
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Uncoiling collagen: a multidimensional mass spectrometry study†
H. J.
Simon
a,
M. A.
van Agthoven
a,
P. Y.
Lam
a,
F.
Floris
a,
L.
Chiron
b,
M.-A.
Delsuc
bc,
C.
Rolando
d,
M. P.
Barrow
 a and
P. B.
O'Connor
*a
a and
P. B.
O'Connor
*a
aDepartment of Chemistry, University of Warwick, Gibbet Hill Road, Coventry, CV4 7AL, UK. E-mail: p.oconnor@warwick.ac.uk
bCASC4DE, Le Lodge, 20, Avenue du Neuhof, 67100 Strasbourg, France
cInstitut de Génétique et de Biologie Moléculaire et Cellulaire, INSERM, U596, CNRS, UMR7104, Université de Strasbourg, 1 rue Laurent Fries, 67404 Illkirch-Graffenstaden, France
dUniversité de Lille, CNRS, USR 3290, MSAP, Miniaturisation pour la Synthèse l'Analyse et la Protéomique, FR 3688, FRABIO, Biochimie Structurale & Fonctionnelle des Assemblages Biomoléculaires, and FR 2638, Institut Eugène-Michel Chevreul, F-59000 Lille, France
First published on 3rd November 2015
Abstract
Mass spectrometry can be used to determine structural information about ions by activating precursors and analysing the resulting series of fragments. Two-dimensional Fourier transform ion cyclotron resonance mass spectrometry (2D FT-ICR MS) is a technique that correlates the mass-to-charge (m/z) ratio of fragment and precursor ions in a single spectrum. 2D FT-ICR MS records the fragmentation of all ions in a sample without the need for isolation. To analyse specific precursors, horizontal cross-sections of the spectrum (fragment ion scans) are taken, providing an alternative to conventional tandem mass spectrometry (MS/MS) experiments. In this work, 2D FT-ICR MS has been used to study the tryptic digest of type I collagen, a large protein. Fragment ion scans have been extracted from the 2D FT-ICR MS spectrum for precursor m/z ratios: 951.81, 850.41, 634.34, and 659.34, and 2D FT-ICR MS spectra are compared with a set of 1D MS/MS spectra using different fragmentation methods. The results show that two-dimensional mass spectrometry excells at MS/MS of complex mixtures, simplifying spectra by eliminating contaminant peaks, and aiding the identification of species in the sample. Currently, with desktop computers, 2D FT-ICR MS is limited by data processing power, a limitation which should be alleviated using cluster parallel computing. In order to explore 2D FT-ICR MS for collagen, with reasonable computing time, the resolution in the fragment ion dimension is limited to 256k data points (compared to 4M data points in 1D MS/MS spectra), but the vertical precursor ion dimension has 4096 lines, so the total data set is 1G data points (4 Gbytes). The fragment ion coverage obtained with a blind, unoptimized 2D FT-ICR MS experiment was lower than conventional MS/MS, but MS/MS information is obtained for all ions in the sample regardless of selection and isolation. Finally, although all 2D FT-ICR MS peak assignments were made with the aid of 1D FT-ICR MS data, these results demonstrate the promise of 2D FT-ICR MS as a technique for studying complex protein digest mixtures.
Introduction
Collagens are the most abundant proteins in vertebrates, representing around a third of the total protein content.1,2 At least 29 different types of collagen have been identified,3 forming the major component of structurally important tissues such as bone, tendon, and cartilage. The collagen molecule is stabilised by an extensive network of hydrogen bonds and cross-links,4 which give it remarkable strength and longevity, and lead to applications in fields that range from medicine5 to archaeology.6,7 The large size of the protein, combined with its unique post-translational modifications (PTMs) – oxidation of proline, P, and lysine, K, residues to hydroxyproline, *P, and hydroxylysine, *K – result in dense mass spectra that are challenging to interpret. To simplify analysis, novel techniques are required.Though it was first conceived in the late 1980s,8–10 experiments using two-dimensional Fourier transform ion cyclotron resonance mass spectrometry (2D FT-ICR MS) were limited11–13 until advances in computer technology allowed rapid 2D Fourier transforms on multi gigabyte datasets. The technique promises a rapid, efficient, and easy to use method for correlating the mass-to-charge (m/z) ratio of precursor and fragment ions in a complex sample.14 Only recently has this potential begun to be explored, made possible by improvements in computational calculations and storage capacity, such as the development algorithms to remove scintillation noise.15,16 These advances have seen 2D FT-ICR MS grow into a promising new analytical method.
