
Cancer cell-selective killing polymer/copper combination†
Huacheng
He
a,
Diego
Altomare
a,
Ufuk
Ozer
b,
Hanwen
Xu
a,
Kim
Creek
a,
Hexin
Chen
b and
Peisheng
Xu
*a
aDepartment of Drug Discovery and Biomedical Sciences, University of South Carolina, Columbia, SC 29208, USA. E-mail: xup@sccp.sc.edu; Fax: +1 803-777-8356; Tel: +1 803-777-0075
bDepartment of Biological Sciences, University of South Carolina, Columbia, SC 29208, USA
First published on 16th November 2015
Abstract
Chemotherapy has been adopted for cancer treatment for decades. However, its efficacy and safety are frequently compromised by the multidrug-resistance of cancer cells and the poor cancer cell selectivity of anticancer drugs. Hereby, we report a combination of a pyridine-2-thiol containing polymer and copper which can effectively kill a wide spectrum of cancer cells, including drug resistant cancer cells, while sparing normal cells. The polymer nanoparticle enters cells via an exofacial thiol facilitated route, and releases active pyridine-2-thiol with the help of intracellularly elevated glutathione (GSH). Due to their high GSH level, cancer cells are more vulnerable to the polymer/copper combination. In addition, RNA microarray analysis revealed that the treatment can reverse cancer cells’ upregulated oncogenes (CIRBP and STMN1) and downregulated tumor suppressor genes (CDKN1C and GADD45B) to further enhance the selectivity for cancer cells.
Introduction
Although various anticancer drugs have been developed to conquer cancer, a large number of cancer patients have ultimately still lost their battle against it.1 There are two major causes for the failure of these drugs, the drug resistance of cancer cells and the side effects of anticancer drugs.2 Because of the inherited or acquired multidrug resistance, cancer cells survive after receiving the original effective drug,3 which results in the recurrence of the cancer and eventually kills cancer patients. The selectivity of anticancer drugs used in chemotherapy is predominantly relying on the proliferation rate difference between normal cells and cancer cells.4 Most cancer cells are fast growing.5 However, besides cancer cells, normal cells in the digestive tract, bone marrow, hair follicles, and reproductive system are also vulnerable to anticancer drugs that target quickly proliferating cells due to their fast renewal nature. Furthermore, some anticancer drugs even compromise the function of the heart, nervous system, and kidneys.To increase the selectivity of anticancer drugs for cancer cells, various approaches have been explored, including utilizing specific receptors’ expression level difference between normal and cancer cells,6 as well as the unique physiological properties of the tumor such as low pH,7 high GSH,8 and altered metal ion concentrations.9 Among them, copper concentration in tumors has attracted extreme high interest recently. Copper is an important trace metal that plays critical roles in maintaining normal biological functions. Elevated copper concentration (up to 2–3 fold) is frequently observed in a wide spectrum of tumors including ovarian, breast, cervical, prostate and leukemia.10 High copper concentration facilitates tumor angiogenesis. Depletion of copper by copper chelators such as D-penicillamine, trientine, and disulfiram has been proven to be effective in inhibiting angiogenesis and killing cancer cells both in vitro and in vivo.11 Chelators can form a complex with copper via a thiol, an amine, and a pyridine ring to reduce copper concentration in the tumor, which eventually results in the death of the cancer cells.12 Thus, several clinical trials involving copper chelators have been conducted.13 However, due to the non-specific tissue distribution and rapid clearance of chelators, none or only little benefit was observed in those trials.
Our preliminary study found that the addition of copper ions to 2,2′-dithiodipyridine (DTP) could significantly boost its cytotoxicity for cancer cells and normal cells (Fig. S1†). To endow a high density of pyridine-2-thiol, the intracellular metabolite of DTP, we designed a poly[(2-(pyridin-2-yldisulfanyl)ethyl acrylate)-co-[poly(ethylene glycol)]] (PDA-PEG) polymer. PDA-PEG was synthesized through free radical polymerization according to our published method.8 The pyridine rings in PDA can complex with Cu2+, turning the polymer into a multi-chelator system.
Results and discussion
The successful synthesis of the polymer was verified using 1H-NMR (Fig. S2A†) and gel permeation chromatography (GPC) (Fig. S2B†). The 1H NMR result revealed that the actual ratio between PDA and mPEG in the final PDA-PEG polymer was close to their feeding ratio (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1). GPC showed that the molecular weight of the PDA-PEG polymer was 41.8 kDa. Due to the co-existence of hydrophobic PDA and hydrophilic PEG, the amphiphilic PDA-PEG self-assembles into a nanoparticle in aqueous solution. Zetasizer analysis revealed that the PDA-PEG nanoparticles had a hydrodynamic size of 87.64 ± 2.06 nm and carried a negative surface charge (−15.4 ± 2.05 mV). The morphology of the PDA-PEG nanoparticle was also confirmed using TEM, which showed a spherical shape with a size around 80 nm (Fig. 1A). After the addition of Cu2+ (CuCl2, 10 μM), the hydrodynamic size of the nanoparticles increased to 196.4 ± 0.07 nm (Fig. S3†), while becoming less negatively charged (−5.47 ± 0.86 mV). The interaction between Cu2+ and the pyridine ring made the PDA segment more hydrophilic so that the core became less condensed (Fig. 1B), which led to the increase of the particle size.14 The formation of the nanoparticle will endow two advantages for cancer therapy. First, due to the existence of the PEG corona, the circulation time of the polymer in the blood stream can be greatly extended. Second, by taking advantage of the leaky structure of the capillaries in the tumor tissue, the formed PDA-PEG/Cu2+ nanoparticle can be enriched in the tumor through the so-called enhanced permeability and retention (EPR) effect.
