
Graphene oxide inhibits hIAPP amyloid fibrillation and toxicity in insulin-producing NIT-1 cells†
Praveen
Nedumpully-Govindan
a,
Esteban N.
Gurzov
bc,
Pengyu
Chen
d,
Emily H.
Pilkington
e,
William J.
Stanley
bc,
Sara A.
Litwak
b,
Thomas P.
Davis
ef,
Pu Chun
Ke
*e and
Feng
Ding
*a
aDepartment of Physics and Astronomy, Clemson University, Clemson, SC 29634, USA. E-mail: fding@clemson.edu
bSt Vincent's Institute of Medical Research, 9 Princes Street, Fitzroy, VIC 3065, Australia
cDepartment of Medicine, The University of Melbourne, St. Vincent's Hospital, Melbourne, Australia
dDepartment of Mechanical Engineering, University of Michigan, Ann Arbor, MI 28109, USA
eARC Center of Excellence in Convergent Bio-Nano Science and Technology, Monash Institute of Pharmaceutical Sciences, Monash University, 381 Royal Parade, Parkville, VIC 3052, Australia. E-mail: pu-chun.ke@monash.edu
fDepartment of Chemistry, University of Warwick, Gibbet Hill, Coventry, CV4 7AL, UK
First published on 24th November 2015
Abstract
Human islet amyloid polypeptide (hIAPP or amylin) aggregation is directly associated with pancreatic β-cell death and subsequent insulin deficiency in type 2 diabetes (T2D). Since no cure is currently available for T2D, it is of great benefit to devise new anti-aggregation molecules, which protect β-cells against hIAPP aggregation-induced toxicity. Engineered nanoparticles have been recently exploited as anti-aggregation nanomedicines. In this work, we studied graphene oxide (GO) nanosheets for their potential for hIAPP aggregation inhibition by combining computational modeling, biophysical characterization and cell toxicity measurements. Using discrete molecular dynamics (DMD) simulations and in vitro studies, we showed that GO exhibited an inhibitory effect on hIAPP aggregation. DMD simulations indicated that the strong binding of hIAPP to GO nanosheets was driven by hydrogen bonding and aromatic stacking and that the strong peptide–GO binding efficiently inhibited hIAPP self-association and aggregation on the nanosheet surface. Secondary structural changes of hIAPP upon GO binding derived from DMD simulations were consistent with circular dichroism (CD) spectroscopy measurements. Transmission electron microscopy (TEM) images confirmed the reduction of hIAPP aggregation in the presence of GO. Furthermore, we carried out a cell toxicity assay and found that these nanosheets protected insulin-secreting NIT-1 pancreatic β-cells against hIAPP-induced toxicity. Our multidisciplinary study suggests that GO nanosheets have the potential to be utilized as an anti-aggregation nanomedicine itself in addition to a biosensor or delivery vehicle for the mitigation of T2D progression.
Introduction
Aberrant aggregation of peptides and proteins into insoluble fibrils is associated with a number of age-related diseases and medical conditions, including Alzheimer's and Parkinson's diseases and type 2 diabetes (T2D).1 Human islet amyloid polypeptide (hIAPP or human amylin), which is secreted together with insulin by pancreatic islets into the blood circulation, is highly aggregation prone.2 hIAPP oligomers formed en route to aggregation are toxic to insulin-secreting pancreatic β-cells3 and are believed to be responsible for pancreatic β-cell death and insulin deficiency which are commonly observed in T2D patients.4 Furthermore, a strong correlation exists between the extent of pancreatic amyloid deposition and the severity of T2D.5 Hence, inhibition of hIAPP aggregation is proposed as a viable medical intervention to control or prevent T2D