Traditionally, tandem mass spectrometry (MS/MS or MSn) is performed by isolating the ion of interest prior to fragmentation. Activation of the parent ion is achieved through a fragmentation technique, such as collisionally activated dissociation (CAD),17 infrared multiphoton dissociation (IRMPD),18 or electron capture dissociation (ECD).19 Analysis of the product ions allows structural information to be determined about the precursor. Mixtures containing several species at a similar m/z ratio can be difficult to analyse if multiple ions are co-isolated and simultaneously fragmented. By coupling to gas or liquid chromatography (GC or LC), components may be separated.20 For tandem MS, however, the ions must then be sequentially isolated and fragmented. This results in a peak selection bias, which usually provides MS/MS data limited to the selected ions of interest.21 In addition, similar species may co-elute, giving inefficient precursor isolation and leading to extra peaks in MS/MS spectra from each co-isolated precursor ion.21 2D FT-ICR MS overcomes these issues by fragmenting ions without isolation, separating species according to their m/z ratio, with precursor m/z along the vertical axis and fragment m/z on the horizontal axis.
Analogous to 2D NMR spectroscopy, 2D FT-ICR MS is based on a similar sequence of radio frequency (RF) pulses and delays, followed by a two-dimensional Fourier transform.8 At the start of a 2D FT-ICR MS experiment, ions are transferred from the ion source to the centre of the ICR cell. Applying an RF pulse, P1, to the excitation plates coherently excites the ions into a small cyclotron radius [excitation pulse]. P1 is followed by a variable delay, t1. During t1, the ion packets rotate freely before being re-excited by a second, identical RF pulse, P2 [encoding pulse]. After t1, if the ion is out of phase with P2, it will be de-excited to the centre of the cell; if the ion is in-phase with P2, it will be excited to double the radius, but most ions are excited somewhere in-between.10,22 By the end of P2, the ions are distributed over different cyclotron radii depending on their cyclotron frequency. A second delay follows P2. This delay is used as a fragmentation period, tm, when ions can interact with neutral molecules (CAD),9 a laser pulse (IRMPD),23–25 or electron beam (ECD).26 After fragmentation, all ions are excited to about 60% of the cell radius using a third RF pulse, P3 [observe pulse] and both precursors and fragments are detected. The delay t1 is regularly incremented at each iteration of the pulse sequence, thus ‘programming’ the precursor ions with a modulation frequency, and each fragment ion will be generated with the precursor ion's modulation frequency. After Fourier transformation along the t2 acquisition time and the t1 delay, the fragmentation pattern of all the ions injected in the ICR cell can be visualized in a single two-dimensional mass spectrum, and all fragments are correlated to their precursors via the modulation frequencies.
Recently, the technique was applied to the study of cholesterol,27 demonstrating its potential for the structural analysis of small molecules and differentiation of fragmentation pathways. So far, two-dimensional mass spectrometry has been applied to a handful of small peptides and proteins. In this work, the most complex sample studied by 2D FT-ICR MS to date is presented: the tryptic digest of collagen. The results show that 2D FT-ICR MS successfully reproduces conventional MS/MS of protein digest mixtures, increasing the number and quality of peak assignments, and providing an abundance of information in one location, making the technique a viable new tool for proteomics investigations.
Experimental procedure
Materials
Type I collagen from bovine Achilles tendon, trypsin from bovine pancreas, and ammonium bicarbonate (NH4HCO3) were purchased from Sigma Aldrich (Gillingham, UK). HPLC grade methanol and formic acid (HCOOH) were obtained from Fisher Scientific (Leicestershire, UK). All aqueous solutions were prepared using water from a Milli-Q water system (Millipore Inc., Durham, UK).Collagen tryptic digest
30 mg of collagen type I was weighed out and denatured in 5 mL of 50 mM ammonium bicarbonate (pH 7.84) for 1 hour at 62 °C. The sample was centrifuged at 14, 475g for 20 minutes at room temperature. The supernatant (∼350 μL) was removed and 12 μL of trypsin solution (2 μg μL−1) added, using a ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :40 enzyme:protein. The mixture (approximate collagen concentration: 16 μM) was allowed to react for 4 hours at 37 °C before being halted with 5% formic acid and dried in a SpeedVac system (Savant SPD121P) at 35 °C overnight. Dried samples were stored at −80 °C until analysis.
:40 enzyme:protein. The mixture (approximate collagen concentration: 16 μM) was allowed to react for 4 hours at 37 °C before being halted with 5% formic acid and dried in a SpeedVac system (Savant SPD121P) at 35 °C overnight. Dried samples were stored at −80 °C until analysis.
