by UV/O3 yield highly stable (for up to 24 days) functional surfaces that retain immobilised biomolecules after several extended wash steps. In contrast, films attached to surfaces pre-treated with oxygen plasma discharge lose their functionality within 5 days of oxidation. Direct DNA and sandwich antibody assays were successfully demonstrated on the UV/O3 functionalised surfaces, showing a low level of non-specific binding. Furthermore, the quenching of fluorescently labelled biomolecules bound to PMMA-coated gold-coated slides is shown to be dependent on the PMMA thickness, indicating potential usage in surface-plasmon resonance-based assays.
Introduction
Biomedical diagnostics and adjunct therapeutics are generating keen interest and intense activity, particularly in the realm of rapid, self-contained medical systems suitable for use at the point-of-care (POC). Combining microfluidics and microarrays is one example of advantageous integration that supports portability, shorter analysis time, and reduced consumption and waste of expensive biological reagents.1 The functional recognition-and-capture moieties of chip-based biosensors are generally attached to glass, oxidised or nitrided silicon, noble metal, or polymer surfaces.2–4 Although glass possesses many favourable characteristics e.g. chemical resistance, high electrical insulating properties, optical transparency, high thermal stability, biocompatibility, glass chips also have some limitations, including the requirement for specialised fabrication procedures.5 Synthetic polymers are a promising alternative substrate for biosensors because of their low specific gravity, a selectable range of mechanical and surface properties, and a number of well-established high-volume, low-cost manufacturing techniques.6 A subset of synthetic polymers also possess excellent optical properties such as low intrinsic auto-fluorescence and high transparency.5 Poly (methyl methacrylate) (PMMA) is a particularly attractive material for the fabrication of low-cost micro-total analysis systems (μ-TAS) as it possesses excellent optical, thermal, chemical and biocompatibility properties.7 PMMA can be used to fabricate microchips with micro-channels by injection moulding, hot embossing/imprinting,8,9 laser cutting, or laser ablation.10,11 Pristine PMMA is a relatively inert and moderately hydrophobic material; it does not possess suitable surface chemical functionalities to enable covalent immobilisation of biomolecules. Consequently, without pre-treatment, bio-recognition elements such as oligonucleotides and antibodies can only be non-covalently adsorbed on the methyl ester surface, typically resulting in poor device longevity and/or poor performance.Previous work from our group showed that through appropriate surface treatment, PMMA can be oxidised using simple methods to form a carboxylic acid surface to enable the covalent attachment of biomolecules for bioassay development.7 Other procedures for the chemical functionalisation of PMMA involve amination in a N-lithioethylenediamine solution followed by the addition of a homo bi-functional cross-linker molecule such as glutaraldehyde to enable attachment of aminated biomolecules, followed by capping of the unreacted aldehyde functional groups with a reducing agent.12,13 Wet chemistry is also applied to generate reactive functional groups on the surface and base/acid hydrolysis of PMMA can be used to generate carboxylic acid groups.8,13–16 Alternatively, PMMA can be activated by plasma treatment or UV (e.g. deep UV, vacuum UV).17–20 The surface activation by UV/O3 differs significantly from the low-pressure oxygen plasma discharge. Plasma treatment can chemically modify the top few nanometres of a polymer surface without using solvents or generating chemical waste, and with less degradation and roughening of the material than many wet chemical treatments.21,22 UV/O3 has been extensively used to pre-treat surfaces for fluorescence-based biomolecular detection techniques.7 Modification of polymers by UV/O3 treatment is also a very attractive method for providing carboxylic functionality as it offers the ability to tailor the depth of surface reactivity by varying the wavelength and thus the controlling absorption coefficient.17–19,23–28 Oxygen plasmas are very effective at oxidising organic substrates such as PMMA.29,30
Although spin coating PMMA is a relatively well-known process,31,32 to date there has been no direct comparison of the different oxidation methods to activate the surface, nor has oxidised spin coated PMMA been used as a platform for DNA hybridisation or immunoassays. In this paper, a comparison of the stability, functionality and fabrication process of spin coating and surface activating a thin film of PMMA on a variety of underlying substrates is described. Activation by two different methods, UV/O3 and oxygen plasma, is compared, with a focus on the stability of functionalisation over 24 days. Adhesion studies and robustness of binding of immobilised biomolecules to each surface is also reported and a direct comparison to commercially available epoxy and bulk PMMA surface is performed. Finally, three applications of the activated, spin coated PMMA films are demonstrated: (a) fluorescence quenching of dye-labelled molecules bound to thin PMMA layers on gold, (b) a DNA direct binding assay, and (c) a sandwich immunoassay.
Results and discussion
Thin PMMA spin coated film process
The thicknesses of the films spin coated on glass and silicon substrates at different spin speeds, using solutions with different concentrations of PMMA are presented in Fig. 1(a). A difference in film thickness of less than 1% is measured between the centre of the substrate and a region close to its edge, which confirms uniformity of the coating. A number of different protocols were used to produce the thinnest coatings. Not surprisingly, it was found that the lowest concentration (0.1% w/v) at the highest speed (3000 rpm) resulted in the thinnest film, which measured less than 8 nm.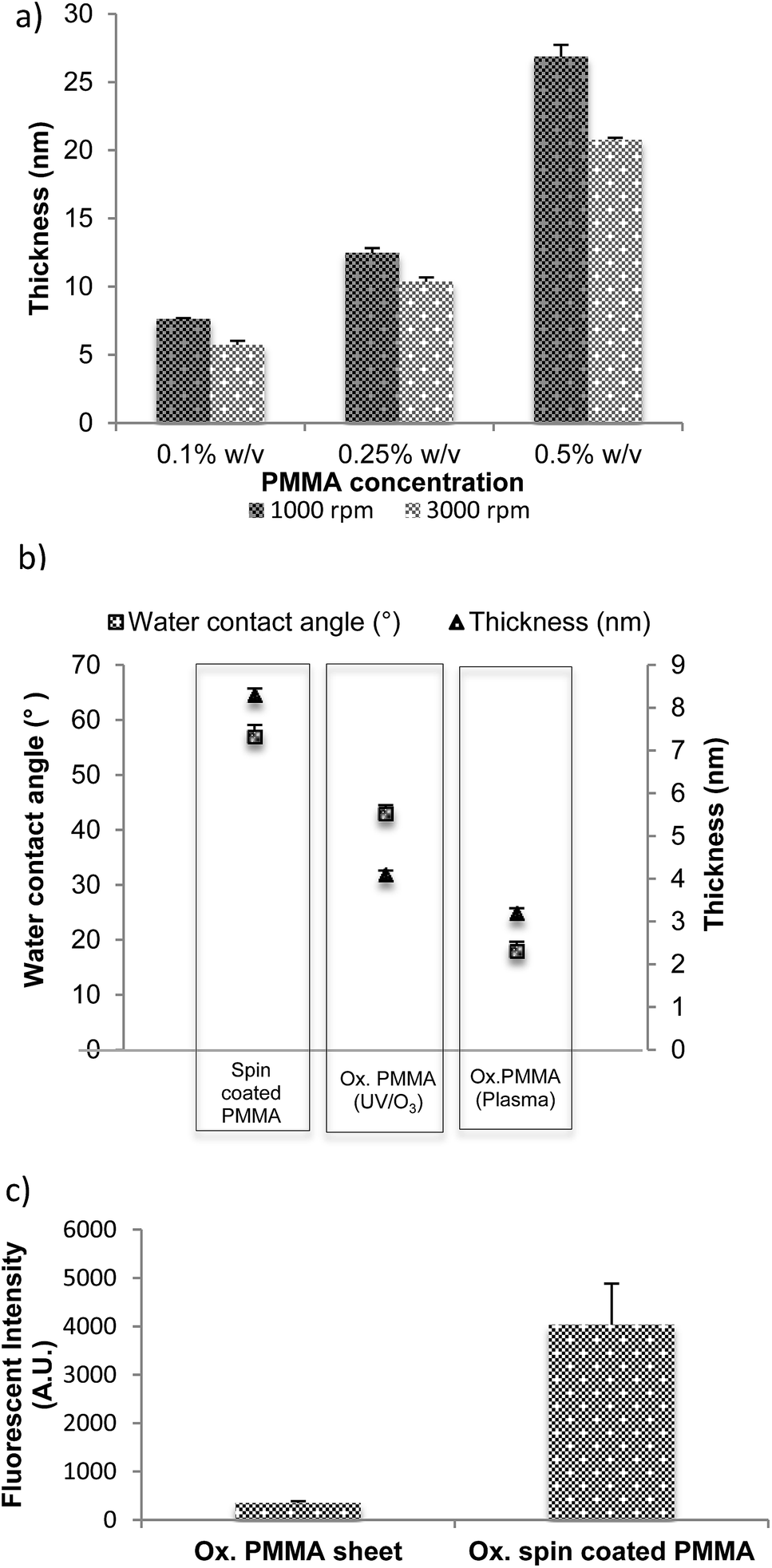 | ||
| Fig. 1 (a) PMMA film thickness (nm) as a function of spin coating speed and concentration. (b) Average water contact angle of spin coated PMMA, UV/O3-oxidised PMMA (ox. PMMA) and oxygen plasma-oxidised PMMA (c) fluorescent intensity of signal from Cy5-labeled DNA probe immobilised to ox. PMMA sheet vs. ox. spin coated PMMA, both treated with UV/O3 for 8 minutes. Error bars are equal to the standard deviation of three fluorescence intensity measurements. Fluorescent intensity was measured at instrument gain of 90. | ||
Tight control over the thickness of films that support capture moieties is very important for a particularly sensitive bioanalytical method, fluorescent quenching by proximity to a metal surface. In this method, the distance between surface-immobilised biomolecules and an underlying metal layer can be related to the strength of the quenching phenomenon: thicker PMMA spacer films reduce the quenching that occurs when a fluorophore is close to a metallic surface; thinner films increase it.33,34 Control of the thickness of the layer allows optimisation of the distance between the metallic surface and the fluorophore in order to enhance the change in the fluorescence signal.35,36 We have studied this phenomenon below.