progression.A number of studies have targeted different stages of hIAPP aggregation with an ultimate goal of preventing the aggregation and reducing its corresponding cytotoxicity. For example, naturally occurring polyphenols such as EGCG (green tea), curcumin (turmeric) and resveratrol (grape skin) display anti-oxidation and anti-aggregation properties.6–8 Zinc ions, which are present in hIAPP-storing β-cell granules, exhibit concentration-dependent promotion and an inhibition effect on hIAPP aggregation,9 implicating that carefully determined zinc supplementation may be useful for managing T2D.10 Similarly, insulin, which is co-stored and co-secreted with hIAPP, has an inhibitory effect on peptide aggregation.11 Some amylin variants and mutants are not only non-aggregating, but also inhibitors of wild type hIAPP.12–14 We have recently used computer simulations combined with experimental methods to delineate the effects of many of these agents on hIAPP aggregation.15–17
Another promising avenue of anti-aggregation research is the use of engineered nanoparticles. Nanoparticles’ large surface-to-bulk ratio and easiness for surface modification make them attractive anti-aggregation agents or carriers of such agents. It is known that nanoparticles exhibit both aggregation promotion and inhibitory effects, depending on their chemical composition, physicochemical properties and the nature of the proteins involved.18–20 One of the most promising nanoparticles for biomedical applications is graphene oxide (GO) nanosheets.21 The effects of carbon-based nanoparticles including GO nanosheets on amyloid-β peptide have been studied by several groups,22–25 demonstrating the inhibitory effect of such nanostructures on amyloid-β peptide aggregation. Since the inhibitory effects vary from case to case, we propose to investigate whether GO exhibits any inhibitory effect on hIAPP aggregation. Recently, molecular dynamics simulations indicated that carbon nanoparticles including graphene (but not GO) strongly bind and inhibit the aggregation of amyloidogenic motif of hIAPP (residues 22–28).26,27 However, the hydrophobic graphene often needs to be functionalized/oxidized in order to achieve bioavailability. To our best knowledge, there are no reported experimental or computational studies on the effect of GO on the aggregation of full-length hIAPP which has direct biological and medicinal relevance. Herein, we combined computer simulations with biophysical and in vitro studies, which synergistically demonstrated that GO strongly bound to hIAPP and inhibited its aggregation. The strong binding was governed by hydrogen bonding, and to a lesser extent, hydrophobic interaction between GO and hIAPP. Furthermore, our toxicity assay on insulin-secreting NIT-1 cells revealed that GO protected the cells against hIAPP-induced toxicity. Our results suggest that the inhibitory effect of GO on peptide aggregation is rather generic and not limited to amyloid-β alone, and that GO has the potential for use not only as a nanoparticle carrier,28,29 but also as an hIAPP aggregation inhibitor.
Results
Inhibition of hIAPP aggregation by GO
