
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Probing DNA-stabilized fluorescent silver nanocluster spectral heterogeneity by time-correlated single photon counting†
Miguel R.
Carro Temboury
a,
Valentina
Paolucci
b,
Emma N.
Hooley
a,
Loredana
Latterini
b and
Tom
Vosch
*a
aNano-Science Center/Department of Chemistry, University of Copenhagen, Universitetsparken 5, 2100 Copenhagen, Denmark. E-mail: tom@chem.ku.dk
bDipartimento di Chimica, Biologia e Biotecnologie, Università Degli Studi di Perugia, Via Elce di Sotto, 8, 06123 Perugia, Italy
First published on 13th October 2015
Abstract
DNA-stabilized silver nanoclusters (DNA-AgNCs) are promising fluorophores whose photophysical properties and synthesis procedures have received increased attention in the literature. However, depending on the preparation conditions and the DNA sequence, the DNA-AgNC samples can host a range of different emitters, which can influence the reproducibility of the optical response and the evolution over time of the populations of these emitters. We have developed a simple method to characterize the spectral heterogeneity and time evolution of these emissive species at any given point in time after preparation, by plotting the average decay time as a function of emission wavelength. These so-called average decay time spectra were acquired for different excitation wavelengths of AgNCs stabilized by an oligonucleotide containing 24 cytosines (C24-AgNCs). The average decay time spectra allowed the comparison of sample preparation and the judgment of reproducibility. Therefore, we propose the use of the average decay time spectra as a robust and easy tool to characterize and compare different as-synthesized DNA-AgNC samples. The average decay time spectra can in general also be used to characterize the spectral heterogeneity of other fluorophores, such as luminescent colloidal nanoparticles, and to assess the reproducibility of a synthetic procedure containing an unknown distribution of emissive species.
Introduction
Since the pioneering work of Petty and Dickson, the use of DNA for stabilizing small silver nanoclusters (AgNCs) has been growing rapidly in the last ten years.1 DNA-AgNCs have been used as sensors for a myriad of different compounds ranging from microRNA, DNA, metal ions, small organic molecules and more.2–11 For these applications, the versatile nature of DNA, the ease of preparation and read out (a change in the fluorescence properties, e.g. quenching, increase, spectral shift…) have been the enabling factors. A significant amount of research has also been carried out on characterizing the photophysical properties of these DNA-AgNCs in order to try to unravel the nature of the emission.12–21 Nowadays, there are many different recipes and procedures to prepare different emissive AgNCs which span the whole visible range.15,22–24 However, many different factors can influence their formation and time evolution; in particular the distribution of emitters produced from the same DNA sequence can be affected by parameters, e.g. the exact DNA to silver ratio and/or concentration, pH, the solvent and other ions present.15,25,26 This can lead to sample heterogeneity or significant differences between samples. This heterogeneity in the distribution of the produced AgNC emitters is not limited to DNA only, but can also be found in other stabilizing ligands, e.g. PS-PMMA copolymers27 and other scaffolds such as zeolites.28 DNA-AgNCs are usually characterized and their evolution is tracked using steady state fluorescence and UV-Vis absorption methods. These techniques show that while some sequences can produce spectrally well-defined emitters (e.g. regarding their emission maxima or fluorescence decay time), many sequences stabilize a variety of emissive species with emission maxima over the entire visible range.16,29,30 In this paper we propose an easy method to probe the spectral heterogeneity and time evolution of the as-synthesized DNA-AgNC emitters by creating the so-called average decay time spectra. As a model system we used C24 (a single stranded oligonucleotide containing 24 cytosines) stabilized AgNCs (C24-AgNC). It is well established that polycytosine oligonucleotides stabilize a large range of emissive AgNCs with emission spectra covering the visible range, and some of these emitters show spectral evolution over time.6,12,25,26,31 High performance liquid chromatography (HPLC) is one method to separate and investigate in detail what was produced during the synthesis. Gwinn et al. showed that HPLC in combination with absorption, fluorescence and mass spectrometry gives information on the DNA structure (monomers, dimers, hairpins, etc.), the number of attached silver atoms and whether the clusters are emissive or non-emissive.15 In this paper, we aim to present a method to directly characterize the as-synthesized solutions (without purification and/or separation steps) and follow their evolution by time-correlated single photon counting (TCSPC). Decay curves are recorded over the entire emission range and the average decay time derived from a multi-exponential fit is plotted as a function of emission wavelength. We show that the average decay time spectra can be used as a characteristic readout of a particular distribution of emissive species at a given point in time. Using the average decay times, the reproducibility, distribution and evolution of the emissive species in the samples can be monitored at various emission wavelengths. This analysis method enables investigation of the composition and evolution of heterogeneous fluorescent samples avoiding tedious separation procedures.Experimental section
C24-AgNC synthesis
C24 single stranded DNA (an oligonucleotide containing 24 cytosines, RP-Cartridge-Gold™ purification, Eurogentec) was diluted in Milli-Q water (MQ) or in a 0.1 M citrate buffer (pH 6.2) to give [DNA] = 2 mM. The diluted DNA was heated to 80–85 °C in order to start with a homogeneous dilution of single stranded C24.32 The solution was then cooled to room temperature. AgNO3 (99.9999%, Sigma Aldrich) and NaBH4 (99.99%, Sigma Aldrich) were separately diluted in MQ to concentrations of [AgNO3] = [NaBH4] = 2 mM and sequentially added to the DNA in the ratio DNA![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :AgNO3:NaBH4 of 1:12:12. This sample was further diluted to a [DNA] = 10 μM concentration for all the spectroscopic measurements. See the ESI† for further details.
:AgNO3:NaBH4 of 1:12:12. This sample was further diluted to a [DNA] = 10 μM concentration for all the spectroscopic measurements. See the ESI† for further details.
