
Charge site assignment in native proteins by ultraviolet photodissociation (UVPD) mass spectrometry†
Lindsay J.
Morrison
and
Jennifer S.
Brodbelt
*
Department of Chemistry, University of Texas, Austin, TX 78712, USA. E-mail: jbrodbelt@cm.utexas.edu
First published on 13th November 2015
Abstract
Characterization of all gas-phase charge sites of natively sprayed proteins and peptides is demonstrated using 193 nm UVPD. The high sequence coverage offered by UVPD is exploited for the accurate determination of charge sites in protein systems up to 18 kDa, allowing charge site to be studied as a function of protein conformation and the presence of disulfide bonds. Charging protons are found on both basic sidechains and on the amide backbone of less basic amino acids such as serine, glutamine, and proline. UVPD analysis was performed on the 3+ charge state of melittin, the 5+ to 8+ charge states of ubiquitin, and the 8+ charge state of reduced and oxidized β-lactoglobulin. The location of charges in gas-phase proteins is known to impact structure; molecular modeling of different charge site motifs of 3+ melittin demonstrates how placement of protons in simulations can dramatically impact the predicted structure of the molecule. The location of positive charge sites in ubiquitin and β-lactoglobulin are additionally found to depend on the presence or absence of salt-bridges, columbic repulsion across the length of the peptide, and protein conformation. Charge site isomers are demonstrated for ubiquitin and β-lactoglobulin but found to be much less numerous than previously predicted.
Introduction
Mass spectrometry has rapidly expanded as a structural biology tool capable of providing structure and sequence information for proteins and other large biomolecules. The development of electrospray ionization (ESI) and its buffered aqueous solution analog, native electrospray ionization, permit facile ionization of denatured proteins and proteins in native or native-like states.1,2 Ion mobility,3–11 gas-phase hydrogen deuterium exchange,12–15 and ion spectroscopy16,17 have all been used to characterize the structures of native-like proteins in the gas phase. Ion mobility studies in particular have demonstrated that the collisional cross sections (CCS) of proteins ionized using native ESI and gentle source conditions are highly similar to cross sections predicted from the crystal structures, suggesting that native ESI may preserve native-like structures.10,11,18–22 The CCS of proteins have been examined as a function of charge state,22 spray conditions,23,24 solvent additives,26–29 and following gas-phase ion-neutral collisions.8,30–32 However, despite numerous advances, many structural features of protein ions remain poorly characterized. For example, one of the most important features of protein ions that has proven difficult to elucidate is the location of protons or charge sites. Williams et al. modeled the maximum charge states of proteins in the gas phase, as well as predicted the locations of protons for different charge states.33 As expected, the most basic sites (Arg, Lys, His) were predominantly protonated for low charge states, but less basic amino acids (Pro, Trp, Gln) were frequently protonated for the higher charge states.In theory, charge sites could be localized by monitoring the charge states of fragment ions produced upon dissociation of proteins. In practice, however, this strategy can be difficult to employ due to proton transfer events that occur after ion energization, particularly for collision-based activation methods. For peptides or proteins with one or more mobile protons (i.e. having a greater number of protons than basic sites, thus allowing facile proton migration), protonation on the backbone amide groups promotes cleavage to produce b and y ions that do not reflect specific protonation sites.34–40 At the same time, CID results in low sequence coverage for proteins lacking sufficient mobile protons, resulting in large gaps in series of fragment ions and again preventing localization of charge sites. In electron transfer and electron capture dissociation (ETD and ECD), the transfer of the electron to a multi-protonated protein or peptide promotes homolytic dissociation, typically at the N–Cα bonds to produce N-terminal c and C-terminal z ions. In 2006, Zubarev and co-workers utilized electron capture dissociation to characterize n − 1 protonation sites in peptides with n charges.41 Analysis of the charge states of the fragment ions produced by ECD suggested that protonation could occur at less basic sites, and this outcome was attributed to stabilization of charge sites via secondary interactions involving backbone carbonyls.41 Application of this approach was limited to small peptides, however, and the largest system studied was the 3.5 kDa peptide melittin, presumably because ECD also suffers from poor sequence coverage, particularly for proteins with low charge states. McLafferty and co-workers examined the distributions of c and z ions generated from ubiquitin (6+ to 13+) by ECD and surmised hydrogen bonding interactions at some sites using ECD fragment abundance as a function of protein charge state.42 ECD was found to result in preferential cleavage within a few residues of the electron capture site. They also suggested that the ECD data supported the existence of multiple protonation isomers with different protonation sites.42 Neutralization at the site of capture and poor sequence coverage for low charge states, however, made direct assignment of protonation sites for some charge states difficult. Both of these ECD-based studies offered compelling evidence that information about locations of charge sites could be obtained from strategic analysis of fragmentation of multi-charged ions.
193 nm ultraviolet photodissociation (UVPD), has recently been demonstrated to achieve up to 100% sequence coverage of intact proteins.43 Fragmentation by UVPD is unique in that a and x type ions are formed in addition to the b, y, c, and z type ions that are formed by CID and ETD. In 157 nm UVPD fragmentation, an amide electron is excited into a Rydberg orbital, inducing homolytic cleavage of the C–Cα bond to generate a and x type ions.44 Based on the evaluation of singly charged peptides, the Reilly group demonstrated that it was possible to differentiate the position of N-terminal vs. C-terminal arginines based on the presence or absence of a and x ions.44 The specific mechanisms for 193 nm UVPD have not been determined, but a mixture of pathways involving direct dissociation from excited states and those occurring after internal conversion and intramolecular vibrational redistribution may coexist as a, b, c, x, y, and z ion are all commonly observed. Given the ongoing interest in understanding the fragmentation patterns observed for intact proteins obtained by different activation methods and correlating them with structural models in the gas phase, a better means to predict the charge sites would be a significant step. In the present study, we use the charge states of the a/x fragment ions produced by 193 nm UVPD to assign charge sites to triply charged melittin, the 5+ to 8+ charge states of ubiquitin, and 8+ oxidized and reduced β-lactoglobulin. We demonstrate that the charge state distributions of the a/x ions created by UVPD provide a unique means to extend charge site predictions to other proteins.