One-dimensional FT-ICR MS measurements
Prior to analysis, dried samples were remade up to a 350 μL (∼16 μM) solution with 50:50 MeOH:H2O and diluted into 2.5 and 5 μM samples in 49.5:49.5:1 MeOH:H2O:formic acid. All concentrations were calculated assuming no loss of the collagen starting material. Experiments were carried out on a 12 Tesla solariX FT-ICR MS (Bruker Daltonik GmbH, Bremen, Germany). Samples were introduced to the instrument using a nano electrospray (nESI) glass capillary tip and ionised in positive-ion mode (capillary voltage −0.8 to −1.5 kV). CAD and IRMPD experiments were performed in the ICR cell, following isolation of the ions using a quadrupole and subsequent accumulation for 1–5 s using a collision cell. CAD experiments used argon gas with collision voltages of 13 to 32 V to activate precursor ions. IRMPD experiments were performed with a 10.6 μM CO2 laser (Model 48-2, 25 W laser from Synrad Inc, Mulkiteo, Washington, USA) at 50% power with a pulse length of 0.35 s. For ECD experiments, ions were irradiated with electrons from a heated hollow cathode (1.7 A) for 100 ms (lens voltage: 10 V, bias 2 V). An average of 100 or 200 scans were taken for each MS/MS spectrum. The mass spectrometer was externally calibrated with HP tuning mixture (Sigma-Aldrich, Gillingham, UK) before sample injections. A small amount of HP mix was introduced to the sample and used as an internal calibrant for the one-dimensional mass spectrum. Internal calibration and data analysis were performed with Data Analysis software 4.1 (Bruker Daltonics).
Two-dimensional FT-ICR MS measurements
Excitation and encoding pulses (P1 and P2) were swept over a frequency range between 1.25 MHz to 61.5 kHz, corresponding to m/z 147–3000. The two identical RF pulses had an overall duration of 760 μs each, and pulse amplitude 72 Vp–p. A frequency range of 500 kHz to 61.5 kHz was used, giving an m/z range of 368–3000 in both the horizontal and vertical direction. Experiments were performed with a Nyquist frequency fN = 500 kHz, equivalent to a delay increment of Δt1 = 1 μs. The observe pulse, P3 had an amplitude of 72 Vp–p and lasted 20 μs per frequency step. 4096 × 256k data points were collected, with 1 scan per increment and a total acquisition time of 1 hour. Precursor ions were fragmented with IRMPD using a CO2 laser at 50% power, pulse length 0.35 s. Data processing was performed using the SPIKE data processing programme,28 which has been developed at the University of Strasbourg and is a complete rewrite of the NMR Processing Kernel (NPK).29 The programme is available at http://www.bitbucket.org/delsuc/spike. To process the spectrum, a sin(0.5) apodisation function was used,30 zerofilling 1× in each dimension. The spectrum is presented in magnitude mode giving a final resolution at m/z 850 of 13000 horizontally and 630 vertically (FWHM = 1.3 Da). The data was denoised using the urQRd16 (uncoiled random QR denoising) algorithm prior to visualisation.Results and discussion
All collagens have a characteristic triple helical structure, containing three parallel polypeptide strands, each in a left-handed polyproline II helical conformation and coiled with a one-residue stagger to form a right-handed triple helix.31,32 Type I bovine collagen is a heterotrimeric triple helix, made from the maturation of soluble procollagen consisting of two α1-[1] chains (UniProt entry P02453) and one α2-[1] chain (UniProt entry P02465). The two polypeptide chains contain 1463 and 1364 amino acids respectively, corresponding to a theoretical molecular weight of over 406 kDa. In silico digestion of the collagen chains leads to 681 tryptic peptides, assuming no missed cleavages and a variable number of oxidised prolines.Individual collagen triple helices have three regions: the N-terminal non-helical domain (N-telopeptide), the central triple helical domain, and the C-terminal helical domain (C-telopeptide).4 Following synthesis in the endoplasmic reticulum,33 procollagen molecules are secreted into the extracellular space. The N- and C-propeptides are cleaved, generating the collagen molecule. Type I collagen is heavily glycosylated34–36 and contains several cross-links, including immature, divalent cross-links, and mature, trivalent cross-links.37–39
Fig. 1 shows the FT-ICR mass spectrum of the collagen tryptic digest mixture (assignment table: Table S1†). The naming system refers to the full polypeptide chain, α1 or α2, and amino acid position of the tryptic peptide, in the format [α1/2(xxx–xxx) + nH]n+. Collagens has a distinctive amino acid sequence, owing to the tight packing of helices, which requires every third amino acid to be glycine, Gly or G; this pattern generates the repeating sequence X-Y-Gly, where X and Y can be any amino acid. Frequently, X and Y are proline, Pro or P, and hydroxyproline, Hyp or *P, making the Pro-Hyp-Gly triplet the most commonly observed and accounting for 10.5% of the protein sequence.40 In the two telopeptide regions, the X-Y-Gly pattern is not observed, and these areas have been highlighted in grey in the sequences at the bottom of Fig. 1. In the helical region, 43% of α1-[1] and 34% of α2-[1] has been assigned by manually interpreting the 1D mass spectrum. Peak assignments were made with errors routinely <1 ppm, and have an RMS mass error of 0.79 ppm (see ESI Table S1†). The corresponding positions in the sequence have been underlined in Fig. 1; solid line for peptides sequenced by MS/MS, dashed line for peptides identified from the MS spectrum, but not sequenced by tandem MS. From the zoomed-in regions around m/z 1100–1125 (right) and m/z 790–850 (left), many species can be seen in a relatively small range, demonstrating the complexity of the sample. MS/MS experiments were performed by isolating ions prior to fragmentation. However, the density of peaks in Fig. 1 resulted in the isolation of multiple species, which, in turn, lead to ‘contaminant’ peaks in the tandem mass spectra from fragmentation of the co-isolated species. To overcome this, a 2D FT-ICR MS experiment was performed and all ions in the sample were fragmented without isolation.