:1). GPC showed that the molecular weight of the PDA-PEG polymer was 41.8 kDa. Due to the co-existence of hydrophobic PDA and hydrophilic PEG, the amphiphilic PDA-PEG self-assembles into a nanoparticle in aqueous solution. Zetasizer analysis revealed that the PDA-PEG nanoparticles had a hydrodynamic size of 87.64 ± 2.06 nm and carried a negative surface charge (−15.4 ± 2.05 mV). The morphology of the PDA-PEG nanoparticle was also confirmed using TEM, which showed a spherical shape with a size around 80 nm (Fig. 1A). After the addition of Cu2+ (CuCl2, 10 μM), the hydrodynamic size of the nanoparticles increased to 196.4 ± 0.07 nm (Fig. S3†), while becoming less negatively charged (−5.47 ± 0.86 mV). The interaction between Cu2+ and the pyridine ring made the PDA segment more hydrophilic so that the core became less condensed (Fig. 1B), which led to the increase of the particle size.14 The formation of the nanoparticle will endow two advantages for cancer therapy. First, due to the existence of the PEG corona, the circulation time of the polymer in the blood stream can be greatly extended. Second, by taking advantage of the leaky structure of the capillaries in the tumor tissue, the formed PDA-PEG/Cu2+ nanoparticle can be enriched in the tumor through the so-called enhanced permeability and retention (EPR) effect.
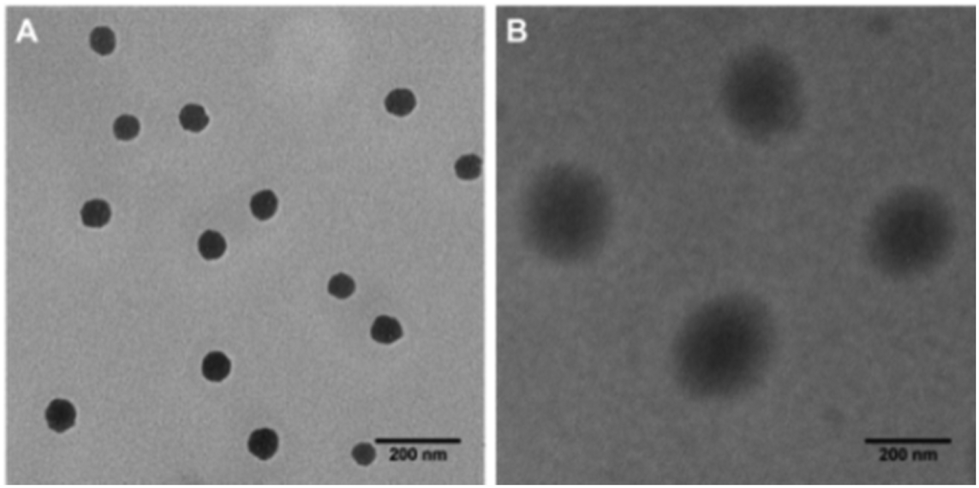 | ||
| Fig. 1 TEM images of nanoparticle fabricated from PDA-PEG alone (A), and the PDA-PEG/Cu2+ combination (B). Scale bars are 200 nm. | ||
To validate whether the polymer form of the DTP chelator possesses similar cell killing capacity to its small molecular counterpart, a cell proliferation assay was employed in 7 cancer cell lines, 5 normal cell lines, and a NIH 3T3 cell line, an immortalized cell line derived from normal cells. As expected, the PDA-PEG nanoparticles did not show obvious toxicity up to the equivalent DTP concentration of 40 μM for all tested cells (Fig. S4†). Similar to DTP, the addition of Cu2+ dramatically enhanced the potency of the PDA-PEG nanoparticles for cancer cells (Fig. 2). The IC50 values of PDA-PEG/Cu2+ for SKOV-3, NCI/ADR-Res, MDA-MB-231, and UMSCC 22A cells were less than 6 μM (Table S1†), and with an IC95 less than 20 μM. Contrary to its small molecule counterpart (Fig. S1B†), the cell killing effect of PDA-PEG/Cu2+ for cancer cells and normal cells are significantly different. Fig. 2 also shows that the PDA-PEG/Cu2+ combination is non-toxic to normal cells, including keratinocytes, fibroblasts, breast epithelial cells, colon cells, and hepatocytes, up to 80 μM. The IC50 values for normal cells were 10–70 fold higher than those for cancer cells (Table S1†). All these suggested that PDA-PEG/Cu2+ could selectively kill cancer cells, including drug resistant cancer cells, while sparing normal ones. In addition, Fig. S5† also revealed that the cytotoxicity of PDA-PEG/Cu2+ for cancer cells increases with the increase of Cu2+ concentration, while not showing any influence on normal cells.