The zeta potential of the hIAPP and GO was determined to be +4.7 (±0.4) mV and −42.0 (±5.6) mV, respectively, in Milli-Q water at room temperature. Transmission electron microscopy (TEM) imaging showed the formation of amyloid fibrils after overnight incubation of hIAPP in water at 0.1 mg mL−1 (25 μM), ranging over 1 μm in length (Fig. 1A) and 7–14 nm in width. This is consistent with the understanding that hIAPP monomers and oligomers at micromolar concentrations are prone to fibrillation through molecular assembly. GO nanosheets (0.1 mg mL−1), on the other hand, were seen as irregularly shaped flakes each of ∼2 μm in size (Fig. 1B), consistent with the specifications provided by the manufacturer. In the sample where 0.1 mg mL−1 solutions of freshly prepared hIAPP and GO were mixed overnight with an equal volume, few to no fibrils were spotted in most cases (Fig. 1C and D), while occasionally short fibrils on the order of 100 nm in length were seen (Fig. 1E). The existence of hIAPP fibrils in the presence of GO was possibly due to the fact that hIAPP is highly aggregation prone and the GO-unbound peptides in solution were still able to nucleate the amyloid fibrils. These observations suggest that GO nanosheets could indeed inhibit the formation of hIAPP fibrils in the aqueous phase. | ||
| Fig. 1 TEM imaging of hIAPP aggregation and its inhibition by GO nanosheets. (A) hIAPP amyloid fibrils, at ∼1 μm or longer. (B) GO nanosheets, each of an irregular flake-like shape. hIAPP fibrils: green arrows. GO: orange arrows. GO creases: red arrows. (C–E) TEM images of hIAPP incubated with GO, showing no fibrils most of the time (C). Occasionally, much shortened fibrils were spotted on the surfaces of the GO (D and E). (F) Raman spectra of hIAPP, GO, and hIAPP incubated with GO at different mass ratios. The purple line denotes the hIAPP amide peak (1642.9 cm−1). Incubation times for (A–F) overnight. hIAPP concentrations in (A) and (C–F) 0.1 mg mL−1 (25 μM). GO concentration in (B) and (C–F): 0.1 mg mL−1. | ||
Thioflavin T (ThT) assay is a major tool for quantifying amyloid fibril formation, but it was not used in this study due to the stacking between the aromatic moieties of ThT and GO to skew experimental observations. The binding of hIAPP and GO was instead further confirmed by Raman spectroscopy. As shown in Fig. 1F, hIAPP (purple trace) displayed the prototypical amide peak30 at 1642.9 cm−1, while the D (impurity) and G (primary mode due to sp2 bonded carbon atoms) bands of GO,31 were determined at 1337.7 cm−1 and 1613.9 cm−1, respectively (gray trace). In comparison, mixing GO and hIAPP at a weight ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 rendered a spectrum characteristic of both the D band of GO and the amide peak of hIAPP (orange trace). Upon mixing GO with hIAPP at a GO:hIAPP weight ratio of 1:5, the amide peak was lost, while the spectrum of the mixture more closely resembled the D band of GO (blue trace). The loss of the hIAPP spectral identity with the increasing percentage of the peptide suggests a change in the hIAPP structure, which was evidently induced by GO–hIAPP binding.
:1 rendered a spectrum characteristic of both the D band of GO and the amide peak of hIAPP (orange trace). Upon mixing GO with hIAPP at a GO:hIAPP weight ratio of 1:5, the amide peak was lost, while the spectrum of the mixture more closely resembled the D band of GO (blue trace). The loss of the hIAPP spectral identity with the increasing percentage of the peptide suggests a change in the hIAPP structure, which was evidently induced by GO–hIAPP binding.
The inhibition mechanism of GO derived from DMD simulations