 | ||
| Fig. 1 FT-ICR mass spectrum of collagen tryptic digest (top) and bovine type I collagen amino acid sequences (bottom). Peaks marked with a circle were used to calibrate the spectrum, green – HP mix, blue – tryptic peptide. Insets show zoomed in regions around m/z 1100–1125 (right) and m/z 790–850 (left). Sequences shaded in grey correspond to telopeptide regions, underlined peptides have been identified in the spectrum; solid – sequenced by MS/MS, dashed – no MS/MS data. | ||
Fig. 2 shows the 2D mass spectrum of the collagen digest mixture. Four precursor ions at m/z 951.81, 850.41, 634.34, and 659.34, have been highlighted in the spectrum and their horizontal fragment ion scans are extracted from the 2D FT-ICR MS data (Fig. 2a–d). These cross-sections show all the fragment m/z ratios that appear from a particular, user-defined precursor m/z. The resulting profile is a spectrum analogous to an MS/MS experiment. Instead of performing multiple, sequential, time and sample consuming MS/MS experiments, fragmentation data about all precursor ions was produced using 2D FT-ICR MS in a single, hour long experiment. The four regions highlighted have been chosen to illustrate the advantages of using a two-dimensional approach. An in-depth discussion of these areas will be the focus of the rest of the paper, but it should be noted that there is much more information contained in the spectrum which is not discussed.
 | ||
| Fig. 2 2D FT-ICR mass spectrum of the collagen tryptic digest mixture. Orange/yellow lines show the regions of the spectrum corresponding to the fragment ion scans in the four insets at precursor m/z (a) 951.81, (b) 850.41, (c) 634.34, and (d) 659.34. | ||
Cleavage coverage of region α1(763–795): precursor m/z 951.81
The peak with the highest magnitude in the MS spectrum, a triply charged peptide at m/z 951.80926, was isolated and fragmented with CAD, Fig. 3a. De novo sequencing of the peptide led to an assignment of the region α1(763–795), with the third and fifth prolines oxidised to hydroxyproline. Additional MS/MS spectra were obtained using ECD, Fig. 3b, and IRMPD, Fig. 3c, as the fragmentation modes (assignment tables: Tables S2–4†). The peptide is 33 amino acids long and there are 32 possible cleavages along the backbone. From the two dimensional mass spectrum in Fig. 2, a horizontal fragment ion scan, Fig. 3d, was taken at precursor m/z 951.81. The resulting profile is analogous to the MS/MS experiments and allows a direct comparison between the fragmentation achieved with the four methods, Table 1 and Fig. 4. | ||
| Fig. 3 Four methods used to fragment the tryptic digest peptide [α1(763–795) + 3H]3+ at m/z 951.80926. (a) CAD, (b) ECD, (c) IRMPD, (d) 2D IRMPD FT-ICR MS fragment ion scan. | ||
 | ||
| Fig. 4 Comparison of the cleavage coverage obtained using four methods to activate precursor [α1(763–795) + 3H]3+, m/z 951.80926. | ||
| CAD | ECD | IRMPD | 2D FT-ICR MS | |
|---|---|---|---|---|
| Number of cleavages | 24 | 20 | 17 | 9 |
| % Cleavage coverage | 75 | 63 | 53 | 28 |
Of the four techniques, CAD gave the highest coverage, cleaving 75% of the bonds in the backbone. For many proteins, ECD leads to more fragment ions than CAD. However, ECD does not cleave N-terminal to proline because the N–Cα bond is constrained in a 5 membered ring. As is typical with collagen tryptic peptides, α1(763–795) is proline rich, and has 8 P residues. If these 8 amino acids are taken out of the comparison, ECD still cleaves 20 bonds, but CAD now only breaks 16, an observation which is consistent with prediction. Unlike the ECD spectrum, both CAD and IRMPD – which activate ions through excitation of the molecule's vibrational modes – show a large number of internal fragments (Tables S2 and S4†), which form when two or more bonds are cleaved in the peptide backbone. Such internal fragments occur if a b or y ion in the primary sequence undergoes further fragmentation.41 No internal fragments have been observed in the 2D FT-ICR MS of α1(763–795), but they are seen for other peptides, as shown in Fig. 2c, the fragment ion scan of precursor m/z 634.34.
