
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Endohedral dynamics of push–pull rotor-functionalized cages†
Marcel
Krick
a,
Julian
Holstein
a,
Christian
Würtele
b and
Guido H.
Clever
*a
aFaculty for Chemistry and Chemical Biology, TU Dortmund University, Otto-Hahn Str. 6, 44227 Dortmund, Germany. E-mail: guido.clever@tu-dortmund.de
bInstitute for Inorganic Chemistry, Georg-August-University Göttingen, Tammannstr. 4, 37077 Göttingen, Germany
First published on 26th July 2016
Abstract
A series of [Pd2L4] coordination cages featuring endohedral functionalities in central backbone positions was synthesized. Although attached via C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C double bonds, the substituents behave as molecular rotors. This is explained by their pronounced donor–acceptor character which lowers rotational barriers and allows for electronic control over the spinning rates inside the cage. The dynamic behaviour of the free ligands, assembled cages and host–guest complexes is compared with the aid of NMR experiments, X-ray structure analysis and molecular modelling.
C double bonds, the substituents behave as molecular rotors. This is explained by their pronounced donor–acceptor character which lowers rotational barriers and allows for electronic control over the spinning rates inside the cage. The dynamic behaviour of the free ligands, assembled cages and host–guest complexes is compared with the aid of NMR experiments, X-ray structure analysis and molecular modelling.
Confined molecular environments in proteins or folded oligonucleotides are often lined with a combination of rigidly fixed functionalities as well as flexible structural elements, such as dangling loops, amino acid residues or nucleobases.1 The controlled motion of such flexible attachments may gate the entrance of substrates, induce chemical conversions or transmit information in the form of allosteric regulation.2 In the field of supramolecular assembly, countless nanoscale cavities have been synthesized over the last decades and examined for guest exchange,3 selective recognition4 and catalytic conversions.5 In recent years, strategies for the incorporation of defined functionalities such as redox units,6 photo switches7 or specific binding sites have been developed in order to further increase the hosts' capabilities and equip the systems with advanced features such as stimuli-responsiveness,8 allosteric regulation9 or tuneable reactivity.10 Furthermore, the metal-mediated self-assembly of structures such as helicates,11 entangled architectures12 and coordination cages13 has turned out to be a particularly successful strategy for the generation of a large degree of structural and functional variety.14 Amongst those architectures, the installation of endohedral functionality remains a challenge and only few examples have been reported including inward-pointing hydrogen bond donors15 and acceptors,16 reactive residues inside helicates, cages and spheres,17,18 organocatalysts,19 metal-coordination sites,20 and others.21
With respect to the incorporation of dynamic functionality in self-assembled cavities, noteworthy examples from the area of solid materials include rotaxane–MOF hybrids22 and the family of flexible porous coordination compounds.23 In the context of discrete self-assemblies, combinations of rings24 and cages25 with mechanically interlocked substructures have been reported. We have recently introduced a sterically overcrowded [Pd2L4] host consisting of four ligands carrying bulky adamantyl ligands in a central position that were shown to flip between two degenerate conformations both in the free ligand as well as the assembled cage.26
