
Halogen mediated voltammetric oxidation of biological thiols and disulfides
Edelmira
Valero-Ruiz
ab,
María I.
González-Sánchez
ab,
Christopher
Batchelor-McAuley
a and
Richard G.
Compton
*a
aDepartment of Chemistry, Physical and Theoretical Chemistry Laboratory, University of Oxford, South Parks Road, Oxford OX1 3QZ, UK. E-mail: Richard.compton@chem.ox.ac.uk
bDepartment of Physical Chemistry, University of Castilla-La Mancha, Campus Universitario s/n, 02071, Albacete, Spain
First published on 30th October 2015
Abstract
The electrochemical generation of the halides, bromine and iodine, in the presence of biologically relevant organosulfur is demonstrated to result in an analytically useful response. In the case of the iodide/iodine redox couple only the thiol causes an increase in the electrochemical oxidative peak current. Conversely, the formed bromine may catalytically oxidise both thiols and disulfides. Hence, the differing reactivities of the halide ions readily allow discrimination between the closely related thiol and disulphide species. For all of the organosulfur species investigated (glutathione, cysteine and homocysteine) micromolar limits of detection are attainable. In the case of the bromine mediated oxidation this sensitivity at least partially arises from the large catalytic amplification, such that, for each disulphide molecule up to ten electrons may be transferred. Ultimately this bromine oxidation results in the formation of the sulfonate species. For the iodine mediated oxidation of the thiols the oxidation proceeds no further than to the formation of the associated disulfide.
Introduction
Organosulfur in its various forms takes a pivotal role in a host of different biological systems1 and more specifically, the thiol/disulfide redox couple is found to be omnipresent in the cellular environment. Of the non-protein sulfhydryl species present glutathione (GSH) is the most abundant; in both mammalian and plant tissue glutathione concentrations are reported to range between 1 to 10 mM.2,3 In combination with its oxidized form (glutathione disulfide, GSSG) this redox couple is imperative for maintaining cellular homeostasis.4,5 Moreover, the analytical determination of the ratio of oxidized to reduced glutathione is of distinct importance for a variety of medical diagnosis.6 Of the methods available HPLC is by far the most commonly employed technique for measuring this redox ratio.7 However, HPLC is not without its detractions, not least, due to the fact that no ‘best’ protocol has been defined,8 the common requirement to derivatise thiol groups9 or the high cost of the equipment required. Consequently, there is a desire for the production of alternate analytical detection methodologies.Over the preceding decades significant effort has been placed upon finding electrochemical methods by which either the glutathione content and/or the thiol/disulfide ratio can be analytically determined.10 The vast majority of these methods utilize a quinone or quinone analog (e.g. aminophenols) as an electrochemical mediator that either results in disulfide or adduct formation.11–15 This route can be viewed, to an extent, as being biomimetic in as much that analogous reaction pathways occur in some glutathione oxidoreductases, such as the DsbB Escherichia coli plasma membrane protein.16 Other electrochemical approaches have sought to produce biosensors directly employing enzymes such as glutathione reductase and glutathione peroxidase.17,18 Away from these organic and biologically based procedures the direct oxidation of both thiols and disulfides has been investigated; however, such methods are commonly hindered by the requirement of the use of high overpotentials for analytically useful signals to be obtained.19
The reactivity of halogens towards organosulfur compounds is well documented.20 Under non-aqueous conditions bromine leads to the near total conversion of thiols to the corresponding disulfide.21 In comparison under aqueous conditions the reaction of thiols with bromine is reported to lead to the formation of the associated sulfonate.22 In comparison the lower reactivity of iodine means that even under aqueous conditions many thiols are terminally oxidized to the corresponding disulfide.23 However, further oxidation is feasible depending upon the neighboring groups present.24 Until recently the use of this halogen/organosulfur reaction for analytical purposes, where the halogen has been electrochemically formed, has been limited to a select number of articles. Here the main method of detection in conjunction with the use of chromatography, has been to record the decrease in the halogen concentration due to reaction with the associated thiol or disulfide.25,26 However, recently work by our group demonstrated that the use of electrochemically formed bromine can be used for the detection and quantification of organosulfur containing molecules contained in garlic extract.27–29 In this new methodology the presence of disulfide and disulfide derivatives (such as thiosulfinates), leads to a clear amplification of the oxidative voltammetric signal.