2D FT-ICR MS resulted in the smallest number of cleavages of the four methods investigated. To obtain one MS/MS spectrum, many scans are accumulated to improve the signal-to-noise ratio of the ions. Accumulation allows low magnitude peaks to rise above the noise and be identified. On the other hand, the 2D FT-ICR MS experiment took 4096 time-domain transients; each of which consisted of 256k data points. The Fourier transform is calculated for each time-domain transient as a function of the detection interval, t2 and a second Fourier transform is calculated as a function of t1. Time-domain transients in 2D FT-ICR MS are currently shorter than in MS/MS, 256k instead of 4M. With more data points, there is an increase in the signal-to-noise ratio of a Fourier transform-based method. As a result of using shorter transients, fewer fragments are expected in the 2D mass spectrum, and those that are observed are likely to correspond to the most abundant fragments, i.e.: the lowest energy bond cleavages. As computational power continues to increase, it is expected that this will allow longer transients to be taken in 2D FT-ICR MS, improving the sensitivity of the method and eventually overtaking MS/MS because of the Fellgett signal-averaging advantage.42
Comparing the IRMPD MS/MS with the 2D-IRMPD fragment ion scan, just over half the cleavages seen in IRMPD MS/MS have been reproduced in the 2D experiment. During the MS/MS experiment, the IRMPD parameters were carefully tuned to give the best possible spectrum for that particular precursor. To tune the fragmentation parameters in 2D FT-ICR MS, one ion is taken as a reference, so it will not be tuned to each individual ion. Instead, each ion experiences a range of fragmentation conditions as they go in and out of the fragmentation zone. Most ions can interact with the laser beam and their fragmentation pattern is recorded.23 Only four of the ions that appear above a signal-to-noise ratio of 3 (y5, y8, y9, and y11) fail to appear in the 2D spectrum. Interestingly, the y262+ peak has a relatively higher magnitude than expected in the 2D spectrum, and appears at a higher magnitude than y13. In both CAD and IRMPD, y13 is more abundant than y262+. The y262+ ion corresponds to cleavage N-terminal to P*P. The P–*P bond has not been cleaved in the 2D experiment, but it is broken in 1D MS/MS. Absence of this cleavage may be the source of the unusually abundant y262+ ion. Other than the exceptions which have been highlighted, the 2D mass spectrum gives a result that reproduces a conventional tandem MS spectrum.
Elimination of multiple candidates: precursor m/z 850.41
