
A new type of biocompatible fluorescent probe AFN for fixed and live cell imaging of intracellular lipid droplets†
Ashutosh
Sharma
a,
Shahida
Umar
a,
Parmita
Kar
b,
Kavita
Singh
c,
Monika
Sachdev
b and
Atul
Goel
*ad
aFluorescent Lab, Medicinal and Process Chemistry Division, CSIR-Central Drug Research Institute, Lucknow 226031, India. E-mail: atul_goel@cdri.res.in; Fax: +91-522-2771941; Tel: +91-522-2772450
bDivision of Endocrinology, CSIR-Central Drug Research Institute, Lucknow 226031, India
cElectron Microscopy Unit, CSIR-Central Drug Research Institute, Lucknow 226031, India
dAcademy of Scientific and Innovative Research, New Delhi 110001, India
First published on 21st October 2015
Abstract
We discovered a new class of nontoxic, highly fluorogenic and biocompatible probe AFN for selective staining of intracellular Lipid Droplets (LDs) in both fixed and live human cervical cancer cells (HeLa) and 3T3-L1 pre-adipocytes without any background artifacts. The salient features of the probe are its visible excitation maximum, aqueous compatibility, selectivity and remarkable stability (for more than a week) in live cells, even better than commercially available Nile red.
Introduction
Lipid Droplets (LDs) are acknowledged as dynamic cytoplasmic organelles composed of a hydrophobic core of neutral lipids (primarily sterol esters and triacylglycerols with their relative amount varying between cell types) surrounded by a phospholipid monolayer and specific proteins.1,2 As opposed to being inert bodies storing excess cellular fat, cellular lipids perform a host of functions such as membrane trafficking, fusion, endocytosis, and signal transduction, and are a source of energy in crisis. Almost all cells have the ability to form LDs3 and understanding lipid biology has assumed enormous importance since excessive lipid storage in LDs has been associated with the etiology of several metabolic disorders such as obesity, diabetes and atherosclerosis.4,5 Therefore the discovery of a new selective and stable fluorescent probe as a tool to visualize, quantify and/or study the molecular dynamics of LDs is essentially required to enhance our understanding in the area of lipid cell biology.6Fluorescence techniques are powerful tools for studying LDs in living cells and tissues due to their advantages such as high sensitivity and selectivity.7 A limited number of LD specific fluorescent dyes have been reported,8–12 in which Nile red8 and BODIPY dyes are commonly employed.9 Nile red stains most intracellular structures,10 thus visualizing poor discernment of LDs from other cellular components and also suffers from low sensitivity because of the high background noise. BODIPY dyes, on the other hand, usually have a small Stokes shift and thus induce nonradiative energy transfer causing fluorescence quenching of the excited chromophore and interference from scattered light ultimately leading to weak fluorescence and background artifacts.11 To improve their selectivity and staining stability for LDs, significant efforts have been made to identify a new type of dyes, however Nile red is still the choice of many researchers.10–12 Our ongoing efforts in this direction led to the identification of the fluoranthene dye FLUN-550 for selective staining of intracellular LDs, however the dye exhibited an excitation maximum in the ultraviolet region leading to photostability only for a few hours.12d We envisaged that D/A fluorene and related aromatic scaffolds exhibit remarkable photophysical and optical properties13,14 but their aqueous incompatibility and weak/no fluorescence in water snag their biological applications.
To overcome these issues and to make them biocompatible while retaining their useful photophysical characteristics, we tailored unique donor–acceptor based azafluorene and azafluorenone by replacing one of the phenyl rings of the fluorene scaffold with its bioisostere pyridine moiety to induce effective charge transfer with improved aqueous solubility (Fig. 1). The incorporation of donor–acceptor functionalities to modulate the electronic properties of diverse skeletons has been quite successful.15–17 Henceforth, in this paper we report the design and synthesis of a new type of biocompatible fluorescent probes with the excitation maximum in the visible region and their application in selective and stable staining of cytoplasmic lipid droplets in HeLa and 3T3-L1 pre-adipocyte cells without any background noise.