Absorbance, steady state fluorescence and TCSPC
Absorbance spectra were recorded on a Cary 300 UV-Vis spectrometer (Agilent Technologies) and on a Lambda 1050 UV/Vis/NIR spectrometer (Perkin Elmer). Steady state fluorescence measurements were performed by using a Cary Eclipse Fluorescence Spectrophotometer (Agilent) (Fig. 1) and by using a Fluorescence Lifetime Spectrometer FluoTime 300 (PicoQuant) (Fig. 2). The spectra from the FluoTime 300 instrument were corrected for the detector efficiency, while the spectra from the Cary Eclipse were not. This does not present a problem, since the information retrieved from Fig. 1 does not depend on this correction. Fluorescence decay times were also measured by using the Fluorescence Lifetime Spectrometer FluoTime 300, using their compatible LDH-P-C-510, LDH-P-635 and LDH-D-TA-560 laser heads. The decay curves were acquired over the entire emission range by varying the emission monochromator in steps of 5 nm and fitted with the FluoFit software (PicoQuant) using a four exponential reconvolution model, and a global fitting of the decay times. Additionally some individual decay curves were fitted with Gaussian distributions of decay times. The instrument response function (IRF) measured under 510 nm, 560 nm and 635 nm excitation gives a Full Width at Half Maximum (FWHM) of 230 ps, 160 ps and 180 ps, respectively. Fig. SI1† shows an example of a decay curve fitted with a 1 to 4 exponential model, which clearly indicates that a four exponential model leads to a better fit. The quality of the fitting was assessed by using χ2 and visual inspection of the residuals.33 More information is available in the ESI.† For all spectroscopic measurements, 10 × 10 mm light path quartz cuvettes 111-QS (Hellma) were used.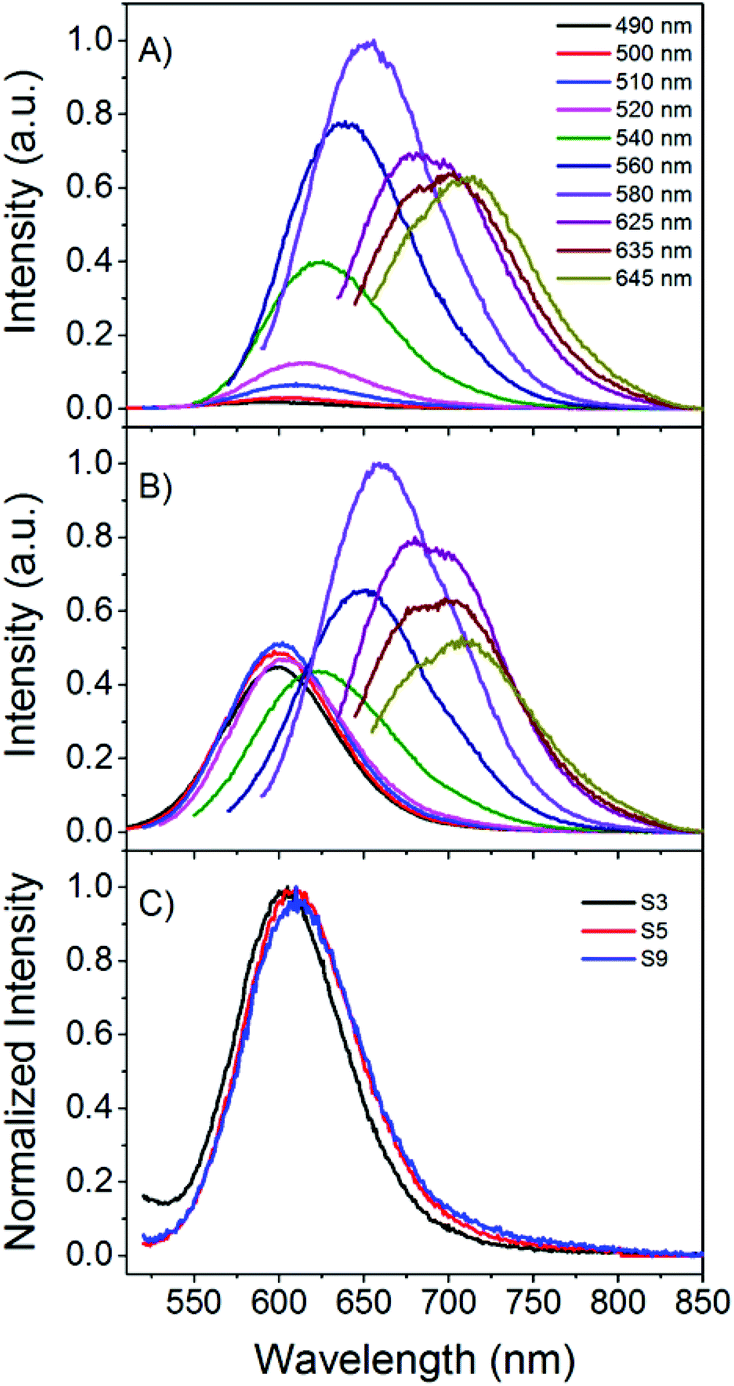 | ||
| Fig. 1 (A, B) Emission spectra at different excitation wavelengths (from 490 nm to 645 nm) of a C24-AgNC solution in citrate buffer (sample S9). (A) Scan started 27 min after preparation. (B) 23 h 6 min after preparation. (C) Normalized emission spectra (λexc = 510 nm) for samples prepared in MQ (S3) and in buffer (S5, S9). S3 measured 40 min after preparation, S5 and S9 measured 27 min after preparation. | ||
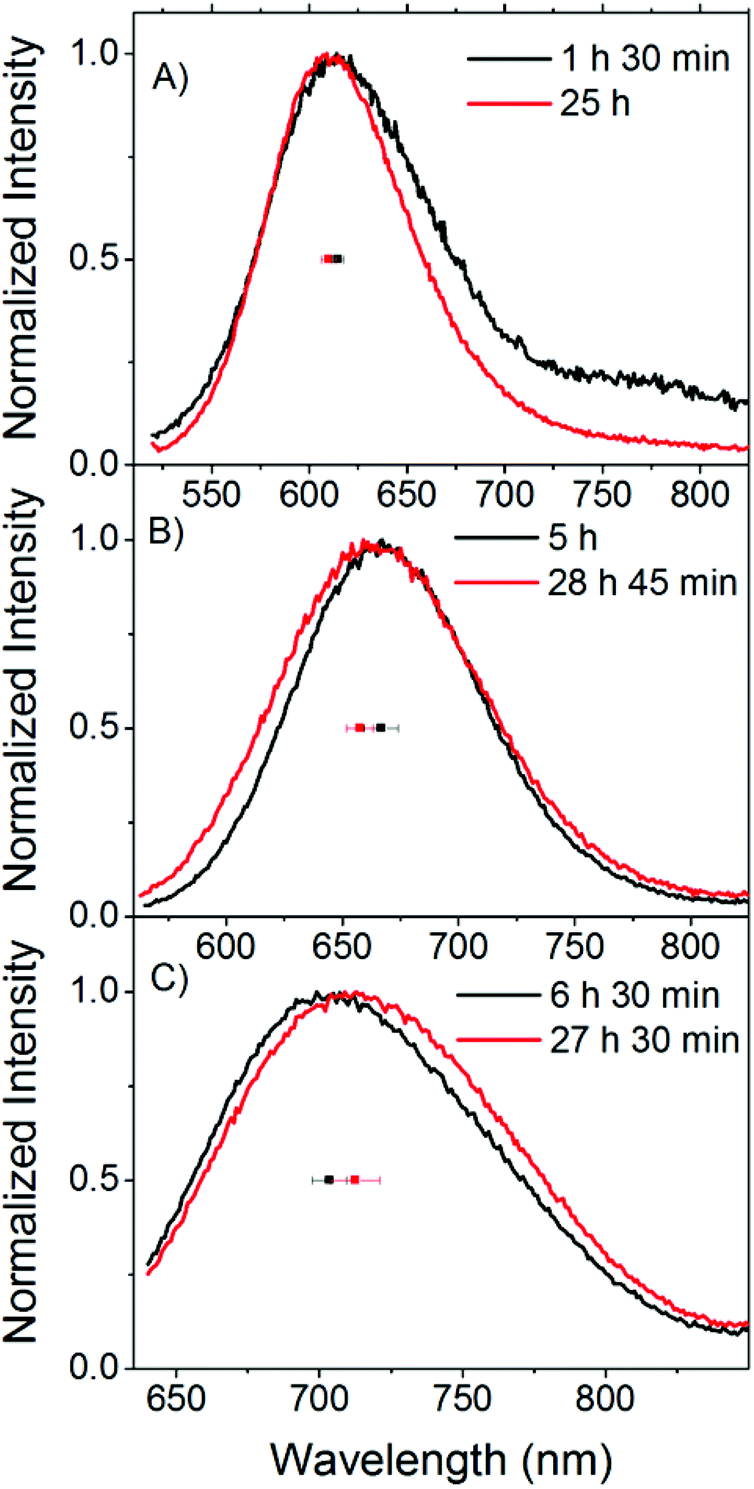 | ||
| Fig. 2 Time evolution of the emission spectra and emission maxima of a C24-AgNC solution in buffer (sample S8). Spectra recorded upon excitation at (A) 510 nm, (B) 560 nm and (C) 635 nm. The individual data points represent the average emission maxima of all the samples in buffer on the day of preparation (black) and approximately one day later (red), and the error bar represents the standard deviation. These emission spectra were recorded right before the time resolved measurements. Therefore, the aging time of the samples when the corresponding time resolved measurements (shown in Fig. 3) were acquired is shown for simplicity. | ||