Comparable to our previous dynamic [Pd2L4] system,26 the one under scrutiny here is synthetically derived from a flat acridone ligand that we have studied before in the context of stimuli responsive double-cages.27 Herein we report the installation of endohedral groups consisting of two electron-withdrawing substituents (COOR and/or CN) attached to an electron-rich backbone via a CC double bond (Fig. 1a).28 In contrast to the chemical drawing which suggests a totally flat system, steric demands (repulsion between backbone protons and the substituents of the double bond) cause the endohedral group to bend to one side of the backbone (Fig. 1b and 3a–c). Compounds based on the same structural motif have been previously examined as luminescent materials,29 conductive polymers30 and EPR labels for DNA.31 We were interested in the structural dynamics of the system in the context of supramolecular self-assembly, since the push–pull character of the underlying conjugated system (Fig. 1c) was expected to allow for rotation around the CC double bond in a kinetic regime comparable to the dynamics of supramolecular host–guest systems. Interestingly, rotation around these systems' CC double bond was already proposed by Dimroth and Criegee in 1957.28a Experimental evidence supporting this rotation, however, has remained scarce since the primarily studied symmetrically substituted systems (2 × CN or 2 × COOR) interconvert between degenerate conformations that cannot be distinguished by NMR methods. For one- or two-electron reduced molecules based on the same backbone, however, Akutagawa and Sutherland reported rotation.32
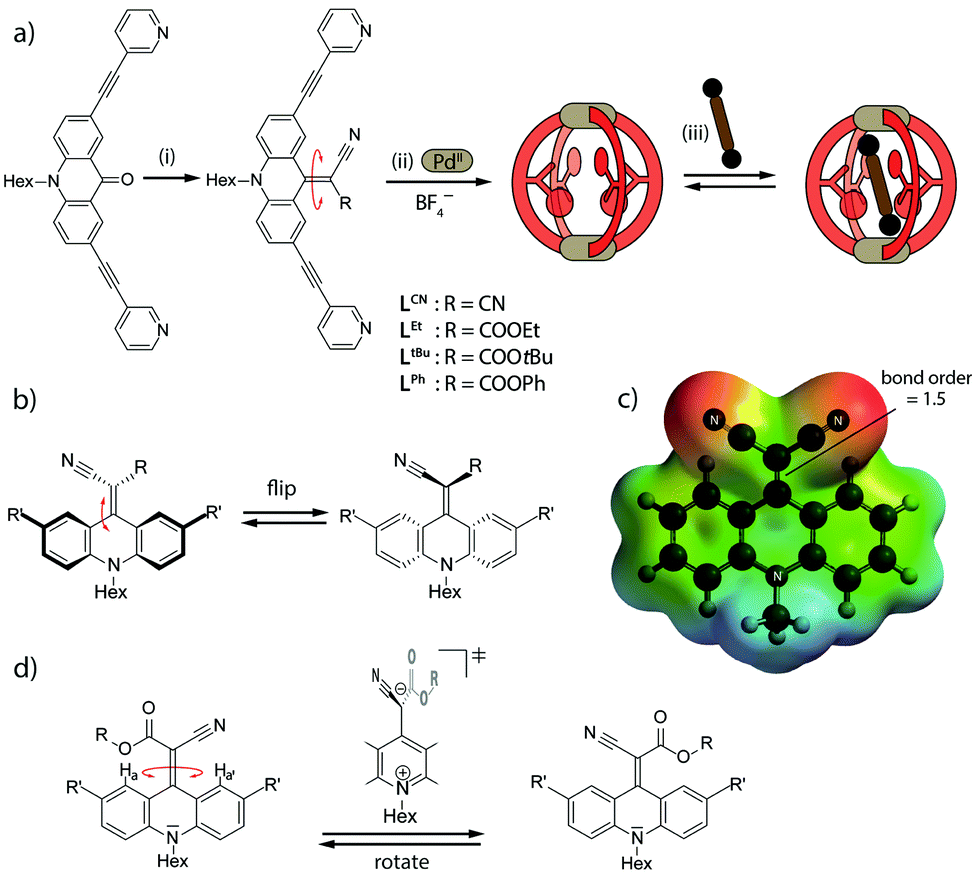 | ||
| Fig. 1 (a) Ligand synthesis, cage assembly and guest uptake: (i) malonic acid derivative, TiCl4, base, CH2Cl2 (details see ESI†) (ii) LR, [Pd(CH3CN)4](BF4)2, CD3CN, (iii) bissulfonate guest (NBu4 salt). (b) Flipping of the endohedral attachments, (c) electrostatic potential map of the dicyano ligand LCN highlighting the partial charge separation and CC double-bond weakening in the push–pull backbone, (d) rotation of the endohedral functionality via a charge-separated transition state. | ||
On the other hand, an internal motion that has already been described for such double-bonded compounds is flipping of the bent-out substituent between both faces of the backbone (Fig. 1b).32a We have observed a similar effect in the previously reported, electron rich adamantyl system, but ruled out any thermally induced rotation around the CC bond as evidenced by experimental and theoretical results.26
To the best of our knowledge, the rotation around the CC double bond in non-reduced, unsymmetrically acceptor-substituted acridinylidene systems has never been reported (Fig. 1d). Furthermore, the use of such a push–pull endohedral functionality inside a self-assembled molecular cavity is unprecedented. In order to compare the dynamics of the rotor functionality in discrete ligands, self-assembled cages and their host–guest complexes we synthesized one symmetrically substituted (2 × CN) ligand LCN and three unsymmetric derivatives LR (R = Et, tBu or Ph esters; Fig. 1a). Variable temperature NMR studies in different solvents revealed that the unsymmetrically substituted ligands indeed show rotational dynamics around the CC double bond depending on the conditions as evidenced by the splitting or coalescence of backbone protons Ha and Ha′ (Fig. 2 and ESI†). As expected, rotation is accelerated at higher temperatures and in more polar solvents (MeOH > DMSO > MeNO2 > MeCN > THF) that better stabilize the charge-separated, twisted transition state (Fig. 1d and ESI†). Exemplarily, Fig. 2 compares NMR spectra of ligand LtBu measured in THF at 198 K (Fig. 2a) and in acetonitrile at 298 K (Fig. 2b) with the former spectrum showing signal splitting for all aromatic protons, i.e. sharp resonances for protons Ha and Ha′, assignable to a locked conformation, and the latter spectrum revealing only one set of aromatic signals and complete broadening of Ha and Ha′ protons, indicating fast rotation in the NMR coalescence regime.