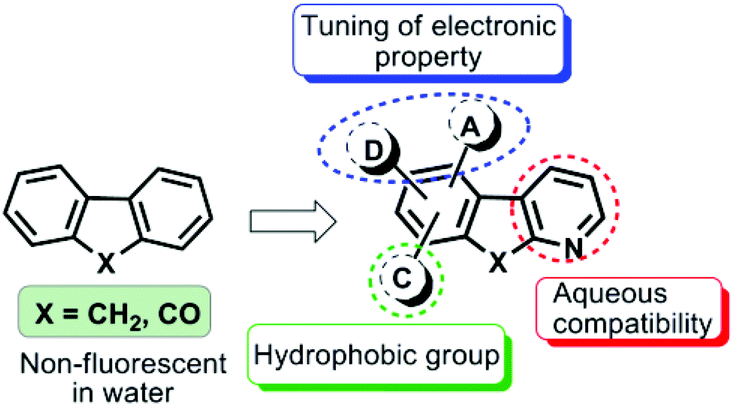 | ||
| Fig. 1 Design of new donor/acceptor-based azafluorenes and azafluorenone as fluorescent bioprobes. | ||
Results and discussion
There is a paucity of literature reports for the synthesis of azafluorenes and azafluorenones. Few synthetic methodologies are available for these compounds, which include three-component [5 + 1] heterocyclization of 3-indanone with aryl glyoxal,18a cross-coupling reaction between pyridylboronic acid and ethyl iodobenzoate followed by intramolecular cyclization,18b,c photocyclisation of 2-benzoylpyridine,18d one-pot DoM–boronation–Suzuki cross coupling of azabiaryls,18e and by generating a diazopyridine compound followed by photolyzation in poor yield.18f Recently Baran et al.18g developed an efficient method to prepare a mixture of azafluorenones through Pschorr-type cyclization of organotrifluoroborates in the presence of silver nitrate. In order to prepare designed donor–acceptor functionalized azafluorene (AF) and azafluorenone (AFN), we developed here a concise and highly rapid synthetic route as depicted in Scheme 1. The key intermediate 6-phenyl-2-oxo-4-methylsulfanyl-2H-pyran-3-carbonitrile (3) was prepared from the easily accessible precursor α-oxo-ketene-S,S-acetal (1) and acetophenone (2) in good yield. Synthesis of azafluorenes with an amine donor and a nitrile acceptor group was achieved by preparing 6-phenyl-2-oxo-4-piperidin-1-yl-2H-pyran-3-carbonitrile (4) from 3 and then reacting with 6,7-dihydro-5H-cyclopenta[b]pyridin-5-one19 (7) in the presence of sodium hydride in dry THF, which furnished 8-phenyl-6-(piperidin-1-yl)-9H-indeno[2,1-b]pyridine-5-carbonitrile (8) in 68% yield. The azafluorene 8 was oxidized to azafluorenone 9 in the presence of potassium hydroxide in air at room temperature. Further reduction of azafluorenone 9 with NaBH4 furnished 9-hydroxy-azafluorene 10 in 73% yield (Scheme 1). | ||
| Scheme 1 Synthesis of azafluorene, azafluoren-9-one and 9-hydroxyazafluorene. | ||
The photophysical properties of the synthesized compounds 8–10 in phosphate buffered saline (PBS, pH = 7.4) were examined by using UV-vis absorption, excitation and fluorescence spectra (Fig. 2, S1,† and Table S1, ESI†). Azafluorenes 8 and 10 showed absorption maxima at 385 nm and 352 nm respectively and fluorescence maxima at 478 nm and 465 nm respectively. To our delight, azafluorenone 9 exhibited an absorption maximum at 430 nm and a fluorescence maximum at 575 nm with a Stokes shift of 145 nm (Fig. 2a). The quantum yields of the azafluorenes 8, 10 and azafluorenone 9 (AFN) were found in the range of 1.4–2.6% (in PBS) and 17–31% (in DMSO, Table S1, ESI†).
 | ||
| Fig. 2 (a) Absorption and emission spectra of 8–10 in PBS (pH = 7.4). (b) Emission spectrum of AFN in solvents of varying polarity. | ||
We next investigated the solvatochromic behaviour of compounds 8–10 in a series of solvents with varying polarity index to examine the effect on the excited state. Azafluorenes 8 and 10 did not show solvatochromic properties while azafluorenone 9 (AFN) exhibited large solvatochromic shifts with increasing polarity from cyclohexane (λPL,max = 510 nm) to dioxane (λPL,max = 535 nm) to DCM (λPL,max = 555 nm) to ACN (λPL,max = 570 nm) to DMSO (λPL,max = 592 nm) as shown in Fig. 2b. This positive solvatochromism is mostly perceived when the dipole moment of a dye in the excited state is greater than that in the ground state during a photo-induced electronic transition, which implicates the intramolecular charge transfer (ICT) characteristic of the AFN. We observed that AFN showed high emission intensity and good fluorescence quantum yield in a nonpolar solvent like cyclohexane in comparison with a polar aprotic solvent like DMSO (Fig. S1, ESI†).
To gain an insight into the electronic properties of AFN, time-dependent density functional theory (TD-DFT) calculations were employed using a B3LYP/6-311++G(d,p) method with a Gaussian 09 package.20 Molecular orbitals in the ground state for AFN are presented in Fig. 3. TD-DFT calculations showed two strong transitions at 349 and 447 nm with an oscillator strength of f = 0.1007 and 0.0906 respectively, which correspond to HOMO−1 → LUMO+1 and HOMO → LUMO respectively. The transition band at a higher wavelength may be assigned to an ICT band due to charge transfer from the piperidine donor moiety (HOMO) to the azafluorenone acceptor moiety (LUMO). The energies of the HOMO and LUMO levels and the band gap of AFN were found to be −5.71, −3.56 and 2.15 eV respectively.