Results and discussion
In this paper we investigate the spectral heterogeneity in DNA-AgNC solutions and their time evolution. For this purpose, 11 samples were prepared using C24. These 11 samples of C24-AgNC (denoted as S1–11, see Table SI1† in the ESI† for specific details) were studied shortly after preparation and the following days by means of steady-state absorption, fluorescence and TCSPC. C24 was selected since it is known that polycytosine oligonucleotides can stabilize a whole range of emitters that span the visible and near infrared (NIR) region.12,25,26 As a consequence, minor changes in the preparation conditions can have an impact on the overall outcome. The effect of using a buffered solution was also investigated; samples S1–3 were prepared in MQ and samples S4–11 in citrate buffer.Steady state characterization
Fig. 1A presents the emission spectra of sample S9 prepared in citrate buffer and gives an example of spectral heterogeneity. In Fig. 1A, the emission behavior of the sample 27 minutes after reagent mixing is presented. The emission maxima shift to longer wavelengths as the excitation wavelength is increased, which is a common behavior for DNA-AgNCs and it has also been reported when other stabilizing matrices, such as polymers, are used.5,27 Looking at one excitation wavelength and comparing the emission spectra of different samples, minor differences in the emission maximum or FWHM are observed. The latter feature can be seen in Fig. 1C where the emission spectra recorded upon excitation at 510 nm of three samples (S3 (MQ), S5 (buffer) and S9 (buffer)) are shown. At first glance one might conclude that the minor differences in the normalized emission spectra are negligible and indicate a very similar distribution of emissive species. However, this can be misleading. We will show later that samples with similar global emission spectra can have different average decay time spectra and time evolution, indicating that the populations of the emissive species can be different from synthesis to synthesis.Fig. 1B shows the emission spectra of sample S9 recorded almost a day after preparation and when compared to the spectra recorded from the freshly made sample (Fig. 1A), the contribution of the emission centered around 600 nm appears higher. This evolution of the emission spectra in polycytosine oligonucleotides has been observed before and the large spectral shifts from red/NIR to blue/green have been assigned to oxidation.25,34 Improving the chemical stability has been the subject of some research efforts.35 However, smaller spectral changes and shifts at one specific excitation wavelength have not been studied in detail to the best of our knowledge.