Methods
Sample preparation
Melittin, ubiquitin, β-lactoglobulin, and solvents and chemicals not otherwise specified were purchased from Sigma-Aldrich (St. Louis, MO) and used without additional purification. Buffer exchange was performed using BioRad (Hercules, CA) P-6 micro bio-spin columns. Acetylation of primary amine functionalities of melittin was achieved by incubation of a protein in 2500-fold excess of acetic anhydride in 150 mM ammonium bicarbonate buffer at 298 K. The reaction was quenched after thirty minutes by buffer exchange into ammonium acetate spray buffer. Reduction of disulfide bonds of lactoglobulin was carried out via incubation with 5 mM dithiothreitol at 55 °C for 2 hours in 50 mM ammonium acetate.Mass spectrometry
Proteins were purchased from Sigma-Aldrich (St. Louis, MO) and solubilized or buffer exchanged into 50 mM ammonium acetate (ubiquitin, β-lactoglobulin) or other appropriate spray solvent (methanol/water for melittin) and introduced into the gas phase via a custom nanospray source comprised of a glass tip pulled in-house to a have tip apertures of less than one micron. The pulled tip was filled with the protein solution and spray was achieved by applying 1–2 kV potential to a platinum wire, which was inserted in the pulled tip. For ubiquitin, the 5+ and 6+ charge states were generated from a 50 mM ammonium acetate solution, and the 7+ and 8+ charge states were produced from a 50/50 water/methanol solution. Oxidized β-lactoglobulin was transmitted using gentle source conditions or applying 50 V source activation for partial unfolding. Partially reduced β-lactoglobulin was subjected to 50 V source activation to sufficiently desolvate it and obtain sufficient signal for UVPD analysis. All mass spectrometry experiments were performed on a Thermo Fisher Scientific Orbitrap Elite mass spectrometer (San Jose, CA) previously coupled in-house with a 193 nm Coherent excimer laser.43 UVPD was typically performed using a single laser pulse with the measured output of the laser being 1.2–2.0 mJ. The laser was not collimated or focused. Spectra were interpreted both manually and in conjunction with the ProSightPC software package, modified by the Kelleher group for use with UVPD data. Raw spectra were deconvolved using the Thrash algorithm and searched against the known protein sequences for melittin, ubiquitin, and β-lactoglobulin.Modeling
Molecular dynamics simulations were performed using the Amber–Cornell forcefield and NAMD software.45 The topology and parameter files were modified to include amide oxygen protonated amino acids. The addition of the positive charge required adjustment of the partial charges across the residue. The proton was given a partial positive charge of 0.589 and the charge on the amide oxygen and carbon raised by a combined 0.41. The values for these alterations were determined from ab initio modeling of protonated and deprotonated diketopiperazine dialanine. These computations were performed using the B3LYP/6-31G* basis set and the Gausssian09 and Macromodel software packages.46,47 Candidate salt bridges were determined using the salt-bridge tool in VMD. The distance between charge centers was determined for ubiquitin and beta-lactoglobulin based on the 1UBQ and 1BSY crystal structures.Results and discussion
The UVPD strategy was developed using two model proteins: melittin and ubiquitin. Melittin is a helical peptide found in honey bee venom and has been extensively examined by ion mobility and solution phase hydrogen/deuterium exchange.48 The experimental collisional cross section was found to vary as a function of methanol content, and higher methanol concentrations yielded higher cross sections for the 3+, 4+ and 5+ charge states.48 From tandem MS and energy-resolved experiments, the 3+ charge state has been speculated to most retain characteristics of the solution phase structure. Interestingly, however, reports for the collisional cross section of melittin 3+ vary greatly; Barran and co-workers reported values ranging from 523 to 566 Å2 as a function of solvent condition,48 Bush and co-workers reported 581 Å2,49 and May and McLean reported two minor populations centered at 410 and 490 Å2, and one major at 523 Å2 that did not change substantially with solvent conditions.50Ubiquitin has been examined in detail as a function of solvent system and collisional activation conditions.25,51–53 Ubiquitin has three solution-phase states: the helical A state, the native N state, and the unfolded U state, which can be accessed by changing the solution conditions. The charge state distribution of ubiquitin has been shown to vary with electrospray solvent conditions; spraying from aqueous solutions generates charge states ranging from 5+ to 7+, and the collisional cross sections of these states are largely consistent with the N state. In contrast, electrospray from 50![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :50 H2O:methanol solutions results in population of charge states 7+ to 9+, which have collisional cross sections more similar to the A state.25,52,54 The 8+ charge state has been studied in detail, and the collisional cross section of the charge state has been shown to vary with solution conditions from which it is sprayed.25
:50 H2O:methanol solutions results in population of charge states 7+ to 9+, which have collisional cross sections more similar to the A state.25,52,54 The 8+ charge state has been studied in detail, and the collisional cross section of the charge state has been shown to vary with solution conditions from which it is sprayed.25
Melittin
Triply charged melittin (26 amino acids) has been studied by ion mobility and hydrogen-deuterium exchange, and all reports indicate it having a helical structure in the gas phase.48 Kjeldsen and co-workers have additionally studied the 3+, 4+, and 5+ charge states by ECD and assigned n − 1 charge sites for these states.41 For simplicity, we focused on the 3+ charge state here, as it was expected to be the most helical. Upon analysis by ECD, two of the three charge sites have been previously assigned: one localized between Ala4 and Val5 and a second between Lys23 and Arg24.41 Following electrospray, the 3+ charge state of melittin was mass selected and activated by a single 1.2 mJ pulse of 193 nm photons. The resulting fragment ions were predominantly a type, with low abundances of b, c, x, y, and z ions also observed. The abundances of each a and x ion, per charge state, were tabulated, and the relative abundances of each charge state of each an or xn ion were calculated as a function of the total an or xn population. The fraction of the an population in a particular charge state was calculated as , where
, where 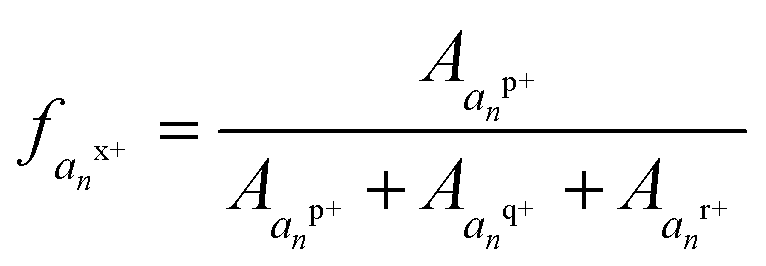 , where p+, q+, and r+ are the observed charge states of a given a ion an and the abundance of a given charge state of that a ion is
, where p+, q+, and r+ are the observed charge states of a given a ion an and the abundance of a given charge state of that a ion is 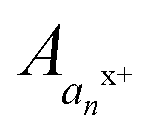 . This value is termed the fractional abundance of each charge state. An analogous equation was used to quantify the charge states of the x ions.
. This value is termed the fractional abundance of each charge state. An analogous equation was used to quantify the charge states of the x ions.
The fractional abundance of each charge state per a and x ion is shown in Fig. 1, and the raw abundances of each of these an and xn fragment ions are shown in ESI Fig. S1.† The charge state distributions of the fragments feature strikingly sharp transitions; for example, the a20 ion is exclusively observed in the 1+ charge state, whereas the a21 ion is exclusively observed in the 2+ charge state. The sharp transition from 1+ for a20 to 2+ for a21 is taken as evidence for the localization of one proton at residue 21 (Lys21). The complementary transition between singly charged x5 and doubly charged x6 confirms this assignment. A similarly sharp transition is observed between doubly charged a23 and triply charged a24, suggesting a second protonation site on Arg24. This transition too is reasonable, particularly because columbic repulsion between positive charges on adjacent amino acids could drive protonation to the less basic Lys21 rather than Arg22. Thus, the charge states of the a and x ions generated by UVPD appear to allow assignment of charge sites of proteins. The b/y and c/z ions were also examined by this approach, but resulted in less distinctive fragment ion charge state transitions (presumably due in part to their different and competing mechanisms of formation which may involve mobile protons and/or hydrogen migration affiliated with radical sites).
