A charged iridophosphor for time-resolved luminescent CO2 gas identification†
Received
9th September 2014
, Accepted 3rd October 2014
First published on 10th October 2014
Abstract
A phosphorescent CO2 gas probe based on an iridium(III) complex with 2-phenylimidazo-[4,5-f][1,10]phenanthroline ligand has been developed. Its phosphorescence is quenched by the addition of CH3COO−. The quenched phosphorescence can be recovered by bubbling CO2 into the detecting solution. This phosphorescent CO2 probe exhibits higher photostability and reduced photobleaching than some of the reported organic fluorescent probes. A time-resolved photoluminescence experiment was performed for CO2 gas detection, which could effectively remove the background fluorescence and improve the sensitivity and signal-to-noise ratio of the sensor in complicated media.
Introduction
Carbon dioxide gas identification is of great importance in various fields, such as public health, food packaging, agricultural production, and medicine.1 Quantification of the environmental release of CO2 also needs significant attention because of its pivotal role in controlling global climate change. Many analytical methodologies have been developed for CO2 detection, including infrared spectroscopy, electrochemical and gas chromatography-mass spectrometry (GC-MS) techniques.2 However, these methods are generally complicated, time-consuming, and require expensive and bulky equipment, and some of them cannot tolerate the interference of carbon monoxide.3 Compared with other traditional methods, optical CO2 sensors have attracted considerable interest because of their low cost, simple construction, rapid response, high tolerance to interfering agents, and efficient visibility to the naked eyes.4
Recently, optical sensors have proven to be useful for the study of many analytes because of their inherent ease of manufacture and use, and their relatively high chemical and mechanical stability. It is not uncommon to make use of the acidic properties of CO2 to design such type of optical sensors, but there are only few fluorescent pH indicators reported in the literature that can simultaneously meet the requirements of pKa, photostability and brightness.5 To date, several alternative detection approaches have been developed. By bubbling CO2 into an amine solution, it can generate carbamate ionic liquid, which is accompanied by an increase in viscosity, and such change can be detected with the help of aggregation-induced emission (AIE).6 Recently, an optical CO2 sensor that utilizes tetrapropyl benzobis(imidazolium) salt has been reported for both fluorescence and colorimetric detection.7a
Although excellent CO2 detection has been realized by exploiting fluorescent sensors,7 the interference of background fluorescence is always a significant problem, which might be due to some organic dyes in the solvents. Phosphorescent heavy-metal complexes typically with long emission lifetimes (in the microsecond to millisecond range) offer an effective way to solve this problem using the time-resolved photoluminescence technique (TRPT).8 In addition, their excellent photophysical properties, such as evident Stokes shifts and high photostability, make them a good candidate for sensing applications.9 Because of these reasons, many chemical sensors based on heavy-metal complexes, such as iridium(III), ruthenium(II) and platinum(II) complexes, have been reported for targeting small molecules, anions and metal ions.10 Among these, iridium(III) complexes are regarded to be one of the most promising phosphorescent materials because of their high photoluminescence quantum yields and excellent color tunability. The utilization of these iridium(III) dyes in chemosensing systems has been well-highlighted in the literature.11 To the best of our knowledge, research on the phosphorescent probes for CO2 gas detection is still quite rare.12
The –NH unit on 2-phenylimidazo-[4,5-f][1,10]phenanthroline can interact with CH3COO− to form a strong hydrogen bond, leading to a quenching effect on the luminophore.13 It is well-known that CO2 can readily react with the amine (–NH) group.14 Therefore, it was expected that CO2 could react with the –NH group to destroy the hydrogen bond of (N–H⋯OOCCH3), resulting in the recovery of phosphorescence. In this work, a phosphorescent CO2 sensor 1, in which 2-phenylimidazo-[4,5-f][1,10]phenanthroline is used as an ancillary ligand in a cyclometalated iridium(III) species, has been designed and synthesized (Scheme 1). After bubbling CO2 gas into the solution, the phosphorescence quenched by CH3COO− can be recovered. This phosphorescent CO2 probe displayed a higher photostability and reduced photobleaching compared to some of the reported organic probes. More importantly, the time-resolved photoluminescence technique demonstrates that 1 can be used to detect CO2 in the presence of strong background fluorescence, which improves the sensitivity and signal-to-noise ratio of the sensor in a complicated media.
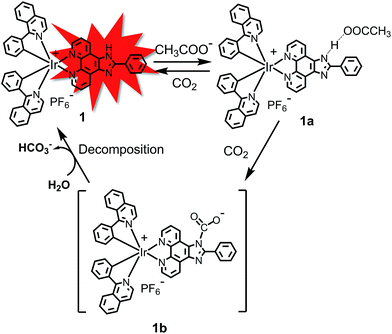 |
| | Scheme 1 Proposed detection mechanism of CO2 gas by complex 1. | |
Results and discussion
Synthetic procedures
The 2-phenylimidazo-[4,5-f][1,10]phenanthroline and 1-phenylisoquinoline ligands were synthesized according to the reported literature.15 Complex 1 was prepared in two steps: firstly, the chloro-bridged iridium(III) precursor, [Ir(piq)2Cl]2 (piq = 1-phenylisoquinoline), was prepared according to the published method.9 Complex 1 was then synthesized through the bridge-splitting reaction of [Ir(piq)2Cl]2 and subsequent complexation with 2-phenylimidazo-[4,5-f][1,10]phenanthroline. The desired complex was characterized by 1H and 13C NMR spectroscopy and MALDI-TOF mass spectrometry.
Photophysical properties
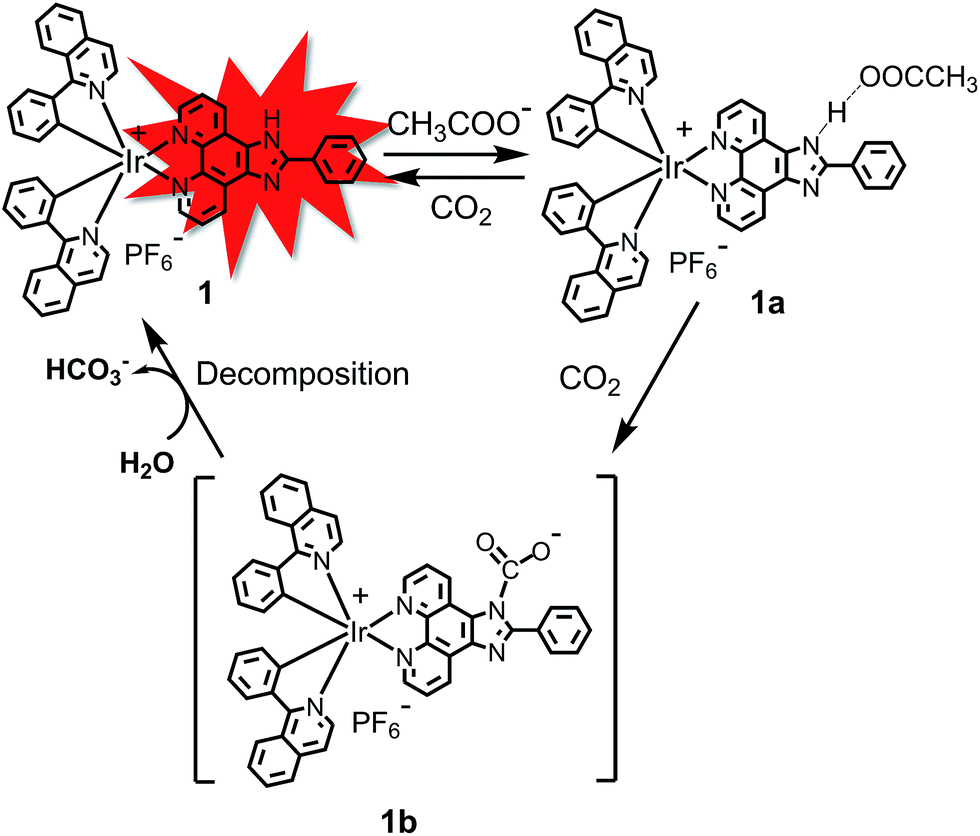
The UV-vis absorption spectrum of 1 in CH3CN displayed an intense band at 281 nm (ε = 3.2 × 104 M−1 cm−1), which was attributed to the spin-allowed ligand-centered transition (1LC). Weaker broad bands at 382 nm (ε = 4.75 × 103 M−1 cm−1) and 444 nm (ε = 2.4 × 103 M−1 cm−1) were attributed to the singlet metal-to-ligand charge-transfer transition (1MLCT), triplet metal-to-ligand charge-transfer transition (3MLCT) and the spin-forbidden ligand-centered transition (3LC) (Fig. 1). The photoluminescence (PL) spectra of 1 in different solutions (CH2Cl2, EtOH, CH3OH, CH3CN and THF) are shown in Fig. 1. The PL maximum of 1 is located at ∼596 nm, which is independent of the solvent changes, suggesting that the emission mainly occurs from the LC excited state.