To investigate the mechanism of hIAPP aggregation inhibition by GO nanosheets we carried out atomistic discrete molecular dynamics (DMD) simulations of an hIAPP molecule with and without the introduction of a GO nanosheet (Fig. 2A and B). The framework of DMD is similar to conventional molecular dynamics (MD), except that in DMD discrete potential energy functions are used instead of continuous potential energy functions. Interatomic interactions in DMD simulations include van der Waals, solvation, hydrogen bonding, and screened electrostatics (see in the ESI,† for details). The DMD method has been successfully applied by our group and others to study protein self-association and amyloid aggregation.15,32–36 In the control simulations without a GO nanosheet, hIAPP mainly sampled α-helix and random coil conformations (Fig. 2). About 30% of the residues, mostly those near the N-terminal, formed an α-helix (Fig. 2A and B), in agreement with previous studies,15,37–39 while about 50% of the residues formed random coils.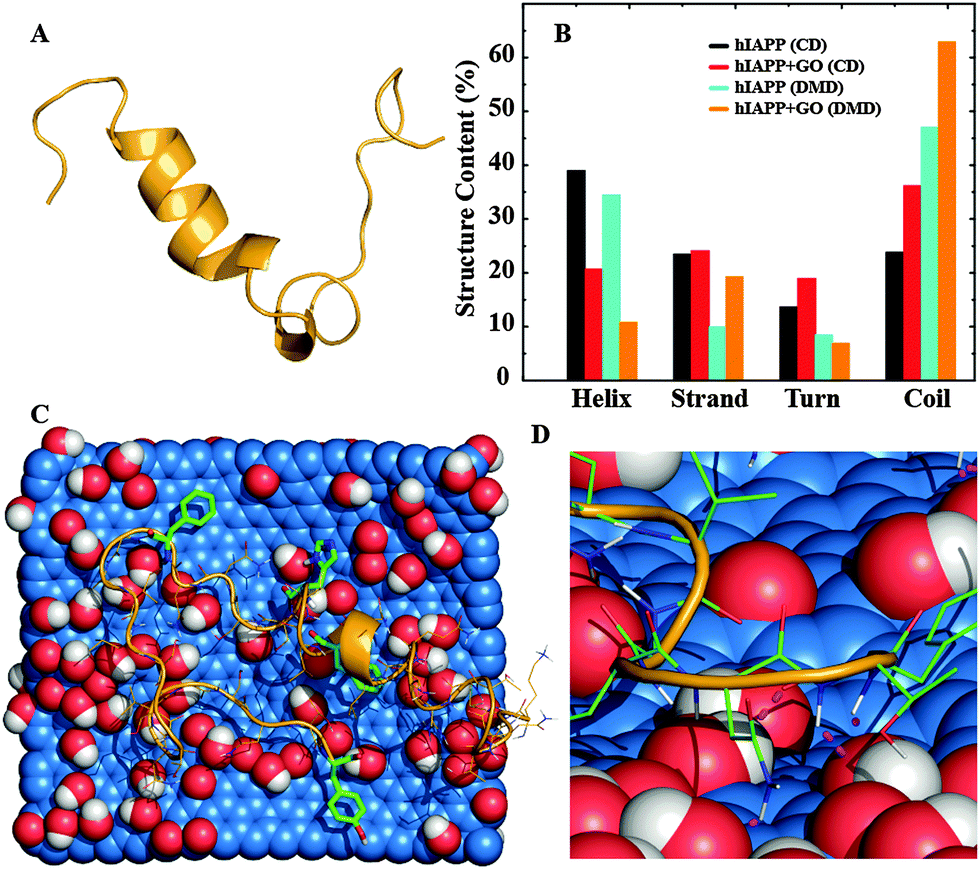 | ||
| Fig. 2 The effect of GO binding on hIAPP. (A) A representative snapshot of hIAPP after 106 steps of DMD simulation. (B) The estimates of secondary structure contents of hIAPP without or with GO nanosheets, obtained from circular dichroism spectra (CD) and DMD simulations. The helical content is reduced due to GO binding. (C) A snapshot showing the typical hIAPP–GO structure. The carbon, oxygen and hydrogen atoms of GO are shown as spheres in blue, red and white, respectively. Aromatic residues of hIAPP, highlighted using stick representation, occasionally formed pi–pi stacking with the GO carbon atoms. (D) The binding is strengthened by hydrophilic interactions. Hydrogen bonds between GO and hIAPP (both main chain and side chains) are shown using pink dotted lines. | ||
When simulations were initiated by placing the hIAPP randomly around a 32.6 Å × 38.1 Å GO nanosheet in a 100 Å cubic simulation box under periodic boundary conditions, the peptide was found to bind the GO nanosheet in ten independent simulations within 50 ns, suggesting a strong binding affinity between the two species. Even though the time hIAPP chains took to make an initial contact with the GO in different simulations varied due to diffusion, hIAPPs remained bound until the end of the simulations. Noticeably, GO binding considerably reduced the helical content of hIAPP to ∼10%, accompanied by an increase of coil structure to ∼62% (Fig. 2). We carried out circular dichroism (CD) spectroscopy measurements to experimentally examine the protein secondary structure in the presence or absence of nanomaterials.40 A reduction of the helical content from ∼39% to ∼21% with a corresponding increase of the coil content from ∼24% to ∼36% was observed when GO nanosheets were added to hIAPP (Fig. 2B and Fig. S1 in the ESI†), matching the same trends as observed in the DMD simulations. Therefore, our experimental and computational studies suggested that the GO nanosheets indeed strongly bound hIAPP and caused losses in the peptide secondary structure. To understand how the peptide bound the nanosheet, we computed the binding frequency of each hIAPP residue with GO over the last quarter of the simulations where the peptides were GO-bound (Fig. S2 in the ESI†). Both N- and C-terminal residues, 1–4 and 32–37 respectively, displayed higher binding frequencies than other residues in the middle. Many of these terminal residues as well as the residues in the middle that displayed high binding frequencies (e.g. Q10, N14, H18, N22, S28) were polar, which could bind the hydroxyl groups of GO via hydrogen bonds (Fig. 2D). Importantly, aromatic residues including F15, H18, F23, and Y37 also displayed high GO-binding frequencies, albeit lower than hydrophilic residues. Therefore, hydrogen bonding and aromatic-stacking were determined as the driving force for hIAPP–GO binding.