 | ||
| Fig. 1 Fractional abundances of a (left) and x (right) ion charge states of melittin (3+). The color codes for the fragment ion charge states are reversed for the two plots to facilitate comparison of trends between the two complementary ion series. | ||
Collectively the trends in Fig. 1 suggest that two of the charge sites are Lys21 and Arg24 and the third is located near the N-terminus. The first a ion observed in the UVPD data is a5 (1+); for the x ions a complementary transition between the doubly and triply charged series is observed between x21 and x22. Together, these observations suggest that the fifth amino acid is protonated. However, the fifth residue, valine, is non-basic and the sidechain is not capable of carrying a positive charge. Protonation of the amide backbone has been shown in small peptide systems using ab initio modeling,55 and Williams and co-workers have modeled the maximum charge states of proteins as a function of gas-phase basicity and suggested that non-basic amino acids, particularly Gln, Pro, and Trp, may be protonated when the charge state of the ion is greater than 60% of the predicted maximum charge state.33 Because a ions arise from cleavage of the Cα–C bond, the transition in a and x ion charge sites at the fifth cleavage site (i.e. resulting in a5/x21) indicates protonation on the preceding amide oxygen. However, given that the nearby N-terminus and Lys7 are both basic sites, the localization of a proton on the amide oxygen of Ala4 is surprising and raises a question as to whether the observed fragment ion charge state transition arises because the amide of Ala4 is protonated with a mobile proton, or because a charge transfer event or other process occurs during dissociation of the UV activated ions, thus confounding the interpretation.
As UVPD is a fast electronic process that does not rely on the presence of a mobile proton or the capture of an electron, scission of the backbone without transfer of the charging protons to generate a/x ions is possible. The transitions of the charge states for fragment ions formed by UVPD contrast with the trends exhibited by HCD and ETD, as shown for melittin (3+) in Fig. 2. Both HCD and ETD are known to facilitate frequent proton transfer or hydrogen transfer processes. In addition to showing several missed cleavage sites, the distributions in Fig. 2 do not display clear transitions between charge states. Moreover, because ETD is initiated by a charge transfer process, only two fragment ion charge states were observed (1+, 2+), thus prohibiting mapping of the location of the third charge site.
 | ||
| Fig. 2 Fractional abundance per charge state of the b and y ions from HCD of 3+ melittin are shown in (a) and (b) and the fractional abundance per charge state of the c and z ions from ETD are shown in (c) and (d). | ||
In order to confirm that Ala4 is protonated, leaving the N-terminus and Lys7 deprotonated, melittin was acetylated at the N-terminus and all three lysine residues. Following acetylation, the only basic sites on the peptide are the two arginine residues. Thus, the formation of the triply charged species serves as an indicator as to whether protonation on a non-basic site is favorable for melittin. ESI Fig. S2† shows the mass spectrum of tetra-acetylated melittin, in which the dominant charge state is 3+, suggesting that protonation at a non-basic site is both possible and energetically favorable.
Based on the a/x ion charge states described above upon UVPD of melittin (3+), the impact of the protonation site (Ala4) on gas-phase structure was examined by molecular dynamics simulations. The Jarrold group has previously shown that the location of basic sites in polyalanine-based peptides has a profound impact on the gas-phase structures of the peptides, with helical structures being favored for peptides protonated at the C-terminus and globular structures being favored for peptides protonated at the N-terminus.4,6,7,56 The starting structure of monomeric melittin was taken from the protein data bank, entry 1MLT, and modified such that only three amino acids were positively charged. Because the C-terminus of melittin is naturally amidated and acidic residues are not present in the peptide, zwitterions were not considered. Three protonation schemes were considered; the first is based on UVPD data and is labeled A4K21R24 to denote charges residing on Ala4, Lys21, and Arg24. Charge site isomers in which more the more typical basic sites near the N-terminus were protonated were also considered and are denoted G1K21R24 and K7K21R24 to indicate location of the third charge on either the N-terminus or Lys7, respectively. The three charge state isomers were subjected to simulated annealing, and the collisional cross sections of the resulting twenty lowest energy conformations of each charge site isomer calculated using the projection superposition approximation (PSA).57–60 The collisional cross sections and relative energies of the twenty lowest energy conformers of each charge site isomer are shown in Fig. S3.† In general, the protonation scheme was found to have a strong impact on conformation, and protonation at Ala4 tended to result in helical structures with two kinks rather than the single kink observed for structures protonated at the N-terminus or Lys7. The lowest energy conformer of the A4K21R24 charging scheme was found to have a CCS of 515 Å2, which falls within 1.5% of the CCS measured by the Barran and McLean groups.48,50 Based collectively on the relative energy values, the UVPD charge site assignments, and collisional cross sections, we assign the observed 3+ charge state of melittin to conformation A of A4K21R24, shown in Fig. S4.† Low energy configurations of the G1K21R24 and K7K21R24 charge site isomers are also shown in Fig. S4.† In conformation A of A4K21R24, the charging proton located on the amide carbonyl of Ala4 is hydrogen bonded to Lys7, and the overall structure is a helix-break-helix-break-helix. UVPD assignment of charge sites in combination with molecular dynamics modeling and ion mobility thus offers a means to reduce the number of reasonable candidate structures from MD simulations and thereby refine the construction of models based on strategic placement of charges.