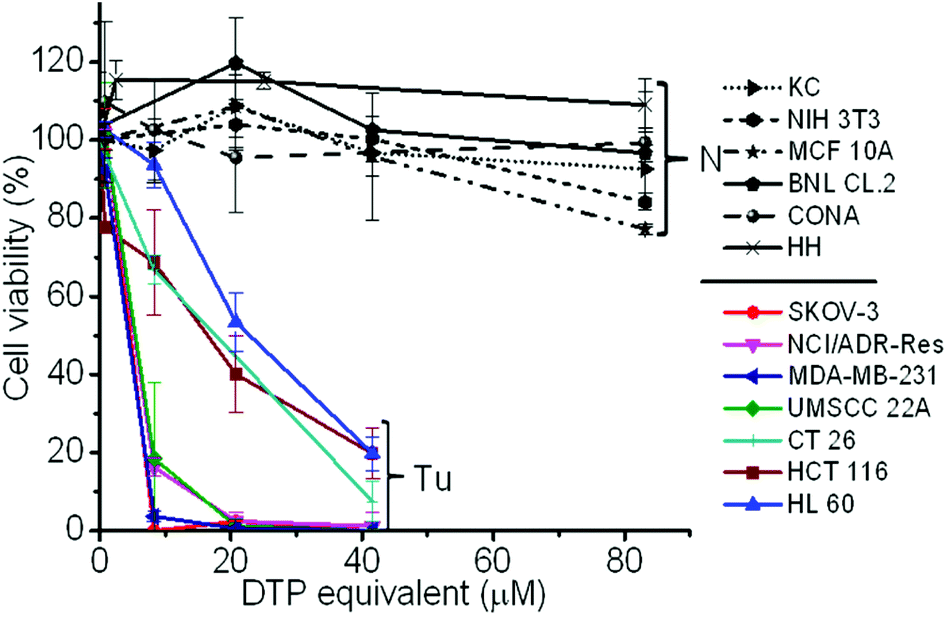 | ||
| Fig. 2 Cytotoxicity of the PDA-PEG/Cu2+ combination for normal (N) and cancer (Tu) cells. Normal cells include KC (human keratinocyte), NIH 3T3 (murine fibroblast), MCF 10A (human breast epithelial cell), BNL CL.2 (murine liver cell), CONA (CCD 841 CoN, human colon cell), and HH (human hepatocyte). Data represent the means ± SD, n = 3. | ||
To further validate that the PDA-PEG/Cu2+ combination can specifically kill cancer cells versus normal ones, NIH 3T3, SKOV-3, NCI/ADR-Res, and UMSCC 22A cells were stained with different color fluorescent dyes. The NIH 3T3 cells were selected as a representative of normal cells because of their comparable growth rate and suitability for co-culture with cancer cells in DMEM medium. Fig. 3 shows that all cells except NIH 3T3 were rounded up after treatment with 8.3 μM of PDA-PEG/Cu2+, indicating that these cells were not in a healthy condition. On the contrary, NIH 3T3 cells still kept their original stretched cell shape at 20.79 μM. All these images visually confirmed the MTT results in Fig. 2. This phenomenon was also observed in the multiple cell line co-culture model (bottom row of Fig. 3), where only NIH 3T3 cells kept their original spindle morphology, indicating that the PDA-PEG/Cu2+ combination could selectively kill cancer cells while sparing normal cells in a more disease relevant co-culture model.
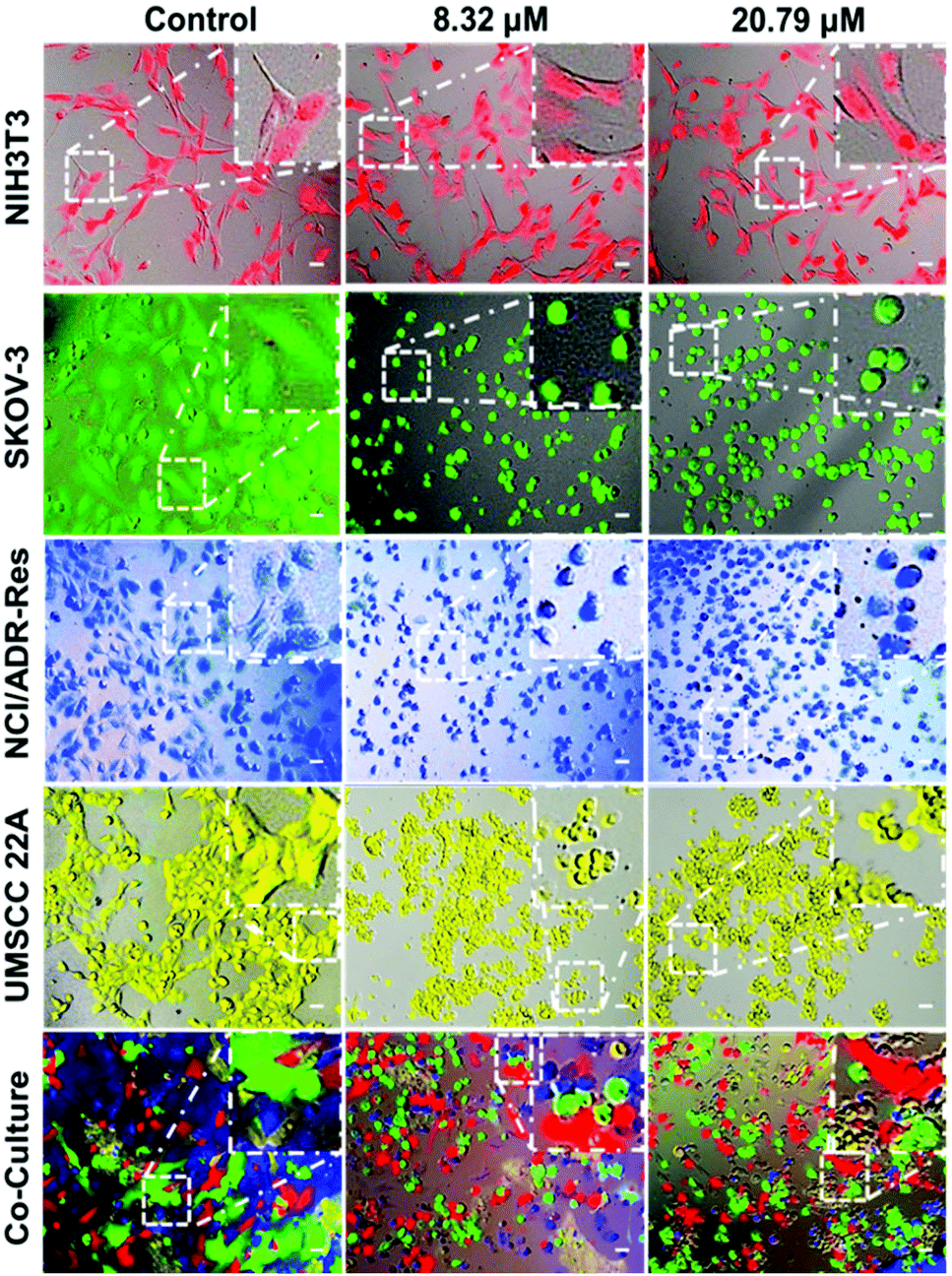 | ||
| Fig. 3 Fluorescence images of cancer cells after culturing with different concentrations of the PDA-PEG/Cu2+ combination. NIH 3T3, NCI/ADR-Res, SKOV-3 and UMSCC 22A were pre-stained with Cell Tracker™ deep red, blue, green CMFDA and orange CMTMR dyes, respectively, and imaged 24 h after the treatment. The scale bars are 40 μm. | ||
To probe the mechanism of PDA-PEG/Cu2+ selectively killing cancer cells, we first investigated the cellular uptake of PDA-PEG and PDA-PEG/Cu2+ nanoparticles using flow cytometry and confocal microscopy. Fig. 4A shows that the nanoparticles fabricated from Cy5-labeled PDA-PEG and PDA-PEG/Cu2+ entered cells in identical manners, suggesting that the addition of copper ions did not affect the entering into cells. In addition, these nanoparticles showed similar efficiency in entering normal cells (NIH 3T3) and cancer cells (SKOV-3). Therefore, the uptake of PDA-PEG/Cu2+ was not the reason for its cancer cell selectivity. 