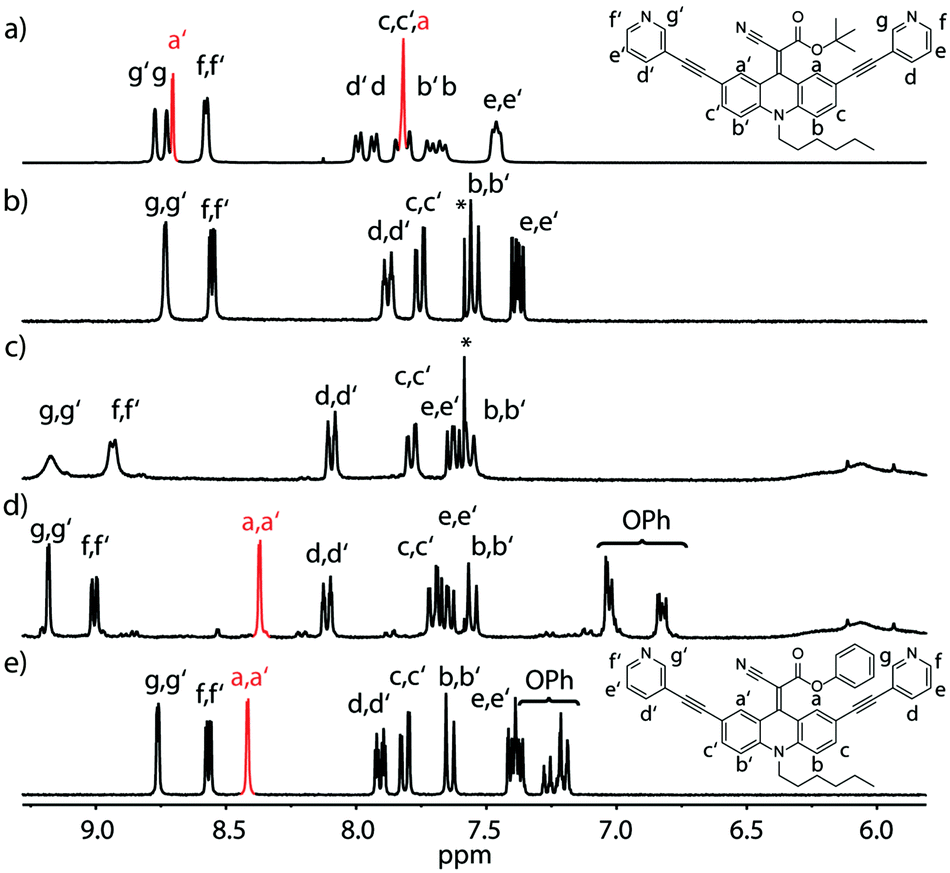 | ||
| Fig. 2 1H-NMR spectra of ligands LtBu and LPh and their corresponding [Pd2L4] cages. (a) Ligand LtBu in THF (400 MHz, 198 K, d8-THF), (b) ligand LtBu in acetonitrile, (c) cage [Pd2LtBu4], (d) cage [Pd2LPh4] and (e) ligand LPh (400 MHz, 298 K, CD3CN). Aromatic protons closest to the spinning rotor are marked in red (a/a′). Fast rotation results in one set of aromatic proton signals (e), slow rotation in two sets (a) and rotation with rates close to the NMR timescale in coalescence and signal broadening (b). * = CHCl3. | ||
Likewise, the rotation of the phenylester was found to be faster than the spinning observed in the more electron rich ethyl- and t-butyl-substituted ligands, as can be seen by comparing the spectra shown in Fig. 2b and e. The latter spectrum contains a single sharp signal assigned to the rapidly interchanging Ha and Ha′ positions in the phenyl-substituted ligand. In addition, the low-temperature NMR measurements of ligand LEt in THF reveal a splitting of the rotor's OC![[H with combining low line]](https://www.rsc.org/images/entities/char_0048_0332.gif) 2CH3 signal due to desymmetrization into a diastereotopic pair of protons sitting next to an unusual element of chirality (a bent double bond carrying distinguishable substituents). Around 213 K, the onset of signal splitting hints at a rapid flipping motion of the endohedral substituent while the discussed rotation is already extremely slow at these low temperatures (Fig. 1b and ESI†).