The present work studies the halogen mediated oxidation of both biologically relevant thiols and disulfides. Even on the voltammetric timescale the halogen mediated oxidation of thiols and disulfides is rapid enough for attainment of an analytically useful signal with micro-molar limits of detection. Moreover, the use of either bromine or iodine allows for facile discrimination between the two organosulfur moieties. It is demonstrated how the iodide/iodine redox couple is only sensitive to the presence of thiols whereas, in contrast, the bromide/bromine redox couple is able to oxidise both thiols and disulfides. Consequently, this work evidences the potential of voltammetric halogen mediated organosulfur oxidation for application to the detection, discrimination and quantification of biologically relevant organosulfur.
Experimental
All chemicals were purchased at the highest grade available and used directly without any further purification. NaClO4 (ACS Reagent, 98%) was sourced from Sigma Aldrich, UK, NaBr was sourced from May and Baker Ltd, UK and NaI was supplied by BDH chemical limited, Poole, UK, all other chemicals were sourced from Sigma Aldrich UK. All solutions were prepared with deionised water of resistivity not less than 18.2 MΩ cm at 298 K (Millipore UHQ, Vivendi, UK).All voltammetric measurements were recorded using an Autolab PGSTAT30 computer-controlled potentiostat (Metrohm, Utrecht, The Netherlands). Experiments were performed using a three-electrode set-up in a Faraday cage, with a graphite rod and a Saturated Calomel Electrode (SCE +0.244 V vs. SHE, BASi inc, Japan) as counter and reference, respectively. A platinum macro-electrode (BASi, West Lafayette, IN, USA, radius: 0.8 mm) was used as the working electrode. Renewal of the platinum surface was achieved by polishing with alumina slurries in the size sequence 1.0 μm, 0.3 μm and 0.05 μm, Buehler Ltd, USA). A beaker containing a total of ∼25 mL of electrolyte was employed and thermostated using a water bath at 25.0 ± 0.3 °C.
Results and discussion