5,5′-Dithio-bis-(2-nitrobenzoic acid) (DTNB) is a compound that binds the free thiol groups on the surface of the cell membrane. The addition of DTNB significantly inhibited the cellular uptake of PDA-PEG (Fig. 4B), suggesting that PDA-PEG entered cells via exofacial thiol-mediated endocytosis. We think this is because the PDA-PEG polymer contains a high density of thiol-reactive PDA segments, which can react with the exofacial thiols through the thiol-disulfide exchange reaction to facilitate cellular uptake. A similar blocking effect was also observed in NIH 3T3 cells treated with DTNB (Fig. S6†).
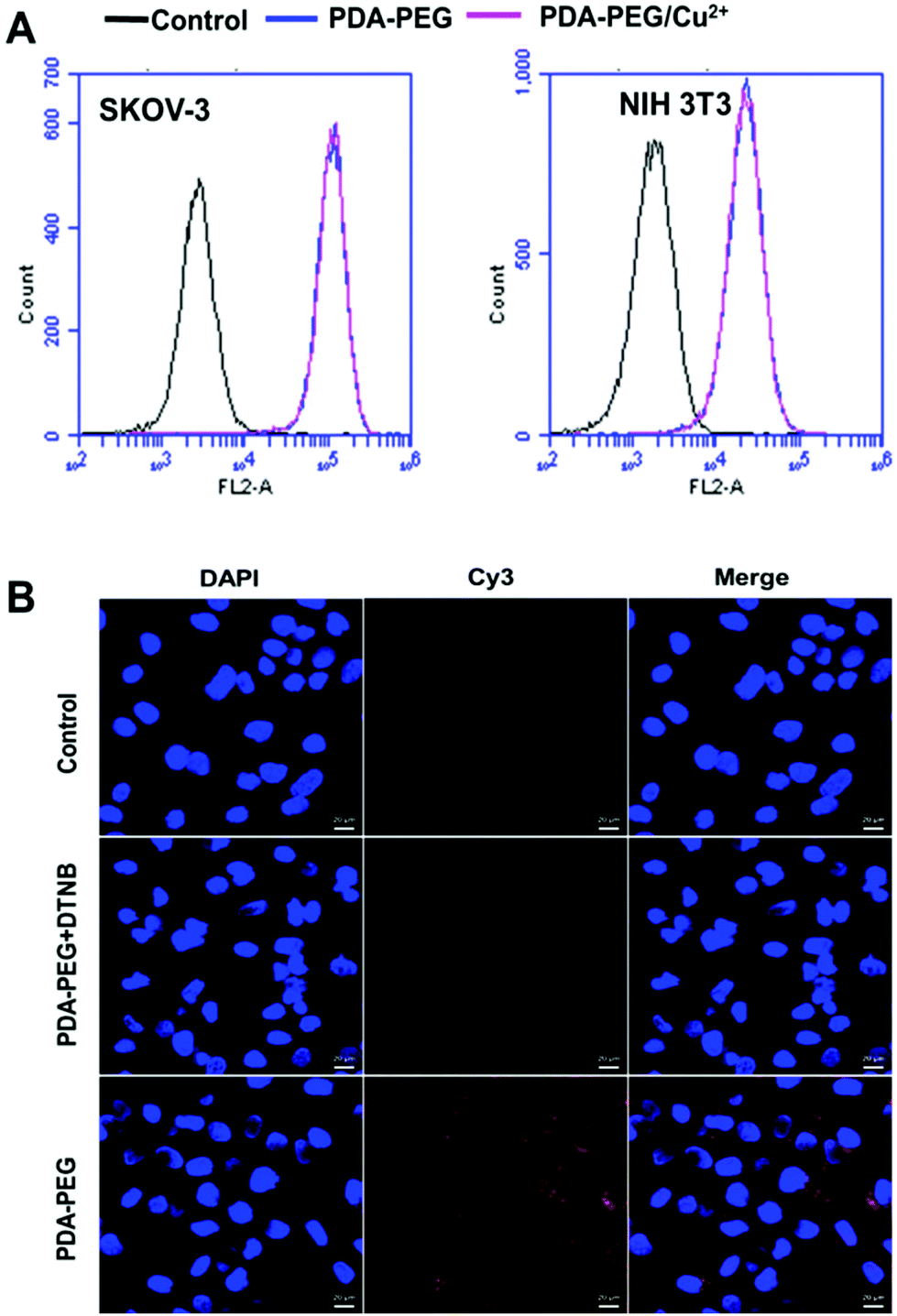 | ||
| Fig. 4 Flow cytometry spectra of SKOV-3 and NIH 3T3 cells (A) and confocal images of the cellular uptake of nanoparticles in SKOV-3 cells (B). Cellular uptake assays were carried out 1 h after the addition of the nanoparticles. Scale bars were 20 μm. | ||
Our previous study found that polymer–drug conjugates linked through a disulfide bond could quickly release their payload by cleaving the disulfide bond with the help of intracellular elevated glutathione (GSH).6 As pyridine–Cu complex analogues have been reported as highly toxic and extensively studied as anticancer drugs, the release of the pyridine–Cu complex probably induced the cell death.14 The high cytotoxicity of the DTP/copper combination also suggested that the pyridine-2-thiol/copper combination could be the active segment for its cytotoxicity. To study the release kinetics of the PDA-PEG/Cu2+ nanoparticles, samples were dispersed in phosphate buffers supplemented with different levels of GSH as well as serum-containing media to mimic the plasma and intracellular environment. Fig. 5A shows that PDA-PEG/Cu2+ is extremely stable in a low reducing environment ([GSH] < 0.1 mM), such as the plasma, which has a GSH level less than 5 μM.15 However, almost all pyridine-2-thiol segments could be instantly released from PDA-PEG at the GSH level of 10 mM, indicating its super-responsiveness to the intracellular reducing conditions. Furthermore, the polymer/copper combination was very stable in 50% serum-containing medium, only 12.92% pyridine-2-thiol was released after 7 days of incubation (Fig. 5B), suggesting its great stability during blood circulation.
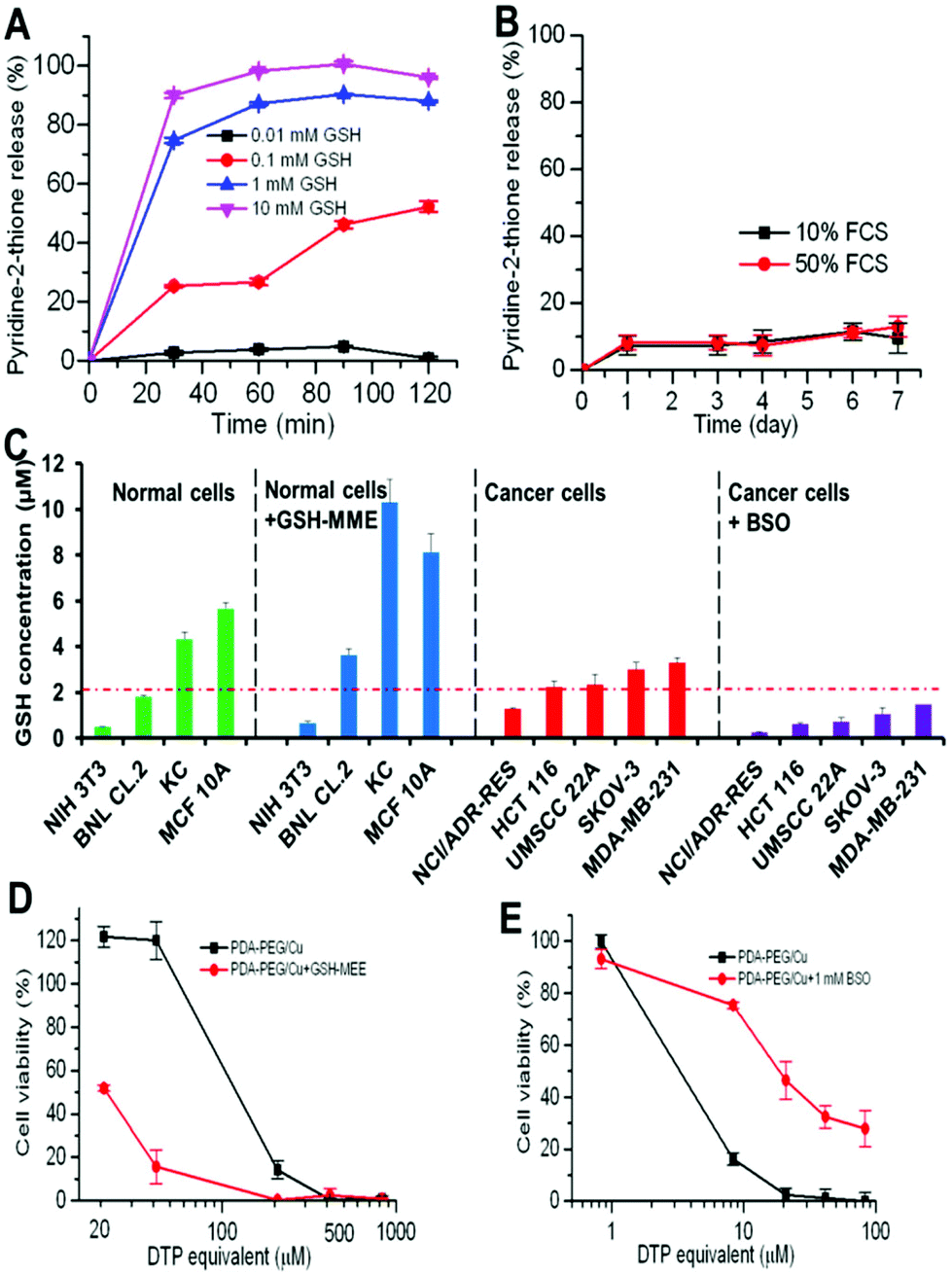 | ||