Spin coated PMMA shows good adhesion on all surfaces tested except for glass slides. Good adhesion of PMMA layer was observed on silicon substrates, which typically develop a thin ∼2 nm silicon oxide layer on the surface. This result suggested that coating the glass slides with a thin oxide layer might improve adhesion. For this reason, the surface is activated with a thin oxide layer using hexamethyldisiloxane (HMDSO) as a precursor (see Materials and methods). Adhesion to plastics such as Zeonor, as well as silicon, does not require any further modifications of the substrate.
Surface morphology of spin coated PMMA films on COP substrates pre- and post-activation by oxidation
Cyclic olefin copolymers (COP) (e.g. Zeonor) are often used in lithography and micro/nanofabrication applications as less-expensive alternatives to silicon substrates, typically as injection-moulded slides, chips, or microfluidic devices. While lacking the ultra-smooth surface characteristic of polished Si wafers, COP surfaces are generally suitable for conducting bioassays, with careful control of surface morphology/roughness being advisable to provide reproducible immobilisation of capture moieties and consistent binding of target analytes.Atomic Force Microscopy (AFM) was used to characterise the overall roughness and morphology of the PMMA-film-coated substrates before and after the activating treatment. Uncoated Zeonor was compared to three types of PMMA-coated Zeonor: as-deposited PMMA, PMMA oxidised by oxygen plasma, and PMMA oxidised by UV/O3. All four surfaces appeared featureless with low average roughness, <1.4 nm (see Fig. S1 in ESI†). It was observed that the deposition of PMMA increases the RMS roughness from ∼0.6 nm (uncoated Zeonor) to ∼1.3 nm (spin coated PMMA); treatment by either UV/O3 or oxygen plasma oxidation then reduces the roughness to <1.0 nm. This suggests that both oxidation processes effectively smooth the deposited PMMA layer, presumably by etching away protruding asperities. Etching involves detachment of loosely bound chemical groups from a top layer and leaving all the finely packed and attached groups to the substrate, resulting in a smoother layer. In terms of the RMS roughness, plasma oxidised surfaces also have a very similar roughness to surfaces treated with UV/O3. The surface roughening is possibly the result of a difference in the mechanical properties between the oxidised layer near the surface (caused by atomic oxygen or ozone) and the underlying UV radiated substrate (caused by UV lamp or radiation generated at the plasma discharge). To utilise spin coated PMMA films in fabrication methods that include photolithography, and for some optical detection modalities, knowledge of the film thickness and wettability in relation to the spin coating and post-processing parameters is important. These parameters were measured with such applications in mind, and to discover any correlation between film surface morphology, film thickness and wettability.
Ellipsometry is an optical technique and it measures the change of polarization upon reflection, hence the reflective material, silicon, was used here instead of transparent glass or plastic. A thin PMMA film was spin coated from a concentration of 0.1% w/v PMMA in toluene onto a silicon wafer at 3000 rpm for 1 minute and cured at room temperature under vacuum overnight, followed by in an oven at 90 °C for 120 minutes. Fig. 1(b) presents thickness and water contact angle measurements for the as-deposited film (spin coated PMMA) as well as after UV/O3 (Ox. PMMA UV/O3), and oxygen plasma treatment (Ox. PMMA plasma). The thickness of the film oxidised by plasma is lower than that oxidised by UV/O3. The water contact angle of the non-oxidised surface was measured at 57°, considerably higher than that of the oxidised surfaces (43° for UV/O3 treated and 18° for plasma treated). This showed that the oxidation did indeed take place. The surface activation by low-pressure oxygen plasma discharge is mainly due to oxygen ion bombardment of the polymer substrate where the energy of the impacting ions, typically greater than 10 eV (230 kcal mol−1), is sufficient to break any bond in the polymer. The bombardment is random and the process may involve the scission of polymer chains by cleavage of backbone bonds or the functionalisation of polymer by group removal and/or oxidation of the methyl or methyl-ester group in PMMA. The data in Fig. 1(b) are consistent with etching that is more aggressive during plasma oxidation compared to the UV/O3 process, which relies on ester group dissociation and methyl/backbone scissioning and oxidation.20
The binding potential of oxidised spin coated PMMA was compared to an oxidised PMMA sheet. The same concentration of a dye-labelled, amino-modified single-stranded DNA fragment (22 bases long) was spotted on each surface in the presence of EDC (1-ethyl-3-(3-dimethylaminopropyl)-carbodiimide), incubated for 30 minutes, after which one wash was carried out. The washing steps are defined as 3 stages: 10 minutes in 0.2XSSC + 0.1% SDS followed by two 10 minutes washes in 0.2XSSC. These solutions were chosen as they reduce the amount of non-specifically (i.e. physically) bound DNA on the surface.