Fig. 2 shows examples of these small spectral shifts that can be observed as a function of aging time. Here the normalized emission spectra recorded 1–7 hours after preparation and approximately one day later are compared for sample S8. Exciting at 510 and 560 nm, a blue shift can be observed upon aging, while a red shift is observed following excitation at 635 nm. These trends were observed for the majority of the solutions in buffer (S4–S11), although small differences from sample to sample are observed. An average shift for the buffer samples is shown in Fig. 2 upon excitation at 510 (blue shift of 5 nm), 560 (blue shift of 9 nm) and 635 nm (red shift of 9 nm). From the fluorescence spectra alone, it is difficult to determine what is causing the small spectral changes over time. One assumption could be that we have a number of emissive species covering the entire visible range and different syntheses give slightly different ratios of these emitters with different evolution patterns, leading to differences in the spectral changes. In this context we consider an emissive species as having a specific central emission maximum and fluorescence decay time, with a specific width around these values. The presence of different emitters could be due to differences in the number of Ag atoms, charge, and geometry of the AgNC itself or different positions of the AgNC in the DNA or different DNA conformations that affect the spectral properties of the stabilized AgNC.
On the other hand, minor changes in the DNA conformation (due to some intrinsic flexibility) might manifest themselves also by increasing the width of the specific central emission maximum and the fluorescence decay time of a specific emitter.36 Some emitters have been shown to evolve into other ones over time, for example after oxidation processes, usually resulting in large spectral shifts.6,25 Additionally, changes in the DNA conformation can lead to changes in the angle between the silver atoms, which was shown theoretically to have an effect on the HOMO–LUMO gap.37,38 Recently Gwinn et al. also proposed that the specific number of neutral silver atoms gives rise to a specific emission color, but that the number of silver ions in the proximity can have an effect on the HOMO–LUMO gap.16 These phenomena motivate the use of a tool to characterize the spectral heterogeneity of the sample.
TCSPC characterization
We found that the average time in the excited state39 (or average decay time) as a function of emission wavelength (average decay time spectra) is a powerful tool to compare different AgNC samples and/or different sample preparation methods in a rather straightforward way. In the simplest case of a mixture of two independent fluorophores (with different emission maxima and different decay time values which are a constant as a function of emission wavelength) the average decay time spectrum profile will feature a flat profile39 where there is no spectral overlap of the two fluorophores and a slope where there is an overlap (unless the relative contribution of the fluorophores to the fluorescence intensity remains constant in the overlapping region). This control experiment, using the method presented here, is shown in Fig. SI4† for Rhodamine B and Rhodamine 6G. Besides multiple emissive species, solvent relaxation effects on the time scale of the excited state lifetime can also lead to an emission wavelength dependent decay time. Recently, it was shown that for AgNC stabilized in different DNA sequences, solvent relaxation could play a role in explaining the observed emission wavelength dependent decay time.40 Regardless of the mechanism involved in the wavelength dependence of the fluorescence decay, average decay time spectra could be used as a tool to visualize the spectral heterogeneity. To calculate the average decay time spectra, we used the formula given in eqn (1).39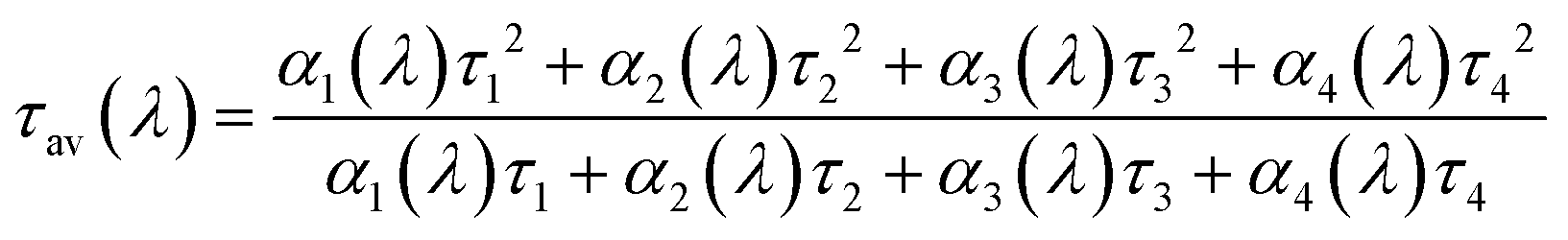 | (1) |
Here αi(λ) are the amplitudes and τi are the globally fitted decay times of the four exponential fit used here. The multi-exponential model gives an easy expression to calculate τav(λ), hence it was used in this work. We fitted the dataset globally, linking the decay times and allowing the amplitude to change freely. It is obvious that from a theoretical point of view the average time τav(λ) spent in the excited state should not depend on the model used to fit the data.39
The average decay time spectra of all our samples are presented in Fig. 3, where they are classified according to their reproducibility and time evolution. Fig. 3 shows the average decay time spectra of the samples in MQ (S1, S2, and S3) and in buffer (S4–S11) for three excitation wavelengths (510, 560 and 635 nm), obtained after preparation and approximately one day later. The average decay time spectra of the buffer samples S4, S7, S8 and S9 have similar properties and therefore were classified as group 1. The rest of the samples in buffer, S5, S6, S10 and S11, show a slightly different behavior from group 1 and were classified as group 2.