Ubiquitin
The absence of a free acid C-terminus and acidic residues in the peptide melittin make it impossible for salt bridges and zwitterionic motifs to exist. Native proteins, however, typically have numerous salt bridges in solution, and some or all of these may be retained following transfer to the gas phase. In order to develop a better understanding of the complex relationship between gas-phase protonation sites and salt bridges, the 5+ to 8+ charge states of ubiquitin (containing 76 residues) were studied by 193 nm UVPD. Fig. 3 shows the fractional abundance of the a and x ions for these charge states, from which it is possible to assign charge sites based on the procedure described earlier. The 5+ and 8+ charge states of ubiquitin feature the sharpest transitions in a/x fragment ion charge states whereas the 6+ and 7+ charge states exhibited more anomalies.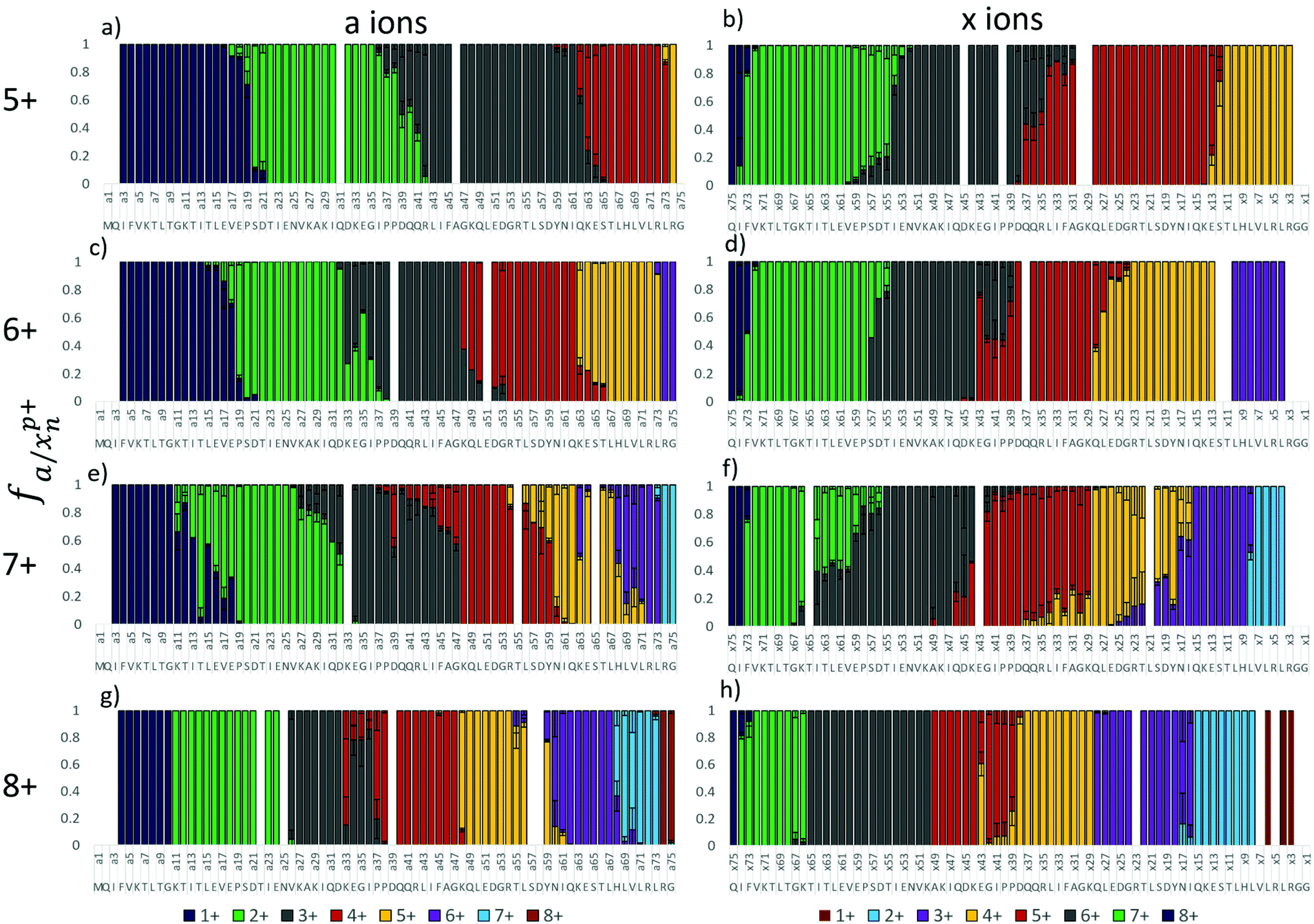 | ||
| Fig. 3 Fractional abundance per charge state of the a and x ions of ubiquitin for 5+ (a and b), 6+, (c and d), 7+ (e and f), and 8+ (g and h). | ||
The a ions that arise from 193 nm UVPD of the 5+ charge state of ubiquitin begin at a3 (which terminates in Ile3) (Fig. 3a), suggesting a possible protonation site on the amide backbone at Gln2. Production of a/x ions by UVPD entails cleavage C-terminal to the amide carbonyl, resulting in the glutamine carbonyl being included with the isoleucine a3 fragment. The N or native state of ubiquitin is well documented to have four strong salt bridges and has been speculated to have several weaker bridges.61 ESI Fig. S5† lists the ion pairs that are within 15 Å of each other. The closest partners for each acidic/basic residue are marked by a connecting line, and the heaviness of the line denotes the strength of the salt bridge. Our analysis of the 1UBQ crystal structure resulted in identification of five ion pairs separated by less than 6 Å, two of which were separated by less than 4 Å. Although the canonical salt-bridge distance is 4 Å, we use 6 Å as the salt bridge threshold for the present study in order to examine the effect of all reasonably close ion pairs in the transition to the gas phase. The five ubiquitin salt bridges are: the N-terminus and Glu18, Lys11 and Glu34, Lys27 and Asp52, Lys29 and Asp21, Arg54 and Glu51. Protonation at Gln2 rather than the N-terminus is therefore in agreement with the native ubiquitin structure. It is not clear why the charge does not localize to Lys6, which is not expected to be engaged in a salt bridge and was predicted by Williams and co-workers to be one of the most basic residues.33 The singly charged a ions transition to the doubly charged a ions at a20, suggesting Pro19 is the second protonation site, (Fig. 3a), which is also consistent with the work done by the Williams group.33 The transition from the 2+ a ions to the 3+ a ions (and in the complementary x ions) occurs in two steps, suggesting there may be two populations, one with the third proton localized at Pro38 and the other with the third proton at Arg42 (Fig. 3a and b). The fourth and fifth protonation sites can similarly be assigned to Lys63 and the Arg74, respectively, based on the charge states of the complementary a and x ions. Interestingly, none of the basic residues predicted to be in an ion pair from the 1UBQ crystal structure were found to be protonation sites predicted by UVPD, (Gln2, Pro19, Pro38/Arg42, Lys63, and Arg74).
Based on similar analysis, the 6+ charge state of ubiquitin is predicted to be protonated at Gln2, Glu18, Lys33/Pro38, Lys48, Lys63, and Arg74 (Fig. 3c and d). Similar to the 5+ charge state, there is one pair of competitive protonation sites for the 6+ charge state of ubiquitin: the third protonation site at Pro38 or Lys33. With the exception of a shift in protonation from Arg42 to Lys33 and the addition of one charge site at Lys48, all other charge sites are similar for the 5+ and 6+ charge states of ubiquitin. The Pro19 charge site was, in fact, observed to shift by one residue to Glu18 between the 5+ and 6+ charge states. This minor yet reproducible change perhaps suggests that electrostatic repulsion involving nearby charges has an effect on charge localization, particularly for non-basic charge sites, or perhaps is indicative of other mitigating factors that cause slight ambiguities in determining charge locations. The 5+ and 6+ charge states are expected to have similar structures based on ion mobility studies of the different charge states,25,52,54,62 and UVPD is consistent with those previous studies.
The 7+ charge state features a large degree of tailing in the charge state transitions of the fragment ions and may be representative of two competing or complementary charging schemes (Fig. 3e and f). Minor and major transitions in fragment ion charge states are observed for both the a and x ion series. Based on the UVPD data, the major charge site isomer is protonated at Gln2, Glu18, Lys33, Lys48, Asn60, His68, and Arg74, and the minor charge site isomer is protonated at Gln2, Lys11, Lys27/29, Arg42, Arg54, His68, and Arg74. Some of these assignments are made with less confidence, and multiple basic sites are listed if they are proximal (e.g. Lys27 and Lys29) because transitions in the minor population are inherently less distinct. The minor charge state population of ubiquitin (7+) contains very different charge sites than does the major 7+ population, the 6+ population, and the 5+ population of ubiquitin, all of which contained protonation sites not included in known salt bridges in the N (native) state of ubiquitin. In contrast, the minor 7+ population is protonated at three basic sites thought to be engaged in salt bridges (Lys11, Lys27 or Lys29, and Arg54). This change, particularly in regards to the native structure, may be an indication that the minor 7+ population has a different conformation than the 5+, 6+, and major 7+ population. As Clemmer and co-workers have shown that the 7+ charge state is mixture of the A and N states of ubiquitin,54,63 it is possible that this minor population is associated with a gas-phase conformation of ubiquitin in the A state. Interestingly, the theoretical analysis performed by the Williams group predicted the 7+ charge state to be most likely protonated at: K7, K11, K27, R42, R54, K63, and R74, although some probability existed for protonation at K33, Q2, H68, and R72.33 Of the charge sites of the major 7+ population found by UVPD, only Arg74 is consistent with these predictions, although Arg42 was found to be protonated for the 5+ charge state. The minor population, on the other hand, has protonation sites predicted by UVPD to be consistent with six of the sites (including H68) proposed by Williams and co-workers.33 McLafferty et al. suggested locations for 5 of the 7 charges of 7+ ubiquitin: Lys7, Lys11, Lys33, Lys63, and Arg72.42 These assignments are largely dissimilar to those based on the UVPD data; however, the sequence coverage provided by ECD of the 7+ species was less than 50 percent, and many of the charge site assignments had to be inferred from one or two fragment ions. This comparison highlights the utility of UVPD, with its impressive top-down sequence coverage of native proteins, for the assignments of charge sites in native proteins.