| Fig. 5 The release kinetics of pyridine-2-thiol liberating from PDA-PEG at different GSH levels (A) and in serum-containing media (B), the GSH level in different cell lines and the response to the addition of GSH-MME and BSO (C), GSH-MME effect on the cytotoxicity of PDA-PEG/Cu2+ for MCF10A cells (D), and the effect of BSO on the cytotoxicity of the PDA-PEG/Cu2+ combination for NCI/ADR-Res cells (E). Data represent the means ± SD, n = 3. | ||
Since there was no significant difference between normal cells and cancer cells in uptaking PDA-PEG/Cu2+ nanoparticles, we postulate that the observed cancer-cell-selective-killing effect was due to the intrinsic difference between normal and cancer cells. It has been reported that GSH levels in tumor tissues, such as ovarian, head and neck, breast, and lung cancer are higher than in normal tissues.16 To probe whether the high GSH level also prevails in cancer cells in vitro, the GSH-Glo™ Glutathione Assay was employed. Interestingly, Fig. 5C reveals that the GSH level range in normal cells is relatively broad, ranging from 0.48 to 5.65 μM, while most tested cancer cell lines displayed a relatively high GSH level (>2 μM), suggesting that the intracellular GSH level could be a valid target for cancer targeted therapy. To prove that, glutathione-monomethylester (GSH-MME, 5 mM) and buthionine sulfoxamine (BSO, 1 mM) were employed to boost or deplete the intracellular GSH level in normal cells (NIH 3T3, BNL CL.2, KC, and MCF 10A) or cancer cells (NCI/ADR-Res, HCT 116, UMSCC 22A, SKOV-3, and MDA-MB-231), respectively.17 After the addition of GSH-MME, all normal cells exhibited a higher intracellular GSH level (Fig. 5C). As expected, these cells became more vulnerable to the polymer/copper combination treatment (Fig. 5D and S7†). On the contrary, BSO treated NCI/ADR-Res, UMSCC 22A, and SKOV-3 cells displayed a declined GSH level. Interestingly, only NCI/ADR-Res cells became more tolerant to the polymer/copper combination treatment (Fig. 5E), while the other two cancer cell lines kept their sensitivity to the treatment (Fig. S8†). Based on these results shown in Fig. 5, we conclude that the intracellular GSH level is not the sole cause for the cancer-cell-selective-killing effect of the PDA-PEG/Cu2+ nanoparticles.