To study how GO binding affected the hIAPP–hIAPP interaction and thereby hIAPP aggregation, we simulated multiple (2, 4 and 6) hIAPPs in the presence and also in the absence of a GO nanosheet. For each molecular system, we performed ten independent simulations where the peptides were randomly positioned around the nanosheet initially. A representative snapshot of six hIAPPs with a GO nanosheet from the simulations is illustrated in Fig. 3A. As the number of hIAPP chains increased, the total number of GO–hIAPP contacts was increased as expected, but the per chain contact number was decreased (Fig. S3 in the ESI†). More importantly, with increasing number of hIAPPs, the helical structure content loss due to GO binding was partially recovered and correspondingly the random coil content was reduced (Fig. 3B). For the largest system that we simulated (six hIAPP molecules with GO), the average helical content was ∼25% compared to ∼10% in the hIAPP1–GO and ∼30% in the hIAPP alone systems. Interestingly, the secondary structure contents upon multiple peptides binding with GO in the DMD simulations were more consistent with the corresponding values in the experimental measurement (Fig. 2B). There are two reasons for this recovery of the secondary structure content. First, as more hIAPPs were added, not all of them had room to bind GO as suggested by the decreased average number of GO-contacts per chain (Fig. S3 in the ESI†). Secondly, the hIAPP–hIAPP association may also stabilize the α-helix as observed previously.16,41
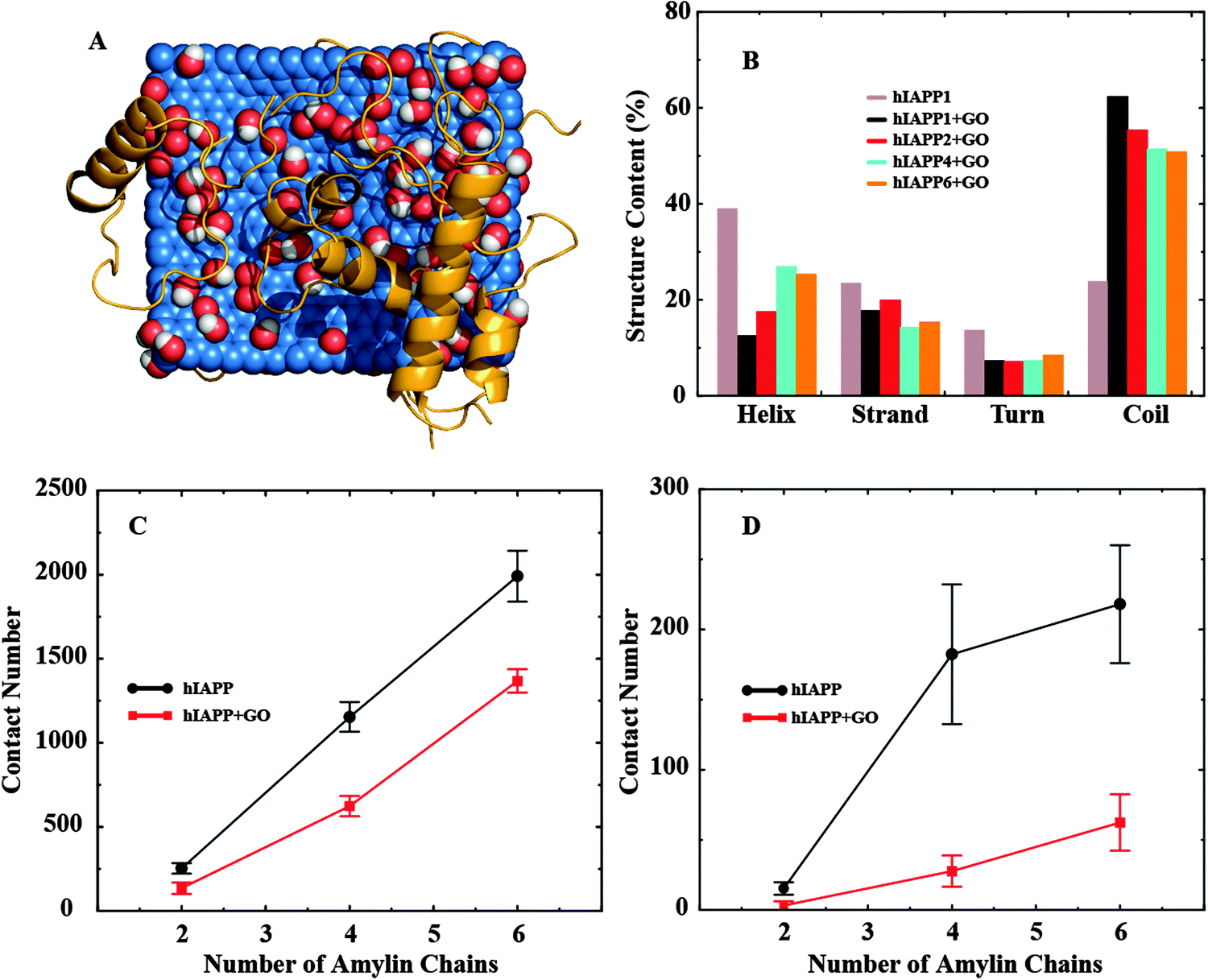 | ||