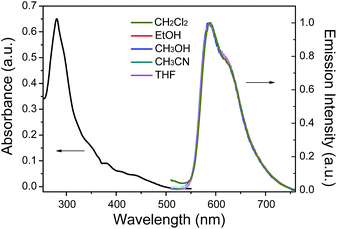 |
| | Fig. 1 Absorption spectra of 1 in CH3CN and PL spectra of 1 in CH2Cl2, EtOH, CH3OH, CH3CN and THF. | |
CO2 detection
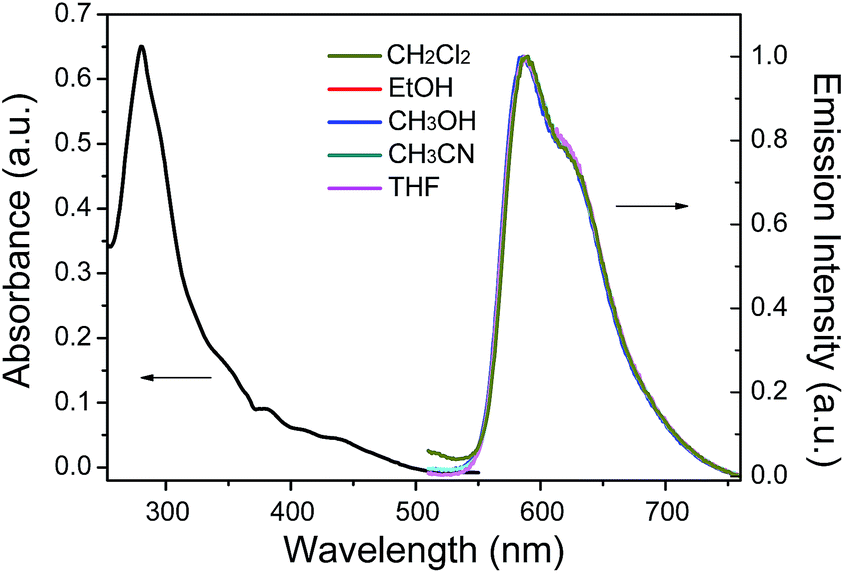
We first investigate the photoluminescence changes of 1 with different anions. Upon the addition of CH3COO− and F−, the emission of 1 was significantly quenched. The quenching of 1 is believed to be due to an electron transfer process from the nitrogen atom of the imidazolyl group, which possesses a partial negative charge after the formation of a hydrogen bond (Scheme 1), to the excited iridium center. However, there were no obvious spectral variations for 1 upon the addition of other anions (such as Cl−, Br−, I−, NO3−, ClO4−, and HSO4−) (Fig. 2).
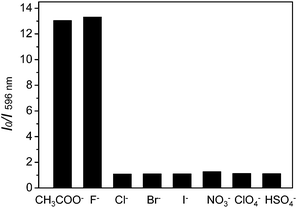 |
| | Fig. 2 Photoluminescence responses of complex 1 (10 μM) with the addition of 3.0 equiv. of various anions, Cl−, Br−, I−, NO3−, ClO4− and HSO4− in CH3CN. The black bars represent the I0/I ratios at 596 nm after the addition of various anions to the solutions of complex 1. | |
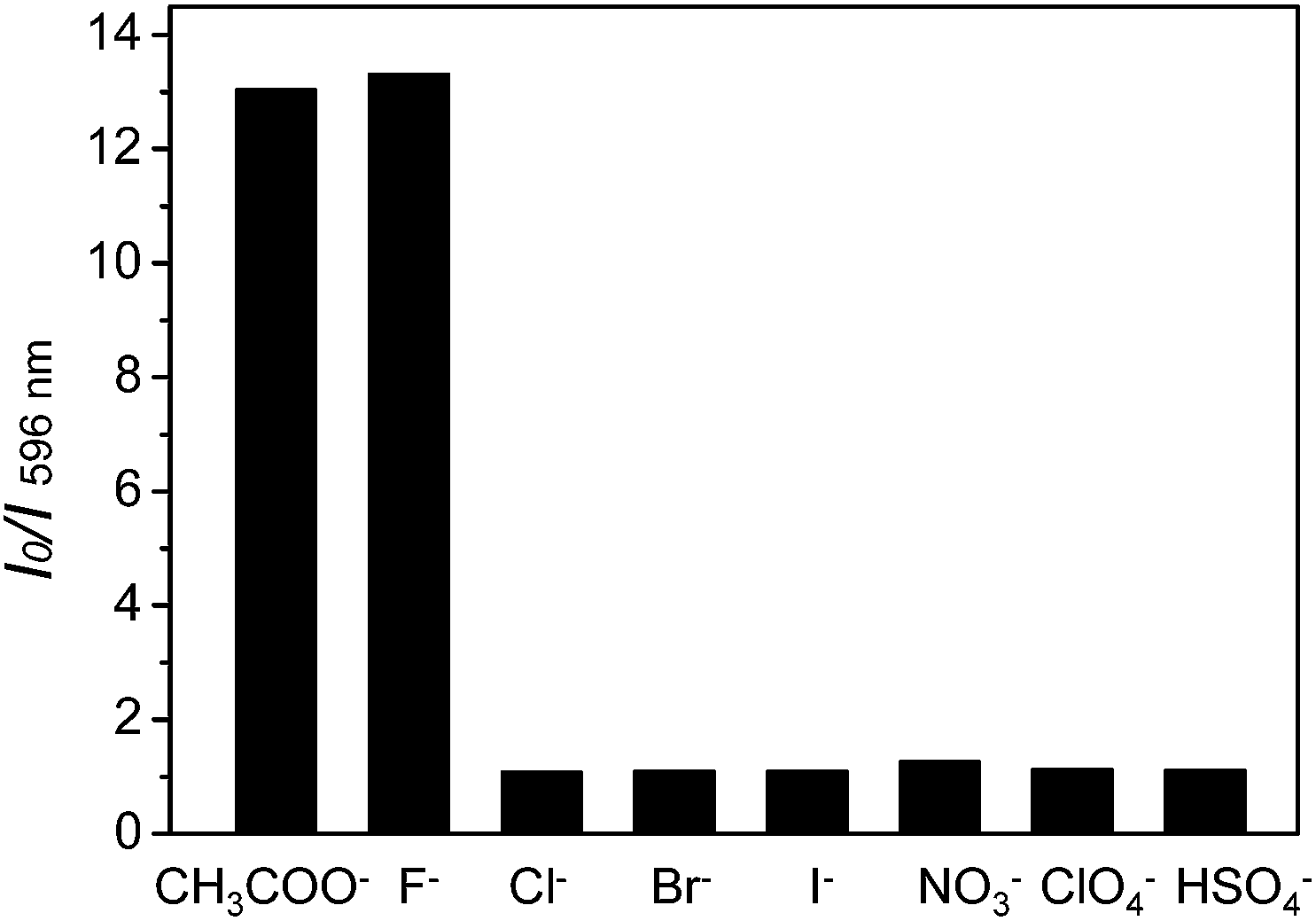
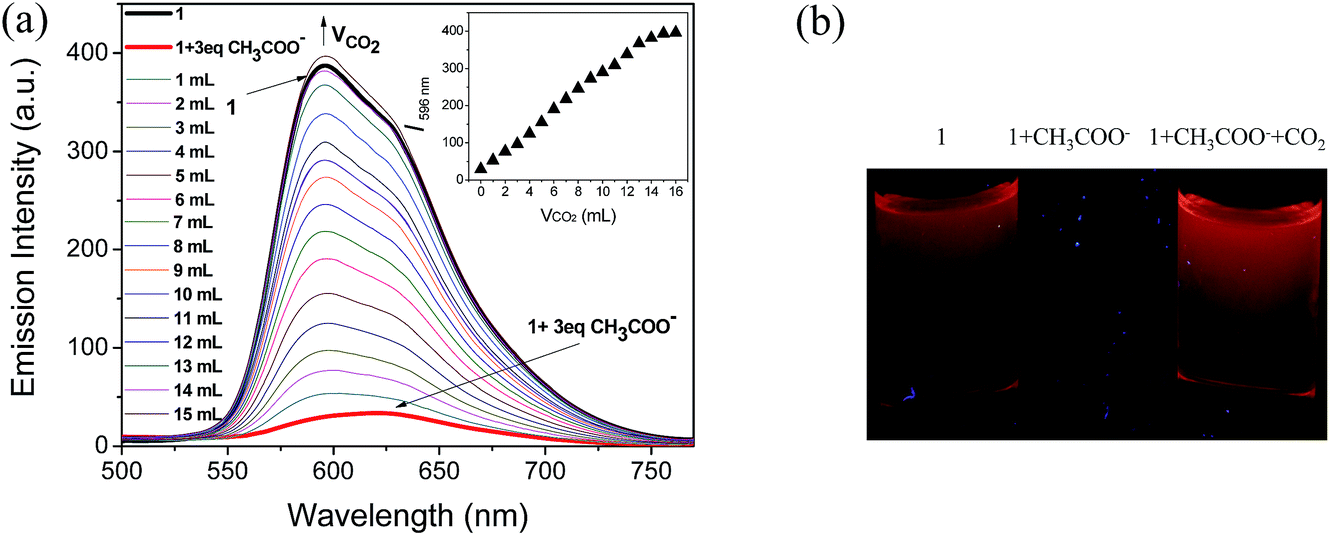
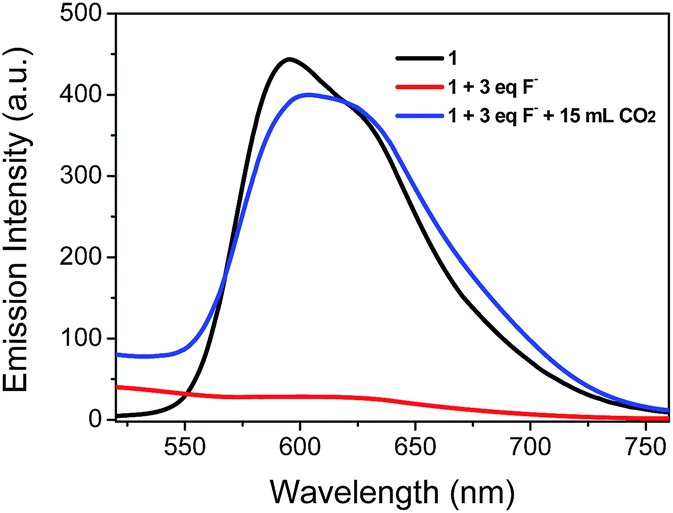
Because CH3COO− shows relatively low toxicity to the environment compared to F−, it was selected to interact with complex 1 to quench its luminescence. As shown in Fig. 3, when CH3COO− (3 equiv.) was added to a CH3CN solution of 1 (10 μM), the absorption band at 281 nm was red-shifted to 295 nm and a new band appeared at 346 nm, indicating that a strong N–H⋯OOCCH3 bond had been formed and the phosphorescence emission was significantly quenched (Fig. 4). Subsequently, it was expected that the bubbling of CO2 gas into the solution of 1 containing CH3COO− would destroy the hydrogen bond by interacting CO2 with the –NH unit on the imidazolyl group, and then recover the luminescence. With the increasing volume of CO2 (as governed by a mass flow controller) bubbled into the solution, the absorbance at 295 and 346 nm gradually