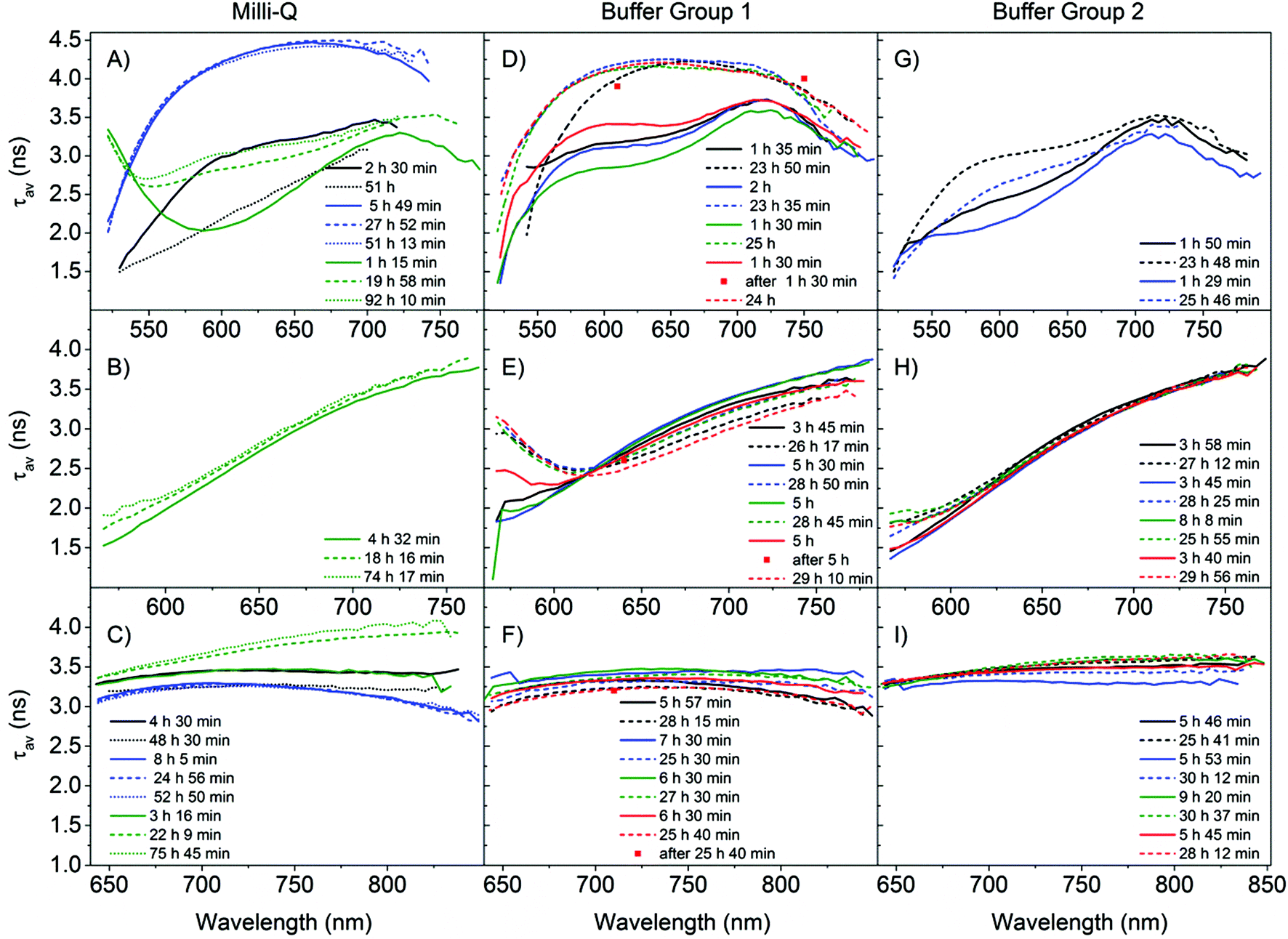 | ||
| Fig. 3 Time evolution of the average decay time spectra (τav(ns)) for all the different samples S1–11. The samples are divided into three columns, the first one for samples stabilized in MQ (A–C) and the second (D–F) and third (G–I) for citrate buffer. The different rows represent the different excitation wavelengths. (A, D, G) 510 nm excitation; (B, E, H) 560 nm excitation; (C, F, I) 635 nm excitation. The samples in buffer were separated into two columns (group 1 and group 2) according to the time evolution of the different average decay time spectra. Within a column, a colour is used to identify a sample. (A, B, C) S1 (black), S2 (blue) and S3 (green); (D, E, F) S4 (black), S7 (blue), S8 (green), and S9 (red); (G, H, I) S5 (black), S6 (blue), S10 (green) and S11 (red). Solid lines represent the data obtained on the same day as the sample preparation (1–10 h after preparation), dashed lines represent data approximately one day after preparation (18–31 h), and the dotted lines data at an even later point in time (>48 h). The legend in the figure indicates the exact time after preparation when the measurements were started. Larger versions of all the panels can be found in the ESI.† | ||
We start by comparing the samples from the MQ group with the samples from the buffer group 1. From the three samples measured in MQ water, three different trends were observed upon 510 nm excitation. The average decay time of sample S1 decreases, for S3 it increases and for S2 it stays approximately the same (Fig. 3A), with time. This is in contrast to the buffered samples from group 1 where all show a clear increase of the average decay time one day after preparation (Fig. 3D). However there are also some similarities between the samples prepared in MQ and those in buffer group 1. In particular, S2 closely resembles the samples prepared in buffer when measured after one day of aging. For 510 nm excitation, samples S1 and S3 resemble the buffer solutions when measured directly after synthesis. Sample S3, excited at 560 nm (Fig. 3B), resembles the buffer samples in group 1 (Fig. 3E), however it shows an increase in the average decay time over time, as opposed to the overall decrease observed in the buffer group 1 samples. Another difference between the samples in MQ and buffer group 1 is the range of average decay time spectra upon 635 nm excitation (Fig. 3C and F), which is between 2.8 and 3.5 ns for the samples in buffer group 1, whereas for MQ the values have a larger range from 2.8 to 4.1 ns. Additionally, under 635 nm excitation, for the samples in MQ the same trend in the time evolution of the average decay time was observed as in the 510 nm excitation case (sample S1 decreases, S3 increases and S2 stays approximately the same), while for all the samples in buffer group 1 the average decay time drops. For the buffer group 1 samples we see that under 635 nm excitation (Fig. 3F, SI10†) the average decay time curves have a fairly flat profile, which could indicate a limited number of emitters. These profiles remain nearly parallel with time for the different samples in group 1. However, in Fig. 2C a red shift of the spectra is shown. This could indicate that the different emitters have very similar decay times but slightly different emission maxima and chemical stabilities. In a recent single molecule characterization study of C24-AgNCs upon 635 nm excitation by Hooley et al.,36 no apparent correlation between the emission maxima and the fluorescence decay time was observed (which is in good agreement with the flat average decay time profile observed here). Additionally, the distribution of decay times for different single emitters showed a good agreement with a Gaussian distribution of decay times centered around 2.7 ns with a FWHM of 1.4 ns. Based on this result we fitted the decay curves of sample S7 at the peak emission wavelength with Gaussian distributions (Fig. SI2†). Upon 635 nm excitation, the decay profile can be fitted with a broad distribution centered at 3.2 ns with a FWHM of 1.8 ns and a sharp distribution centered at 0.22 ns (which could be interpreted as a single exponential component). The distribution centered at 3.2 ns with a FWHM of 1.8 ns resembles the distribution obtained at the single molecule level (more details in the ESI†), whereas the sharp distribution at 0.22 ns requires further investigation and a contribution from the experimental set-up cannot be excluded. The satisfactory fits obtained with Gaussian distributions warrant further exploration as a physical model in future studies.