This work first investigates the oxidation of bromide at a platinum electrode, followed by the bromine mediated oxidation of glutathione and glutathione disulphide. Second, the use of the iodide/iodine redox couple is studied; demonstrating that the lower reactivity of the formed iodine leads to a differing response, in that only the thiol oxidation is mediated on the voltammetric timescale.The oxidation of 1.0 mM bromide (0.2 M NaClO4 electrolyte) to bromine was recorded at a platinum macro-electrode (Fig. 1 – blue line), a well-defined quasi-reversible diffusional redox wave occurred at +0.95 V (mid-point potential vs. SCE). The redox process for this voltammetric peak is taken to correspond to the one-electron oxidation of the bromide ion as given by:
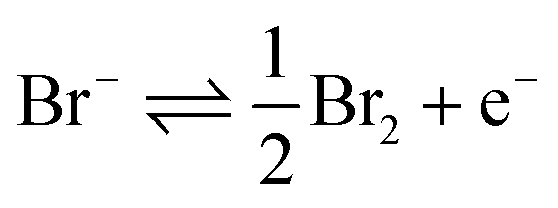 | (1) |
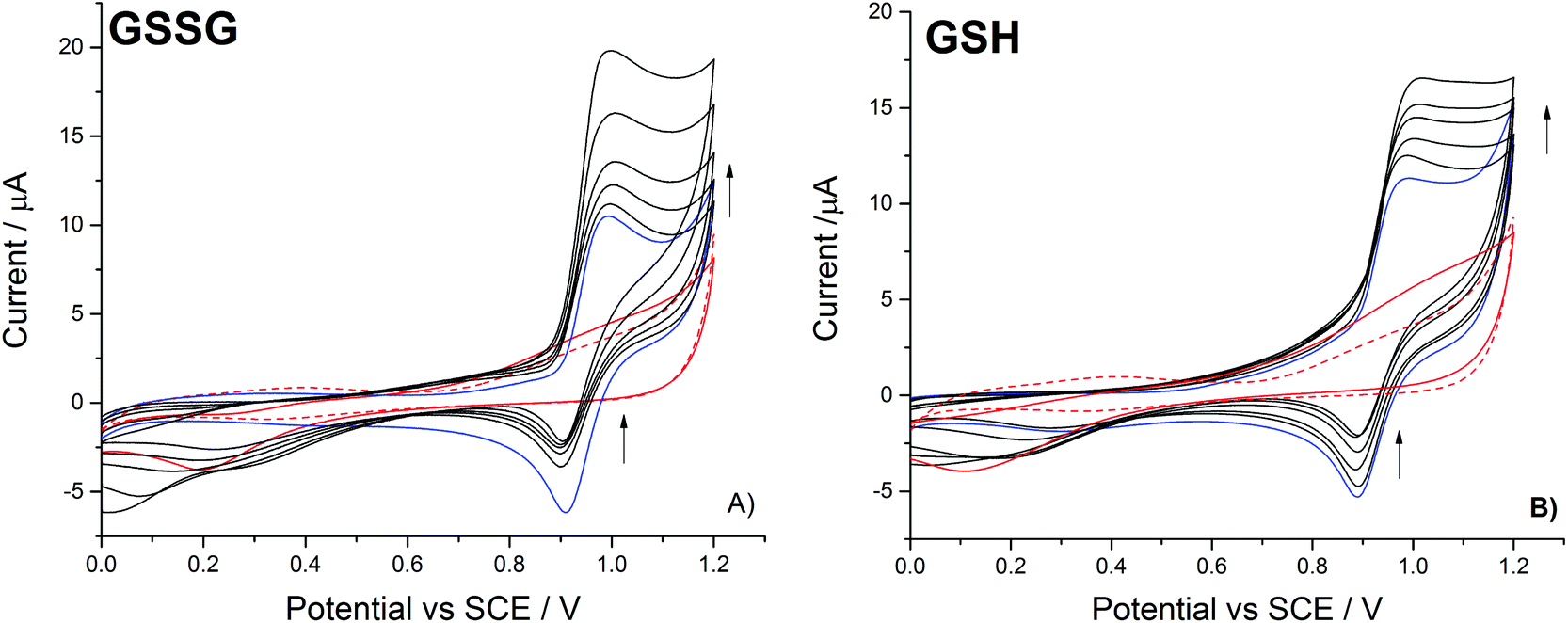 | ||
| Fig. 1 (A) The voltammetric oxidation (scan rate = 100 mV s−1) of 1.0 mM Br− in the absence (blue line) and the presence of increasing concentrations of GSSG (black lines: 49.0, 77.5, 105.4, 163.5 and 247.8 μM). Also depicted is the voltammetric response in the absence of the bromide with (150 μM, red solid line) and without (red dashed line) GSSG. (B) The voltammetric oxidation of 1 mM Br− in the absence (blue line) and the presence of increasing concentrations of GSH (black lines: 19.9, 39.7, 117.2, 164.4 and 210.7 μM). The response in the absence of the bromide with (150 μM, red solid line) and without (red dashed line) GSH is also shown. | ||
At higher bromide concentrations or in non-aqueous environments the formation of tribromide, as given by the following expression;
| Br− + Br2 ⇌ Br3− | (2) |
The partially distorted diffusional tail of the voltammetric bromide oxidation signal relates to the onset of the platinum oxide peak31 – as evidenced by the scans recorded in the absence of bromide (Fig. 1). With the addition of the glutathione disulfide and in the presence of bromide, the forward voltammetric peak current associated with the bromide oxidation is observed to increase with a concomitant decrease in the magnitude of the reductive peak (Fig. 1a). This voltammetric response is indicative of