decreased (Fig. 3), suggesting that CO2 reacted with the lone electron pair in 1. Furthermore, a dramatic increase in the phosphorescence intensity at 596 nm was observed upon exposure to an increasing volume of CO2 gas (Fig. 4a). As shown in Fig. 4b, after being bubbled with approximately 15 mL of CO2 gas, the mixture emitted bright orange light, and no additional change in the emission spectrum was observed. The same phenomenon was observed by the addition of F− followed by bubbling with CO2 gas (Fig. 5). The selectivity of complex 1 to CH3COO− for CO2 is extremely high because there are no apparent changes in the phosphorescence intensity upon treatment with CO, N2, O2 or argon gas. The CO2 detection limit of this complex was calculated to be ca. 6.45 × 10−6 M (Fig. S1†).
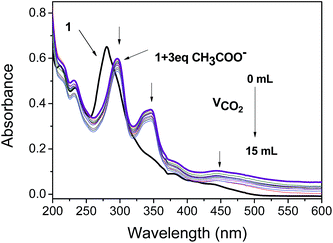 |
| | Fig. 3 Changes in the UV-vis spectra of complex 1 in CH3CN solution (10 μM) containing CH3COO− (3 equiv.) with different volumes of CO2 gas (0–15 mL). | |
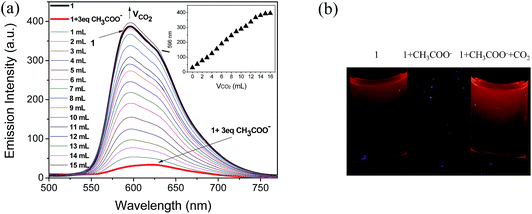 |
| | Fig. 4 (a) Changes in the phosphorescence spectra of 1 in CH3CN solution (10 μM) containing CH3COO− (3 equiv.) with different volumes of CO2 gas (0–15 mL). Inset: titration curve of 1 solution (10 μM) containing CH3COO− (3 equiv.) with CO2 gas (0–15 mL). (b) Images of the emission of 1, 1 + CH3COO−, and 1 + CH3COO− with CO2 bubbling in CH3CN. | |
 |
| | Fig. 5 Changes in the phosphorescence spectra of complex 1 in CH3CN solution (10 μM) containing F− (3 equiv.) with 15 mL of CO2 gas. | |
The CO2–N2 mixtures with different percentages of CO2 were bubbled into the detecting solution to determine the response of PL of 1 to variations in the CO2 fraction (fCO2). As shown in Fig. S2,† the PL spectrum of 1 was intensified with the increasing percentage of CO2, which further indicated the high sensitivity of 1. In addition, the plot showed a characteristic linear signal over the entire concentration range (Fig. S2†). From this calibration curve, the amount of CO2 in the gas mixture could be quantified.