Under 560 nm excitation, for both MQ and buffer group 1, an overall positive slope can be observed in the average decay time spectra in all the samples (Fig. 3B and E), leading us to postulate that there are at least two independent emissive species: an emitter with a short decay time on the short wavelength side and one emitter with a long decay time on the long wavelength side. The day after preparation, a long decay time tail on the short wavelength side (below 625 nm) appears in the buffer group 1 samples and not in the MQ sample (dashed lines in Fig. 3B and E). The appearance of the tail in the buffer group 1 samples is due to the increasing contribution of a long decay time species in this spectral region (most likely a new species that evolved over time on the short wavelength side).
As previously noted, the samples in buffer group 1 yield more consistent curves (Fig. 3D–F) than in MQ. For the 510 nm excitation (Fig. 3D), the average decay time at 610 nm increases from a value close to 3 ns after preparation to a value of 4 ns one day later. As a matter of fact, we noticed that the increase of the average decay time happens during the measurement, likely because major changes are occurring during this time span. Since the decay times are recorded from short to long emission wavelengths and the full scan takes approximately 1 hour, this artificially increases the average decay time on the red side of the spectrum. However, for the average decay time spectra, if the measurement time after preparation and the acquisition time are the same, the average decay time spectra remain a valid and reproducible characteristic as shown in Fig. 3D. The red squares in Fig. 3D are individual decays recorded after the whole scan was finished which confirms that the average decay time increased. We did not observe this behavior at 560 and 635 nm (see red squares in Fig. 3E and F), but the latter data were measured several hours after preparation, as opposed to 1.5 hours after preparation for the 510 nm excitation. Most likely a significant evolution of the AgNC population took place in the first few hours after preparation. Thus, our results show that the reported decay times for DNA-AgNCs can depend on how long after preparation they are measured22 and that significant dynamics occur at early stages after preparation. Additionally, the same argument can be made for transient quenching species that can affect the emission intensity and the fluorescence decay time. The relevant advantage of the present analytical procedure is that it evidences the populations’ dynamics.
Comparison of the average decay time spectra of the MQ versus the buffer samples in group 1 shows that the results from the MQ samples (Fig. 3A and C) are more heterogeneous. This lack of reproducibility among the MQ samples suggests that control of the pH and the presence of specific ions are important to control the final distribution of DNA-AgNC emitters.
Finally, we will discuss the differences between buffer group 1 and group 2, emphasizing the preparation procedure (see Table SI1†). Under 510 nm excitation the average decay time shows an increase one day after preparation for both groups (Fig. 3D and G), but in group 2 the average decay time does not reach the value of 4 ns. Under 560 nm excitation we observe similar average decay time spectra in both groups (Fig. 3E and H), but the development of the tail on the blue side of the spectrum, one day after preparation, is less pronounced in group 2. For 635 nm excitation, the average decay times in group 2 increase with time, in contrast to group 1. All samples within group 1 were prepared using the heating procedure described in the ESI† and in the case of group 2 only samples S10 and S11 were prepared following the heating procedure. The reason samples S10 and S11 gave different average decay time spectra from the samples of group 1 (and thus were included in group 2) despite following the same preparation procedure is not obvious. One possible explanation is that S10 and S11 were prepared using another batch of DNA and there could have been differences between batch handling and storage over time. As an extra control, we also recorded the absorbance spectra and focused on the DNA absorption band around 270 nm (Fig. SI3†). Most of the samples had similar absorption values around 270 nm in the 1.2–1.6 range, except for sample S6, which had more than twice the amount of DNA bases. Differences in the ratio of DNA to silver could also affect the ratio of emitters formed although based on the absorption spectra presented in Fig. SI3,† this cannot explain the differences in the average decay time spectra observed between groups 1 and 2, but further investigations on differences between oligonucleotide batches might be useful.