2CH3 signal due to desymmetrization into a diastereotopic pair of protons sitting next to an unusual element of chirality (a bent double bond carrying distinguishable substituents). Around 213 K, the onset of signal splitting hints at a rapid flipping motion of the endohedral substituent while the discussed rotation is already extremely slow at these low temperatures (Fig. 1b and ESI†).
A comparison of the UV-Vis spectra of all examined ligands further reveals that an increasing charge stabilization within the push–pull system goes along with increasing red-shift of the longest-wavelength maximum (tBu < Et < Ph < CN; ESI†).
Regardless of their dynamic peculiarities, all ligands readily formed dinuclear [Pd2L4] coordination cages upon treatment with the palladium salt [Pd(CH3CN)4](BF4)2 in acetonitrile as observed in the NMR spectra (Fig. 2c and d) and high-resolution ESI MS results (ESI†). Analogous to the behaviour of the free ligands, the spinning rates of the rotors inside the cages increase with increasing solvent polarity.
Single crystal X-ray structures were obtained for ligands LPh and LEt (Fig. 3a, b and ESI†). In comparison with a DFT calculated model of ligand LtBu (Fig. 3c), a substituent effect on the double-bond twisting and bending of the rotor moiety with respect to the backbone was clearly observed. The phenyl-substituted ester can stabilize negative charge density at the rotor-end of the double bond, hence leading to a rather large double bond twisting of 23° and a quite flat backbone structure (167° internal bending). In contrast, the more electron rich ethyl and tert-butyl substituents lead to twists of only 8° and 10°, respectively, and more pronounced backbone bending (148° and 150°).
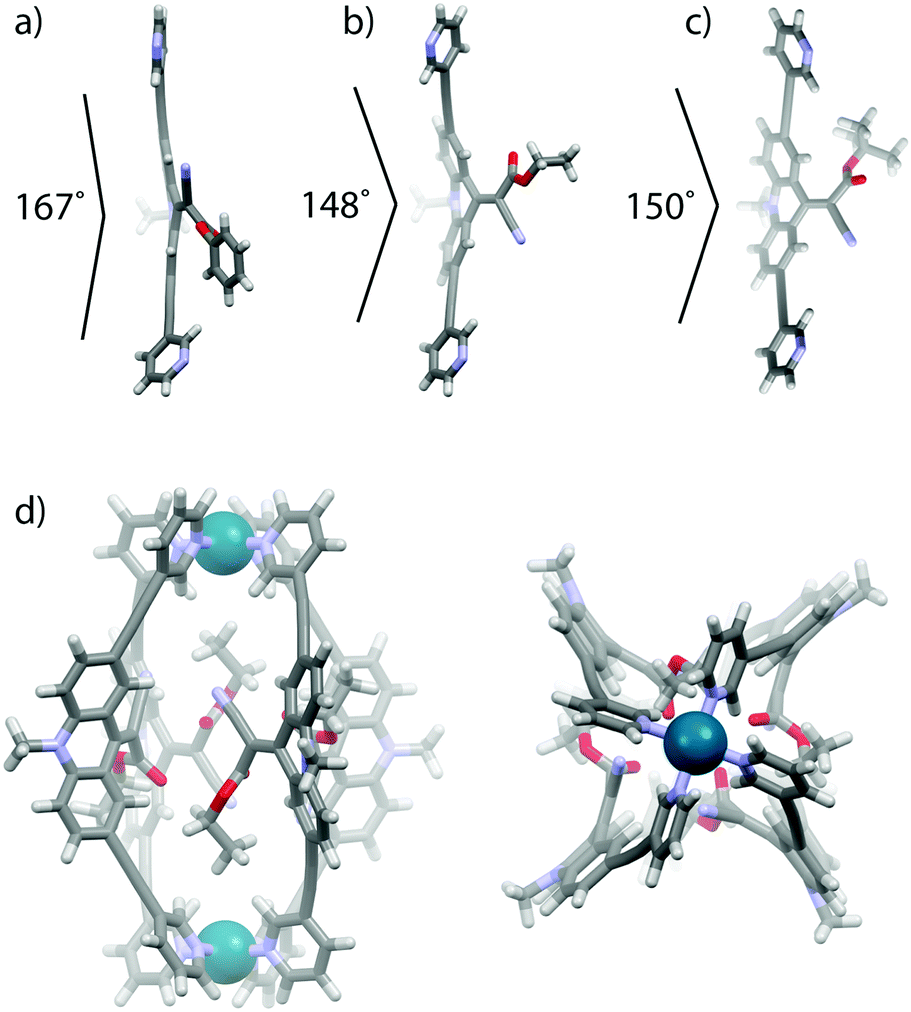 | ||
| Fig. 3 (a) X-Ray crystal structures of ligands (a) LPh and (b) LEt and (c) DFT calculated structures of ligand LtBu and (d) the lowest energy conformer of cage [Pd2LEt4]. | ||
Since no crystals suitable for X-ray structure determination of the self-assembled cages could be obtained, we calculated the lowest energy conformations of cage [Pd2LEt4] by dispersion corrected DFT (B3LYP-D3/def2-SVP in PCM-acetonitrile) geometry optimizations (Fig. 3d, ESI†). In the two lowest energy conformers (differing by less than 1 kcal mol−1), the four unsymmetric endhohedral attachments adopt a trans- or cis-up/down arrangement, respectively, with respect to the Pd–Pd axis. The calculation of the cis-conformer shown in Fig. 3d smoothly converged with a Ci symmetry in which two of the ethyl groups are slightly twisted away from the cage while the other two are oriented inside the cavity. A tentative arrangement with all four substituents showing in the direction of only one of the Pd centres was found to be of significant higher energy according to the calculations (ESI†).