EDC ensures that the binding observed was covalent binding (see later for more detail). The slides were then dried and scanned in order to obtain fluorescence intensity, which was directly proportional to the amount of DNA bound to the surface. Fig. 1(c) shows that the spin coated PMMA was highly active after 8 minutes of oxidation and bound a substantial amount of DNA probe. On the other hand, 8 minutes oxidation was not enough to activate the PMMA sheet; hence, no DNA bound to the surface. It can therefore be concluded that spin coated oxidised PMMA provided a highly activated surface to which biomolecules could bind, when compared the oxidised PMMA in the bulk. We suggest that this increased binding results from spin coated films acting as quasi three-dimensional structures in which there is a lack of close-packing between the functional groups when compared to the material in bulk, thus providing increased functionality on the surface.37
Covalent biomolecule immobilisation onto oxidised PMMA spin coated films
The dye-labelled, 22-mer DNA probe was used to demonstrate the immobilisation efficacy of the activated PMMA polymer surface. DNA binding was examined for activated PMMA produced by both oxidation processes: UV/O3 (8 minutes treatment) and oxygen plasma (1 minute treatment) with and without including a covalent linker, EDC. Fig. 2(a) shows the resultant fluorescence intensity, which is proportional to the quantity of DNA bound to the surface. Non-oxidised PMMA has a minimal DNA probe binding, regardless of the use or lack of the EDC linker. In terms of oxidised PMMA by either UV/O3 or plasma, the amount of DNA bound with EDC linker is much higher than DNA bound/adsorbed without EDC linker. From these results, it is clear that the binding of the probe to the oxidised PMMA is covalent and requires an EDC linker, with very little physical or non-specific binding occurring.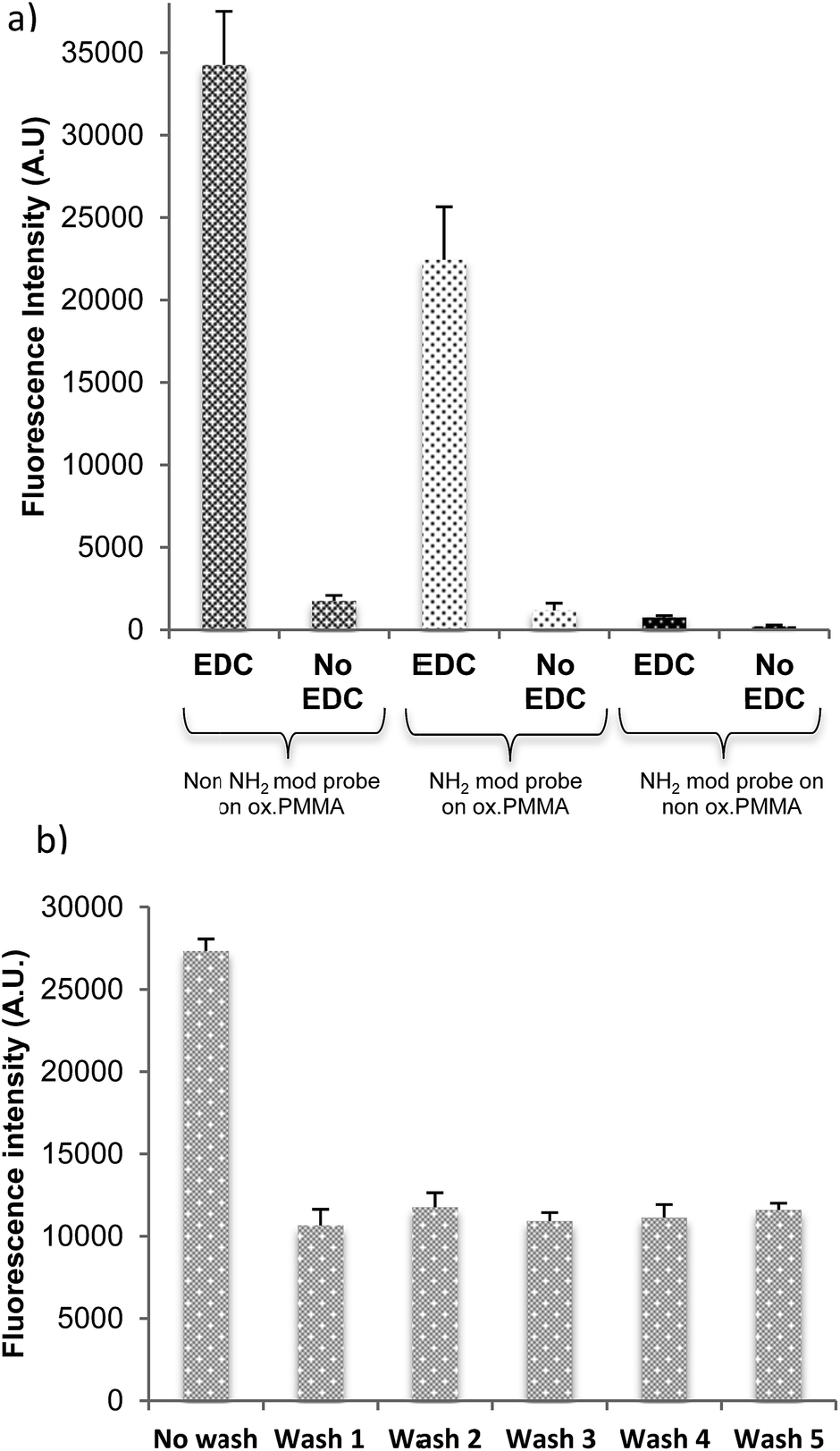 | ||
| Fig. 2 (a) Fluorescence intensity (after one wash) of DNA probe immobilised on as-deposited and oxidised PMMA films, with and without EDC covalent linker. (b) Fluorescence intensity of Cy3-labelled DNA covalently bound via EDC to UV/O3 oxidised PMMA films after the defined wash steps; this wash process was repeated 5 times, as indicated. Error bars are equal to the standard deviation of three fluorescence intensity measurements. Fluorescent intensity was measured at instrument gain of 90. | ||
Stability of the PMMA film upon washing
The long-term stability of spin coated films under aqueous conditions is crucial in the development and fabrication of biosensors and POC devices. Here, adhesion tests of the oxidised PMMA films to the surface as well as its robustness to multiple washes are reported. Adhesion of the PMMA layer onto a particular substrate was tested by checking contact angle of the substrate before and after PMMA spin coating, and by covalently immobilising the DNA probe labelled with a fluorophore. The fluorophore attached to the DNA molecules is used as an indicator of successful binding and surface stability, as DNA will only bind if the oxidised PMMA layer is present; see Fig. 2(a). If the PMMA film is well adhered, a stable fluorescence signal is obtained regardless the washing.The recipe of 0.1% PMMA spin coated at 3000 rpm for 1 minute, followed by UV/O3 oxidation, results in great adhesion and robustness against multiple washing (Fig. 2(b)). A high fluorescence signal is apparent from the Cy3-labelled DNA before wash, suggesting that there are some physically adsorbed DNA strands on the slide. After the wash, the fluorescence signal decreases by half, indicating removal of non-covalently-bound DNA. The amount of DNA present on the surface stabilises after the first wash regardless of how many further washes were carried out (up to 5 were tested).
Stability of the spin coated PMMA against washing was investigated. For measurements of PMMA thickness by ellipsometry, the thin film had to be spin coated on a reflective material (silicon in this instance). Silicon was spin coated with PMMA and oxidised by UV/O3. The thickness was measured after each wash. Each wash consisted of 10 minutes with 0.2XSSC + 0.01% SDS followed by 10 minutes with 0.2XSSC. There was no decrease in thickness of PMMA, which would suggest the PMMA film is robust against multiple washing steps. It also confirms good adhesion of the film to the substrate, silicon in this case.