Conclusions
In conclusion, we compared the spectral heterogeneity and time evolution of different samples of C24-AgNCs by means of TCSPC. We used the average decay time spectra as a readout to characterize the distribution of emissive AgNCs at a given point in time and for different excitation wavelengths, which is an easy and robust tool providing information on the as-synthesized emissive species distribution. Classification of our samples in terms of the average decay time spectra allowed us to compare and comment on the reproducibility between samples prepared under the same and different experimental conditions and from different DNA batches, highlighting the importance of using a buffered medium. Based on our results, we propose the use of the average decay time spectra to compare the as-prepared DNA-AgNCs and not rely on emission spectra as proof of a similar distribution of emissive species. Average decay time spectra are proposed as a tool to characterize spectrally heterogeneous fluorescent samples in which the composition of emissive species and the mechanism behind the decay time dependence on the wavelength are unknown. Besides, the average decay time spectra are useful to compare the reproducibility of a synthesis method. For future experiments, TCSPC on purified samples (e.g. HPLC) or single molecule studies could help in gaining more insight into the actual number of different emissive species, the intrinsic distribution of the spectral heterogeneity of DNA-AgNCs and the specific photophysical properties of each emitter. Despite the further tests and validation experiments necessary to generalize the method, average decay time spectra can be a versatile and robust tool to investigate and compare AgNC emission and hence to extend their application in different sensing fields.Acknowledgements
T.V. gratefully acknowledges financial support from the “Center for Synthetic Biology” at Copenhagen University funded by the UNIK research initiative of the Danish Ministry of Science, Technology and Innovation (Grant 09-065274), bioSYNergy, University of Copenhagen's Excellence Programme for Interdisciplinary Research, the Villum Foundation (Project number VKR023115) and the Danish Council of Independent Research (Project number DFF-1323-00352). L. L. acknowledges the financial support from the Fondazione Cassa di Risparmio di Perugia (Project number 2014.0260.021). V.P. thanks the support of the University of Perugia for the Erasmus fellowship. We would also like to thank Bo Wegge Laursen and Thomas Just Sørensen for use of equipment.References
- J. T. Petty, J. Zheng, N. V. Hud and R. M. Dickson, DNA-Templated Ag Nanocluster Formation, J. Am. Chem. Soc., 2004, 126, 5207–5212 CrossRef CAS PubMed.
- J. Obliosca, C. Liu, R. Batson, M. Babin, J. Werner and H.-C. Yeh, DNA/Rna Detection Using DNA-Templated Few-Atom Silver Nanoclusters, Biosensors, 2013, 3, 185–200 CrossRef CAS PubMed.
- H. C. Yeh, J. Sharma, J. J. Han, J. S. Martinez and J. H. Werner, A DNA-Silver Nanocluster Probe That Fluoresces Upon Hybridization, Nano Lett., 2010, 10, 3106–3110 CrossRef CAS PubMed.
- S. W. Yang and T. Vosch, Rapid Detection of MicroRNA by a Silver Nanocluster DNA Probe, Anal. Chem., 2011, 83, 6935–6939 CrossRef CAS PubMed.
- P. Shah, A. Rorvig-Lund, S. Ben Chaabane, P. W. Thulstrup, H. G. Kjaergaard, E. Fron, J. Hofkens, S. W. Yang and T. Vosch, Design Aspects of Bright Red Emissive Silver Nanoclusters/DNA Probes for MicroRNA Detection, ACS Nano, 2012, 6, 8803–8814 CrossRef CAS PubMed.
- S. Park, S. Choi and J. Yu, DNA-Encapsulated Silver Nanodots as Ratiometric Luminescent Probes for Hypochlorite Detection, Nanoscale Res. Lett., 2014, 9, 129 CrossRef PubMed.
- J. J. Li, X. Q. Zhong, H. Q. Zhang, X. C. Le and J. J. Zhu, Binding-Induced Fluorescence Turn-on Assay Using Aptamer-Functionalized Silver Nanocluster DNA Probes, Anal. Chem., 2012, 84, 5170–5174 CrossRef CAS PubMed.
- L. Deng, Z. Zhou, J. Li, T. Li and S. Dong, Fluorescent Silver Nanoclusters in Hybridized DNA Duplexes for the Turn-on Detection of Hg2+ Ions, Chem. Commun., 2011, 47, 11065–11067 RSC.
- W. W. Guo, J. P. Yuan and E. K. Wang, Oligonucleotide-Stabilized Ag Nanoclusters as Novel Fluorescence Probes for the Highly Selective and Sensitive Detection of the Hg2+ Ion, Chem. Commun., 2009, 3395–3397 RSC.
- Z. Zhou, Y. Du and S. Dong, DNA-Ag Nanoclusters as Fluorescence Probe for Turn-on Aptamer Sensor of Small Molecules, Biosens. Bioelectron., 2011, 28, 33–37 CrossRef CAS PubMed.
- M. Zhang, Y.-Q. Liu, C.-Y. Yu, B.-C. Yin and B.-C. Ye, Multiplexed Detection of MicroRNAs by Tuning DNA-Scaffolded Silver Nanoclusters, Analyst, 2013, 138, 4812–4817 RSC.
- T. Vosch, Y. Antoku, J. C. Hsiang, C. I. Richards, J. I. Gonzalez and R. M. Dickson, Strongly Emissive Individual DNA-Encapsulated Ag Nanoclusters as Single-Molecule Fluorophores, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 12616–12621 CrossRef CAS PubMed.
- S. A. Patel, M. Cozzuol, J. M. Hales, C. I. Richards, M. Sartin, J. C. Hsiang, T. Vosch, J. W. Perry and R. M. Dickson, Electron Transfer-Induced Blinking in Ag Nanodot Fluorescence, J. Phys. Chem. C, 2009, 113, 20264–20270 CAS.
- S. S. R. Oemrawsingh, N. Markešević, E. G. Gwinn, E. R. Eliel and D. Bouwmeester, Spectral Properties of Individual DNA-Hosted Silver Nanoclusters at Low Temperatures, J. Phys. Chem. C, 2012, 116, 25568–25575 CAS.
- D. Schultz and E. G. Gwinn, Silver Atom and Strand Numbers in Fluorescent and Dark Ag:Dnas, Chem. Commun., 2012, 48, 5748–5750 RSC.
- S. M. Copp, D. Schultz, S. Swasey, J. Pavlovich, M. Debord, A. Chiu, K. Olsson and E. Gwinn, Magic Numbers in DNA-Stabilized Fluorescent Silver Clusters Lead to Magic Colors, J. Phys. Chem. Lett., 2014, 5, 959–963 CrossRef CAS PubMed.
- N. Markešević, S. S. R. Oemrawsingh, D. Schultz, E. G. Gwinn and D. Bouwmeester, Polarization Resolved Measurements of Individual DNA-Stabilized Silver Clusters, Adv. Opt. Mater., 2014, 2, 765–770 CrossRef.
- S. M. Swasey, N. Karimova, C. M. Aikens, D. E. Schultz, A. J. Simon and E. G. Gwinn, Chiral Electronic Transitions in Fluorescent Silver Clusters Stabilized by DNA, ACS Nano, 2014, 8, 6883–6892 CrossRef CAS PubMed.