Next, we examined changes of the dynamics of the rotor in dependence of its supramolecular surrounding by comparing the behaviour of the free ligand with the respective self-assembled cage and a host–guest complex. Fig. 4 summarizes the results for the LEt system. At room temperature (298 K) this ligand shows rotational dynamics with a rate close to the NMR timescale, thus the spectrum shows a single, broadened resonance as an average for protons Ha and Ha′ (Fig. 4a). Interestingly, assembly to the coordination cage results in a sharpening of this signal, indicating accelerated rotation, which we assume to be either caused by electronic changes in the conjugated system resulting from the coordination to the Pd(II) cations or by the cavity's special environment. As possible explanations for the latter effect we suggest an altered viscosity or polarity inside the cage confinement as compared to the free solution.
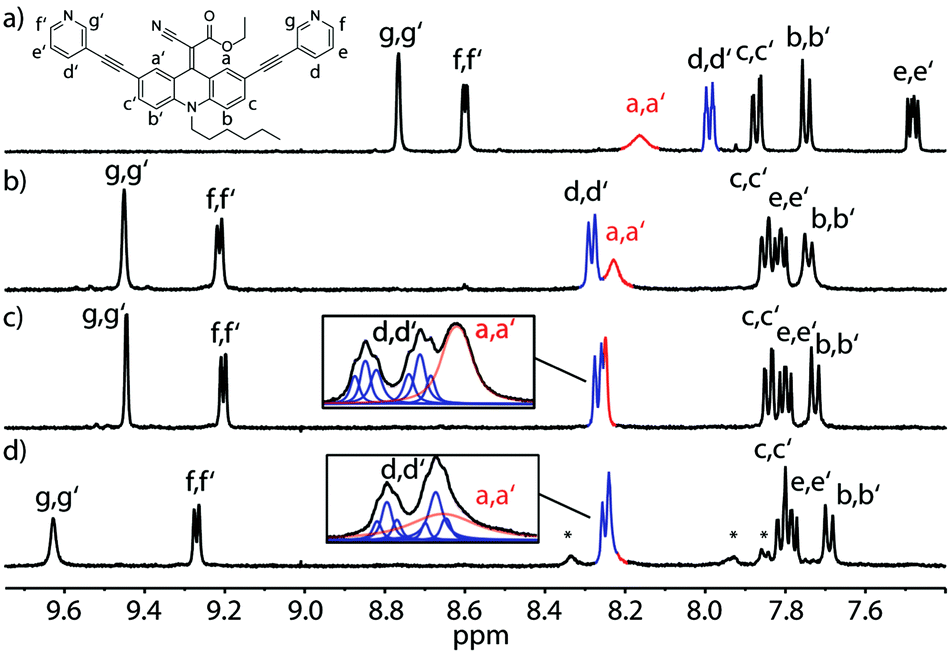 | ||
| Fig. 4 1H-NMR spectra of (a) ligand LEt, (b) cage [Pd2LEt4] at 298 K, (c) the same cage at 333 K and (d) host–guest complex [guest@Pd2LEt4] containing 1.0 eq. 2,7-naphthyl bissulfonate at 333 K (400 MHz, DMSO-d6). Cage formation and temperature increase leads to accelerated rotation and sharpening of the a/a′ signal (red). Guest uptake slows down spinning of the endohedral rotor, as shown by line fitting analysis of deconvoluted signals d/d′ (blue) and a/a′ (red). * = signals of the encapsulated guest. | ||
The signal further sharpens when the cage is heated to 333 K resulting from a thermally induced increase of the spinning frequency (Fig. 4c). Subsequently, we examined cage [Pd2LEt4] for the uptake of several anionic guest molecules (ESI†). A particularly interesting interaction was observed with the guest 2,7-naphthyl bissulfonate. In acetonitrile and nitromethane, this guest showed slow exchange on the NMR timescale, in DMSO it exhibited fast exchange, whilst In methanol, precipitation hampered the analysis. In the presence of the guest, the spectra measured at room temperature suffered from ample signal broadening which was partially resolved by conducting the measurements at 333 K (Fig. 4c, d and ESI†).