| Fig. 3 (A) A snapshot showing the binding of six hIAPPs to GO. hIAPPs clearly are more helical than in Fig. 2A. (B) The secondary structure contents (per chain) of multiple hIAPPs binding to GO nanosheets. The (C) total and (D) amyloidogenic inter-peptide contact number as a function of the number of hIAPPs. The contact numbers are consistently smaller in the presence of GO nanosheets. | ||
We computed the hIAPP–hIAPP contact number as an indicator for hIAPP self-association. We found that, both in the absence and in the presence of GO, the contact number averaged over independent simulations increased as the number of hIAPPs was increased (Fig. 3C and D). However, compared to the hIAPP alone (no GO nanosheet) case, the hIAPP–hIAPP contact number was significantly smaller in the presence of GO nanosheets (Fig. 3C). This is because hIAPP had a strong affinity to bind the GO nanosheets through both hydrogen bonding and aromatic interactions, effectively reducing the hIAPP–hIAPP interactions. Residues 22–29 of hIAPP are particularly prone to aggregation, and are known as the amyloidogenic sites of the peptide.42,43 In addition to the total contact number, we also calculated the amyloidogenic contact number, defined as the inter-chain contact number between residues 22–29 (Fig. 3D). We found that, similar to the total contact number, the average amyloidogenic contact number was also significantly smaller in the presence of GO nanosheets. More importantly, the reduction of the amyloidogenic contact number upon GO-binding (by ∼71%) was much greater compared to the total contact number (∼31%) (Fig. 3D). Our simulations thus indicate that strong binding and sequestering of hIAPPs by GO nanosheets caused a reduction in the hIAPP–hIAPP association, especially in the amyloidogenic region, thereby reducing peptide aggregation on the nanosheet surface.
GO protects β-cells from hIAPP-induced cell death