Longevity studies
Fig. 3(a) summarises the results of a longevity study over a period of 24 days of the spin coated film oxidised using UV/O3. A set of Zeonor microscope slides were treated with a solution of PMMA in toluene, dried, and oxidised with UV/O3 for 8 minutes. The spin coated and oxidised slides were prepared on the first day and stored under vacuum conditions until required. Contact angle and fluorescence intensity of newly bound Cy3-labelled DNA were measured on the first three days in a row, followed by every 2nd/3rd day until day 24. DNA spotting for fluorescent intensity measurements was made on the same day the slide was tested. In Fig. 3(a), slight variations in the fluorescence intensity and contact angle of the bound DNA are observed, which could be caused by variations between each coated slide. The longevity studies were also performed for spin coated PMMA on Zeonor oxidised with oxygen plasma for 1 minute; the data are shown in Fig. 3(b). There was a significant decrease in the fluorescence signal observed in the first 5 days. The intensity of fluorescence decreased after day 5 and remained at this level. The use of EDC linker chemistry in the fluorescence studies, alongside stringent washing protocols confirms to us that the binding occurring is covalent. The results showed that surfaces oxidised by plasma do not remain active for covalent binding for long periods when compared to those oxidised using UV/O3, which provided strong binding for up to 24 days. A change in the water contact angle for films oxidised by plasma over 25 days indicated that the surface was unstable after oxidation.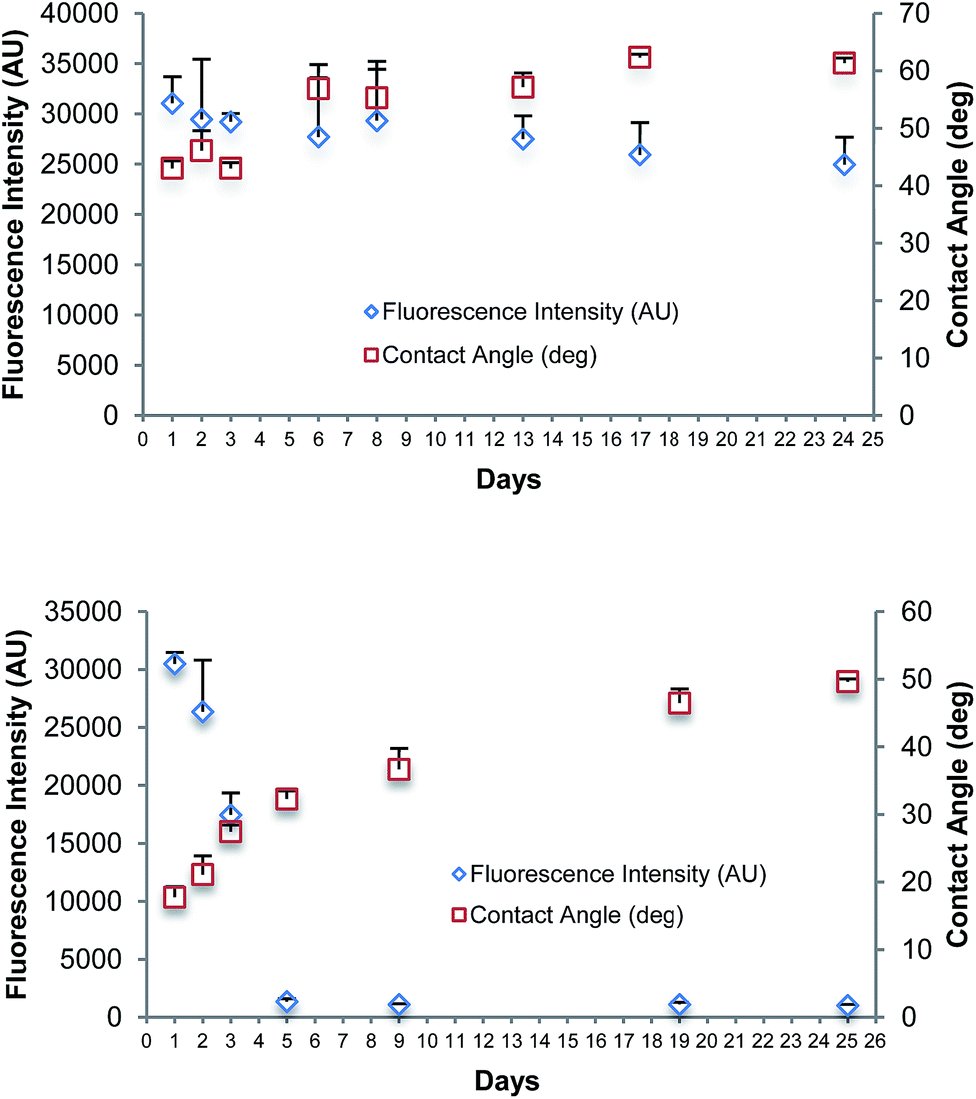 | ||
| Fig. 3 (a) Longevity study of PMMA film spin coated and oxidised by UV/O3 then functionalised by covalent linkage of Cy3-labelled DNA, over a period of 24 days. (b) Longevity study of PMMA film spin coated and oxidised by oxygen plasma, then functionalised by covalent linkage of Cy3-labelled DNA, over a period of 25 days. Fluorescence intensity and water contact angle were measured. Error bars are equal to the standard deviation of three fluorescence intensity measurements. Fluorescent intensity was measured at instrument gain of 90. | ||
Longevity studies indicate that UV/O3 treated substrates retain their functionality over a longer period than those treated by oxygen plasma. The polymer PMMA absorbs UV radiation with wavelengths below 255 nm.17 Short wavelength UV radiation (below 255 nm) is typically absorbed on the top layer of the polymer substrate up to a few hundred nanometres; the penetration depth is wavelength dependent.36 The improved longevity of the UV/O3 treated samples is therefore associated with the modification method where UV radiation penetrates through the entire PMMA film, ∼5–30 nm thick. Thus, the UV radiation generates carboxylic groups within the polymer, not limited to the film surface as in the case of plasma treatment. In this manner, the UV/O3 treated sample is not susceptible to functionality changes on the polymer surface after polymer chain diffusion brings bulk material to the surface over prolonged periods. In the case of oxygen plasma treated samples, only the top polymer layer is modified, but the bottom layer remains unmodified. In addition to the UV radiation, the presence of ozone has to be considered. The effect of ozone or atomic oxygen is localised to the substrate surface, as it cannot penetrate the polymer unlike the UV radiation. Yuan et al. have reported that the combined reaction of VUV and atomic oxygen with PMMA substrate is lower than the sum of individual effects indicating an interfering effect that hinders the mass removal.36
Quenching of fluorophore emission
As shown above, varying the polymer concentration and spinning speed can control the PMMA film thickness. Thickness control is beneficial to control and optimise the strong quenching that occurs when a fluorophore is close to a metallic surface.18,19,27,28Fig. 4 shows the fluorescence signal on a gold-coated glass substrate with thin and thick layers of spin coated PMMA. The layers measure thicknesses of 5.1 ± 0.2 nm, 11.08 ± 0.22, 15.81 ± 0.39, 26.0 ± 0.5 nm and 38.79 ± 0.44 nm respectively. The difference in the fluorescence intensity of an immobilised probe was significant: the quenched signal for the thinnest gold film was negligible compared to an unquenched signal of the thickest film.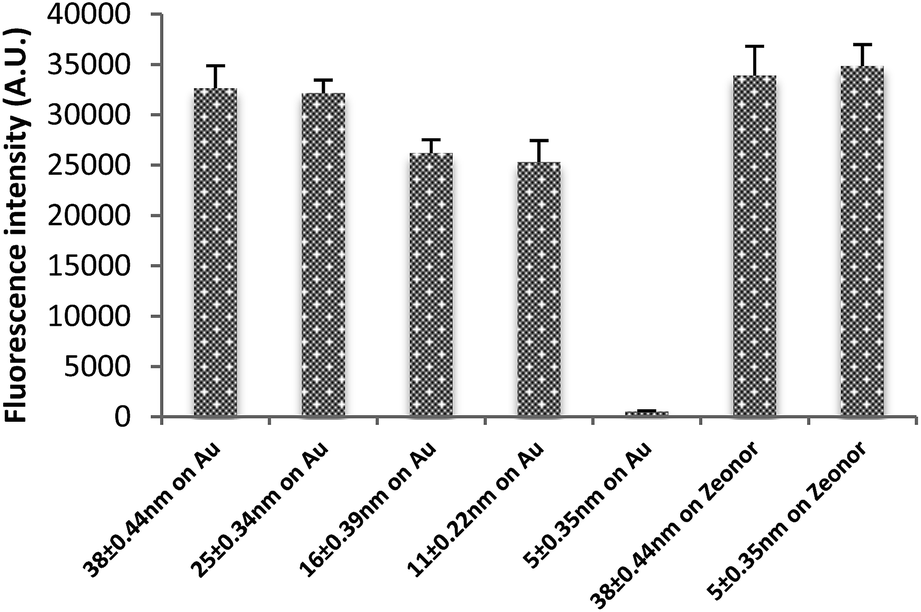 | ||
| Fig. 4 Quenching effect on different thicknesses of spin coated PMMA films on gold-coated glass substrates. The thinnest and thickest films of ox. PMMA on Zeonor (without any gold layer) were used as a control. Error bars are equal to the standard deviation of three fluorescence intensity measurements. Fluorescent intensity was measured at instrument gain of 90. | ||
The same protocol was followed for thick and thin PMMA layers without gold under layers, resulting in similarly large fluorescence signals for both. This control showed that the small signal of DNA bound to the thin PMMA layer on gold was not simply due to a lack of bound DNA.