Most interestingly, a careful analysis of the overlapping NMR signals including the resonance of the a/a′ protons revealed, that the encapsulation of one molecule 2,7-naphthyl bissulfonate slows down the spinning of the endohedral rotors, accompanied by typical downfield shifts for the cage protons in contact with the guest (Fig. 4d). This guest-induced “breaking effect” nicely complements our previous findings concerning the flipping motion in a related adamantyl cage.26
In conclusion, we reported a series of self-assembled coordination cages based on a new type of endohedral push–pull rotor whose rotational dynamics are influenced by the assembly process, solvent polarity and derivatization of the electron-withdrawing moieties. In addition, the presence of an encapsulated guest was found to slow down the rotation. We see potential uses for such dynamic push–pull systems in the construction of stimuli responsive supramolecular systems, catalytic cavities and functional materials. Comparable sterically congested stilbene and imine derivatives based on tricyclic aromatic molecules featuring light-triggered rotation around CC or CN double-bonds have found remarkable application in supramolecular systems.33,34 Currently, we examine the photochemical and redox behaviour of herein reported compounds with the aim to equip them with stimuli-responsive control elements.
This work was supported by the DFG through Grants CL 489/2-1, SPP 1807 and IRTG 1422. We thank Dr H. Frauendorf (Georg-August University Göttingen) for measuring the ESI mass spectra and Rene Rieger, Dr Michael John and Dr Wolf Hiller (TU Dortmund) for their help with the NMR experiments.
Notes and references
- A. Liljas, L. Liljas, J. Piskur, G. Linblom, P. Nissen and M. Kjeldgaard, Textbook of Structural Biology, World Scientific Publishing, Singapore, 2009 Search PubMed.
- J.-P. Changeux and S. J. Edelstein, Science, 2005, 308, 1424–1428 CrossRef CAS PubMed.
- (a) L. Palmer and J. Rebek, Org. Biomol. Chem., 2004, 2, 3051–3059 RSC; (b) M. D. Pluth and K. N. Raymond, Chem. Soc. Rev., 2007, 36, 161–171 RSC.
- E. Persch, O. Dumele and F. Diederich, Angew. Chem., Int. Ed., 2015, 54, 3290–3327 CrossRef CAS PubMed.
- M. Yoshizawa, J. K. Klosterman and M. Fujita, Angew. Chem., Int. Ed., 2009, 48, 3418–3438 CrossRef CAS PubMed.
- V. Croué and S. Goeband M. Sallé, Chem. Commun., 2015, 51, 7275–7289 RSC.