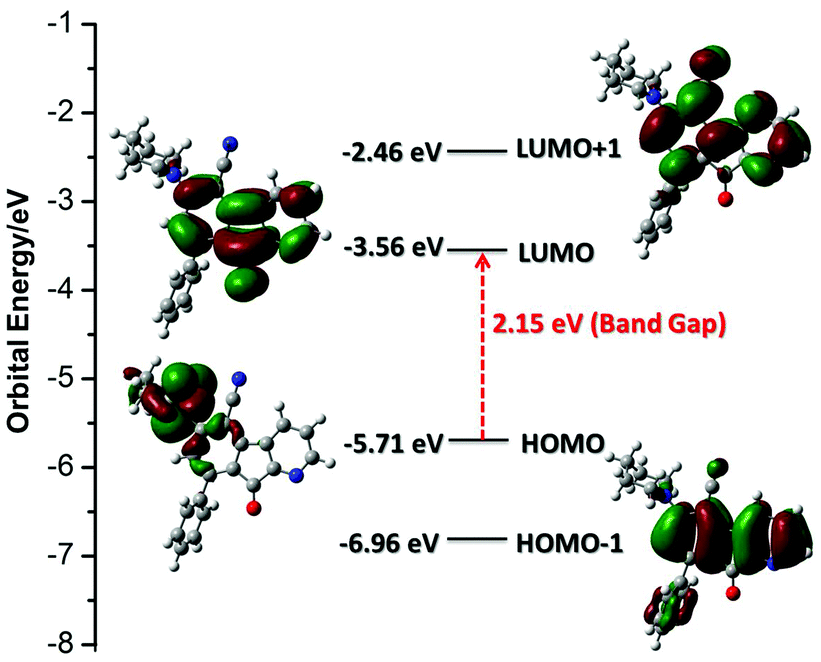 | ||
| Fig. 3 Computed molecular orbital HOMO, LUMO, HOMO−1 and LUMO+1 for AFN. | ||
Owing to their interesting photophysical properties in water, we explored the application of these compounds (8–10) as fluorescent probes in live and fixed cell imaging. These compounds were systematically assessed for their capability to infuse and stain the intracellular space in HeLa cells using confocal microscopy. For this purpose, fixed HeLa cells were permeabilised and incubated with different concentrations (0.1, 0.25, 0.5 and 1 μM) under growth conditions. The excess dye was washed away with phosphate buffered saline. Confocal imaging results revealed that only azafluorenone (AFN) showed permeability and the staining in the cytosol in a specific pattern (Fig. 4A–C). Experimentation with various commercially available organelle-specific dyes revealed that the spherical objects in the cytosol with distinctly bright fluorescence were lipid droplets (LDs), which showed strong co-localization with Nile red, a known commercial LD marker (Fig. 4D–F). In order to reconfirm the LD specific staining by AFN, we performed experiments on fixed 3T3-L1 pre-adipocytes (generally enriched in LDs) and the results are shown in Fig. 4G–I. Interestingly the staining pattern implicated that the dye AFN selectively stained LDs having co-localization with Nile red (Fig. 4J–L). The overall staining pattern of AFN in fixed HeLa cells and 3T3-L1 pre-adipocytes confirmed that AFN is a fluorescent probe for selective staining of LDs without any background noise and is better than Nile red, which showed non-specific staining in the background.21 We next examined the toxicity of AFN to check its biocompatibility. The cytotoxicity of the dye was assessed using the 3-(4,5-dimethyl-2-thiazolyl)-2,5-diphenyltetrazolium bromide (MTT) assay. AFN showed 99% cell viability in both HeLa and 3T3-L1 up to 10 μM concentration (Fig. S2, ESI†). The excellent biocompatibility of AFN motivated us to further investigate its application in live cell imaging, which is important in the perspective of identification of various cellular processes.
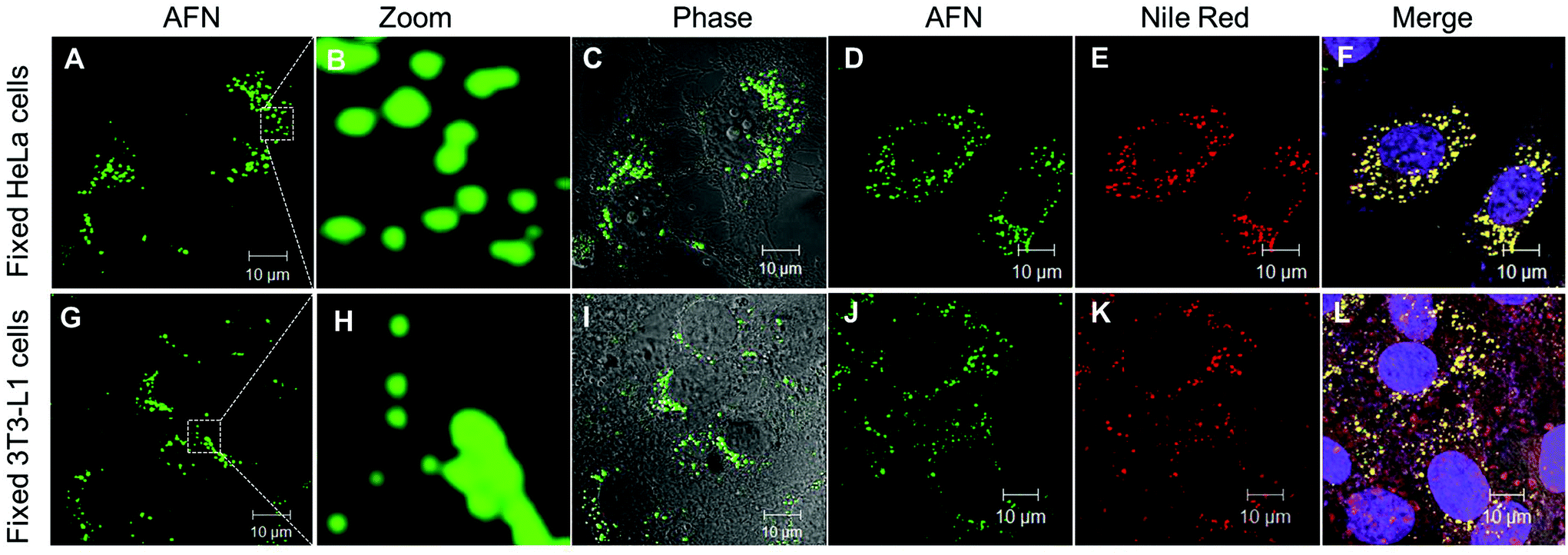 | ||
| Fig. 4 Confocal fluorescence microscopy images: in fixed HeLa cells (A−C) specific localization of LDs with AFN (0.5 μM) alone; (D−F) co-localization of AFN (green channel, λex 405/ λem 505-570) with Nile red (red channel, λex 561/ λem 575) and To-Pro (λex 633/ λem 650). In fixed 3T3-L1 pre-adipocytes (G–I) specific localization of LDs with AFN (0.5 μM) alone; (J–L) co-localization of AFN with Nile red and To-Pro. | ||
Now further experiments were performed in live HeLa cells and 3T3-L1 pre-adipocytes as described in the ESI† and confocal images are shown in Fig. 5. Interestingly these results revealed that AFN is permeable in both these cell lines and is highly selective towards staining LDs present in the cytoplasmic region. The specific staining of LDs with emission in the green region (505–570 nm) was in agreement with good quantum yields observed in hexane. Further dual staining experiments with Nile red clearly showed that compound AFN is highly selective to LDs compared to Nile red (Fig. 5D–F and 5J–L). To the best of our knowledge there has been no literature report available so far to substantiate the application of azafluorenones as organelle specific fluorescent bioimaging probes.
 | ||