DNA and antibody-binding experiments
The suitability of spin coated oxidised PMMA for performing both a DNA direct binding and an antibody sandwich assay (α-hIgG/hIgG/α-hIgG-Cy5) was investigated. A schematic diagram of both assays is shown in Fig. 5 and a full protocol is described in Materials and methods section.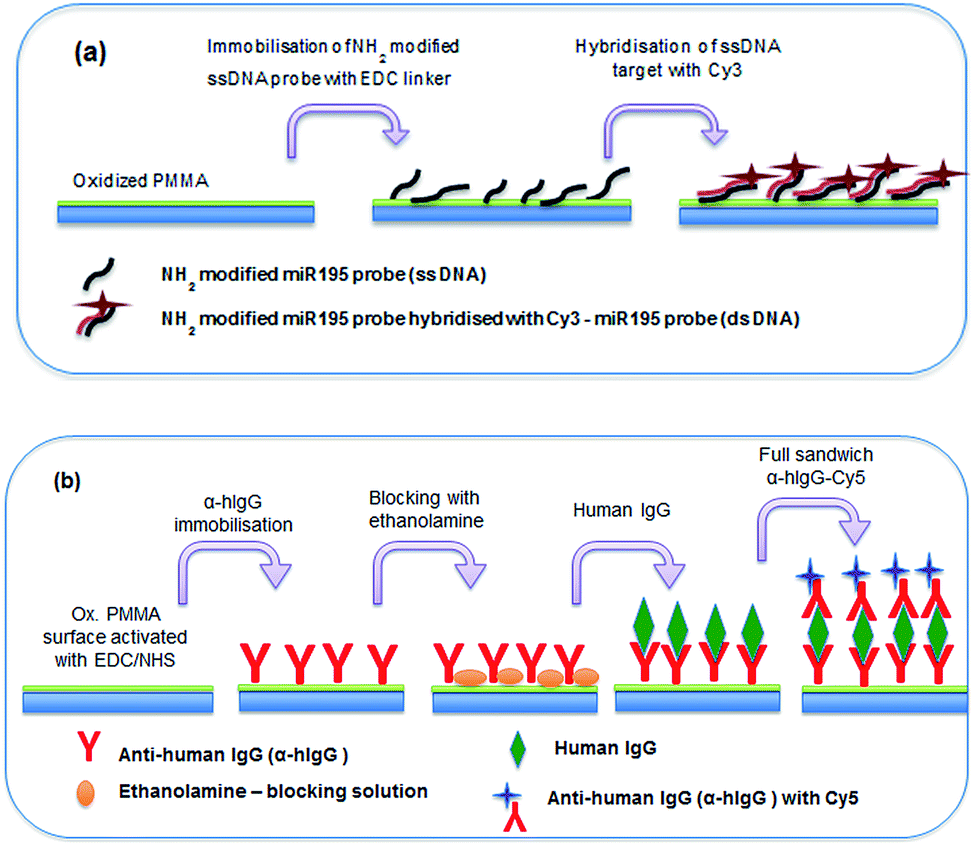 | ||
| Fig. 5 (a) Schematic diagram of DNA binding experiment on glass, spin coated with PMMA and oxidised in UV/O3. (b) Schematic diagram of full IgG sandwich binding experiment on a glass, spin coated with PMMA and oxidised in UV/O3. | ||
For the DNA direct hybridisation assay, capture probes were immobilised using EDC linker chemistry and controls were used to examine non-specific binding (NSB) to the oxidised PMMA surface. Results are shown in Fig. 6(a). The DNA oligomers used were amine-modified, enabling the use of EDC directly in the DNA probe solution. The fluorescence signal for the full hybridisation was significantly high, which confirms a successful hybridisation. A number of controls were carried out to determine if the high signal for DNA hybridisation was specific. All controls showed minimal fluorescence intensity. Control 1 (fluorescent intensity = 13.98 ± 4.2) showed minimal binding of aminated probe to the oxidised PMMA in the absence of EDC linker, which confirmed that EDC linker was crucial to obtain covalent DNA probe immobilisation. Control 2 (fluorescent intensity = 91.3 ± 14.3) was used to determine if the target could bind non-specifically to the surface during the hybridisation step, since there would have been available areas on the surface after probe immobilisation. The fluorescence signal was minimal which proved that the target did not bind to the surface during hybridisation step; hence, the signal obtained for hybridisation was extremely specific. Finally, minimal fluorescence signal in Control 3 (fluorescent intensity = 26.9 ± 3.9) confirmed that the aminated-DNA probe did not bind to non-oxidised PMMA.
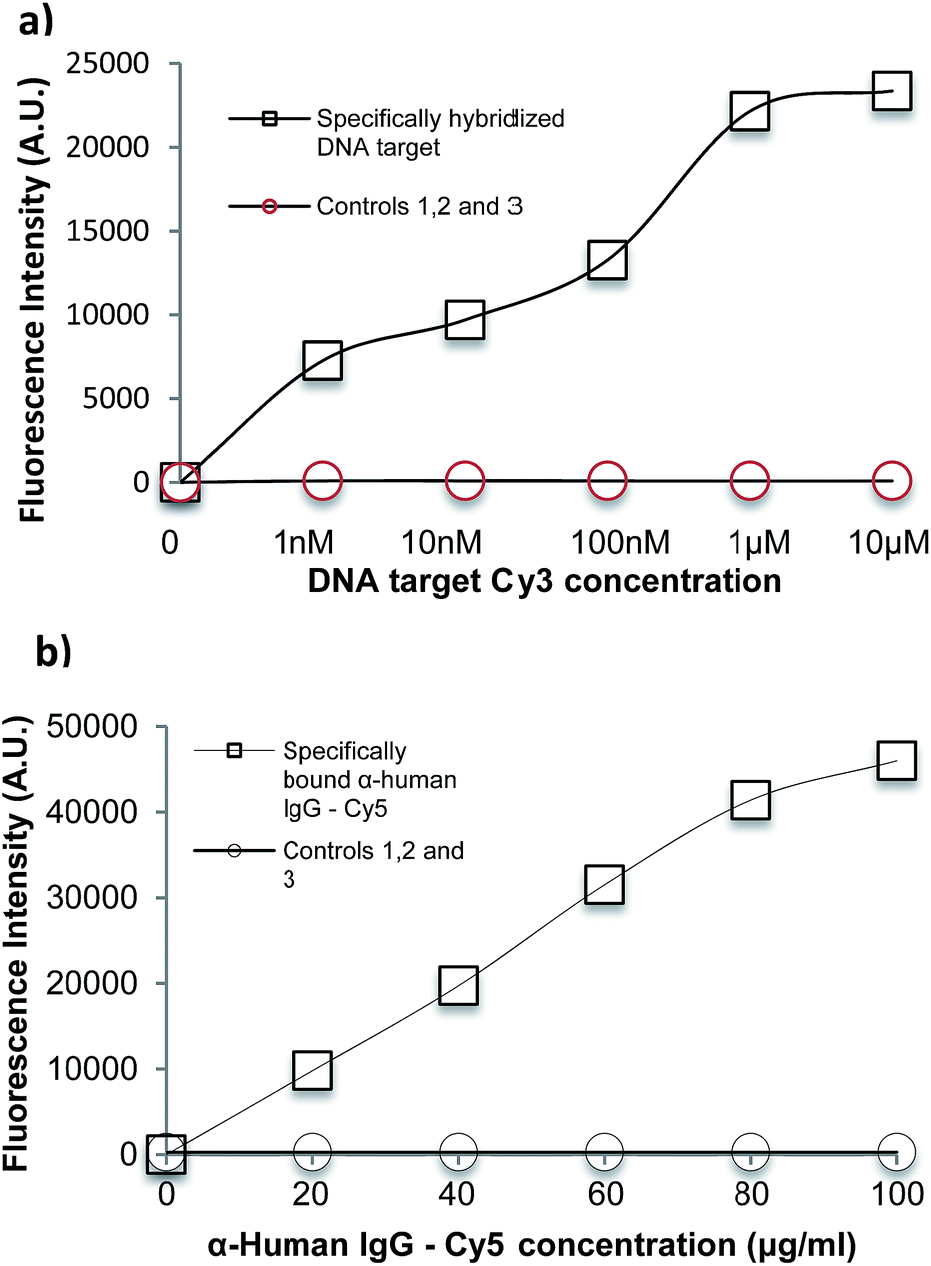 | ||
| Fig. 6 (a) Fluorescence intensity of DNA binding experiment with different DNA target concentrations: 0–10 μM on oxidised PMMA. Control 1 – to determine if NH2 modified probe binds non-specifically with absence of EDC linker, Control 2 – to determine if target can bind non-specifically to the surface during hybridisation step, Control 3 – to investigate if aminated probe can bind non-specifically to non-oxidised PMMA. (b) Fluorescence intensity of IgG binding experiment with different hIgG concentrations: 0–100 μg ml−1 on oxidised PMMA. Control 1 – α-mIgG-Cy3 as the secondary antibody confirms that the binding is specific to α-hIgG. Control 2 – to determine if α-hIgG-Cy5 binds non-specifically to ox. PMMA with absence of EDC linker, Control 3 – to investigate if α-hIgG-Cy5 can bind non-specifically to non-oxidised PMMA. Error bars are equal to the standard deviation of three fluorescence intensity measurements. Fluorescent intensity was measured at instrument gain of 70. | ||
For the immobilisation of antibodies to the surface, EDC/NHS was used to activate the surface before adding any protein as carboxylic acid terminal groups on the protein could bind with amino terminal groups on the protein in the presence of EDC, resulting in crosslinking. Controls were used to determine specificity of the experiment. Results are shown in Fig. 6(b). A full sandwich binding experiment was carried out showing strong and specific binding of hIgG to α-hIgG immobilised on surface, as well as specific binding of the dye-labelled α-hIgG binding to the bound hIgG. The use of Cy3-labelled anti-mouse antibody (α-mIgG-Cy3) in place of the Cy5-labelled α-hIgG as Control 1 confirms the specificity of the binding (fluorescent intensity = 409.83 ± 147.84). Low signal in Control 2 (fluorescent intensity = 287 ± 97) confirmed that α-hIgG-Cy5 did not bind non-specifically when the surface was not activated with EDC/NHS. Control 3 (fluorescent intensity = 254 ± 66) confirmed that α-hIgG did not bind to non-oxidised PMMA. This showed that the oxidised PMMA surface was suitable for immobilisation of proteins followed by a low non-specific binding during sandwich binding experiments.