The charge sites observed for the 8+ population are similar to those observed for the minor 7+ population (Fig. 3g and h). The 8+ charge state featured very sharp fragment ion charge state transitions for the most part, suggesting the presence of only one charge site isomer. Protonation sites can thus be assigned to the N-terminus, Lys11, Lys27, Pro37, Lys48, Asn60, His68, and Arg74. The exception to the sharp transitions for the 8+ charge state is the region from a33 to a37 and the complementary x43 ion. In the a ion series, the 4+ charge state is observed to be dominant for a33 and a37 to a47, whereas the 3+ charge state is dominant for a34, a35, and a36. It is possible that a salt bridge exists between Lys33 and Glu34 and thus cleavage between these residues separates the ion pair such that an additional positive charge is observed on the N-terminal fragment (a33) and a negative charge observed on the corresponding C-terminal fragment (x43). A similar pattern in observed for the 6+ charge state of ubiquitin, and suggests that one or more salt bridges are present in this for ubiquitin in this charge state (though not observed in the 5+ or 7+ charge states). Two similar spikes in charge state to the one observed at x43 of 8+ ubiquitin are observed for a39 (C-terminal Asp) and a63 (C-terminal Lys), which may be evidence for additional ion pairs. Asp39 is only 5-6 Å from Arg72 and Arg74; this C-terminal tail is often omitted from crystal structures due to its flexibility and it is therefore possible that an ion pair exists as a minor population in solution or forms during the desolvation process. Lys63, on the other hand, is only 5.5 Å from Glu64, and by similar reasoning an ion pair may form between these residues for some portion of the solution phase ubiquitin population. This could explain why Lys63 is protonated in the 5+ and 6+ charge states but not in the 7+.
The changes in charge site as a function of ubiquitin charge state are represented in schematic format in Fig. 4. The protonation sites derived from the a/x fragment ions are highlighted in red font. Arrows are used to highlight the specific charge sites that appear to change with the precursor charge state. As noted above, the charge site distributions for the 5+ and 6+ charge states are very similar; the Arg42 protonation site is the only one that changes. This site is effectively “split” into two sites, Lys33 and Lys48, in the 6+ charge state of ubiquitin, both of which are basic sites not engaged in strong salt bridges. The major charge site distribution for the 7+ charge site of ubiquitin also only features a single additional change from the 5+ and 6+ populations as the charge site localized at K63 for the 6+ charge state shifts to charge sites at Asn60 and His68 for the 7+ charge state of ubiquitin. In contrast, the minor charge site distribution for the 7+ charge state and the charge site distribution of 8+ of ubiquitin feature four alternative protonation sites, and is likely indicative of a structural change. Because Lys11, Lys27, and Arg54 are known to engage in very strong salt bridges, protonation at these sites is expected to necessitate significant structural re-organization.
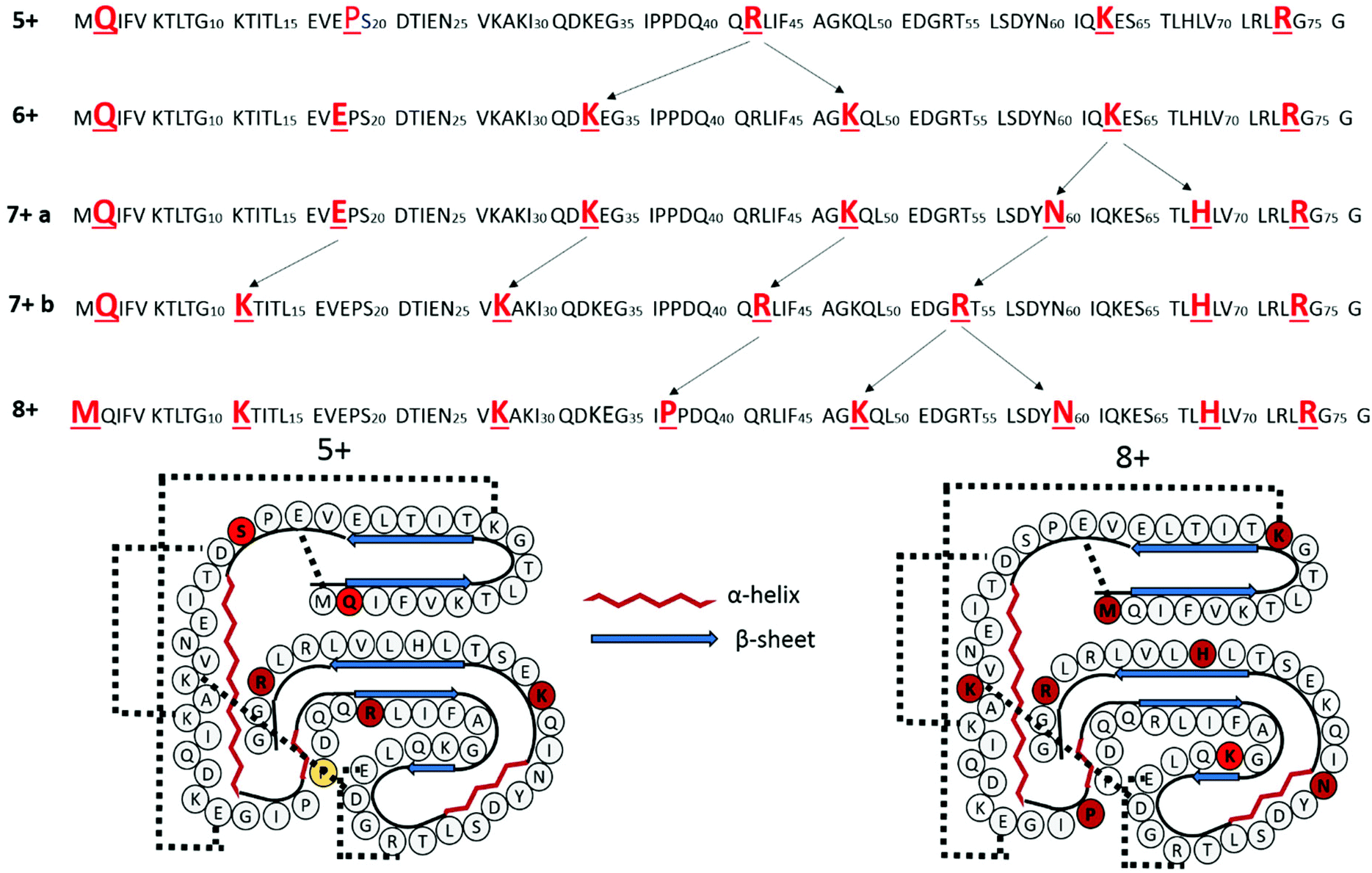 | ||
| Fig. 4 Top: The charge sites of ubiquitin elucidated based on the a/x ions from UVPD are highlighted in bold red font for the 5+, 6+, 7+, and 8+ charge states. The arrows are shown to highlight the changes in protonation sites as a function of increasing charge state. Bottom: the secondary and tertiary structures of ubiquitin (5+ and 8+) are represented with salt bridges denoted by hashed lines. The amino acids that are protonated for the 5+ and 8+ charge states are highlighted in red font. | ||
β-Lactoglobulin
The impact of protein conformation on charge site location was evaluated using oxidized and reduced β-lactoglobulin (BLG), a protein which has two disulfide bonds that cause conformational rigidity. BLG is additionally interesting in that it crystallizes in no less than six forms and has a number of sequence variants, two of which are common. In this study, we focus on variant B, which differs from variant A by D64G and V114A sequence variations. Only minor differences in structure have been reported for the two variants.64,65 Both variants contain two disulfide bonds, one between Cys66 and Cys160 and a second between Cys106 and Cys119. The Cys106–Cys119 disulfide bond is deeply buried in the interior of the protein, whereas the Cys66–Cys160 bond is solvent exposed. The protein was reduced with dithiothreitol in ammonium acetate buffer prior to infusion (see Experimental section). This resulted in reduction of one of the disulfide bonds, evidenced by a 2 Da shift in the mass of the protein. Note that longer reduction times, increased DTT concentration, and elevated temperatures did not result in the reduction of the second disulfide bond. For this reason, it is likely that the reduction of β-lactoglobulin corresponds to exclusive or near exclusive reduction of the exposed Cys66–Cys160 bond.UVPD was used to characterize the 8+ charge state of folded and elongated forms of oxidized BLG and singly-reduced, source activated BLG, and the resulting a ions were used to localize the charge sites of the N-terminal portion of the protein (Fig. 5a–c). C-terminal ions (e.g. x ions) were not observed (for oxidized BLG), presumably due to the disulfide bond connecting Cys66 to Cys160 and thus could not be used to create histograms. Oxidized β-lactoglobulin (Fig. 5a) shows evidence for multiple charge state isomers, featuring tailing in the transitions for the 1+ to 2+ charge site location, and fronting in the transitions for the 3+ to 4+ charge sites. Particularly interesting is the series of a ions that cover the Lys47 to Lys60 stretch, which features two distinct charge site populations: approximately 75% of the a ion population is triply charged in this region while 25% is quadruply charged. Replicates of this experiment performed on different days under similar but non-identical source conditions resulted in a similar pattern but with a 60:40 and 70:30 ratio of the 3+:4+ a ion populations, suggesting that some variation occurs in these populations as a function of instrument settings. The tailing distribution observed for the 1+ to 2+ and 2+ to 3+ charge site transitions are also observed for reduced and source activated β-lactoglobulin. For example, the tailing distribution of singly charged a ions is observed to completely disappear at a21 (which terminates in the Ser21 residue) of the oxidized protein, and this transition is found to be increasingly abundant for source activated and partially reduced/source-activated BLG. In addition, the split 3+/4+ population for the series of a ions from a47 to a60 in the oxidized protein (Fig. 5a) converts solely to the 4+ charge state in the reduced protein (Fig. 5c) with the charge site clearly localized on Lys60. Source activated BLG (Fig. 5b) shows intermediate behavior in this region, featuring a split population with protonation on Lys47 having approximately 10% relative abundance. The 2+/3+ charge transition also features two sites, Arg40 and Tyr42; Arg40 is more dominantly protonated in oxidized, unactivated BLG (Fig. 5a, 60:40 Arg40:Tyr42), and protonation at Tyr42 is increasingly favored with source activation and with partial reduction and source activation (Fig. 5b and c). Interestingly, the abundance of the a ion series increases by 10-fold between a7 and a8 for the three conditions of BLG studied; as an x ion series was not observed for this region, this can be tentatively interpreted as Lys8 being the dominant charge site and the N-terminus or other non-basic site being a secondary site. As none of the other proteins examined in this study featured an increase in the intensity of the a ion series increase at the first basic residue, this is reasonable. Thus, the UVPD data suggest that at least two charge state isomers of the protein are present in the gas phase, and the relative abundance of these varies with the presence of an exterior disulfide bond and the source conditions of the instrument.
 | ||
| Fig. 5 Fractional abundances of the a ions from the N-terminal portion of β-lactoglobulin for (a) completely oxidized, (b) source activated (50V), and (c) partially reduced and source activated β-lactoglobulin. The crystal structure of β-lactoglobulin is shown from two different perspectives in (d) and (e), with the experimental protonation sites associated with the more native structure highlighted in red and the protonation sites associated with the more unfolded structure shown in blue. Dotted spheres indicate the solvent accessibility of the protein and disulfide bonds are highlighted in yellow. *The relative abundance of fragments increased by 10-fold between a7 and a8, suggesting that Lys8 is the major protonation site and possibly suggesting the N-terminus as a minor charge site. | ||
In order to better understand protonation site as a function of protein structure, the 1BSY crystal structure of β-lactoglobulin was examined. The four charge state transitions associated with native BLG in its oxidized state, transferred through the mass spectrometer using gentle source conditions, correspond to protonation sites at basic, highly solvent exposed residues (Lys8, Lys14, Arg40, and Lys47) not engaged in salt bridges. These residues are shown in red in Fig. 5d and e. β-Lactoglobulin has three ion pairs separated by less than 4 Å (Asp98:Lys100, Asp137:Arg148, and Glu62:Lys69) and another five separated by less than 5 Å (Asp129:Lys101, Glu45:Lys47, Glu55:Lys70, Glu108:Lys91, and Glu134:Lys138). Lys47 was found to be situated 4.25 Å from Glu45, suggesting that while these two amino acids may not form a canonical salt bridge, they likely interact to some extent. The distance separating these residues could certainly allow protonation of the Glu45 carboxylic acid moiety during ionization, resulting in the retention of a positive charge site on Lys47.
The three charge sites that are most consistent with reduced and source activated BLG, Tyr20, Tyr42, and Lys60, are highlighted in blue in Fig. 5d and e and correspond to a cluster of closely located residues near the Cys106–Cys117 disulfide bond on the β-sheet. Tyr42, in particular, is not solvent exposed, making protonation at this site unlikely for the native structure. The close proximity of charge required for these residues to be protonated in a native structure, in conjunction with the changing protonation sites observed as a function of source activation and disulfide reduction, indicates that protonation on Tyr20, Tyr42, and Lys60 is consistent with a more unfolded conformation.