- (a) M. Han, R. Michel, B. He, Y.-S. Chen, D. Stalke, M. John and G. H. Clever, Angew. Chem., Int. Ed., 2013, 52, 1319–1323 CrossRef CAS PubMed; (b) M. Han, Y. Luo, B. Damaschke, L. Gómez, X. Ribas, A. Jose, P. Peretzki, M. Seibt and G. H. Clever, Angew. Chem., Int. Ed., 2016, 55, 445–449 CrossRef CAS PubMed.
- A. J. McConnell, C. S. Wood, P. P. Neelakandan and J. Nitschke, Chem. Rev., 2015, 115, 7729–7793 CrossRef CAS PubMed.
- L. Kovbasyuk and R. Krämer, Chem. Rev., 2004, 104, 3161–3187 CrossRef CAS PubMed.
- J. Wang and B. L. Feringa, Science, 2011, 331, 1429–1432 CrossRef CAS PubMed.
- C. Piguet, G. Bernardinelli and G. Hopfgartner, Chem. Rev., 1997, 97, 2005–2062 CrossRef CAS PubMed.
- (a) J. E. Beves, B. A. Blight, C. J. Campbell, D. A. Leigh and R. T. McBurney, Angew. Chem., Int. Ed., 2011, 50, 9260–9327 CrossRef CAS PubMed; (b) R. S. Forgan, J.-P. Sauvage and J. F. Stoddart, Chem. Rev., 2011, 111, 5434–5464 CrossRef CAS PubMed.
- (a) M. Fujita, K. Umemoto, M. Yoshizawa, N. Fujita, T. Kusukawa and K. Biradha, Chem. Commun., 2001, 509–518 RSC; (b) S. J. Dalgarno, P. Power and J. L. Atwood, Coord. Chem. Rev., 2008, 252, 825–841 CrossRef CAS; (c) R. Chakrabarty, P. S. Mukherjee and P. J. Stang, Chem. Rev., 2011, 111, 6810–6918 CrossRef CAS PubMed; (d) R. Custelcean, Chem. Soc. Rev., 2014, 43, 1813–1824 RSC; (e) M. Han, D. M. Engelhard and G. H. Clever, Chem. Soc. Rev., 2014, 43, 1848–1860 RSC.
- M. M. J. Smulders, I. A. Riddell, C. Browne and J. R. Nitschke, Chem. Soc. Rev., 2013, 42, 1728–1754 RSC.
- C. Desmarets, G. Gontard, A. L. Cooksy, M. N. Rager and H. Amouri, Inorg. Chem., 2014, 53, 4287–4294 CrossRef CAS PubMed.
- J. E. M. Lewis, E. L. Gavey, S. A. Cameron and J. D. Crowley, Chem. Sci., 2012, 3, 778–784 RSC.
- (a) M. Tominaga, K. Suzuki, T. Murase and M. Fujita, J. Am. Chem. Soc., 2005, 127, 11950–11951 CrossRef CAS PubMed; (b) K. Suzuki, M. Kawano, S. Sato and M. Fujita, J. Am. Chem. Soc., 2007, 129, 10652–10653 CrossRef CAS PubMed; (c) T. Murase, S. Sato and M. Fujita, Angew. Chem., Int. Ed., 2007, 46, 1083–1085 CrossRef CAS PubMed.
- (a) A. M. Johnson, O. Moshe, A. S. Gamboa, B. W. Langloss, J. F. K. Limtiaco, C. K. Larive and R. J. Hooley, Inorg. Chem., 2011, 50, 9430–9442 CrossRef CAS PubMed; (b) A. M. Johnson and R. J. Hooley, Inorg. Chem., 2011, 50, 4671–4673 CrossRef CAS PubMed; (c) M. C. Young, A. M. Johnson, A. S. Gamboa and R. J. Hooley, Chem. Commun., 2013, 49, 1627–1629 RSC; (d) Q.-Q. Wang, S. Gonell, S. H. A. M. Leenders, M. Dürr, I. Ivanović-Burmazović and J. N. H. Reek, Nat. Chem., 2016, 8, 225–230 CrossRef CAS PubMed.
- P. Howlader, P. Das, E. Zangrando and P. S. Mukherjee, J. Am. Chem. Soc., 2016, 138, 1668–1676 CrossRef CAS PubMed.