the operation of an electrocatalytic system (EC′).32 Moreover, in the absence of the bromide only a small and poorly defined increase (above +0.9 V) in the background oxidative signal is observed. Hence, under the present experimental conditions the direct oxidation of the disulphide is significantly hindered, however the presence of the bromide/bromine redox couple serves to mediate the oxidation process. Fig. 1b) shows the corresponding voltammetric response for the oxidation of bromide in the presence of increasing glutathione (thiol) concentrations, a similar catalytic response is observed. For the present case of the direct thiol oxidation the onset of background voltammetric feature occurs at a lower potential (+0.7 V); however, again no clearly defined voltammetric wave is observed. This earlier onset likely reflects the fact that the oxidation of the thiol is thermodynamically more favourable than the already partially oxidised disulphide. This can be seen from consideration of the thermodynamically calculated standard redox potentials for sulphurous compounds where the disulphide/thiol couple has a redox potential of +0.287 V (vs. SHE), where in contrast the potential for the disulphide/sulfonate couple is +0.399 V (vs. SHE).33 For the organic sulfur compounds currently under investigation their exact redox potentials will be influenced by the presence of the organic side chain, however the relative redox potentials for the thiol and disulphide species will still follow comparable trends.
Under aqueous conditions the bromination of the organosulfur may lead to a host of different products and intermediates,20 however, if the reaction goes to completion the end point for both the oxidation of disulfides and thiols will be the formation of the associated sulfonate. Fig. 2, provides the proposed overall reaction scheme for the mediated oxidation of both thiol and disulphide with bromine. The formed bromine is reduced by the organosulfur back to bromide which may be subsequently re-oxidised. Consequently, due to this electrocatalytic mechanism the magnitude of the voltammetric wave for the oxidation of bromide is sensitive to the concentration of either thiol or disulphide.