Among those tested cells, there were several exceptions. First, MCF 10A and KC exhibited a higher GSH level than other normal cells and cancer cells, while not being sensitive to the PDA-PEG/Cu2+ treatment. Second, NCI/ADR-Res cells showed a lower GSH level than other cancer cells, but were still vulnerable to the treatment. Third, SKOV-3 and UMSCC 22A cells displayed a lower GSH level after BSO treatment, however, the decreased GSH level only slightly abolished their response to PDA-PEG/Cu2+ (Fig. S8†). To solve these puzzles, we further investigated the gene response to PDA-PEG/Cu2+ treatment in MCF 10A, NCI/ADR-Res, and SKOV-3 cells through RNA microarray analysis. The cells were treated with PDA-PEG/Cu2+ for 12 h before RNA extraction. Microarray analysis data revealed that, for untreated cells, the RNA levels of oncogenes (CIRBP and STMN1) were upregulated, while tumor suppressor genes (CDKN1C and GADD45B) were downregulated in both ovarian cancer cell lines (Fig. 6A). Surprisingly, PDA-PEG/Cu2+ treatment reversed the above gene expression pattern by downregulating the RNA level of CIRBP and STMN1 (>3 folds), while upregulating CDKN1C and GADD45B (>5 folds) (Fig. 6B). Interestingly, no obvious expression level change of these genes was detected in the normal breast cell line. Other studies showed that oncogenes (CIRBP18 and STMN119) are upregulated while tumor suppressor genes (CDKN1C20 and GADD45B21) are downregulated in various types of cancers. Since the upregulated oncogenes and downregulated tumor suppressor genes stimulate cancer cell proliferation and promote tumor growth, reversing those malregulated genes through PDA-PEG/Cu2+ treatment would result in cancer cell apoptosis. For MCF 10A cells, although the high level of GSH could release a large amount of pyridine-2-thiol intracellularly, cells still survived because the oncogenes and tumor suppressor genes are not sensitive to the treatment. For NCI/ADR-Res cells, the GSH level is relatively low but still high enough to release the needed pyridine-2-thiol to regulate their oncogenes and tumor suppressor genes due to their high sensitivity. Similarly, BSO decreased the GSH level in SKOV-3 and UMSCC 22A cells, while the resulting GSH level is still high enough to release pyridine-2-thiol to regulate their oncogenes and tumor suppressor genes. Therefore, the cancer-cell-selective-killing property of PDA-PEG/Cu2+ is the combination of the effects of the high intracellular GSH level and the malregulation of oncogenes and tumor suppressor genes in cancer cells.