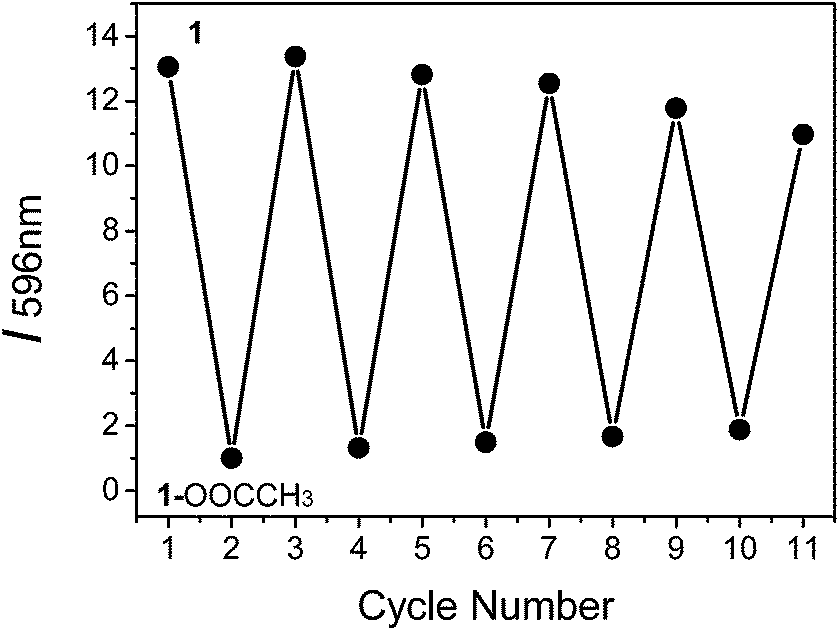
To determine if the phosphorescence of complex 1 can be repeatedly switched on and off by treatment with CH3COO− followed by CO2 bubbling, we conducted experiments with on–off cycling between 1 and 1–OOCCH3. It was observed that complex 1 was not degraded after five cycles of treatment with CH3COO− followed by CO2 bubbling, indicating the good cyclability of the probe (Fig. 6).
 |
| | Fig. 6 Reversible phosphorescence intensity (I596 nm) change between 1–OOCCH3 (bottom points) and 1 (top points), which corresponds to the repeated treatment of 1 by the addition of CH3COO− followed by CO2 bubbling. | |
Time-resolved phosphorescence assay
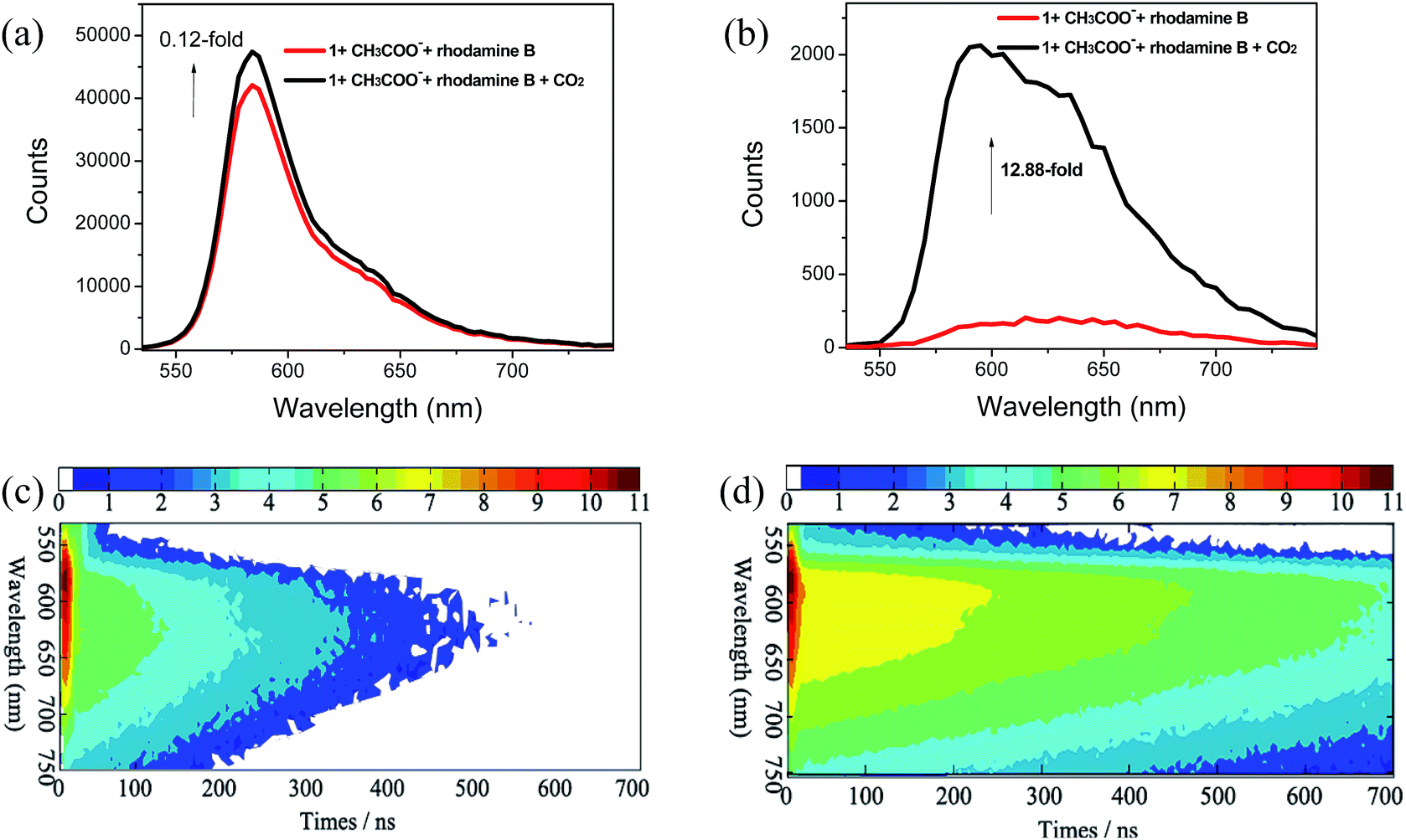
The time-resolved emission spectra (TRES) provide essential information about the luminophore as a function of time. The emission spectra on the time–intensity–wavelength contour plot can be set up by recording the phosphorescence decay at various wavelengths, which can provide insight into the chemical surroundings of the luminophore and their decay profile. In particular, one of the most important features of 1 is the long phosphorescence lifetime (τ), which is beneficial for avoiding the interference of short-lived background fluorescence emission. Therefore, complex 1 is envisioned to be useful for the detection of CO2 gas in the presence of short-lived fluorescence emission by employing the time-resolved PL technique. Here, rhodamine B (RB) with a τ value of 1.67 ns (Fig. S3†) was selected to be a typical source of short-lived background fluorescence because its broad emission band can overlap with that of 1. The emission lifetime of 1 in CH3CN solution was measured to be τ = 227 ns (Fig. S3†), which is sufficiently long to carry out the TRPT experiment. The steady-state PL spectrum for the mixture of complex 1 with CH3COO− and RB was dominated by the strong emission of RB. The emission maximum at 584 nm was assigned to RB (Fig. 7a, red line and S4†), which made it difficult to observe the emission band from 1 with CH3COO−. Subsequently, 15 mL CO2 gas was bubbled into the mixture to react with the complex 1 containing CH3COO−. However, very slight change in the steady-state spectrum (Fig. 7a, black line) was observed, which revealed that the detection of the probe suffered from severe interference due to the background fluorescence. In sharp contrast, the apparent emission variation in the time-gated PL spectrum acquired after a 100 ns delay of the mixture was observed with a 12.9-fold increase in the phosphorescent emission intensity (Fig. 7b). The time-resolved emission spectra of the mixture are portrayed in Fig. 7c and d. As can be seen from Fig. 7c, the emission intensity of the mixture of complex 1 with CH3COO− and RB was very strong at 584 nm within 10 ns because of the strong emission of RB, while the signal rapidly became weaker after about 20 ns because the phosphorescence of complex 1 was quenched by CH3COO−. By bubbling 15 mL CO2 gas into the mixture, distinct emission intensity enhancement at 596 nm was observed in TRES even after 200 ns due to the recovery of the long-lived phosphorescence of complex 1. This clearly suggests that TRES can help to easily distinguish the difference between short-lived fluorescence from RB and long-lived phosphorescence from complex 1 after the time delay between the photoexcitation and acquisition of signals. This result highlights the promising applications of long-lived phosphorescent probes in time-resolved assays.