| Fig. 5 (A–C) Specific localization of LDs with AFN (0.5 μM) in live HeLa cells through confocal microscopy using the laser line of 405 nm. (D–F) Co-localisation of AFN (green channel, 405 laser/505-570 BP) with Nile red (red channel, 561 laser/575 LP) in live HeLa cells. (G–I) Specific localization of LDs with AFN (0.5 μM) in live 3T3-L1 pre-adipocyte. (J–L) Co-localisation of AFN (green channel, 405 laser/505-570 BP) with Nile red (red channel, 561 laser/575 LP) in live 3T3-L1. | ||
The stability of a fluorescent probe is an important parameter in cell imaging experiments particularly under live cell conditions, which allows researchers to get more time for analysing the test samples. Interestingly AFN showed longer staining (up to 7 days of studies) compared to Nile red. Cells co-stained with AFN and Nile red showed quenched Nile red staining whereas the AFN staining was still quite promising. These results implicated a unique property of the AFN dye, which proved to be a more stable probe for LDs compared to the commercially available Nile red (Fig. 6).
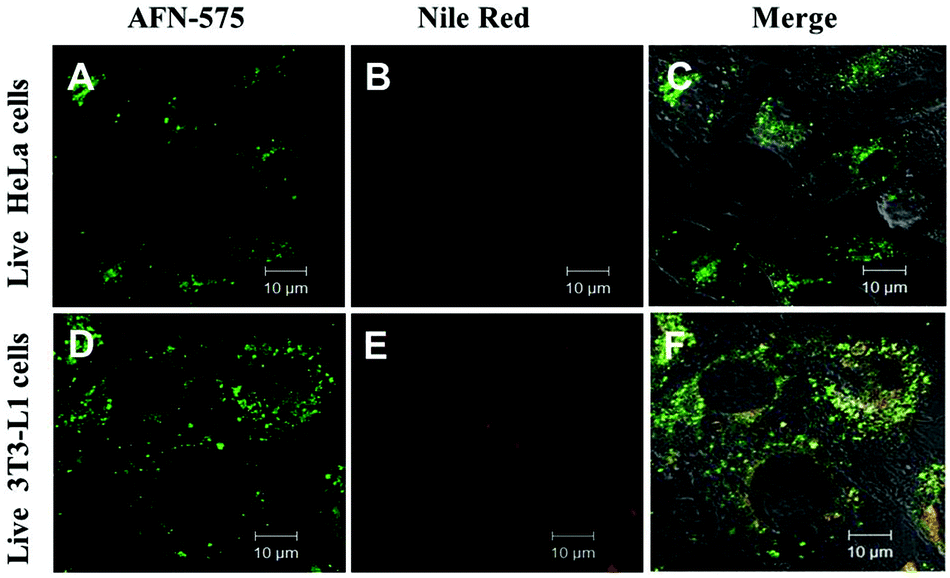 | ||
| Fig. 6 Stability of AFN fluorescence in live cells, after a week through confocal microscopy after staining, the slides were stored at 4 °C for a week: image of live HeLa cells incubated with (A) AFN (0.5 μM); (B) Nile red (0.5 μM); (C) merge of A and B; image of live 3T3-L1 cells incubated with (D) AFN (0.5 μM); (E) Nile red (0.5 μM); (F) merge of D and E. | ||
Experimental
Chemicals
DMEM media (Sigma), FBS (Gibco), DAPI (Sigma), formaldehyde (Sigma), and antibiotic antimycotic (Gibco) solution. The stock solutions of Nile red, and AFN were prepared in analytical grade DMSO and subsequent dilutions were made using Triple Distilled Water (TDW) and PBS. The final concentration of DMSO used in the media for cell studies was merely 0.01% v/v. Assays were performed in triplicate.Cell culture of HeLa and 3T3-L1
The cervical cancer cell line of HeLa and the pre-adipocyte cell line of 3T3-L1 were cultured in Dulbecco's Modified Eagles Medium (DMEM) with phenol red, L-glutamine and 4.8 g L−1D-glucose supplemented with 10% heat inactivated FBS (Gibco) and penicillin–streptomycin (Sigma). These cells were monitored thoroughly for their density and morphology at 37 °C in a CO2 incubator. Once the cells were 70% confluent, they were trypsinized and seeded (∼105) onto a coverslip and incubated overnight in the CO2 incubator. Next day morning, these cells were either fixed with 4% formaldehyde for 20 minutes or used for the treatment of AFN; at this step fixed cells can be stored at 4 °C.Staining of AFN in fixed HeLa and 3T3-L1 cells
Fixed HeLa and 3T3-L1 cells were washed with PBS and subjected to permeabilization buffer (0.1% Tween 20, 0.05% NP-40 in PBS) for 10 min. Now these cells were washed with PBS and incubated with either 500 nM of compound AFN or 500 nM of Nile red or both and incubated in a rocker overnight at 4 °C. After the incubation, the coverslips were washed with PBST thrice and once with PBS. The cells were then incubated with the nuclear stain To-Pro (1 μM) for 25 minutes. After the nuclear staining, washing steps were performed again with PBST thrice and PBS twice. Now the coverslips having the stained adhered cells were finally mounted up-side down with Prolong gold antifade reagent (Invitrogen) on glass slides. Images were acquired on a confocal microscope with a 63×Plan Apochromat Oil Phase II 1.4 objective. Lasers used were Diode 405 nm for the compound AFN, DPSS 561 nm for Nile red and Ar/ML 633 nm for the nuclear stain To-Pro. Z-Stacking of images was also done.Staining of AFN in live HeLa and 3T3-L1 cells