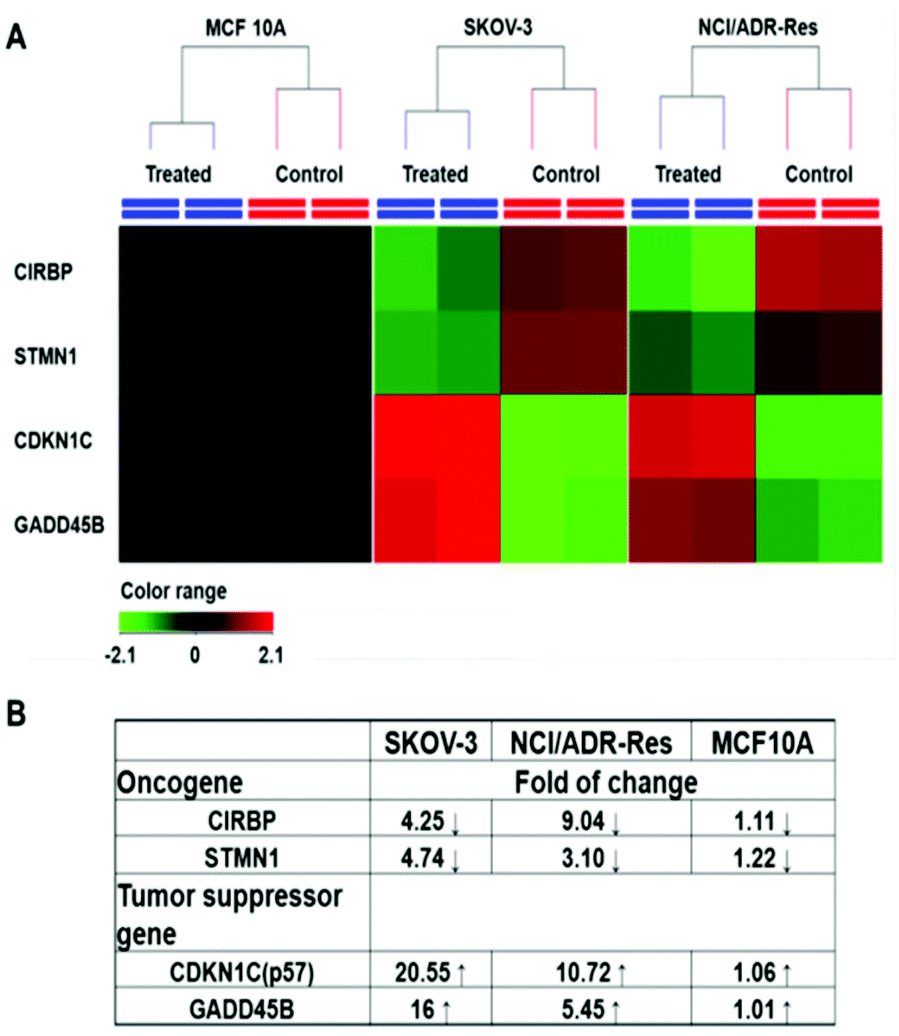 | ||
| Fig. 6 RNA expression in response to PDA-PEG/Cu2+ treatment. Cells were treated with 41.58 μM of PDA-PEG/Cu2+ for 12 h. (A) Heatmap RNA level with and without drug treatment. Red: upregulation; green: downregulation; black: no change. (B) Gene alteration fold after treatment. | ||
CDKN1C is an inhibitor for G1 cyclin/Cdk complexes and causes cell arrest in G1 phase.20 To investigate the effect of CDKN1C up-regulation after PDA-PEG/Cu2+ treatment, cell cycle analysis was employed. Fig. 7 shows that PDA-PEG/Cu2+ inhibited cell division and arrested cancer cell growth in the G1 phase, which could induce cell apoptosis. As expected, the PDA-PEG polymer or Cu2+ ion alone did not show any effect on the cell cycle distribution of SKOV-3 cells.
 | ||
| Fig. 7 Cell cycle analysis of SKOV-3 cells after treatment with 41.58 μM of PDA-PEG/Cu2+ for 12 h. Flow cytometry spectra (A) and quantitative analysis (B) of the cell cycle. Data represent the means ± SD, n = 3. *p < 0.01. | ||
Conclusions
In summary, we developed a cancer-cell-selective-killing nanoparticle from a PDA-PEG/Cu2+ combination aiming for safe and effective cancer therapy. The polymer/Cu2+-based nanoparticle entered cells through the interaction with exofacial thiols, and subsequently released a pyridine-2-thiol–Cu complex to kill cells. Due to the difference in the intracellular GSH level as well as the expression level of oncogenes (CIRBP and STMN1) and tumor suppressor genes (CDKN1C and GADD45B) between normal and cancer cells, PDA-PEG/Cu2+ exhibited high selectivity in killing a broad spectrum of cancer cells, including drug resistant ones, while sparing normal cells. Therefore, the PDA-PEG/Cu2+ nanoparticles will provide a new paradigm for the cure of ovarian and other cancers. The ongoing research will investigate the cell killing mechanism of PDA-PEG/Cu2+ on the molecular level and test whether the cancer-cell-selective-killing effect of PDA-PEG/Cu2+ can be translated into a safe and effective cancer treatment tool in vivo.Acknowledgements
The authors want to thank the ASPIRE award from the Office of the Vice President for Research of The University of South Carolina and National Institutes of Health (5P20GM109091-02 and 1R15CA188847-01A1) for financial support of the research.Notes and references
- R. Siegel, J. Ma, Z. Zou and A. Jemal, CA-Cancer J. Clin., 2014, 64, 9–29 CrossRef PubMed.
- C. Holohan, S. Van Schaeybroeck, D. B. Longley and P. G. Johnston, Nat. Rev. Cancer, 2013, 13, 714–726 CrossRef CAS PubMed; X.-J. Liang, C. Chen, Y. Zhao and P. Wang, in Multi-Drug Resistance in Cancer, ed. J. Zhou, Humana Press, 2010, vol. 596, ch. 21, pp. 467–488 Search PubMed.
- E. K. Pauwels, P. Erba, G. Mariani and C. M. Gomes, Drug News Perspect., 2007, 20, 371–377 CAS.
- K. Masui, B. Gini, J. Wykosky, C. Zanca, P. S. Mischel, F. B. Furnari and W. K. Cavenee, Carcinogenesis, 2013, 34, 725–738 CrossRef CAS PubMed.