 |
| | Fig. 7 PL spectra of the mixture of complex 1 solution containing CH3COO− and rhodamine B with bubbling of 15 mL CO2 gas (a) in a steady state (red line, 1 containing CH3COO− and rhodamine B; black line, treatment of mixture by bubbling 15 mL CO2) and (b) at delay time of 100 ns (red line, 1 containing CH3COO− and rhodamine B; black line, treatment of the mixture by bubbling 15 mL CO2) excited at 365 nm. (c) Time-resolved emission spectra of the mixture of complex 1 solution containing CH3COO− and rhodamine B. (d) Time-resolved emission spectra of the mixture of complex 1 solution containing CH3COO− and rhodamine B with bubbling of 15 mL CO2 gas. Scale bars in (c) and (d) refer to the emission intensity. | |
Photostability
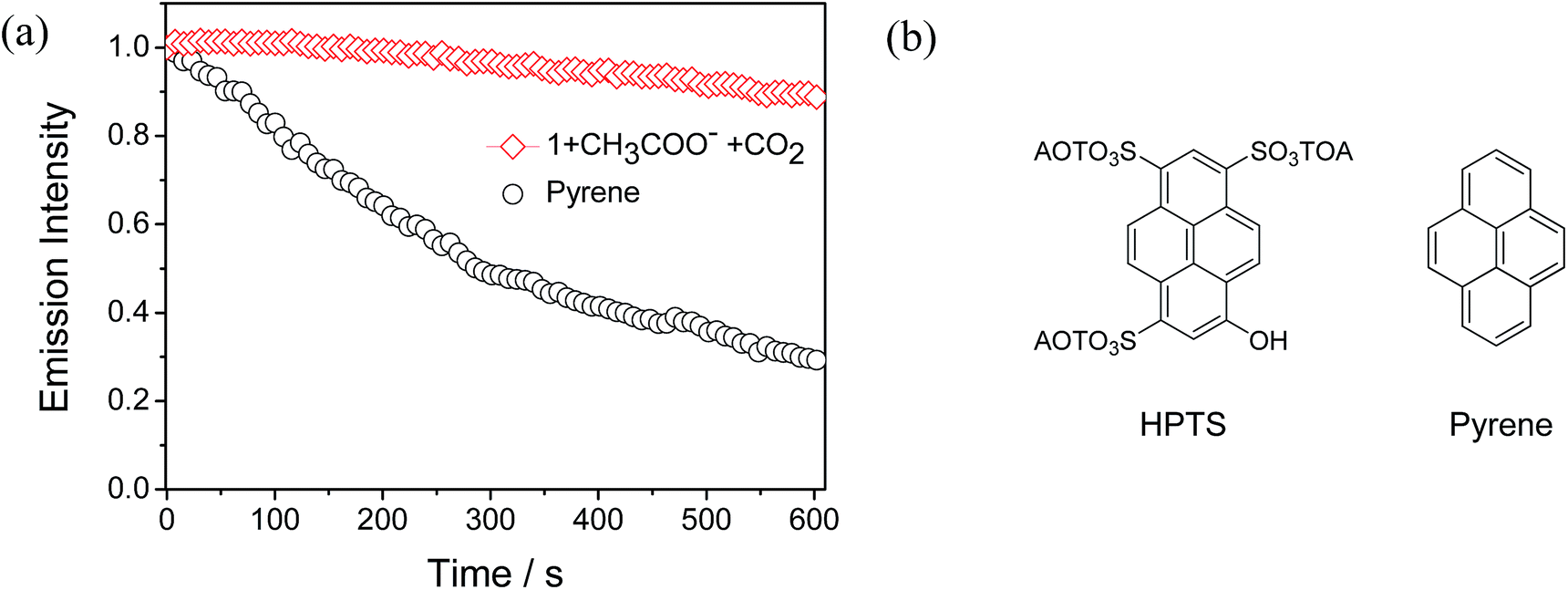
Considering that continuous excitation often results in the photobleaching of fluorescent sensors, and thus high photostability becomes a very important issue for fluorescent sensors. In particular, in real-life applications, high photostability makes a sensor suitable for long-duration measurements and validates the accuracy of quantitation. Therefore, photobleaching experiments were carried out between complex 1 and pyrene under the same excitation conditions (405 nm, laser as the excitation source) in CH3CN solution. Pyrene has a similar structure to 8-hydroxypyrene-1,3,6-trisulfonate (HPTS) (Fig. 8b), which is the state-of-the-art sensor for CO2 gas.4d As shown in Fig. 8a, after the continuous excitation at 405 nm for 600 seconds, which is sufficiently long to perform the identification in practical applications, the emission intensity of complex 1 only decreased to 90% of its initial value. This high photostablility of complex 1 suggests that it is suitable for long-duration measurements and accurate quantitation. In contrast, the emission intensity of pyrene decreased to 29% of the original value due to photobleaching. This result demonstrates that our phosphorescent sensor has higher photostability and reduced photobleaching than that of an organic pyrene, which is very important for the “real-world” application of a sensor.