HeLa and 3T3-L1 cells were seeded on coverslips and incubated overnight at 37 °C in a CO2 incubator. Next day morning these live cells were treated with either 500 nM of compound AFN (dissolved AFN in DMSO, the stock was made with 100 μM concentration and the final concentration of DMSO in the media was merely 0.01% v/v) or 500 nM of Nile red or both, and incubated further for 4 hours in the CO2 incubator. After that these coverslips were washed with 10% FBS (Gibco) in PBST thrice and twice with PBS. After the staining, these cells were fixed now with 4% formaldehyde and mounted up-side down with Prolong gold antifade reagent (Invitrogen) on glass slides. The images were acquired on a confocal microscope with a 63×Plan Apochromat Oil Phase II 1.4 objective. Lasers used were Diode 405 nm for the compound AFN and DPSS 561 nm for Nile red.Cell viability assay after the treatment of AFN through MTT
Yellow MTT [3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl-tetrazolium bromide] (Sigma), a tetrazolium compound is reduced to purple formazan in the mitochondria of living cells. The absorbance of this colored solution can be quantified at a certain wavelength (usually between 500 and 600 nm) through a spectrophotometer. Intensity of purple formazan produced by cells treated with an agent is compared with the amount of formazan produced by untreated control cells, if the effectiveness of the agent is causing death of cells can be deduced, through the production of a dose–response curve. To check the cytotoxicity of the compound AFN, the MTT assay was performed in triplicate on 96 well viable cells treated with AFN in the range of 50 nM to 10 μM. The cells were trypsinized and counted in a haemocytometer to seed about 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 cells per well and incubated overnight in a CO2 incubator. Next day morning, these cells were treated with the compound AFN and incubated further for 24 h in the CO2 incubator. Now 20 μl of 5 mg ml−1 MTT was added to each well and the plate was incubated for 4 hours at 37 °C in a culture hood. After that the media were carefully removed and the reaction was stopped using 200 μl MTT solvent. The plate was covered and agitated on an orbital shaker for 15 minutes; then the absorbance was read at 570 nm. The Optical Density (O.D.) of the MTT containing wells was used to obtain the percent viability of the cells in the presence of various concentrations of the A261 dye to access the toxic effect of AFN. To calculate the percent viability of the cells, the O.D. for 0 nM of AFN was considered as 100% viability and the corresponding values for other AFN concentrations were calculated.
000 cells per well and incubated overnight in a CO2 incubator. Next day morning, these cells were treated with the compound AFN and incubated further for 24 h in the CO2 incubator. Now 20 μl of 5 mg ml−1 MTT was added to each well and the plate was incubated for 4 hours at 37 °C in a culture hood. After that the media were carefully removed and the reaction was stopped using 200 μl MTT solvent. The plate was covered and agitated on an orbital shaker for 15 minutes; then the absorbance was read at 570 nm. The Optical Density (O.D.) of the MTT containing wells was used to obtain the percent viability of the cells in the presence of various concentrations of the A261 dye to access the toxic effect of AFN. To calculate the percent viability of the cells, the O.D. for 0 nM of AFN was considered as 100% viability and the corresponding values for other AFN concentrations were calculated.