Going beyond simply reproducing an MS/MS investigation, the 2D spectrum has been used to assign peaks in the one-dimensional mass spectrum with more than one candidate with an error margin <2 ppm. This demonstrates that there are advantages to using 2D FT-ICR MS over multiple 1D MS/MS experiments, besides reducing the amount of time taken to obtain data. For example, a triply charged ion at m/z 850.41468 was isolated and fragmented with CAD, Fig. S1.† Unfortunately, multiple ion species with m/z ratios in the quadrupole isolation window were isolated and fragmented, and the resulting spectrum had too many overlapping sequences to determine a fragmentation ladder by manual inspection. From the m/z ratio in the MS spectrum, Fig. 1, two tryptic peptides were narrowed down as possible candidates: α1(442–468) and α1(422–448). Though only two cleavages were seen in the 2D fragment ion scan at precursor m/z 850.41, Fig. 2b, this was sufficient to eliminate α1(442–468) as a possibility and, in conjunction with the MS/MS data, locate the positions of the *P modifications.Removal of contaminant peaks: precursor m/z 634.34 and 659.34
Two major advantages of using 2D FT-ICR MS are that it allows unassigned, contaminant peaks in MS/MS to be eliminated, and that it provides information about all fragmenting ions in the sample, in one convenient location. To show that this is repeated across a range of precursor m/z ratios, horizontal fragment ion scans have been pulled out of the 2D mass spectrum at m/z 634.34, Fig. 2c, and 659.34, Fig. 2d. The corresponding CAD mass spectra, Fig. 5 and 6, both contain a number of contaminant peaks, presumably from multiple precursor ion isolation, marked with a “?” in the spectra and assignment tables (Tables S5 and S6†). None of these unassigned peaks were present in the 2D fragment ion scans.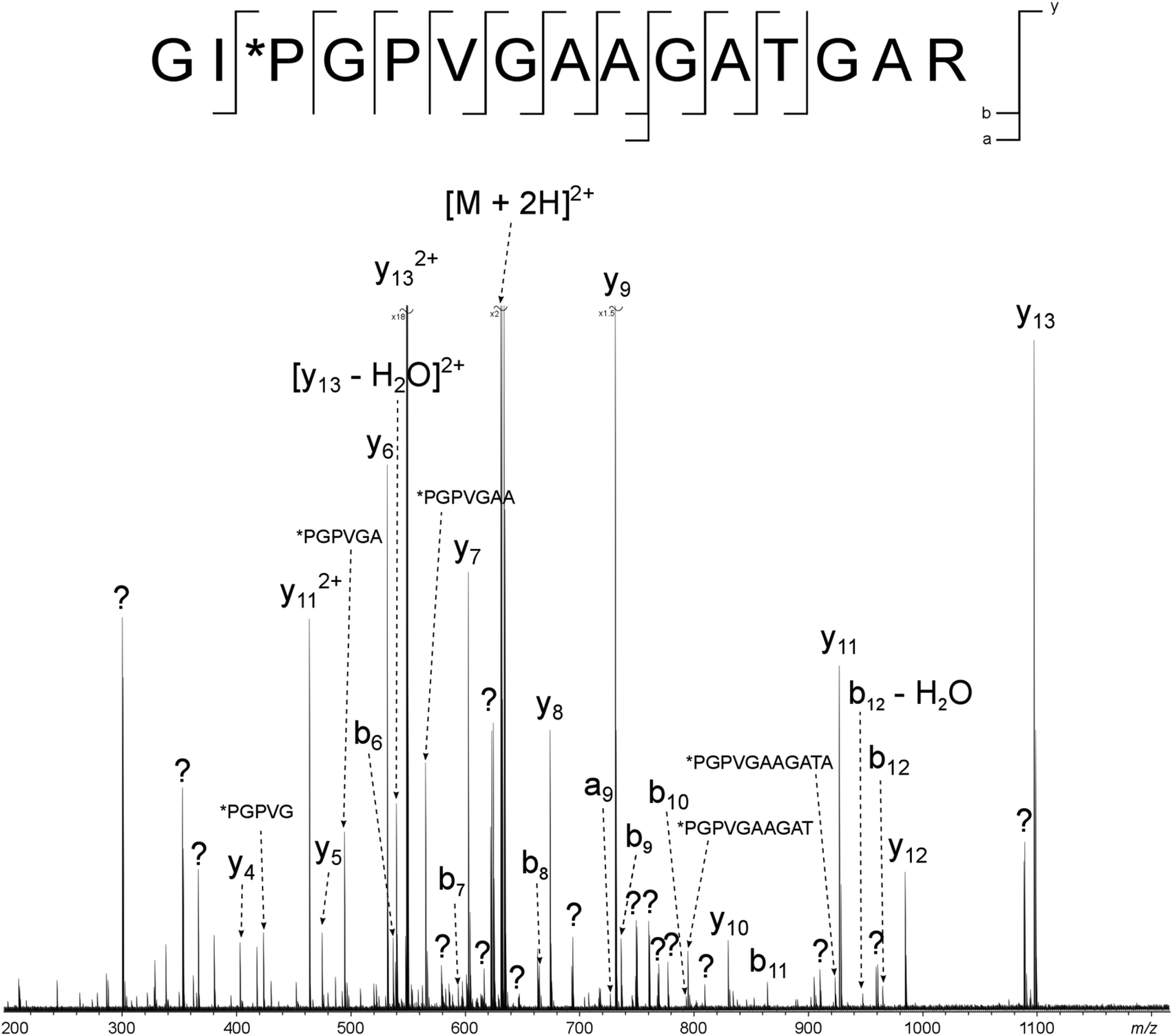 | ||
| Fig. 5 CAD spectrum of the tryptic digest peptide [α2(326–340) + 2H]2+ at m/z 634.34193. ? = unassigned peak. | ||
 | ||
| Fig. 6 CAD spectrum of the tryptic digest peptide ion [α1(1062–1083) + 3H]3+ at m/z 659.33679. ? = unassigned peak. | ||
Fig. 7 shows the cleavage coverage obtained using CAD and 2D FT-ICR MS for the three peptides at precursor m/z 951.81, 659.34, and 634.34. The two larger peptides at precursor m/z 952.81 (33 amino acids) and 659.34 (22 amino acids), show a very similar fragmentation efficiency, with 76% of bonds broken in CAD and 29% in 2D FT-ICR MS. The smaller 15 amino acid peptide at precursor m/z 634.34 shows remarkably extensive fragmentation in the fragment ion scan extracted from the 2D mass spectrum. 64% cleavage coverage has been obtained with 2D FT-ICR MS, compared to 79% with CAD. This corresponds to just 2 fewer cleavages than in the CAD mass spectrum, but without any of the contaminant peaks, a striking demonstration of the promise of 2D FT-ICR MS as a method for studying complex protein mixtures.