 |
| | Fig. 8 (a) Emission intensity of a solution of complex 1 containing CH3COO− with bubbling of CO2 gas and pyrene under continuous laser excitation (at 405 nm). (b) Chemical structures of HPTS and pyrene. | |
Detection mechanism of CO2 gas
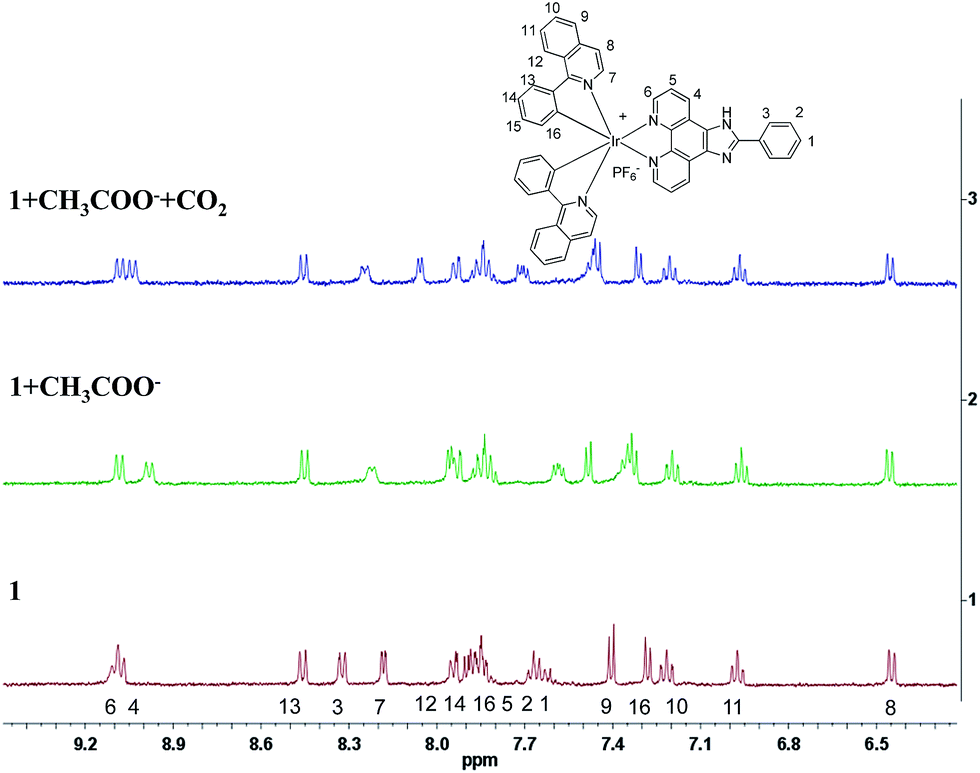
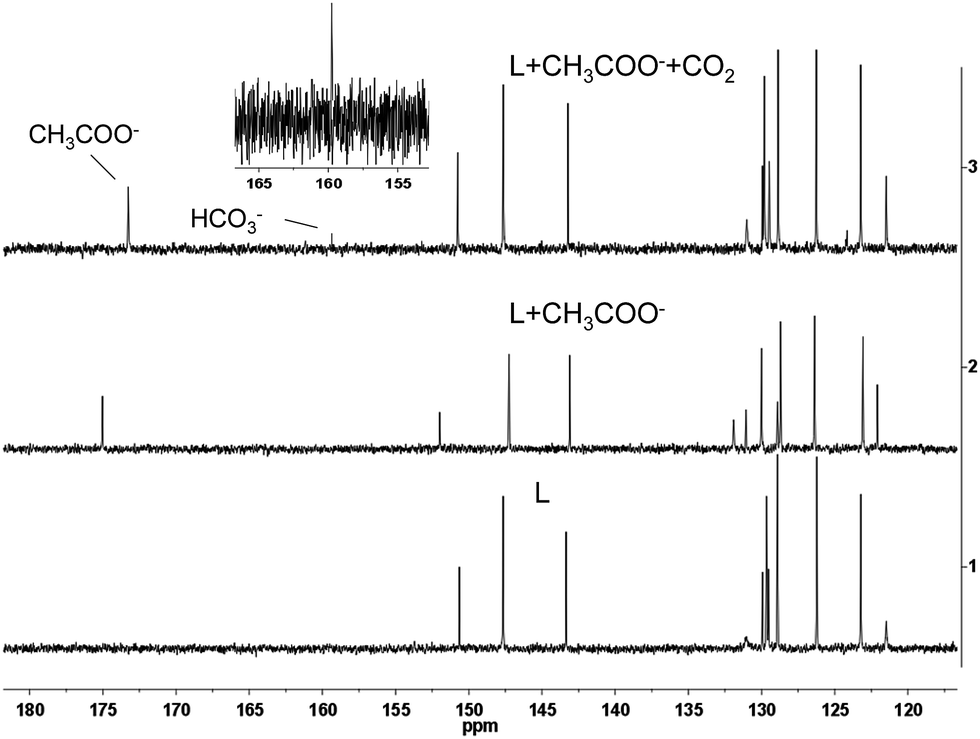
To confirm the detection mechanism of CO2 gas by 1, the interactions of 1 with CH3COO− and CO2 gas were investigated by 1H NMR. Fig. 9 shows the change in the chemical shifts of 1 upon the addition of 3 equiv. of CH3COO−. As observed in the 1H NMR spectra, the resonance signals of protons on the cyclometalated ligands showed little change. In contrast, the partial negative charge on nitrogen, which was generated by the interaction between the CH3COO− and –NH units, led to the proton signals on 2-phenylimidazo-[4,5-f][1,10]phenanthroline ligand exhibiting an obvious upfield shift. After bubbling CO2 gas into a solution of complex 1 containing CH3COO−, the chemical shifts of the protons on the ancillary ligand were recovered to certain extent because of the destruction of the hydrogen bond. However, this restoration was not complete, which might be caused by the presence of acetate, acidic conditions and the increase in viscosity after the bubbling of CO2 gas into the solution. As a control study, 2-phenylimidazo-[4,5-f][1,10]phenanthroline ligand was used to react with CH3COO− and CO2 gas to further confirm the detection mechanism. Compared with complex 1, the same variation trend in chemical shifts was observed in DMSO-d6 after the addition of CH3COO− in the 1H NMR spectra. The exposure of L–OOCCH3 to CO2 gas induced almost complete recovery of the chemical shifts of all the protons (Fig. S5†). Fig. 10 shows the 13C NMR spectrum of 2-phenylimidazo-[4,5-f][1,10]phenanthroline that was first exposed to CH3COO− and then bubbled with CO2 in DMSO-d6. The chemical shifts of the carbon atoms due to the formation of a hydrogen bond can be well recovered by the exposure of 2-phenylimidazo-[4,5-f][1,10]phenanthroline to CO2 gas. In addition, a new characteristic signal at ∼160 ppm was observed, which can be assigned to HCO3− anion, based on the literature results.16 These results are consistent with the observed photoluminescence changes and support the abovementioned proposal that a hydrogen bond is formed upon CH3COO− treatment, which is followed by the destruction of the bond via interaction with CO2.
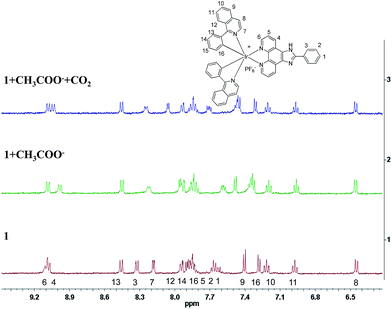 |
| | Fig. 9 Partial 1H NMR spectra of 1, 1 + CH3COO− and 1 + CH3COO− treated with CO2 in CD3CN-d3. | |
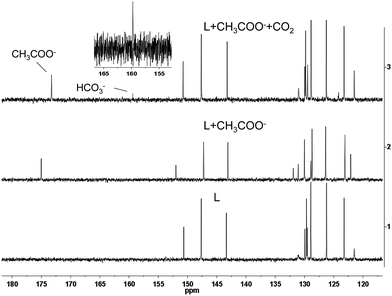 |
| | Fig. 10 Partial 13C NMR spectra of L, L + CH3COO− and L + CH3COO− treated with CO2 in DMSO-d6. | |
As a further control study, 2-phenylimidazo-[4,5-f][1,10]phenanthroline ligand was used to prepare a corresponding N-dithiolate derivative by interaction with carbon disulfide, which is an isoelectronic analogue of CO2. The N-dithiolate derivative was prepared using 2-phenylimidazo-[4,5-f][1,10]phenanthroline ligand, which was treated with KOH for 4 h followed by reaction with CS2 in DMSO solution (Fig. S6†). 13C NMR spectroscopy showed a new carbon signal at ∼216 ppm (Fig. S6†), indicating that a stable N-dithiolate adduct was formed. Collectively, these results support the conclusions that complex 1 reacts with CO2 gas to form the intermediate complex 1b, and then the original complex 1 can be recovered after the decomposition of 1b.