Experimental
Materials and chemicals
PMMA sheets (0.25 mm thick, impact modified, MW = 120![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000) were sourced from Goodfellow Cambridge Limited (Huntingdon, England). Zeonor® substrates, injection-molded cyclic olefin polymer (COP) slides (Zeonor® 1060R, 25 mm × 75 mm, 1 mm thick) were sourced from Sigolis (Uppsala, Sweden). Gold-coated standard glass microscope slides (Ti/Au, 5 nm/30 nm, 25 mm × 75 mm, 1.1 mm thick) were sourced from PhasisSarl (Geneva, Switzerland). Universal microscope glass slides were sourced from VWR (Dublin 15, Ireland). 1-Ethyl-3-(3-dimethylaminopropyl) carbodiimide (EDC), 2-(N-morpholino)ethanesulfonic acid buffer (MES), sodium dodecyl sulfate (SDS), saline-sodium citrate (SSC) and toluene (anhydrous, 99.8%) were purchased from Sigma Aldrich (Arklow, Ireland). N-Hydroxysulfo-succinimide (Sulfo-NHS) was sourced from ThermoFisher Scientific (Dublin, Ireland). Amino-modified oligonucleotide DNA probes (5′-GCC-AAT-ATT-TCT-GTG-CTG-CTA-3′) (miR-195 probe, 21-mer) and synthetic oligonucleotide target DNAs (5′-TAG-CAG-CAC-GTA-AAT-ATT-GGCG-3′) (miR-16 target, 22-mer) with Cy3 label and without the label and (5′-TAG-CAG-CAC-AGA-AAT-ATT-GGC-3′) (miR-195 target, 21-mer) with Cy3 label and without the label were sourced from Eurofins MWG Operon (Ebersberg, Germany). Human IgG (hIgG), anti-human IgG (α-hIgG), Cy5-labelled anti-human IgG (α-hIgG-Cy5), and Cy3-labelled anti-mouse IgG (α-mIgG-Cy3) were sourced from Biomeda Corp. (Burlingame, California). All reagents were used as received without further purification.
000) were sourced from Goodfellow Cambridge Limited (Huntingdon, England). Zeonor® substrates, injection-molded cyclic olefin polymer (COP) slides (Zeonor® 1060R, 25 mm × 75 mm, 1 mm thick) were sourced from Sigolis (Uppsala, Sweden). Gold-coated standard glass microscope slides (Ti/Au, 5 nm/30 nm, 25 mm × 75 mm, 1.1 mm thick) were sourced from PhasisSarl (Geneva, Switzerland). Universal microscope glass slides were sourced from VWR (Dublin 15, Ireland). 1-Ethyl-3-(3-dimethylaminopropyl) carbodiimide (EDC), 2-(N-morpholino)ethanesulfonic acid buffer (MES), sodium dodecyl sulfate (SDS), saline-sodium citrate (SSC) and toluene (anhydrous, 99.8%) were purchased from Sigma Aldrich (Arklow, Ireland). N-Hydroxysulfo-succinimide (Sulfo-NHS) was sourced from ThermoFisher Scientific (Dublin, Ireland). Amino-modified oligonucleotide DNA probes (5′-GCC-AAT-ATT-TCT-GTG-CTG-CTA-3′) (miR-195 probe, 21-mer) and synthetic oligonucleotide target DNAs (5′-TAG-CAG-CAC-GTA-AAT-ATT-GGCG-3′) (miR-16 target, 22-mer) with Cy3 label and without the label and (5′-TAG-CAG-CAC-AGA-AAT-ATT-GGC-3′) (miR-195 target, 21-mer) with Cy3 label and without the label were sourced from Eurofins MWG Operon (Ebersberg, Germany). Human IgG (hIgG), anti-human IgG (α-hIgG), Cy5-labelled anti-human IgG (α-hIgG-Cy5), and Cy3-labelled anti-mouse IgG (α-mIgG-Cy3) were sourced from Biomeda Corp. (Burlingame, California). All reagents were used as received without further purification.
Preparation of spin coated thin PMMA films
A PMMA sheet was cut into small pieces and dissolved by sonication in toluene at concentrations ranging from 0.1% to 0.5% w/v or, for spin coating films on Zeonor slides, in 80% ethanol. Ethanol was used instead of toluene, if spin coated on Zeonor. Water contact angle and roughness measurements between samples prepared using ethanol and toluene were carried out (see ESI†). The PMMA solution was then sonicated for 2 hours at 50 °C to completely dissolve the PMMA pieces, followed by filtration through a PTFE filter (pore size 0.25 mm, Chroma-filXtra PTFE-20/25 Macherey-Nagel, Duren, Germany) to eliminate precipitates and dust particles. The PMMA solution was then spin coated onto the required substrate slide at 1000 to 3000 rpm for 1 minute. The acceleration and deceleration to and from the desired spin speed occurred in 5 seconds. The samples were then cured at room temperature overnight and then in an oven for 2 hours at 90 °C. This slow annealing approximately 15 °C below the glass-transition temperature of PMMA was employed in order to provide a smoother coating.7 To determine the dependence of film thickness upon spin rate, the PMMA solution was spin coated onto glass slides at 1000, 2000, and 3000 rpm. Fig. 7 represents a schematic of PMMA preparation.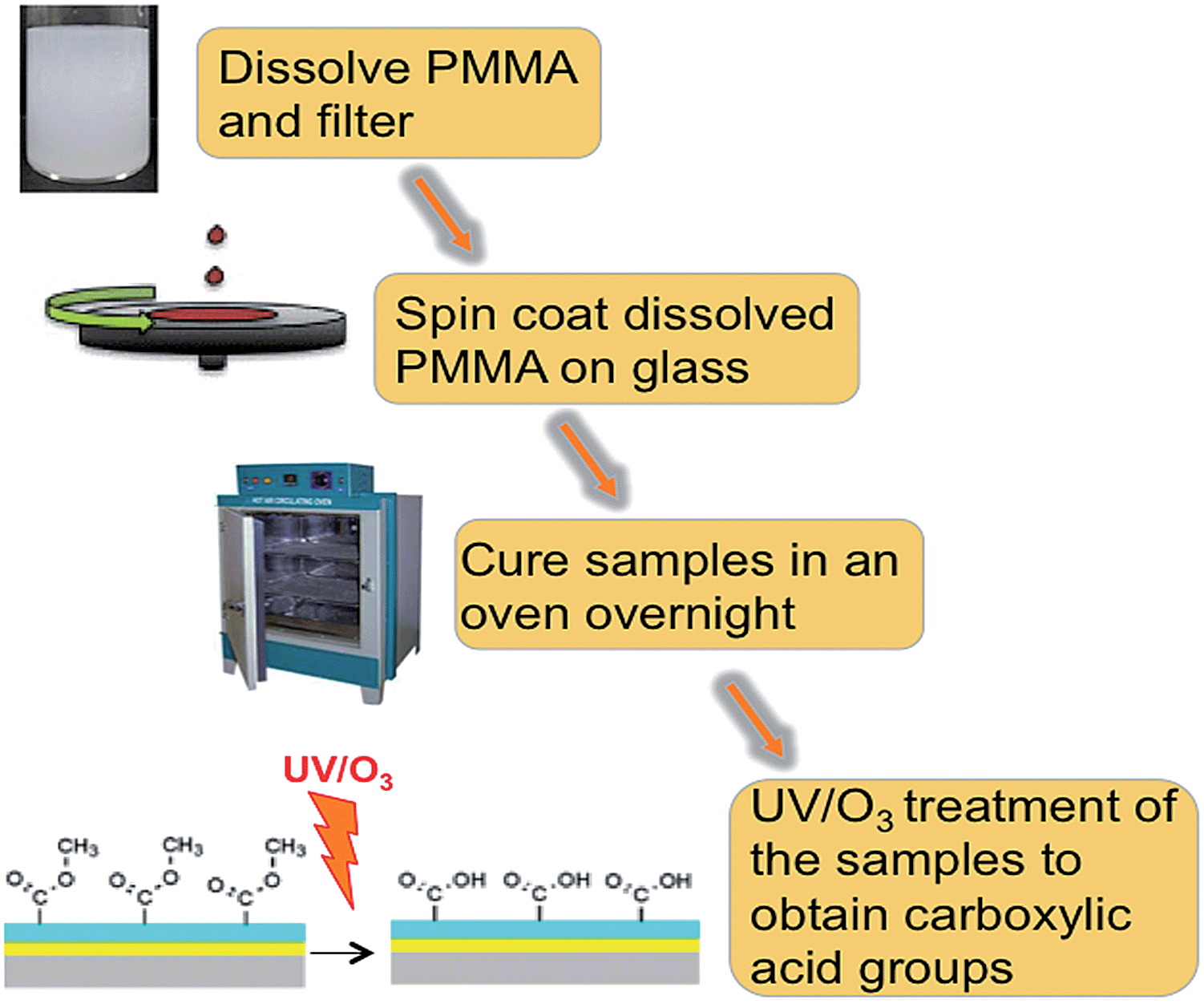 | ||
| Fig. 7 Schematic representation of PMMA film fabrication and activation. | ||
The coating of PMMA films onto glass slides required an additional step, due to the hydrophobicity of the glass. The water contact angle of glass is very low; hence, the PMMA solution could not gain contact with it during spin coating. In order to modify the hydrophobicity of the glass, the deposition of a silicon oxide layer was applied to ensure adhesion of the PMMA. The silicon oxide was deposited with a plasma enhanced chemical vapour deposition (PECVD) method using a Plasmalab 100 system. The precursor reagents for this process were oxygen gas and hexamethyldisiloxane (HMDSO). The silicon oxide layer thickness was approximately 30 nm.
PMMA surface activation
UV/O3 treatment was performed using a commercial ozone cleaning and activation system (PSD-UV, Novascan Technologies, Ames, IA, USA). According to the manufacturer specifications, at the 50 W power setting, approximately 50% of the total lamp output power is delivered around the 254 nm peak and 5% around the 185 nm peak. The optical power was kept constant but the treatment time was varied. A period of 8 minutes of UV/O3 treatment was optimal for spin coated thin PMMA films. When the treatment time was too short, insufficient numbers of carboxylic functionalities were generated, while if the treatment time was too long, the thin PMMA film was etched away. Direct plasma activation was carried out in an Oxford Instruments Plasmalab 100 PECVD system, which consists of a parallel-plate reactor with a radio-frequency (13.56 MHz) driven top electrode, which incorporates a showerhead for uniform gas feed into the vacuum chamber. Substrates are supported on a 200 mm-diameter quartz wafer, which is placed on the bottom electrode. This electrode and the chamber walls are electrically grounded. Plasma activation is performed under the following conditions: oxygen gas flow 50 standard cm3 min−1 (SCCM), pressure 200 mTorr, radio-frequency power 50 W, bottom plate temperature 20 °C, processing time 1 minute.Fluorescence scanner
Fluorescence spectroscopy was carried out using a Perkin Elmer LS 45 fluorescence spectrometer. Fluorescence images were analysed using ScanArray Gx software (Perkin Elmer). PMMA-coated substrates were scanned at 633 nm, the emission wavelength of Cy5, or at 543 nm, the emission wavelength of Cy3, at 10 μm lateral resolution.Water contact angle