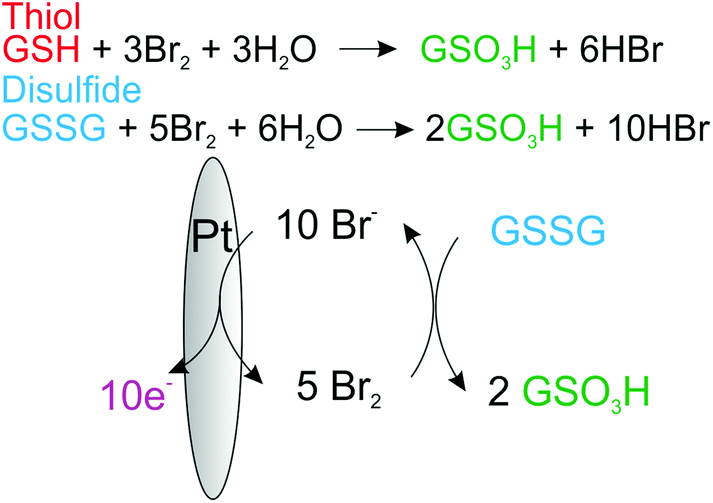 | ||
| Fig. 2 Schematic of the proposed electrocatalytic reaction mechanism for the oxidation of thiols and disulfides by the electrochemically generated bromine at a platinum electrode. | ||
Fig. 3 depicts the normalised peak current versus the concentration of disulphide (GSSG), thiol (GSH) or the amine control (glycine). The normalised peak current (Ip,norm) is given by;
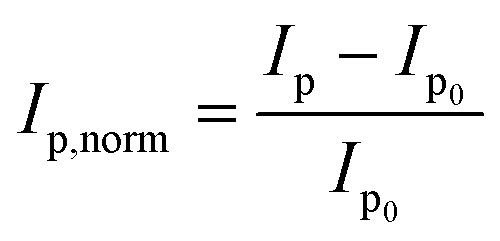 | (3) |
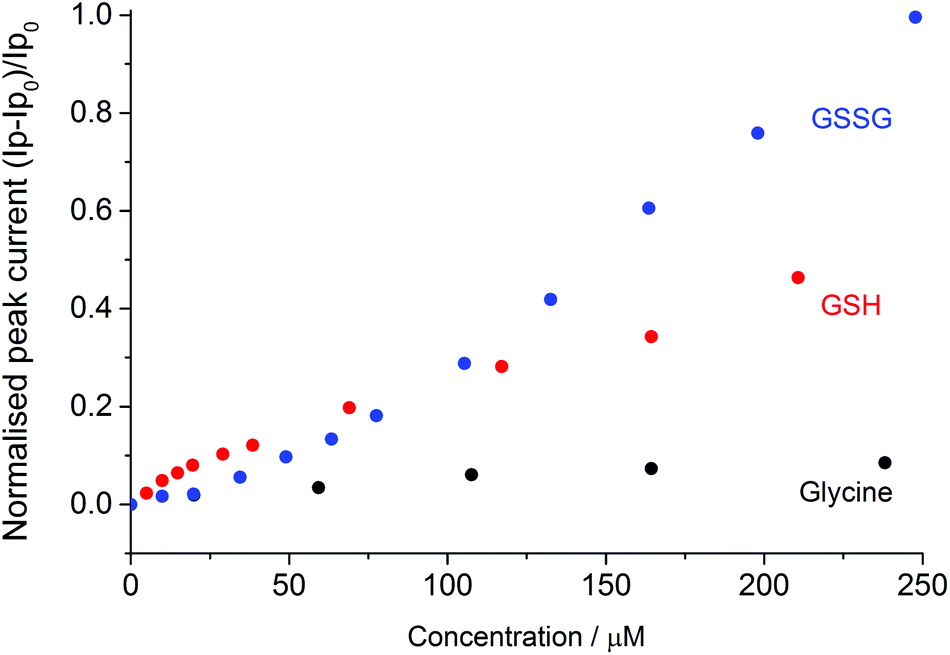 | ||
| Fig. 3 Normalized peak current versus the glycine (black), GSH (red) and GSSG (blue) concentration, where Ip0 is the oxidative peak height of the bromide in the absence of the compound. | ||
As can be seen at higher organosulfur concentrations (>100 μM), the electrocatalytic response of the bromine mediated system is more sensitive to the presence of the glutathione disulphide (GSSG) as compared to the thiol (GSH). This enhanced sensitivity likely reflects the differing reaction stoichiometries of the two species where for each disulphide molecule up to five bromines (ten electrons) may be consumed as given by the following reaction;
| GSSG + 5Br2 + 6H2O ⇌ 2GSO3H + 10HBr | (4) |
In contrast for thiols the oxidation results in the associated consumption of up to three bromine molecules (six electrons transferred) as given by;
| GSH + 3Br2 + 3H2O ⇌ GSO3H + 6HBr | (5) |
Interestingly at the lower concentrations (<100 μM) the mediated system exhibits a greater sensitivity towards the presence of the thiol, it is speculated that this result reflects the pre-adsorption of the thiol to the platinum surface. Importantly, the bromide oxidative peak is shown to be far less sensitive to the presence of the amino acid glycine. The presence of ∼250 μM of glycine results in less than a nine percent increase in the bromine oxidation peak current. Consequently, it is concluded that over the voltammetric timescale the glycine is not significantly oxidised by the electrochemically generated bromine.