Conclusions
In summary, we have developed a novel phosphorescent probe based on an iridium(III) complex with 2-phenylimidazo-[4,5-f][1,10]phenanthroline for CO2 detection. After bubbling CO2 into the detecting solution, the phosphorescence quenched by the addition of CH3COO− could be recovered. Photobleaching experiment demonstrates that this phosphorescent CO2 probe exhibits higher photostability than some of the reported organic probes. More importantly, the time-resolved PL experiment demonstrated that 1 can be used to detect CO2 in the presence of strong background fluorescence, which improves the sensitivity and signal-to-noise ratio of the sensor in complicated media. Our on-going work is focused on the development of novel luminescent sensors to detect CO2 gas in living cells.
Experimental
General
Commercially available chemical reagents were used without further purification. The solvents were carefully dried and distilled from appropriate drying agents prior to use. NMR spectra were obtained on a Bruker Ultrashield 400 MHz FT-NMR spectrometer. Mass spectra were obtained on a Bruker Autoflex matrix-assisted laser desorption/ionization time of flight mass spectrometer (MALDI-TOF MS). UV-vis absorption spectra were recorded using an HP-8453 spectrophotometer. Photoluminescence spectra were measured on an Edinburgh LFS920 fluorescence spectrophotometer. Time-resolved emission spectra (TRES) and emission lifetimes were obtained through a time-correlated single photon counting (TCSPC) technique using an Edinburgh FL 920 instrument with a laser source (365 nm) as the excitation source. The time-gated acquisition of photoluminescence spectra were carried out employing the TRES technique.
Synthesis of complex 1
Ligand, 1-phenylisoquinoline, was synthesized according to a previous report.15b Cyclometalated iridium(III) chloro-bridged dimer [Ir(piq)2Cl]2 was prepared according to the literature method.17 A solution of [Ir(piq)2Cl]2 (0.079 mmol) and 2-phenylimidazo-[4,5-f][1,10]phenanthroline (0.158 mmol) in CH2Cl2/MeOH (15 mL, 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 v/v) was heated to reflux. After 4 h, the red solution was cooled to room temperature, and a 10-fold excess of potassium hexafluorophosphate was then added. The suspension was stirred for 1 h and filtered to remove the insoluble inorganic salts. The solution was evaporated to dryness under reduced pressure. The crude product was applied to a silica gel column and eluted with CH2Cl2/acetone (10:1, v/v) to afford a red solid in 69% yield. 1H NMR (400 MHz, CD3CN), δ (ppm): 9.12 (t, 4H, J = 17.2 Hz, phenanthroline), 8.47 (d, 2H, J = 8.0 Hz, isoquinoline), 8.33 (d, 2H, J = 7.2 Hz, phenyl), 8.19 (d, 2H, J = 4.0 Hz, isoquinoline), 7.95 (d, 2H, J = 7.2 Hz, isoquinoline), 7.83–7.91 (m, 6H, isoquinoline), 7.69 (t, 3H, J = 12.0 Hz, phenyl), 7.41 (d, 2H, J = 4.0 Hz, isoquinoline), 7.29 (d, 2H, J = 4.0 Hz, isoquinoline), 7.24 (t, 2H, J = 14.0 Hz, isoquinoline), 6.99 (t, 2H, J = 16.0 Hz, isoquinoline), 6.46 (d, 2H, J = 8.0 Hz, isoquinoline). 13C NMR (100 MHz, CD3CN), δ (ppm): 168.6, 153.7, 153.0, 149.1, 145.8, 144.6, 141.0, 137.1, 132.2, 132.1, 131.9, 130.8, 130.5, 129.4, 129.2, 128.9, 127.5, 126.8, 126.6, 126.2, 122.3, 121.8 (aromatic). MALDI-TOF MS (m/z): 899.32 (M − PF6)+. Anal. calcd for C49H33N6PF6Ir: C, 56.43; H, 3.19; N, 8.06. Found: C, 56.65; H, 3.44; N, 7.90%.
:1 v/v) was heated to reflux. After 4 h, the red solution was cooled to room temperature, and a 10-fold excess of potassium hexafluorophosphate was then added. The suspension was stirred for 1 h and filtered to remove the insoluble inorganic salts. The solution was evaporated to dryness under reduced pressure. The crude product was applied to a silica gel column and eluted with CH2Cl2/acetone (10:1, v/v) to afford a red solid in 69% yield. 1H NMR (400 MHz, CD3CN), δ (ppm): 9.12 (t, 4H, J = 17.2 Hz, phenanthroline), 8.47 (d, 2H, J = 8.0 Hz, isoquinoline), 8.33 (d, 2H, J = 7.2 Hz, phenyl), 8.19 (d, 2H, J = 4.0 Hz, isoquinoline), 7.95 (d, 2H, J = 7.2 Hz, isoquinoline), 7.83–7.91 (m, 6H, isoquinoline), 7.69 (t, 3H, J = 12.0 Hz, phenyl), 7.41 (d, 2H, J = 4.0 Hz, isoquinoline), 7.29 (d, 2H, J = 4.0 Hz, isoquinoline), 7.24 (t, 2H, J = 14.0 Hz, isoquinoline), 6.99 (t, 2H, J = 16.0 Hz, isoquinoline), 6.46 (d, 2H, J = 8.0 Hz, isoquinoline). 13C NMR (100 MHz, CD3CN), δ (ppm): 168.6, 153.7, 153.0, 149.1, 145.8, 144.6, 141.0, 137.1, 132.2, 132.1, 131.9, 130.8, 130.5, 129.4, 129.2, 128.9, 127.5, 126.8, 126.6, 126.2, 122.3, 121.8 (aromatic). MALDI-TOF MS (m/z): 899.32 (M − PF6)+. Anal. calcd for C49H33N6PF6Ir: C, 56.43; H, 3.19; N, 8.06. Found: C, 56.65; H, 3.44; N, 7.90%.
Acknowledgements
We thank National Basic Research Program of China (973 Project 2013CB834702), Hong Kong Research Grants Council (HKBU203011 and HKUST2/CRF/10), Areas of Excellence Scheme of HKSAR (AoE/P-03/08), National Natural Science Foundation of China (21171098), Hong Kong Baptist University (FRG2/12-13/083) and Hong Kong Scholars Program (XJ2012053) for financial support. The work was also supported by Partner State Key Laboratory of Environmental and Biological Analysis (SKLP-14-15-P011) and Strategic Development Fund of HKBU.
Notes and references
-
(a) M. D. D. Grandpre, Anal. Chem., 1993, 65, 331 CrossRef;
(b) C. V. Butzingslowen, A. K. McEvoy, C. M. Donagh, B. D. M. Craith, I. Klimant, C. Krause and O. S. Wolfbeis, Analyst, 2002, 127, 1478 RSC.
-
(a) R. N. Dansby-Sparks, J. Jin, S. J. Mechery, U. Sampathkumaran, T. W. Owen, B. D. Yu, K. Goswami, K. L. Hong, J. Grant and Z. L. Xue, Anal. Chem., 2010, 82, 593 CrossRef CAS PubMed;
(b) J. Lin, TrAC, Trends Anal. Chem., 2000, 19, 541 CrossRef CAS;
(c)
M. J. Hitchman and M. Park, U.S. Pat. 6,365,022, 2002.
-
(a) Y. Amao and N. Nakamura, Sens. Actuators, B, 2005, 107, 861 CrossRef CAS PubMed;
(b) O. Oter, K. Ertekin and S. Derinkuyu, Talanta, 2008, 76, 557 CrossRef CAS PubMed;
(c) C. S. Chu and Y. L. Lo, Sens. Actuators, B, 2008, 129, 120 CrossRef CAS PubMed;
(d)
A. Mills and S. Hodgen, Advanced Concepts in Fluorescence Spectroscopy-Part A Small Molecule Sensing, Topics in Fluorescence Spectroscopy, Springer, Berlin, 2005, vol. 9, p. 119 Search PubMed.