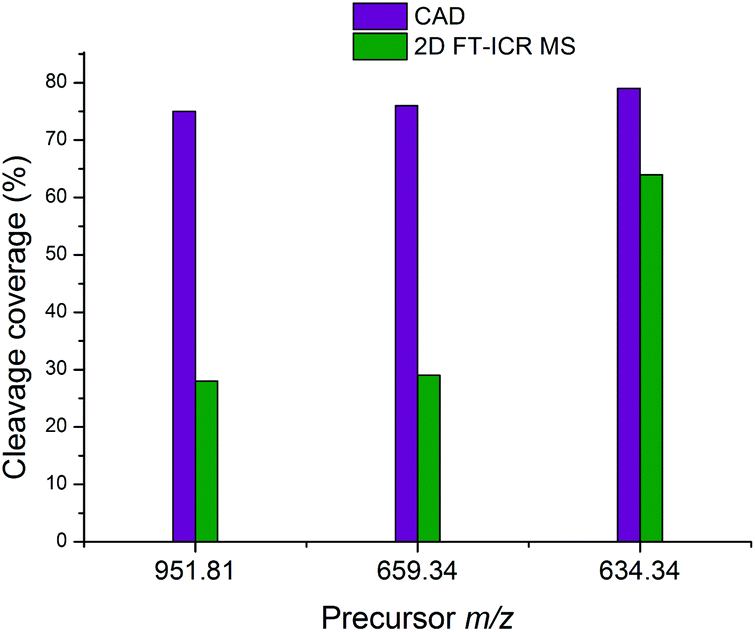 | ||
| Fig. 7 Comparison of the cleavage coverage obtained using CAD and 2D FT-ICR MS for three peptides at precursor m/z 951.81, 659.34, and 634.34. | ||
Conclusions
In this research, 2D FT-ICR MS has been used to study the complex protein digest mixture of collagen. Horizontal fragment ion scans have been shown to give results consistent with MS/MS, going beyond conventional methods to provide an abundance of additional information in a single spectrum. This enabled the removal of unassigned, contaminant peaks, which simplified difficult tandem mass spectra and helped assign peptides in the one-dimensional spectrum. Where more than one candidate was proposed as a peak assignment, 2D FT-ICR MS has been used to eliminate one of the proposed species. Unfortunately, in its current, early form, 2D FT-ICR MS cannot stand alone from conventional mass spectrometry. All the 2D assignments made in this work were done with the aid of 1D FT-ICR MS. The main limitation lies in the computational analysis of spectra. Two-dimensional spectra are very large, with file sizes over 1 GB, resulting in slow computation times for the 2D-FFT and denoising algorithm. Computational times are a current limitation of the technique, but will be largely alleviated by performing these dense calculations on clusters or workstations. Despite the data analysis challenges, this study has shown that 2D FT-ICR MS can produce one data set which may be used to generate and analyse the fragmentation patterns of many precursors in a complex sample. The experiment is less time consuming than taking multiple 1D MS/MS spectra, making it an exciting and promising new technique for analysing complex protein data.Acknowledgements
The authors would like to thank Mr Chris Wootton and Mr Cookson Chiu for their helpful discussions and support. The authors are grateful to Bruker Daltonics, and the Engineering and Physical Sciences Research Council (EP/J000302) for funding this project. LC, MAD, and CR thank the Agence Nationale pour la Recherche (ANR, France, grant Défi de tous le savoirs 2014, ONE_SHOT_FT-ICR_MS_2D) for financial support.Notes and references
-
J. Brinckmann, Collagens at a Glance, Springer, Berlin, Heidelberg, 2005 Search PubMed
.
-
J. Woodhead-Galloway, Collagen: the Anatomy of a Protein, Edward Arnold, Southampton, 1980 Search PubMed
- M. Yamauchi and M. Sricholpech, Essays Biochem., 2012, 52, 113–133 CrossRef CAS PubMed
- M. D. Shoulders and R. T. Raines, Annu. Rev. Biochem., 2009, 78, 929–958 CrossRef CAS PubMed
- J. Myllyharju and K. I. Kivirikko, Ann. Med., 2001, 33, 7–21 CrossRef CAS PubMed
- M. Buckley, M. Collins, J. Thomas-Oates and J. C. Wilson, Rapid Commun. Mass Spectrom., 2009, 23, 3843–3854 CrossRef CAS PubMed
- P. Perez Hurtado and P. B. O'Connor, Anal. Chem., 2012, 84, 3017–3025 CrossRef CAS PubMed
- P. Pfandler, G. Bodenhausen, J. Rapin, R. Houriet and T. Gaumann, Chem. Phys. Lett., 1987, 138, 195–200 CrossRef
- P. Pfandler and T. Gaumann, J. Am. Chem. Soc., 1988, 110, 5625–5628 CrossRef
- S. Guan and P. R. Jones, J. Chem. Phys., 1989, 91, 5291–5295 CrossRef CAS
- C. W. Ross, S. Guan, P. B. Grosshans, T. L. Ricca and A. G. Marshall, J. Am. Chem. Soc., 1993, 115, 7854–7861 CrossRef CAS
- C. W. Ross, W. J. Simonsick and D. J. Aaserud, Anal. Chem., 2002, 74, 4625–4633 CrossRef CAS PubMed
- G. van der Rest and A. G. Marshall, Int. J. Mass Spectrom., 2001, 210–211, 101–111 CrossRef