- P. M. Herst, J. E. Davis, P. Neeson, M. V. Berridge and D. S. Ritchie, Haematologica, 2009, 94, 928–934 CrossRef CAS PubMed; T. J. Mitchison, Mol. Biol. Cell, 2012, 23, 1–6 CrossRef PubMed.
- H. He, A. W. Cattran, T. Nguyen, A.-L. Nieminen and P. Xu, Biomaterials, 2014, 35, 9546–9553 CrossRef CAS PubMed; B. K. Remant, V. Chandrashekaran, B. Cheng, H. Chen, M. M. Pena, J. Zhang, J. Montgomery and P. Xu, Mol. Pharm., 2014, 11, 1897–1905 CrossRef PubMed.
- D. Ling, W. Park, S.-j. Park, Y. Lu, K. S. Kim, M. J. Hackett, B. H. Kim, H. Yim, Y. S. Jeon, K. Na and T. Hyeon, J. Am. Chem. Soc., 2014, 136, 5647–5655 CrossRef CAS PubMed; H.-m. Ding and Y.-q. Ma, Sci. Rep., 2013, 3, 2804 Search PubMed.
- K. C. Bahadur and P. Xu, Adv. Mater., 2012, 24, 6479–6483 CrossRef CAS PubMed; B. Cheng, B. Thapa, K. C. Remant and P. Xu, J. Mater. Chem. B, 2015, 3, 25–29 RSC.
- A. Gupte, S. Wadhwa and R. J. Mumper, Bioconjugate Chem., 2008, 19, 1382–1388 CrossRef CAS PubMed; A. Gupte and R. J. Mumper, Free Radicals Biol. Med., 2007, 43, 1271–1278 CrossRef PubMed.
- A. Gupte and R. J. Mumper, Cancer Treat. Rev., 2009, 35, 32–46 CrossRef CAS PubMed.
- F. Wang, P. Jiao, M. Qi, M. Frezza, Q. P. Dou and B. Yan, Curr. Med. Chem., 2010, 17, 2685–2698 CrossRef CAS PubMed; D. Chen and Q. P. Dou, Expert Opin. Ther. Targets, 2008, 12, 739–748 CrossRef PubMed.
- D. J. Lewis, P. Deshmukh, A. A. Tedstone, F. Tuna and P. O'Brien, Chem. Commun., 2014, 50, 13334–13337 RSC; C. Santini, M. Pellei, V. Gandin, M. Porchia, F. Tisato and C. Marzano, Chem. Rev., 2013, 114, 815–862 CrossRef CAS PubMed.
- S. Brem, S. A. Grossman, K. A. Carson, P. New, S. Phuphanich, J. B. Alavi, T. Mikkelsen, J. D. Fisher and f. t. N. A. t. B. T. T. C. Consortium, Neuro–Oncology, 2005, 7, 246–253 CrossRef CAS PubMed; M. T. Schweizer, J. Lin, A. Blackford, A. Bardia, S. King, A. J. Armstrong, M. A. Rudek, S. Yegnasubramanian and M. A. Carducci, Prostate Cancer Prostatic Dis., 2013, 16, 357–361 CrossRef PubMed.
- I. Kinoshita, L. James Wright, S. Kubo, K. Kimura, A. Sakata, T. Yano, R. Miyamoto, T. Nishioka and K. Isobe, Dalton Trans., 2003, 1993–2003 RSC.
- M. E. Anderson, Methods Enzymol., 1985, 113, 548–555 CAS.
- M. P. Gamcsik, M. S. Kasibhatla, S. D. Teeter and O. M. Colvin, Biomarkers, 2012, 17, 671–691 CrossRef CAS PubMed.
- P. Xu, G. K. Quick and Y. Yeo, Biomaterials, 2009, 30, 5834–5843 CrossRef CAS PubMed.
- T. Sakurai, H. Kashida, T. Watanabe, S. Hagiwara, T. Mizushima, H. Iijima, N. Nishida, H. Higashitsuji, J. Fujita and M. Kudo, Cancer Res., 2014, 74, 6119–6128 CrossRef CAS PubMed; T. Sakurai, N. Yada, T. Watanabe, T. Arizumi, S. Hagiwara, K. Ueshima, N. Nishida, J. Fujita and M. Kudo, Cancer Sci., 2015, 106, 352–358 CrossRef PubMed.
- J. Akhtar, Z. Wang, C. Yu, C.-S. Li, Y.-L. Shi and H.-J. Liu, BMC Cancer, 2014, 14, 28 CrossRef PubMed; B. E. Batsaikhan, K. Yoshikawa, N. Kurita, T. Iwata, C. Takasu, H. Kashihara and M. Shimada, Anticancer Res., 2014, 34, 4217–4221 Search PubMed.
- E. Kavanagh and B. Joseph, Biochim. Biophys. Acta, 2011, 1816, 50–56 Search PubMed; A. Borriello, I. Caldarelli, D. Bencivenga, M. Criscuolo, V. Cucciolla, A. Tramontano, A. Oliva, S. Perrotta and F. Della Ragione, Mol. Cancer Res., 2011, 9, 1269–1284 CrossRef CAS PubMed.
- L. Zhang, Z. Yang and Y. Liu, Exp. Biol. Med., 2014, 239, 773–778 CrossRef CAS PubMed; R. E. Tamura, J. F. de Vasconcellos, D. Sarkar, T. A. Libermann, P. B. Fisher and L. F. Zerbini, Curr. Mol. Med., 2012, 12, 634–651 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Details of the synthesis and characterization of the polymer, nanoparticles, and cell based in vitro assays. See DOI: 10.1039/c5bm00325c |
| This journal is © The Royal Society of Chemistry 2016 |