Lys60 is predominantly protonated in both oxidized and reduced β-lactoglobulin based on the production of the a60 ion nearly exclusively in the 4+ charge state. Without scission of the disulfide bond, this residue can only be solvent exposed by structural changes in which the distal two strands of the β-sheet peel away from the loop containing residues 32–39. Because the disulfide bond is intact in the oxidized protein, this necessitates shifting of the C-terminal region as well. Given that Lys60 is protonated over 25–40% of the time in oxidized BLG, it is likely that this structural change occurs relatively easily even under relatively gentle source and transfer conditions.
Conclusions
193 nm UVPD of native proteins is shown to produce a and x fragment ions with charge states that are consistent with the protonation sites of the intact protein. Interestingly, non-basic amino acids such as glutamine, proline, tyrosine, and serine are frequently found as protonation sites, even in charge states of proteins at less than half of their maximum charge. MD simulations of triply protonated melittin protonated at Ala4, Lys21, and Arg24 are consistent with the collisional cross section of the peptide from methanol/water solutions. The diverse structures discovered for different protonation schemes highlights the role that charge location can play on local structure and demonstrates how experimental determination of charge site can guide gas-phase simulations.The 5+ to 8+ charge states of ubiquitin follow two motifs, one consistent with the native structure in which basic residues engaged in salt bridges are preferentially not protonated, and one in which many of the cationic pairs of known salt bridges are protonated. The protonation scheme consistent with the native structure is observed for the 5+ and 6+ charge states of ubiquitin, the protonation scheme consistent with an alternative structure is observed for the 8+ charge state, and the 7+ shows evidence for a mixture of the two. These results are in agreement with the ion mobility work reported by the Clemmer group in which cross sections most similar to the native N structure were found for lower charge states (6+ and 7+) and cross sections most similar to the helical A state were found for the 8+ charge state.25,54,63 This is also consistent with the ECD fragment ion analysis from McLafferty and co-workers in which the 8+ charge state featured much more abundant fragmentation, particularly in the middle of the protein, than did the 6+ or 7+ charge states, consistent with the 8+ charge state having a much less ordered conformation.42
Reduction of the exterior disulfide bond of β-lactoglobulin and the addition of collisional activation in the source of the mass spectrometer provided an interesting comparison for the natively sprayed, oxidized protein. Charge site analysis by UVPD demonstrated multiple charge site isomers for the native, oxidized 8+ charge state of β-lactoglobulin. Comparison to the charge state analysis with source activated and partially reduced and source activated BLG and examination of the crystal structure provides evidence to suggest that BLG adopts a mostly native conformation in the gas phase under gentle source conditions but may feature some degree of unfolding of the strands of the β-sheet.
Acknowledgements
Funding from the NSF (CHE-1402753), the Welch Foundation (F-1155) and NIH 1K12GM102745 (fellowship to LM) is acknowledged.References
- J. B. Fenn, M. Mann, C. K. Meng, S. F. Wong and C. M. Whitehouse, Science, 1989, 246, 64–71 CAS.
- D. C. Gale and R. D. Smith, Rapid Commun. Mass Spectrom., 1993, 7, 1017–1021 CrossRef CAS.
- D. T. Kaleta and M. F. Jarrold, J. Phys. Chem. B, 2001, 105, 4436–4440 CrossRef CAS.
- M. Kohtani, T. C. Jones, J. E. Schneider and M. F. Jarrold, J. Am. Chem. Soc., 2004, 126, 7420–7421 CrossRef CAS PubMed.
- L. W. Zilch, D. T. Kaleta, M. Kohtani, R. Krishnan and M. F. Jarrold, J. Am. Soc. Mass Spectrom., 2007, 18, 1239–1248 CrossRef CAS PubMed.
- M. Kohtani, J. E. Schneider, T. C. Jones and M. F. Jarrold, J. Am. Chem. Soc., 2004, 126, 16981–16987 CrossRef CAS PubMed.
- M. Kohtani, T. C. Jones, R. Sudha and M. F. Jarrold, J. Am. Chem. Soc., 2006, 128, 7193–7197 CrossRef CAS PubMed.
- M. F. Jarrold, Acc. Chem. Res., 1999, 32, 360–367 CrossRef CAS.
- N. P. Barrera and C. V. Robinson, Annu. Rev. Biochem., 2011, 80, 247–271 CrossRef CAS PubMed.
- M. F. Bush, Z. Hall, K. Giles, J. Hoyes, C. V. Robinson and B. T. Ruotolo, Anal. Chem., 2010, 82, 9557–9565 CrossRef CAS PubMed.
- B. T. Ruotolo, J. L. P. Benesch, A. M. Sandercock, S.-J. Hyung and C. V. Robinson, Nat. Protoc., 2008, 3, 1139–1152 CrossRef CAS PubMed.
- T. Wyttenbach and M. T. Bowers, J. Am. Soc. Mass Spectrom., 1999, 10, 9–14 CrossRef CAS.
- M. Freitas and A. Marshall, Int. J. Mass Spectrom., 1999, 182–183, 221–231 CrossRef.
- M. A. Freitas, C. L. Hendrickson, M. R. Emmett and A. G. Marshall, J. Am. Soc. Mass Spectrom., 1998, 9, 1012–1019 CrossRef CAS.
- M. A. Freitas, C. L. Hendrickson, M. R. Emmett and A. G. Marshall, Int. J. Mass Spectrom., 1999, 185–187, 565–575 CrossRef.
- N. S. Nagornova, T. R. Rizzo and O. V. Boyarkin, Angew. Chem., Int. Ed., 2013, 52, 6002–6005 CrossRef CAS PubMed.
- J. A. Stearns, C. Seaiby, O. V. Boyarkin and T. R. Rizzo, Phys. Chem. Chem. Phys., 2009, 11, 125–132 RSC.
- Z. Hall, A. Politis, M. F. Bush, L. J. Smith and C. V. Robinson, J. Am. Chem. Soc., 2012, 134, 3429–3438 CrossRef CAS PubMed.
- R. Salbo, M. F. Bush, H. Naver, I. Campuzano, C. V. Robinson, I. Pettersson, T. J. Jorgensen and K. F. Haselmann, Rapid Commun. Mass Spectrom., 2012, 26, 1181–1193 CrossRef CAS PubMed.
- M. Zhou and V. H. Wysocki, Acc. Chem. Res., 2014, 47, 1010–1018 CrossRef CAS PubMed.
- M. Zhou, C. Huang and V. H. Wysocki, Anal. Chem., 2012, 84, 6016–6023 CrossRef CAS PubMed.
- B. T. Ruotolo, J. L. P. Benesch, A. M. Sandercock, S.-J. Hyung and C. V. Robinson, Nat. Protoc., 2008, 3, 1139–1152 CrossRef CAS PubMed.
- C. Wu, W. F. Siems, G. R. Asbury and H. H. Hill Jr., Anal. Chem., 1998, 70, 4929–4938 CrossRef CAS PubMed.
- L. Konermann and D. J. Douglas, J. Am. Soc. Mass Spectrom., 1998, 9, 1248–1254 CrossRef CAS PubMed.
- H. Shi, N. A. Pierson, S. J. Valentine and D. E. Clemmer, J. Phys. Chem. B, 2012, 116, 3344–3352 CrossRef CAS PubMed.
- C. J. Hogan Jr., R. R. Ogorzalek Loo, J. A. Loo and J. F. d. l. Mora, Phys. Chem. Chem. Phys., 2010, 12, 13476–13483 RSC.