Having evidenced the voltammetric sensitivity of the bromide/bromine redox couple towards the presence and concentration of both reduced and oxidised glutathione, the work now turns to study the oxidation of iodide. The oxidation of 1.0 mM iodide (0.2 M NaClO4 electrolyte) was recorded at a platinum macro-electrode (Fig. 4 – blue line), a well-defined diffusional redox wave occurred at +0.48 V (mid-point potential vs. SCE). Hence the oxidation of iodide occurs at a potential over half a Volt less positive than that found for bromide (reflecting the change in thermodynamics upon moving down the periodic table). The question of the possible formation of the isopolyhalogen ion (triiodide in this case) is more complex than was found for the oxidation of bromide. The associated formation constant is reported be 102.9 M−1 (ref. 30) and the kinetics of the complexation are reported to be close to diffusion limit.34 Moreover, the standard potentials for the oxidation of iodide to iodine and iodide to triiodide only differ by less than a millivolt.33 Note that although this triiodide formation is more favourable than the previously discussed tribromide ion, under non-aqueous conditions both of these species are far more readily formed.35 Utilising this reported formation constant of 102.9 M−1 for the triiodide ion then it can be estimated that during the course of the electrochemical oxidation of 1 mM iodide the concentration of the formed iodine will be ∼5 times greater than that of the triiodide. However, if higher iodide concentrations were used experimentally (i.e. 10 mM iodide) the dominant species formed is likely to be the triiodide ion. In light of the above discussion it should be commented that in the following section where iodine is referred to as the mediating species, it cannot be ruled out that the triiodide, although likely present in a lower concentration, is the active oxidising species.
 | ||
| Fig. 4 (A) The voltammetric oxidation of 1.0 mM iodide in the absence (blue line) and in the presence of GSSG (168 μM, black line). The response in the absence of the iodide with (150 μM, red solid line) and without (red dashed line) GSSG is also shown. (B) The voltammetric oxidation (scan rate = 100 mV s−1) of 1 mM iodide in the absence (blue line) and in the presence of increasing concentrations of GSH (black lines: 49.5, 229, 335.8, 471 and 629.4 μM). Also depicted is the voltammetric response in the absence of the iodide with (150 μM, red solid line) and without (red dashed line) GSH. Inset: Normalized peak current versus GSH concentration. | ||
Fig. 4 also depicts the voltammetric response of the iodide/iodine redox couple in the presence of both the reduced and oxidised forms of glutathione. In contrast to the results found for bromide oxidation only the thiol containing reduced glutathione leads to an increase in the magnitude of the oxidative voltammetric peak and an associated decrease in the back peak. Again this voltammetric response is indicative of the formed halogen mediating the oxidation of the thiol. At the oxidation potential for iodide neither of the organosulfurs nor glycine are oxidised on the bare electrode to any measurable amount. The change in the voltammetric response of the halogen, such that iodine is unable to oxidise the disulphide, purely represents the lower reactivity of the formed iodine as compared to bromide. Consequently, oxidation of the thiol by iodine will likely lead to the formation of the disulphide (GSSG). This ability to readily differentiate between the thiol and disulphide is not limited to the GSH/GSSG redox couple. Table 1 presents the sensitivity and corresponding limits of detection for various biologically relevant thiols and disulfides. It should be commented that in more complex media the presence of interferents may be need to be considered. Due to chloride being oxidised at significantly high potentials it is unlikely that this species will alter the sensitivity of the methodology. Moreover, the present work has been studied under non-buffered conditions, such that, the success of the technique and the insensitivity of the halide mediator couples to pH, implies that the procedure may be well suited for application to a wide range of differing analytical conditions. Hence, the use of the voltammetrically recorded halogen mediated oxidation of thiols and disulfides may provide a rapid and simple methodology by which these two closely related species may be differentiated.