The wettability of the different surfaces was analysed by measuring their water contact angle (WCA) using a First Ten Angstroms FTA200 (Portsmouth, VA, USA) contact-angle analyser. High-purity (HPLC grade) water (Sigma Aldrich, Arklow, Ireland) was used for the measurement. The water contact angle of each surface was measured three times at different locations.Spectroscopic ellipsometry
Ellipsometry is an optical technique for investigating dielectric properties and it is widely used to characterise the thickness of thin films. As an optical technique, spectroscopic ellipsometry is non-destructive and contactless. Ellipsometry measures the change of polarization upon reflection and compares it to a model. Hence for measurements of PMMA thickness, thin film had to be spin coated on reflective material, silicon in this instance.The thickness of the PMMA films, and subsequently oxidised/treated PMMA films deposited on silicon substrates, was measured by a UVISEL spectroscopic ellipsometer (JobinYvon Horiba, France). Measurements were performed in triplicate in external mode at an angle of incidence of 70° with wavelengths ranging from 350 to 800 nm with a resolution of 5 nm.
Atomic force microscopy
Surface morphology and roughness of each surface were measured with a Digital Instruments BioScopeTM II (Veeco Instruments Inc., Plainview, NY, USA) atomic force microscope (AFM) in tapping mode in air. Silicon cantilevers with integrated tips (TESP, Veeco Probes, Camarillo, CA, USA) with resonant frequencies between 327 and 349 kHz and with 20–80 N m−1 spring constants were used for measurements. Research NanoScope 7.30 software (Veeco Instruments Inc., Plainview, NY, USA) was used to analyse the data and estimate the root mean squared (RMS) roughness.DNA washing protocol
Each washing step for DNA described contains three distinct stages; 10 minutes in 0.2XSSC + 0.1% SDS followed by two 10 minutes washes in 0.2XSSC. After initial immobilisation, slides were washed once (all three stages). The fluorescent signal measured then was taken as the signal from covalently bound DNA (having removed most of the physically bound DNA). Each wash step thereafter contained the three wash stages.DNA direct hybridisation assay
Probe immobilisation: 1 μM of DNA–micro-RNA 195 analogue (miR195) probe in MES and 100 mM EDC was manually spotted onto the oxidised PMMA surface and left to incubate for 40 minutes at room temperature. The spotting solution was removed by vacuum and the slide was extensively washed once to minimise any non-specific binding (NSB) and loosely bound DNA molecules. The slide was then dried by centrifugation. Target hybridisation: 0.1 μM DNA target in 2XSSC was spotted manually onto previously immobilised probe and incubated for 3 hours in a humidity chamber to avoid evaporation of spotted solution. The spotting solution was removed by the vacuum and washed following the wash protocol detailed above. The slide was then dried by centrifugation and scanned using the fluorescence scanner. Three controls were used: Control 1 contained the aminated probe but no EDC linker. Control 2 consisted of exposing the surface of the slide to the target and was used determine if the target can bind non-specifically to the surface. Finally, Control 3 consisted of an NH2-modified probe exposed to non-oxidised PMMA and was used to investigate if the probes can bind non-specifically to non-oxidised PMMA.Antibody sandwich assay
0.01 g EDC + 0.003 g NHS in 500 μL of MES were manually spotted onto the surface for 15 minutes at room temperature. The slide was then dried by centrifugation. 10 μg ml−1 of α-hIgG was manually spotted and left for incubation for 1 hour at 37 °C in a humidity chamber, followed by washing with 0.1 M PBS/1% Tween twice and then PBS only. 0.1 M ethanolamine was spotted for 10 minutes to allow deactivation of EDC/NHS. The above was rinsed with deionised H2O for 1 minute. 4 mg ml−1 of hIgG solution was spotted and incubated at 37 °C for 1 hour in a humidity chamber. The substrate was then rinsed as outlined above, including an extra PBS and Tween wash. Finally, 20 μg ml−1 of the α-hIgG-Cy5 solution was spotted and incubated for an hour at 37 °C. The substrate was then washed with PBS/Tween and PBS only. The substrate was scanned with the fluorescence scanner. Controls used included use of α-mIgG-Cy3 as opposed to α-hIgG-Cy5, in order to determine the amount of NSB to the immobilised antibodies. α-hIgG-Cy5 was added to the oxidised PMMA and non-oxidised surfaces in order to determine if the antibody binds non-specifically.Conclusion
A method to prepare a carboxylic acid functional film on different substrates for use in bioassays is reported here. By varying the condition of the PMMA spin coating process, including PMMA concentration and solvent, thin, smooth and uniform PMMA films were achieved. Two different techniques were used to generate carboxylic functionalities, which enable covalent binding of amino modified biomolecules. Longevity studies were also presented to compare the stability of the films after the oxidation treatment by UV/O3 and plasma. UV/O3 with comparison to oxygen plasma shows excellent stability of reactivity for a period of up to 24 days. This result could be particularly useful in biosensor applications and confirms that PMMA could be prepared in batches before being employed as a platform. A DNA direct binding assay and IgG immunoassay were successfully demonstrated, confirming the applicability for POC devices and bioassays, specifically showing low non-specific binding. It was shown that by manipulating the spin coated PMMA film thickness on a gold substrate, reduction of the strong quenching or enhancing of the fluorescence signal can be achieved. It can be particularly useful in surface plasmon resonance techniques such as Total Internal Reflective Ellipsometry (TIRE), where the substrate is gold and the immobilised biomolecule has a fluorophore attached. To avoid quenching of the dye, a thicker film should be applied to increase the distance between metal and fluorophore. The ease of preparation, robustness and plausibly cost effectiveness of PMMA films can possibly find its application in carboxylic acid functionalisation of substrates such as glass, plastic, silicon and many more. The surfaces developed here are currently being used in the development of a point-of-care breast cancer diagnostics assay.Acknowledgements
The presented work was supported by Science Foundation Ireland (SFI) grant number 10/CE/B1821.Notes and references
- G. A. Diaz-Quijada, R. Peytavi, A. Nantel, E. Roy, M. G. Bergeron, M. M. Dumoulin and T. Veres, Lab Chip, 2007, 7, 856–862 RSC.