- T. Driehorst, P. ONeill, P. M. Goodwin, S. Pennathur and D. K. Fygenson, Distinct Conformations of DNA-Stabilized Fluorescent Silver Nanoclusters Revealed by Electrophoretic Mobility and Diffusivity Measurements, Langmuir, 2011, 27, 8923–8933 CrossRef CAS PubMed.
- S. H. Yau, et al., Bright Two-Photon Emission and Ultra-Fast Relaxation Dynamics in a DNA-Templated Nanocluster Investigated by Ultra-Fast Spectroscopy, Nanoscale, 2012, 4, 4247–4254 RSC.
- I. L. Volkov, P. Y. Serdobintsev and A. I. Kononov, DNA-Stabilized Silver Nanoclusters with High Yield of Dark State, J. Phys. Chem. C, 2013, 117, 24079–24083 CAS.
- S. Choi, R. M. Dickson and J. Yu, Developing Luminescent Silver Nanodots for Biological Applications, Chem. Soc. Rev., 2012, 41, 1867–1891 RSC.
- J. Sharma, H. C. Yeh, H. Yoo, J. H. Werner and J. S. Martinez, A Complementary Palette of Fluorescent Silver Nanoclusters, Chem. Commun., 2010, 46, 3280–3282 RSC.
- A. Latorre and A. Somoza, DNA-Mediated Silver Nanoclusters: Synthesis, Properties and Applications, ChemBioChem, 2012, 13, 951–958 CrossRef CAS PubMed.
- Y. Antoku, Fluorescent Polycytosine-Encapsuled Silver Nanoclusters, Georgia Tech, Atlanta, 2007 Search PubMed.
- C. M. Ritchie, K. R. Johnsen, J. R. Kiser, Y. Antoku, R. M. Dickson and J. T. Petty, Ag Nanocluster Formation Using a Cytosine Oligonucleotide Template, J. Phys. Chem. C, 2007, 111, 175–181 CAS.
- I. Diez, R. H. A. Ras, M. I. Kanyuk and A. P. Demchenko, On Heterogeneity in Fluorescent Few-Atom Silver Nanoclusters, Phys. Chem. Chem. Phys., 2013, 15, 979–985 RSC.
- G. De Cremer, et al., Characterization of Fluorescence in Heat-Treated Silver-Exchanged Zeolites, J. Am. Chem. Soc., 2009, 131, 3049–3056 CrossRef CAS PubMed.
- C. I. Richards, S. Choi, J. C. Hsiang, Y. Antoku, T. Vosch, A. Bongiorno, Y. L. Tzeng and R. M. Dickson, Oligonucleotide-Stabilized Ag Nanocluster Fluorophores, J. Am. Chem. Soc., 2008, 130, 5038–5039 CrossRef CAS PubMed.
- S. M. Copp, P. Bogdanov, M. Debord, A. Singh and E. Gwinn, Base Motif Recognition and Design of DNA Templates for Fluorescent Silver Clusters by Machine Learning, Adv. Mater., 2014, 26, 5839–5845 CrossRef CAS PubMed.
- Y. Antoku, J.-I. Hotta, H. Mizuno, R. M. Dickson, J. Hofkens and T. Vosch, Transfection of Living Hela Cells with Fluorescent Poly-Cytosine Encapsulated Ag Nanoclusters, Photochem. Photobiol. Sci., 2010, 9, 716–721 CAS.
- W. A. Kibbe, Oligocalc: An Online Oligonucleotide Properties Calculator, Nucleic Acids Res., 2007, 35, W43–W46 CrossRef PubMed.
- D. O'Connor and D. Philips, Time-Correlated Single Photon Counting, Academic Press, London, 1984 Search PubMed.
- S. Choi, S. Park, K. Lee and J. Yu, Oxidant-Resistant Imaging and Ratiometric Luminescence Detection by Selective Oxidation of Silver Nanodots, Chem. Commun., 2013, 49, 10908–10910 RSC.
- J. Sharma, R. C. Rocha, M. L. Phipps, H. C. Yeh, K. A. Balatsky, D. M. Vu, A. P. Shreve, J. H. Werner and J. S. Martinez, A DNA-Templated Fluorescent Silver Nanocluster with Enhanced Stability, Nanoscale, 2012, 4, 4107–4110 RSC.
- E. N. Hooley, V. Paolucci, Z. Liao, M. R. Carro-Temboury and T. Vosch, Single Molecule Characterization of Near-Infrared Emitting Silver Nanoclusters, Adv. Opt. Mater., 2015, 3, 1109–1115 CrossRef CAS.
- R. R. Ramazanov and A. I. Kononov, Excitation Spectra Argue for Threadlike Shape of DNA-Stabilized Silver Fluorescent Clusters, J. Phys. Chem. C, 2013, 117, 18681–18687 CAS.
- N. V. Karimova and C. M. Aikens, Time-Dependent Density Functional Theory Investigation of the Electronic Structure and Chiroptical Properties of Curved and Helical Silver Nanowires, J. Phys. Chem. A, 2015, 119, 8163–8173 CrossRef CAS PubMed.
- J. R. Lakowicz, Principles of Fluorescence Spectroscopy, Springer Science & Business Media, 2013 Search PubMed.
- H.-C. Hsu, M.-C. Ho, K.-H. Wang, Y.-F. Hsu and C.-W. Chang, DNA Stabilized Silver Nanoclusters as the Fluorescent Probe for Studying the Structural Fluctuations and the Solvation Dynamics of Human Telomeric DNA, New J. Chem., 2015, 39, 2140–2145 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Details of the C24-AgNC preparation and description of the samples, decay curve fitting and average decay time spectra calculation, average decay time spectra of a mixture of known fluorophores and larger versions of the average decay time spectra and UV-Vis absorption spectra. See DOI: 10.1039/c5an02011e |
| This journal is © The Royal Society of Chemistry 2016 |