-
(a) X. Xie, M. Pawlak, M. Tercier-Waeber and E. Bakker, Anal. Chem., 2012, 84, 3163 CrossRef CAS PubMed;
(b) R. Ali, T. Lang, S. M. Saleh, R. J. Meier and O. S. Wolfbeis, Anal. Chem., 2011, 83, 2846 CrossRef CAS PubMed;
(c) S. Schutting, S. M. Borisov and I. Klimant, Anal. Chem., 2013, 85, 3271 CrossRef CAS PubMed;
(d) Y. Amao and N. Nakamura, Sens. Actuators, B, 2004, 100, 347 CrossRef CAS PubMed.
-
(a) O. S. Wolfbeis, L. J. Weis, M. J. P. Leiner and W. E. Ziegler, Anal. Chem., 1988, 60, 2028 CrossRef CAS;
(b) A. Mills and Q. Chang, Analyst, 1993, 118, 839 RSC.
- Y. Liu, Y. Tang, N. N. Barashkov, I. S. Irgibaeva, J. W. Y. Lam, R. Hu, D. Birimzhanova, Y. Yu and B. Z. Tang, J. Am. Chem. Soc., 2010, 132, 13951 CrossRef CAS PubMed.
-
(a) Z. Q. Guo, N. R. Song, J. H. Moon, M. Kim, E. J. Jun, J. Choi, J. Y. Lee, C. W. Bielawski, J. L. Sessler and J. Yoon, J. Am. Chem. Soc., 2012, 134, 17846 CrossRef CAS PubMed;
(b) M. Ishida, P. Kim, J. Choi, J. Yoon, D. Kim and J. L. Sessler, Chem. Commun., 2013, 49, 6950 RSC;
(c) Q. L. Xu, S. Y. Lee, M. H. Kim, J. Bouffard and J. Yoon, J. Am. Chem. Soc., 2013, 135, 17751 CrossRef CAS PubMed.
-
(a) Y. You, S. Lee, T. Kim, K. Ohkubo, W. S. Chae, S. Fukuzumi, G. J. Jhon, W. Nam and S. J. Lippard, J. Am. Chem. Soc., 2011, 133, 18328 CrossRef CAS PubMed;
(b) Y. Ma, S. J. Liu, H. R. Yang, Y. Q. Wu, H. B. Sun, J. X. Wang, Q. Zhao, F. Y. Li and W. Huang, J. Mater. Chem. B, 2013, 1, 319 RSC;
(c) H. F. Shi, H. B. Sun, H.
R. Yang, S. J. Liu, G. Jenkins, W. Feng, F. Y. Li, Q. Zhao, B. Liu and W. Huang, Adv. Funct. Mater., 2013, 23, 3268 CrossRef CAS;
(d) W. J. Xu, X. Zhao, W. Lv, H. R. Yang, S. J. Liu, H. Liang, Z. Z. Tu, H. Xu, W. L. Qiao, Q. Zhao and W. Huang, Adv. Healthcare Mater., 2014, 3, 658 CrossRef CAS PubMed.
-
(a) W.-Y. Wong and C.-L. Ho, J. Mater. Chem., 2009, 19, 4457 RSC;
(b) P. T. Chou and Y. Chi, Chem.–Eur. J., 2007, 13, 380 CrossRef CAS PubMed;
(c) M. S. Lowry and S. Bernhard, Chem.–Eur. J., 2006, 12, 7970 CrossRef CAS PubMed;
(d) S. J. Liu, Q. Zhao, Q. L. Fan and W. Huang, Eur. J. Inorg. Chem., 2008, 2177 CrossRef CAS;
(e) H. B. Sun, S. J. Liu, W. P. Lin, K. Y. Zhang, W. Lv, X. Huang, F. W. Huo, H. R. Yang, G. Jenkins, Q. Zhao and W. Huang, Nat. Commun., 2014, 5, 3601 Search PubMed.
-
(a) V. Fernandez-Moreira, F. L. Thorp-Greenwood and M. P. Coogan, Chem. Commun., 2010, 46, 186 RSC;
(b) P. Wu, E. L. M. Wong, D. L. Ma, G. S. M. Tong, K. M. Ng and C. M. Che, Chem.–Eur. J., 2009, 15, 3652 CrossRef CAS PubMed;
(c) K. K. W. Lo and J. S. Y. Lau, Inorg. Chem., 2007, 46, 700 CrossRef CAS PubMed;
(d) C.-L. Ho, K.-L. Wong, H. K. Kong, Y. M. Ho, C. T. L. Chan, W. M. Kwok, K. S. Y. Leung, H. L. Tam, M. H. W. Lam, X. F. Ren, A. M. Ren, J. K. Feng and W.-Y. Wong, Chem. Commun., 2012, 48, 2525 RSC.
-
(a) Y. M. Yang, Q. Zhao, W. Feng and F. Y. Li, Chem. Rev., 2013, 113, 192 CrossRef CAS PubMed;
(b) Q. Zhao, F. Y. Li and C. H. Huang, Chem. Soc. Rev., 2010, 39, 3007 RSC;
(c) Q. Zhao, S. J. Liu, F. Y. Li and C. H. Huang, Dalton Trans., 2008, 3836 RSC;
(d) Y. Ma, S. J. Liu, H. R. Yang, Y. Q. Wu, C. J. Yang, X. M. Liu, Q. Zhao, H. Z. Wu, J. C. Liang, F. Y. Li and W. Huang, J. Mater. Chem., 2011, 21, 18974 RSC;
(e) W. J. Xu, S. J. Liu, H. B. Sun, X. Y. Zhao, Q. Zhao, S. Sun, S. Cheng, T. C. Ma, L. X. Zhou and W. Huang, J. Mater. Chem., 2011, 21, 7572 RSC.
-
(a) M. D. Marazuela, M. C. Moreno-Bondi and G. Orellana, Appl. Spectrosc., 1998, 52, 1314 CrossRef CAS;
(b) G. Orellana, M. C. Moreno-Bondi, E. Segovia and M. D. Marazuela, Anal. Chem., 1992, 64, 2210 CrossRef CAS;
(c) N. B. Borcherta, J. P. Kerrya and D. B. Papkovsky, Sens. Actuators, B, 2013, 176, 157 CrossRef PubMed.
-
(a) M. Boiocchi, L. D. Boca, D. E. Gómez, L. Fabbrizzi, M. Licchelli and E. Monzani, J. Am. Chem. Soc., 2004, 126, 16507 CrossRef CAS PubMed;
(b) V. Amendola, D. E. Gómez, L. Fabbrizzi and M. Licchelli, Acc. Chem. Res., 2006, 39, 343 CrossRef CAS PubMed.
- D. L. Boger and M. Patel, J. Org. Chem., 1987, 52, 3934 CrossRef CAS.
-
(a) Q. Zhao, S. J. Liu, M. Shi, F. Y. Li, H. Jing, T. Yi and C. H. Huang, Organometallics, 2007, 29, 1085 CrossRef;
(b) G. J. Zhou, W.-Y. Wong, B. Yao, Z. Y. Xie and L. X. Wang, Angew. Chem., Int. Ed., 2007, 46, 1149 CrossRef CAS PubMed.
- N. J. Bridges, C. C. Hines, M. Smiglak and R. D. Rogers, Chem.–Eur. J., 2007, 13, 5207 CrossRef CAS PubMed.
- K. Nonoyama, Bull. Chem. Soc. Jpn., 1974, 47, 467 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: Details of PL and NMR spectra. See DOI: 10.1039/c4tc02014f |
|
| This journal is © The Royal Society of Chemistry 2015 |
Click here to see how this site uses Cookies. View our privacy policy here.