- (a) M. D. Johnstone, E. K. Schwarze, G. H. Clever and F. M. Pfeffer, Chem. – Eur. J., 2015, 21, 3948–3955 CrossRef CAS PubMed; (b) S. Bandi, A. K. Pal, G. S. Hanan and D. K. Chand, Chem. – Eur. J., 2014, 20, 13122–13126 CrossRef CAS PubMed; (c) R. Saalfrank, V. Seitz, D. L. Caulder, K. Raymond, M. Teichert and D. Stalke, Eur. J. Inorg. Chem., 1998, 1313–1317 CrossRef CAS.
- S. Kubik, Top. Curr. Chem., 2012, 319, 1–34 CrossRef CAS PubMed.
- (a) S. J. Loeb, Chem. Commun., 2005, 1511–1518 RSC; (b) V. N. Vukotic, K. J. Harris, K. Zhu, R. W. Schurko and S. J. Loeb, Nat. Chem., 2012, 4, 456–460 CrossRef CAS PubMed.
- (a) S. Kitagawa and K. Uemura, Chem. Soc. Rev., 2005, 34, 109–119 RSC; (b) A. Schneemann, V. Bon, I. Schwedler, I. Senkovska, S. Kaskel and R. A. Fischer, Chem. Soc. Rev., 2014, 43, 6062–6096 RSC; (c) Z.-J. Lin, J. Lu, M. Hong and R. Cao, Chem. Soc. Rev., 2014, 43, 5867–5895 RSC; (d) X. Jiang, B. Rodriguez-Molina, N. Nazarian and M. A. Garcia-Garibay, J. Am. Chem. Soc., 2014, 136, 8871–8874 CrossRef CAS PubMed.
- H.-B. Yang, K. Ghosh, B. H. Northrop, Y.-R. Zheng, M. M. Lyndon, D. C. Muddiman and P. J. Stang, J. Am. Chem. Soc., 2007, 129, 14187–14189 CrossRef CAS PubMed.
- S. P. Black, A. R. Stefankiewicz, M. M. J. Smulders, D. Sattler, C. A. Schalley, J. R. Nitschke and J. K. M. Sanders, Angew. Chem., Int. Ed., 2013, 52, 5749–5752 CrossRef CAS PubMed.
- S. Löffler, J. Lübben, A. Wuttke, R. A. Mata, M. John, B. Dittrich and G. H. Clever, Chem. Sci., 2016, 7, 4676–4684 RSC.
- S. Löffler, J. Lübben, L. Krause, D. Stalke, B. Dittrich and G. H. Clever, J. Am. Chem. Soc., 2015, 137, 1060–1063 CrossRef PubMed.
- (a) O. Dimroth and R. Criegee, Chem. Ber., 1957, 90, 2207–2215 CrossRef CAS; (b) O. Tsuge, Bull. Chem. Soc. Jpn., 1973, 46, 283–285 CrossRef CAS.
- W. Chen, S. Wang, G. Yang, S. Chen, K. Ye, Z. Hu, Z. Zhang and Y. Wang, J. Phys. Chem. C, 2016, 120, 587–597 CAS.
- W.-C. Chow, G.-J. Zhou and W.-Y. Wong, Macromol. Chem. Phys., 2007, 208, 1129–1136 CrossRef CAS.
- M. Beyer, J. Fritscher, E. Feresin and O. Schiemann, J. Org. Chem., 2003, 68, 2209–2215 CrossRef CAS PubMed.
- (a) T. Takeda, H. Sugihara, Y. Suzuki, J. Kawamata and T. Akutagawa, J. Org. Chem., 2014, 79, 9669–9677 CrossRef CAS PubMed; (b) K. D. Thériault, C. Radford, M. Parvez, B. Heyne and T. C. Sutherland, Phys. Chem. Chem. Phys., 2015, 17, 20903–20911 RSC.
- (a) N. Koumura, R. W. Zijlstra, R. A. van Delden, N. Harada and B. L. Feringa, Nature, 1999, 401, 152–155 CrossRef CAS PubMed; (b) A. A. Kulago, E. M. Mes, M. Klok, A. Meetsma, A. M. Brouwer and B. L. Feringa, J. Org. Chem., 2010, 75, 666–679 CrossRef CAS PubMed.
- L. Greb and J.-M. Lehn, J. Am. Chem. Soc., 2014, 136, 13114–13117 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures, characterization, NMR and mass spectra, X-ray crystallography and computational details. CCDC 1478914 and 1478915. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6cc04155h |
| This journal is © The Royal Society of Chemistry 2016 |