Instrumentation
1H and 13C NMR spectra were obtained at 300 MHz and 400 MHz respectively. CDCl3 and DMSO-d6 were taken as solvents. Chemical shifts are reported in parts per million shift (δ-value) from Me4Si (δ 0 ppm for 1H) or based on the middle peak of the solvent (CDCl3) (δ 77.00 ppm for 13C NMR) as an internal standard. The signal patterns are indicated as s, singlet; brs, broad singlet; d, doublet; t, triplet; m, multiplet. Coupling constants (J) are given in hertz. Infrared (IR) spectra were recorded using KBr discs and reported in wavenumbers (cm−1). The ESI-MS were recorded on a MICROMASS Quadro-II LCMS system. The HRMS spectra were recorded as ESI-HRMS on a mass analyzer system. All the reactions were monitored by TLC and visualization was done with UV light (254 nm).General procedure for the synthesis of 3 and 4
A mixture of methyl 2-cyano-3,3-dimethylsulfanylacrylate 1 (10 mmol), acetophenone 2 (11 mmol) and powdered KOH (12 mmol) in DMSO (50 mL) was stirred at room temperature for 14 h. After completion, the reaction mixture was poured into ice water with constant stirring. The precipitate thus obtained was filtered and purified on a silica gel column using chloroform as the eluent to yield 6-aryl-2-oxo-4-methylsulfanyl-2H-pyran-3-carbonitrile 3. Compound 3 (1 mmol) was refluxed in methanol with a secondary amine (piperidine, 1.2 mmol) for 6 h, the reaction mixture was cooled to room temperature and the solid obtained was filtered to furnish 6-aryl-2-oxo-4-piperidin-1-yl-2H-pyran-3-carbonitrile 4 in good yield.22:9, v/v); mp (ethyl acetate/n-hexane) 166–168 °C; MS (ESI) 352 [M + H]+; IR (KBr) ν = 2216 (CN) cm−1; 1H NMR (300 MHz, CDCl3): δ = 8.76–8.83 (m, 1H), 8.54–8.59 (m, 1H), 7.45–7.54 (m, 5H), 7.34–7.44 (m, 1H), 7.01 (s, 1H), 3.98 (s, 2H), 3.24 (t, J = 5.0 Hz, 4H), 1.83–1.95 (m, 4H), 1.62–1.69 (m, 2H); 13C NMR (50 MHz, CDCl3): δ = 165.3, 157.6, 148.9, 143.9, 142.6, 139.7, 133.0, 132.7, 129.8, 128.8, 128.4, 128.3, 122.0, 118.5, 117.6, 98.5, 53.8, 37.8, 26.2, 24.1 ppm; HRMS calculated for C24H22N3 [M+ + H] 352.1814, found: 352.1805.
:9, v/v); mp (ethyl acetate/n-hexane) 172–174 °C; MS (ESI) 366 [M + H]+; IR (KBr) ν = 2208 (CN), 1718 (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O) cm−1; 1H NMR (300 MHz, CDCl3): δ = 8.62–8.70 (m, 2H), 7.43–7.55 (m, 5H), 7.37–7.46 (m, 1H), 6.71 (s, 1H), 3.46 (t, J = 5.2 Hz, 4H), 1.79–1.85 (m, 4H), 1.68–1.75 (m, 2H); 13C NMR (100 MHz, CDCl3): δ = 187.5, 159.8, 153.5, 151.4, 148.5, 147.3, 136.4, 135.9, 129.5, 129.2, 128.9, 128.0, 126.6, 120.2, 119.8, 116.9, 96.0, 52.5, 25.8, 23.8 ppm; HRMS calculated for C24H20N3O [M+ + H] 366.1606, found: 366.1579.
:9, v/v); mp (ethyl acetate/n-hexane) 186–188 °C; MS (ESI) 368 [M + H]+; IR (KBr) ν = 2209 (CN), 3437 (OH) cm−1; 1H NMR (300 MHz, CDCl3): δ = 8.67 (d, J = 7.7 Hz, 1H), 8.36–8.42 (m, 1H), 7.60–7.66 (m, 2H), 7.46–7.56 (m, 3H), 7.32–7.39 (m, 1H), 6.92 (s, 1H), 5.75 (s, 1H), 3.92 (brs, 1H), 3.20–3.30 (m, 4H), 1.78–1.88 (m, 4H), 1.59–1.69 (m, 2H) ppm; 13C NMR (100 MHz, CDCl3): δ = 153.6, 151.5, 148.9, 147.5, 145.0, 136.5, 129.6, 129.2, 128.9, 128.7, 128.4, 128.1, 126.7, 123.9, 120.3, 119.8, 98.4, 52.6, 25.9, 23.9 ppm; HRMS calculated for C24H22N3O [M+ + H] 368.1763, found: 368.1755.
O) cm−1; 1H NMR (300 MHz, CDCl3): δ = 8.62–8.70 (m, 2H), 7.43–7.55 (m, 5H), 7.37–7.46 (m, 1H), 6.71 (s, 1H), 3.46 (t, J = 5.2 Hz, 4H), 1.79–1.85 (m, 4H), 1.68–1.75 (m, 2H); 13C NMR (100 MHz, CDCl3): δ = 187.5, 159.8, 153.5, 151.4, 148.5, 147.3, 136.4, 135.9, 129.5, 129.2, 128.9, 128.0, 126.6, 120.2, 119.8, 116.9, 96.0, 52.5, 25.8, 23.8 ppm; HRMS calculated for C24H20N3O [M+ + H] 366.1606, found: 366.1579.