- M. A. van Agthoven, M.-A. Delsuc, G. Bodenhausen and C. Rolando, Anal. Bioanal. Chem., 2013, 405, 51–61 CrossRef CAS PubMed
- M. A. van Agthoven, M.-A. Coutouly, C. Rolando and M.-A. Delsuc, Rapid Commun. Mass Spectrom., 2011, 25, 1609–1616 CrossRef CAS PubMed
- L. Chiron, M. A. van Agthoven, B. Kieffer, C. Rolando and M.-A. Delsuc, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 1385–1390 CrossRef CAS PubMed
- J. W. Gauthier, T. R. Trautman and D. B. Jacobson, Anal. Chim. Acta, 1991, 246, 211–225 CrossRef CAS
- D. P. Little, J. P. Speir, M. W. Senko, P. B. O'Connor and F. W. McLafferty, Anal. Chem., 1994, 66, 2809–2815 CrossRef CAS PubMed
- R. Zubarev, N. L. Kelleher and F. W. McLafferty, J. Am. Chem. Soc., 1998, 120, 3265–3266 CrossRef CAS
- R. Aebersold and M. Mann, Nature, 2003, 422, 198–207 CrossRef CAS PubMed
- A. Michalski, J. Cox and M. Mann, J. Proteome Res., 2011, 10, 1785–1793 CrossRef CAS PubMed
- A. G. Marshall, T.-C. L. Wang and T. L. Ricca, Chem. Phys. Lett., 1984, 105, 233–236 CrossRef CAS
- M. van Agthoven, L. Chiron, M.-A. Coutouly, A. A. Sehgal, P. Pelupessy, G. Bodenhausen, M.-A. Delsuc and C. Rolando, Int. J. Mass Spectrom., 2014, 370, 114–124 CrossRef CAS
- M. A. van Agthoven, M.-A. Delsuc and C. Rolando, Int. J. Mass Spectrom., 2011, 306, 196–203 CrossRef CAS
- M. Bensimon, G. Zhao and T. Gaumann, Chem. Phys. Lett., 1989, 157, 97–100 CrossRef
- M. A. van Agthoven, L. Chiron, M.-A. Coutouly, M.-A. Delsuc and C. Rolando, Anal. Chem., 2012, 84, 5589–5595 CrossRef CAS PubMed
- M. A. van Agthoven, M. P. Barrow, L. Chiron, M.-A. Coutouly, D. Kilgour, C. A. Wootton, J. Wei, A. Soulby, M.-A. Delsuc, C. Rolando and P. B. O'Connor, J. Am. Soc. Mass Spectrom., 2015, 1–10 Search PubMed
- SPIKE, http://www.bitbucket.org/delsuc/spike, (accessed July 2015).
- D. Tramesel, V. Catherinot and M.-A. Delsuc, J. Magn. Reson., 2007, 188, 56–67 CrossRef CAS PubMed
- D. D. Traficante and G. A. Nemeth, J. Magn. Reson., 1987, 71, 237–245 Search PubMed
- A. Rich and F. H. C. Crick, J. Mol. Biol., 1961, 3, 483–506 CrossRef CAS PubMed
- J. Bella, M. Eaton, B. Brodsky and H. M. Berman, Science, 1994, 266, 75–81 CAS
- S. R. Lamandé and J. F. Bateman, Semin. Cell Dev. Biol., 1999, 10, 455–464 CrossRef PubMed
- I. Perdivara, L. Perera, M. Sricholpech, M. Terajima, N. Pleshko, M. Yamauchi and K. B. Tomer, J. Am. Soc. Mass Spectrom., 2013, 24, 1072–1081 CrossRef CAS PubMed
- M. Sricholpech, I. Perdivara, M. Yokoyama, H. Nagaoka, M. Terajima, K. B. Tomer and M. Yamauchi, J. Biol. Chem., 2012, 287, 22998–23009 CrossRef CAS PubMed
- M. Terajima, I. Perdivara, M. Sricholpech, Y. Deguchi, N. Pleshko, K. B. Tomer and M. Yamauchi, J. Biol. Chem., 2014, 289, 22636–22647 CrossRef CAS PubMed
- D. R. Eyre, M. A. Paz and P. M. Gallop, Annu. Rev. Biochem., 1984, 53, 717–748 CrossRef CAS PubMed
- D. R. Eyre, M. A. Weis and J. J. Wu, Methods, 2008, 45, 65–74 CrossRef CAS PubMed
- W. Henkel and K. Dreisewerd, J. Proteome Res., 2007, 6, 4269–4289 CrossRef CAS PubMed
- J. A. Ramshaw, N. K. Shah and B. Brodsky, J. Struct. Biol., 1998, 122, 86–91 CrossRef CAS PubMed
- B. Paizs and S. Suhai, Mass Spectrom. Rev., 2005, 24, 508–548 CrossRef CAS PubMed
- P. B. Fellgett, J. Opt. Soc. Am., 1949, 39, 970–976 CrossRef CAS PubMed
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5an01757b |
| This journal is © The Royal Society of Chemistry 2016 |