- M. Lovrinovic and C. M. Niemeyer, Angew. Chem., Int. Ed., 2005, 44, 3179–3183 CrossRef CAS PubMed.
- M. C. Pirrung, Angew. Chem., Int. Ed., 2002, 41, 1276–1289 CrossRef CAS.
- V. G. Cheung, M. Morley, F. Aguilar, A. Massimi, R. Kucherlapati and G. Childs, Nat. Genet., 1999, 21, 15–19 CrossRef CAS PubMed.
- Y. H. Tennico, M. T. Koesdjojo, S. Kondo, D. T. Mandrell and V. T. Remcho, Sens. Actuators, B, 2010, 143, 799–804 CrossRef CAS PubMed.
- Y. Li, Z. Wang, L. M. L. Ou and H.-Z. Yu, Anal. Chem., 2007, 79, 426–433 CrossRef CAS PubMed.
- N. C. H. Le, V. Gubala, E. Clancy, T. Barry, T. J. Smith and D. E. Williams, Biosens. Bioelectron., 2012, 36, 250–256 CrossRef CAS PubMed.
- L. Brown, T. Koerner, J. H. Horton and R. D. Oleschuk, Lab Chip, 2006, 6, 66–73 RSC.
- Y. Wang, B. Vaidya, H. D. Farquar, W. Stryjewski, R. P. Hammer, R. L. McCarley, S. A. Soper, Y.-W. Cheng and F. Barany, Anal. Chem., 2003, 75, 1130–1140 CrossRef CAS.
- A. Zribi and J. Fortin, Functional Thin Films and Nanostructures for Sensors, 2009, ISBN 978-0-387-68609-7 Search PubMed.
- L. Yao, B. Liu, T. Chen, S. Liu and T. Zuo, Biomed. Microdevices, 2005, 7, 253–257 CrossRef CAS PubMed.
- F. Fixe, M. Dufva, P. Telleman and C. B. V Christensen, Lab Chip, 2004, 4, 191–195 RSC.
- F. Fixe, M. Dufva, P. Telleman and C. B. V Christensen, Nucleic Acids Res., 2004, 32, e9 CrossRef CAS PubMed.
- K. Holmberg and H. Hydén, Prep. Biochem., 1985, 15, 309–319 CrossRef CAS PubMed.
- M. Tanahashi, T. Yao, T. Kokubo, M. Minoda, T. Miyamoto, T. Nakamura and T. Yamamuro, J. Appl. Biomater., 1994, 5, 339–347 CrossRef CAS PubMed.
- H. K. Varma, K. Sreenivasan, Y. Yokogawa and A. Hosumi, Biomaterials, 2003, 24, 297–303 CrossRef CAS.
- D. C. Miller, J. D. Carloni, D. K. Johnson, J. W. Pankow, E. L. Gjersing, B. To, C. E. Packard, C. E. Kennedy and S. R. Kurtz, Sol. Energy Mater. Sol. Cells, 2013, 111, 165–180 CrossRef CAS PubMed.
- P. Anger, P. Bharadwaj and L. Novotny, Phys. Rev. Lett., 2006, 96, 113002 CrossRef.
- G. Schneider, G. Decher, N. Nerambourg, R. Praho, M. H. V. Werts and M. Blanchard-Desce, Nano Lett., 2006, 6, 530–536 CrossRef CAS PubMed.
- E. Gonzalez II, M. D. Barankin, P. C. Guschl and R. F. Hicks, Plasma Processes Polym., 2010, 7, 482–493 CAS.
- A. Dinklage, T. Klinger, G. Marx and L. Schweikhard, Plasma Physics: Confinement, Transport And Collective Effects, Springer (India) Pvt. Limited, 2007, vol. 2007 Search PubMed.
- J. M. Lane and D. J. Hourston, Prog. Org. Coat., 1993, 21, 269–284 CrossRef CAS.
- C.-M. Chan, T.-M. Ko and H. Hiraoka, Surf. Sci. Rep., 1996, 24, 1–54 CrossRef CAS.
- S. A. Soper, M. Hashimoto, C. Situma, M. C. Murphy, R. L. McCarley, Y.-W. Cheng and F. Barany, Methods, 2005, 37, 103–113 CrossRef CAS PubMed.
- J. M. Goddard and J. H. Hotchkiss, Prog. Polym. Sci., 2007, 32, 698–725 CrossRef CAS PubMed.
- S. Yoshimura, Y. Tsukazaki, M. Kiuchi, S. Sugimoto and S. Hamaguchi, J. Phys. D: Appl. Phys., 2012, 45, 505201 CrossRef.
- J. Zhang and J. R. Lakowicz, Opt. Express, 2007, 15, 2598 CrossRef CAS.
- T. Mangeat, A. Berthier, C. Elie-Caille, M. Perrin, W. Boireau, C. Pieralli and B. Wacogne, Laser Phys., 2009, 19, 252–258 CrossRef CAS.
- N. Vourdas, A. G. Boudouvis and E. Gogolides, J. Phys.: Conf. Ser., 2005, 10, 405–408 CrossRef CAS.
- M. Zeuner, J. Meichsner and H.-U. Poll, Plasma Sources Sci. Technol., 1995, 4, 406–415 CrossRef CAS.
- C. B. Walsh and E. I. Franses, Thin Solid Films, 2003, 429, 71–76 CrossRef CAS.
- N. G. Semaltianos, Microelectron. J., 2007, 38, 754–761 CrossRef CAS PubMed.
- M. Abouelezz, US Department of Commerce. National Technical Information Srevice, PB 281 828. 1978.
- J. O. Choi, J. Vac. Sci. Technol., B: Microelectron. Nanometer Struct.–Process., Meas., Phenom., 1988, 6, 2286 CrossRef CAS.
- H. Shinohara, T. Kasahara, S. Shoji and J. Mizuno, J. Micromech. Microeng., 2011, 21, 085028 CrossRef.
- H. Yuan, D. R. Killelea, S. Tepavcevic, S. I. Kelber and S. J. Sibener, J. Phys. Chem. A, 2011, 115, 3736–3745 CrossRef CAS PubMed.
- C. L. Feng, Z. Zhang, R. Förch, W. Knoll, G. J. Vancso and H. Schönherr, Biomacromolecules, 2005, 6, 3243–3251 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4tb01748j |
| This journal is © The Royal Society of Chemistry 2015 |