:9, v/v); mp (ethyl acetate/n-hexane) 186–188 °C; MS (ESI) 368 [M + H]+; IR (KBr) ν = 2209 (CN), 3437 (OH) cm−1; 1H NMR (300 MHz, CDCl3): δ = 8.67 (d, J = 7.7 Hz, 1H), 8.36–8.42 (m, 1H), 7.60–7.66 (m, 2H), 7.46–7.56 (m, 3H), 7.32–7.39 (m, 1H), 6.92 (s, 1H), 5.75 (s, 1H), 3.92 (brs, 1H), 3.20–3.30 (m, 4H), 1.78–1.88 (m, 4H), 1.59–1.69 (m, 2H) ppm; 13C NMR (100 MHz, CDCl3): δ = 153.6, 151.5, 148.9, 147.5, 145.0, 136.5, 129.6, 129.2, 128.9, 128.7, 128.4, 128.1, 126.7, 123.9, 120.3, 119.8, 98.4, 52.6, 25.9, 23.9 ppm; HRMS calculated for C24H22N3O [M+ + H] 368.1763, found: 368.1755.
Conclusion
In conclusion we have designed and developed an efficient methodology for the synthesis of donor–acceptor appended azafluorenes and azafluorenone using easily accessible precursors. We also discovered a new type of fluorescent probe azafluorenone AFN, which is a nontoxic, highly selective and stable dye for staining LDs in different cell lines (fixed/live HeLa cells and 3T3-L1 pre-adipocyte). This new LD-specific biocompatible fluorescent probe AFN with a visible excitation and distinct emission band has opened new avenues for analysing LDs in cancer containing tissue samples and real time monitoring of LD functions in lipid cell biology. Current work in this direction is in progress.Acknowledgements
The work was supported by the CSIR, New Delhi under Network project BSC0102 and BSC0114. The communication reference number for the CSIR-CDRI is 9094.Notes and references
- (a) M. R. Wenk and P. D. Camilli, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 8262–8269 CrossRef CAS PubMed; (b) J. C. Fernandez-Checa, Biochem. Biophys. Res. Commun., 2003, 304, 471–479 CrossRef CAS PubMed.
- T. C. Walther and R. V. Farese, Annu. Rev. Biochem., 2012, 81, 687–714 CrossRef CAS PubMed.
- C. W. Roberts, R. M. Leod, D. W. Rice, M. Ginger, M. L. Chance and L. J. Goad, Mol. Biochem. Parasitol., 2003, 126, 129–142 CrossRef CAS PubMed.
- S. Calandra, P. Tarugi, H. E. Speedy, A. F. Dean, S. Bertolini and C. C. Shoulders, J. Lipid Res., 2011, 52, 1885–1962 CrossRef CAS PubMed.
- B. M. Spiegelman and J. S. Flier, Cell, 2001, 104, 531–543 CrossRef CAS PubMed.
- (a) A. B. Neef and C. Schultz, Angew. Chem., Int. Ed., 2009, 48, 1498–1500 CrossRef CAS PubMed; (b) N. P. Damayanti, L. L. Parker and J. M. K. Irudayaraj, Angew. Chem., Int. Ed., 2013, 52, 3931–3943 CrossRef CAS PubMed.
- S. M. Borisov and O. S. Wolfbeis, Chem. Rev., 2008, 108, 423 CrossRef CAS PubMed.
- (a) P. Greenspan, E. P. Mayer and S. D. Fowler, J. Cell Biol., 1985, 100, 965–973 CrossRef CAS PubMed; (b) K. S. Jones, A. P. Alimov, H. L. Rilo, R. J. Jandacek, L. A. Woollett and W. T. Penberthy, Nutr. Metab., 2008, 5, 23 CrossRef PubMed.
- (a) J. Spandl, D. J. White, J. Peychl and C. Thiele, Traffic, 2009, 10, 1579–1584 CrossRef CAS PubMed; (b) Y. Ohsaki, Y. Shinohara, M. Suzuki and T. Fujimoto, Histochem. Cell Biol., 2010, 133, 477480 CrossRef PubMed.
- P. M. Gocze and D. A. Freeman, Cytometry, 1994, 17, 151–158 CrossRef CAS PubMed.
- A. Loudet and K. Burgess, Chem. Rev., 2007, 107, 4891–4932 CrossRef CAS PubMed.
- (a) J. H. Lee, J.-H. So, J. H. Jeon, E. B. Choi, Y.-R. Lee, Y.-T. Chang, C.-H. Kim, M. A. Bae and J. H. Ahn, Chem. Commun., 2011, 47, 7500–7502 RSC; (b) H. J. Yang, C. L. Hsu, J. Y. Yang and W. Y. Yang, PLoS One, 2012, 7, e32693 CAS; (c) E. Kim, S. Lee and S. B. Park, Chem. Commun., 2012, 48, 2331–2333 RSC; (d) A. Goel, A. Sharma, M. Kathuria, A. Bhattacharjee, A. Verma, P. R. Mishra, A. Nazir and K. Mitra, Org. Lett., 2014, 16, 756–759 CrossRef CAS PubMed.
- (a) C. Qin, A. Islam and L. Han, J. Mater. Chem., 2012, 22, 19236–19243 RSC; (b) X.-H. Zhou, Y. Zhang, Y.-Q. Xie, Y. Cao and J. Pei, Macromolecules, 2006, 39, 3830–3840 CrossRef CAS.