- S. H. Lomeli, I. X. Peng, S. Yin, R. R. Ogorzalek Loo and J. A. Loo, J. Am. Soc. Mass Spectrom., 2010, 21, 127 CrossRef CAS PubMed.
- H. J. Sterling, M. P. Daly, G. K. Feld, K. L. Thoren, A. F. Kintzer, B. A. Krantz and E. R. Williams, J. Am. Soc. Mass Spectrom., 2010, 21, 1762–1774 CrossRef CAS PubMed.
- H. Sterling and E. Williams, J. Am. Soc. Mass Spectrom., 2009, 20, 1933–1943 CrossRef CAS PubMed.
- Y. Zhong, L. Han and B. T. Ruotolo, Angew. Chem., Int. Ed., 2014, 53, 9209–9212 CrossRef CAS.
- E. R. Badman, S. Myung and D. E. Clemmer, J. Am. Soc. Mass Spectrom., 2005, 16, 1493–1497 CrossRef CAS PubMed.
- M. Zhou, S. Dagan and V. H. Wysocki, Angew. Chem., Int. Ed., 2012, 51, 4336–4339 CrossRef CAS PubMed.
- P. D. Schnier, D. S. Gross and E. R. Williams, J. Am. Soc. Mass Spectrom., 1995, 6, 1086–1097 CrossRef CAS PubMed.
- A. R. Dongré, J. L. Jones, Á. Somogyi and V. H. Wysocki, J. Am. Chem. Soc., 1996, 118, 8365–8374 CrossRef.
- C. Gu, G. Tsaprailis, L. Breci and V. H. Wysocki, Anal. Chem., 2000, 5804–5813 CrossRef CAS.
- K. Hermann and V. Wysocki, J. Am. Soc. Mass Spectrom., 2005, 1067–1080 CrossRef PubMed.
- O. Burlet, R. S. Orkiszewski, K. D. Ballard and S. J. Gaskell, Rapid Commun. Mass Spectrom., 1992, 6, 658–662 CrossRef CAS PubMed.
- O. Burlet, C.-Y. Yang and S. Gaskell, J. Am. Soc. Mass Spectrom., 1992, 3, 337–344 CrossRef CAS PubMed.
- K. A. Cox, S. J. Gaskell, M. Morris and A. Whiting, J. Am. Soc. Mass Spectrom., 1996, 7, 522–531 CrossRef CAS PubMed.
- S. G. Summerfield and S. J. Gaskell, Int. J. Mass Spectrom. Ion Processes, 1997, 165–166, 509–521 CrossRef.
- F. Kjeldsen, M. M. Savitski, C. M. Adams and R. A. Zubarev, Int. J. Mass Spectrom., 2006, 252, 204–212 CrossRef CAS.
- K. Breuker, H. Oh, D. M. Horn, B. A. Cerda and F. W. McLafferty, J. Am. Chem. Soc., 2002, 124, 6407–6420 CrossRef CAS PubMed.
- J. B. Shaw, W. Li, D. D. Holden, Y. Zhang, J. Griep-Raming, R. T. Fellers, B. P. Early, P. M. Thomas, N. L. Kelleher and J. S. Brodbelt, J. Am. Chem. Soc., 2013, 135, 12646–12651 CrossRef CAS PubMed.
- R. Parthasarathi, Y. He, J. P. Reilly and K. Raghavachari, J. Am. Chem. Soc., 2010, 132, 1606–1610 CrossRef CAS PubMed.
- J. C. Phillips, R. Braun, W. Wang, J. Gumbart, E. Tajkhorshid, E. Villa, C. Chipot, R. D. Skeel, L. Kale and K. Schulten, J. Comput. Chem., 2005, 26, 1781–1802 CrossRef CAS PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery, Jr., J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, N. J. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Ö. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian Inc., 09, Wallingford, CT, 2009 Search PubMed.
- Schrödinger Release 2015-1: MacroModel, version 10.7, Schrödinger, LLC, New York, NY, 2015.
- H. V. Florance, A. P. Stopford, J. M. Kalapothakis, B. J. McCullough, A. Bretherick and P. E. Barran, Analyst, 2011, 136, 3446–3452 RSC.
- M. F. Bush, I. D. G. Campuzano and C. V. Robinson, Anal. Chem., 2012, 84, 7124–7130 CrossRef CAS PubMed.
- J. C. May and J. A. McLean, Proteomics, 2015, 15, 2862–2871 CrossRef CAS PubMed.
- R. A. Jockusch, P. D. Schnier, W. D. Price, E. F. Strittmatter, P. A. Demirev and E. R. Williams, Anal. Chem., 1997, 69, 1119–1126 CrossRef CAS PubMed.
- S. L. Koeniger and D. E. Clemmer, J. Am. Soc. Mass Spectrom., 2007, 18, 322–331 CrossRef CAS PubMed.
- E. R. Badman, C. S. Hoaglund-Hyzer and D. E. Clemmer, J. Am. Soc. Mass Spectrom., 2002, 13, 719–723 CrossRef CAS PubMed.
- H. Shi, N. Atlasevich, S. I. Merenbloom and D. E. Clemmer, J. Am. Soc. Mass Spectrom., 2014, 25, 2000–2008 CrossRef CAS PubMed.
- G. Bouchoux, Mass Spectrom. Rev., 2015, 34, 493–534 CrossRef CAS PubMed.
- R. R. Hudgins, M. A. Ratner and M. F. Jarrold, J. Am. Chem. Soc., 1998, 120, 12974–12975 CrossRef CAS.
- S. E. Anderson, C. Bleiholder, E. R. Brocker, P. J. Stang and M. T. Bowers, Int. J. Mass Spectrom., 2012, 330–332, 78–84 CrossRef CAS.
- C. Bleiholder, S. Contreras and M. T. Bowers, Int. J. Mass Spectrom., 2013, 354–355, 275–280 CrossRef CAS.
- C. Bleiholder, S. Contreras, T. D. Do and M. T. Bowers, Int. J. Mass Spectrom., 2013, 345–347, 89–96 CrossRef CAS.
- C. Bleiholder, T. Wyttenbach and M. T. Bowers, Int. J. Mass Spectrom., 2011, 308, 1–10 CAS.
- O. S. Skinner, K. Breuker and F. W. McLafferty, J. Am. Soc. Mass Spectrom., 2013, 24, 807–810 CrossRef CAS PubMed.
- S. Lee, M. A. Ewing, F. M. Nachtigall, R. T. Kurulugama, S. J. Valentine and D. E. Clemmer, J. Phys. Chem. B, 2010, 114, 12406–12415 CrossRef CAS PubMed.
- H. Shi and D. E. Clemmer, J. Phys. Chem. B, 2014, 118, 3498–3506 CrossRef CAS PubMed.
- B. Y. Qin, M. C. Bewley, L. K. Creamer, E. N. Baker and G. B. Jameson, Protein Sci., 1999, 8, 75–83 CrossRef CAS PubMed.
- K. M. Oliveira, V. L. Valente-Mesquita, M. M. Botelho, L. Sawyer, S. T. Ferreira and I. Polikarpov, Eur. J. Biochem./FEBS, 2001, 268, 477–483 CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5an01819f |
| This journal is © The Royal Society of Chemistry 2016 |