| Compound | Br−/Br2 | I−/I2 | |||||
|---|---|---|---|---|---|---|---|
| Linear range (μM) | LOD (μM) | Sensitivity (μA μM−1) | Linear range (μM) | LOD (μM) | Sensitivity (μA μM−1) | ||
| Thiols | GSH | 19.9–300.7 | 22.80 | 0.019 ± 0.001 | 19.9–629.4 | 21.28 | (6.6 ± 0.1) × 10−3 |
| Cysteine | 9.9–68.1 | 9.18 | 0.056 ± 0.001 | 9.9–34.5 | 2.96 | 0.041 ± 0.002 | |
| Homocysteine | 19.8–109.9 | 17.86 | 0.025 ± 0.002 | 10.0–198.0 | 6.86 | 0.012 ± 0.001 | |
| Disulfides | GSSG | 77.5–247.7 | 7.15 | 0.040 ± 0.001 | — | — | — |
| Cystine | 19.4–81.8 | 16.80 | 0.063 ± 0.006 | — | — | — | |
| Homocystine | 19.8–193.6 | 11.52 | 0.045 ± 0.002 | — | — | — | |
| Amine | Glycine | 107.6–621.7 | 44.78 | (2.9 ± 0.1) × 10−3 | — | — | — |
Conclusions
Organosulfur is of distinct biological importance, glutathione is omnipresent in the cellular environment and the GSH/GSSG redox couple is imperative to the maintenance of homeostasis. Consequently, the quantification of the total and relative concentrations of organosulfurs present in a variety of media is of distinct analytical importance. Challengingly, for electrochemical methods the overpotentials required for the direct oxidation and reduction of these species is commonly prohibitively high. This work presents an alternate electrochemical methodology based upon the mediation of the electrochemical oxidation by the formation of low halide concentrations (cf. millimolar) from the corresponding halide ion at an electrochemical interface. This formed halide may then react with micromolar concentrations of organosulfur, leading to the oxidation of the analyte and the reformation of the halide ions. These halide ions may be then re-oxidised resulting in an electrocatalytic amplification of the oxidative signal. The increase in the voltammetric oxidative peak is found to be analytically useful, such that micromolar limits of detection for the studied biologically relevant organosulfurs (glutathione, cysteine, homocysteine) may be readily attained. Importantly, it is demonstrated that the differing reactivities of iodine and bromine facilitates discrimination between organo-thiols and -disulfides. This ability to resolve between the two species arises, as demonstrated, due to the formed iodine being insufficiently oxidizing, on the voltammetric timescale, to lead to the oxidation of disulfide and only reacts with thiols present; conversely, bromine is shown to be able to react with both thiols and disulfides.Acknowledgements
The research leading to these results has received partial funding from the European Research Council under the European Union's Seventh Framework Programme (FP/2007-2013)/ERC Grant Agreement n. [320403] and from the Ministerio de Economía y Competitividad (MINECO, Spain; BFU2013-44095-P with FEDER co-funding). EVR would like to acknowledge her financial support provided by the University of Castilla-La Mancha, which is partially supported by FEDER funds. MIGS would like to thank to the Regional Ministry of Education and Science of Castilla-La Mancha for her postdoctoral grant, co-funded by the European Social Fund (Programa Operativo FSE 2007-2013 de Castilla-La Mancha).References
- S. Oae and T. Okuyama, Organic Sulfur Chemistry: Biochemical Aspects, Taylor & Francis, 1992 Search PubMed.
- G. Wu, Y.-Z. Fang, S. Yang, J. R. Lupton and N. D. Turner, J. Nutr., 2004, 134, 489–492 CAS.
- T. Leustek and K. Saito, Plant Physiol., 1999, 120, 637–644 CrossRef CAS PubMed.
- G. Noctor, A. Mhamdi, S. Chaouch, Y. I. Han, J. Neukermans, B. Marquez-Garcia, G. Queval and C. H. Foyer, Plant, Cell Environ., 2012, 35, 454–484 CrossRef CAS PubMed.
- E. Valero, M. I. González-Sánchez, H. Maciá and F. García-Carmona, Plant Physiol., 2009, 149, 1958–1969 CrossRef CAS PubMed.
- V. I. Lushchak, J. Amino Acids, 2012, 2012 Search PubMed.
- D. Giustarini, I. Dalle-Donne, R. Colombo, A. Milzani and R. Rossi, Free Radicals Biol. Med., 2003, 35, 1365–1372 CrossRef CAS PubMed.
- D. Giustarini, I. Dalle-Donne, A. Milzani, P. Fanti and R. Rossi, Nat. Protoc., 2013, 8, 1660–1669 CrossRef CAS PubMed.
- G. Chwatko, E. Kuzniak, P. Kubalczyk, K. Borowczyk, M. Wyszczelska-Rokiel and R. Glowacki, Anal. Methods, 2014, 6, 8039–8044 RSC.