- (a) A. Goel, S. Chaurasia, M. Dixit, V. Kumar, S. Prakash, B. Jena, J. K. Verma, M. Jain, R. S. Anand and S. S. Manoharan, Org. Lett., 2009, 11, 1289–1292 CrossRef CAS PubMed; (b) A. Goel, S. Chaurasia, V. Kumar, R. S. Anand and S. Manoharan, PCT, WO/2009/122445, 2009 Search PubMed.
- (a) Z. M. Hudson and S. Wang, Acc. Chem. Res., 2009, 42, 1584–1596 CrossRef CAS PubMed; (b) H. J. Kim, C. H. Heo and H. M. Kim, J. Am. Chem. Soc., 2013, 135, 17969–17977 CrossRef CAS PubMed; (c) X. H. Jin, C. Chen, C. X. Ren, L. X. Cai and J. Zhang, Chem. Commun., 2014, 50, 15878–15881 RSC; (d) Y. I. Park, C. Y. Kuo, J. S. Martinez, Y. S. Park, O. Postupna, A. Zhugayevych, S. Kim, J. Park, S. Tretiak and H. L. Wang, ACS Appl. Mater. Interfaces, 2013, 5, 4685–4695 CrossRef CAS PubMed.
- (a) Q. Li, M. Peng, H. Li, C. Zhong, L. Zhang, X. Cheng, X. Peng, Q. Wang, Q. Jingui and Z. Li, Org. Lett., 2012, 14, 2094–2097 CrossRef CAS PubMed; (b) J. Dhuguru, W. Liu, W. G. Gonzalez, W. M. Babinchak, J. Miksovska, R. Landgraf and J. N. Wilson, J. Org. Chem., 2014, 79, 4940–4947 CrossRef CAS PubMed; (c) G. Feng, J. Liu, R. Zhang and B. Liu, Chem. Commun., 2014, 50, 9497–9500 RSC.
- (a) A. Goel, S. Umar, P. Nag, A. Sharma, L. Kumar, Shamsuzzama, Z. Hossain, J. R. Gayen and A. Nazir, Chem. Commun., 2015, 51, 5001–5004 CAS; (b) A. Goel, A. Sharma, M. Rawat, R. S. Anand and R. Kant, J. Org. Chem., 2014, 79, 10873–10880 CrossRef CAS PubMed; (c) A. Goel, V. Kumar, S. P. Singh, A. Sharma, S. Prakash, C. Singh and R. S. Anand, J. Mater. Chem., 2012, 22, 14880–14888 RSC; (d) A. Goel, V. Kumar, P. Nag, V. Bajpai, B. Kumar, C. Singh, S. Prakash and R. S. Anand, J. Org. Chem., 2011, 76, 7474–7481 CrossRef CAS PubMed; (e) A. Goel, V. Kumar, S. Chaurasia, M. Rawat, R. Prasad and R. S. Anand, J. Org. Chem., 2010, 75, 3656–3662 CrossRef CAS PubMed.
- (a) Y. Li, W. Fan, H.-W. Xu, B. Jiang, S.-L. Wang and S.-J. Tu, Org. Biomol. Chem., 2013, 11, 2417–2420 RSC; (b) A.-S. Rebstock, F. Mongin, F. Tricourt and G. Queguiner, Tetrahedron, 2003, 59, 4973–4997 CrossRef CAS; (c) D. Tilly, A.-S. Castanet and J. Mortier, Tetrahedron Lett., 2006, 47, 1121–1123 CrossRef CAS; (d) Y. Du, J. Xue, M.-De Li, X. Guan, D. W. McCamant and D. L. Phillips, Chem. – Eur. J., 2010, 16, 6961–6972 CrossRef CAS PubMed; (e) M. Alessi, A. L. Larkin, K. A. Ogilvie, L. A. Green, S. Lai, S. Lopez and V. Snieckus, J. Org. Chem., 2007, 72, 1588–1594 CrossRef CAS PubMed; (f) E. P. Kyba, S.-T. Liu, K. Chockalingam and B. R. Reddy, J. Org. Chem., 1988, 53, 3513–3521 CrossRef CAS; (g) J. W. Lockner, D. D. Dixon, R. Risgaard and P. S. Baran, Org. Lett., 2011, 13, 5628–5631 CrossRef CAS PubMed.
- Y. Huang and R. W. Hartmann, Synth. Commun., 1998, 28, 1197–1200 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr., J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, T. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09 (Revision D.01), Gaussian, Inc., Wallingford CT, 2013 Search PubMed.
- E. J. O'Rourke, A. A. Soukas, C. E. Carr and G. Ruvkun, Cell Metab., 2009, 10, 430–435 CrossRef PubMed.
- N. Mizuyama, Y. Murakami, S. Kohra, K. Ueda, K. Hiraoka, J. Nagaoka, K. Takahashi, Y. Shigemitsu and Y. Tominaga, J. Heterocycl. Chem., 2007, 44, 115–132 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Spectroscopic characterization data, 1H, 13C NMR spectra and absorption, excitation and fluorescence data of all the compounds 8–10 in PBS and DMSO. Computed TDDFT data and Cartesian coordinates, cytotoxicity data, materials and methods. See DOI: 10.1039/c5an01623a |
| This journal is © The Royal Society of Chemistry 2016 |