- J. C. Harfield, C. Batchelor-McAuley and R. G. Compton, Analyst, 2012, 137, 2285–2296 RSC.
- P. C. White, N. S. Lawrence, J. Davis and R. G. Compton, Electroanalysis, 2002, 14, 89–98 CrossRef CAS.
- P. T. Lee, D. Lowinsohn and R. G. Compton, Analyst, 2014, 139, 3755–3762 RSC.
- D. Lowinsohn, P. T. Lee and R. G. Compton, J. Braz. Chem. Soc., 2014, 25, 1614–1620 CAS.
- N. S. Lawrence, J. Davis and R. G. Compton, Talanta, 2001, 53, 1089–1094 CrossRef CAS PubMed.
- P. C. White, N. S. Lawrence, J. Davis and R. G. Compton, Anal. Chim. Acta, 2001, 447, 1–10 CrossRef CAS.
- K. Inaba and K. Ito, Biochim. Biophys. Acta, Mol. Cell Res., 2008, 1783, 520–529 CrossRef CAS PubMed.
- D. Bhattacharyay, K. Dutta, S. Banerjee, A. P. F. Turner and P. Sarkar, Electroanalysis, 2008, 20, 1947–1952 CrossRef CAS.
- L. Rover, L. T. Kubota and N. F. Höehr, Clin. Chim. Acta, 2001, 308, 55–67 CrossRef CAS.
- C. Terashima, T. N. Rao, B. V. Sarada, Y. Kubota and A. Fujishima, Anal. Chem., 2003, 75, 1564–1572 CrossRef CAS PubMed.
- S. Oae and J. Doi, Organic Sulfur Chemistry: Structure and Mechanism, Taylor & Francis, 1991 Search PubMed.
- X. Wu, R. D. Rieke and L. Zhu, Synth. Commun., 1996, 26, 191–196 CrossRef CAS.
- H. A. Young, J. Am. Chem. Soc., 1937, 59, 811–812 CrossRef CAS.
- J. P. Danehy and M. Y. Oester, J. Org. Chem., 1967, 32, 1491–1495 CrossRef CAS.
- J. T. Doi and W. K. Musker, J. Am. Chem. Soc., 1984, 106, 1887–1888 CrossRef CAS.
- L. A. Holland and S. M. Lunte, Anal. Chem., 1999, 71, 407–412 CrossRef CAS PubMed.
- X. Huang and W. Kok Th, J. Liq. Chromatogr., 1991, 14, 2207–2221 CrossRef CAS.
- B. C. M. Martindale, L. Aldous, N. V. Rees and R. G. Compton, Analyst, 2011, 136, 128–133 RSC.
- E. M. Hall, C. Batchelor-McAuley, K. Tschulik and R. G. Compton, 2015, submitted.
- R. G. Compton, N. V. Rees, L. Aldous and B. C. M. Martindale, Electrochemical detection method, WO2012052755A1, 2012 Search PubMed.
- L. G. Sillén, A. E. Martell and J. Bjerrum, Stability constants of metal-ion complexes, Chemical Society London, 1964, vol. 17 Search PubMed.
- B. E. Conway, Y. Phillips and S. Y. Qian, J. Chem. Soc., Faraday Trans., 1995, 91, 283–293 RSC.
- J. M. Savéant, Elements of Molecular and Biomolecular Electrochemistry: An Electrochemical Approach to Electron Transfer Chemistry, Wiley, 2006 Search PubMed.
- A. J. Bard, R. Parsons and J. Jordan, Standard Potentials in Aqueous Solution, Taylor & Francis, 1985 Search PubMed.
- D. H. Turner, G. W. Flynn, N. Sutin and J. V. Beitz, J. Am. Chem. Soc., 1972, 94, 1554–1559 CrossRef CAS.
- I. V. Nelson and R. T. Iwamoto, J. Electroanal. Chem., 1964, 7, 218–221 CAS.
| This journal is © The Royal Society of Chemistry 2016 |