To evaluate whether the inhibition of hIAPP aggregation by GO may result in reduced cytotoxicity, we performed a cellular study using NIT-1 cells as the model system.44 This β-cell line shows β-granulation and insulin secretory responsiveness to glucose. NIT-1 cells were treated for 24 h with 10 μM hIAPP, 100 μg mL−1 GO, or a combination of both. To study viability, the cells were incubated with the DNA binding dyes Hoechst 33342 and propidium iodide. Hoechst 33342 freely diffuses and enters cells with intact or damaged membranes (live cells), staining DNA blue. Propidium iodide is a highly polar dye impermeable to cells with preserved membranes and only stains DNA of dying/death cells as red. Viable cells were identified by their intact nuclei with blue fluorescence, whereas cell death was quantified by blue–red fluorescence or by fragmented blue nuclei.45 hIAPP showed significant cytotoxicity with ∼60% cell death after 24 h of exposure (Fig. 4). Pre-exposure of hIAPP to GO protected NIT-1 cells from hIAPP-mediated toxicity, reducing from ∼60% to ∼12% (Fig. 4). GO alone showed minimal cytotoxicity to NIT-1 cells with only ∼2% cell death observed. Therefore, the inhibition of hIAPP aggregation by GO indeed reduced the aggregation-induced cytotoxicity in cultured β-cells.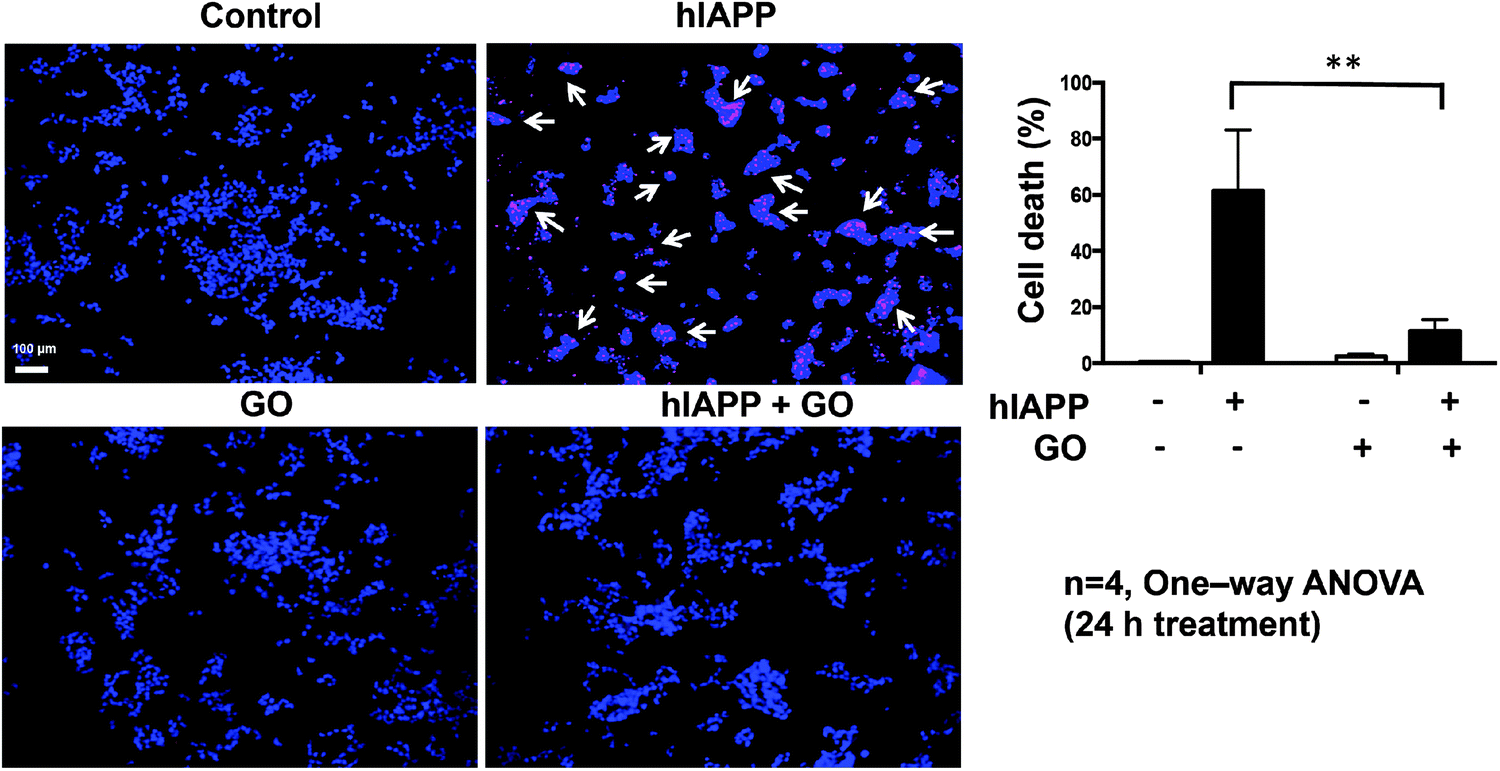 | ||
| Fig. 4 GO protects β-cells from hIAPP-induced cell death. NIT-1 cells were untreated (control) or incubated with GO, hIAPP or combination of both for 24 h. Cell death was evaluated by Hoechst-33342 (blue)/propidium iodide (red). White arrows indicate propidium iodide positive cells. Data shown are means ± SEM of 4 independent experiments. **p < 0.01. Scale bar: 100 μm. | ||
Discussion
The effects of nanoparticles on protein aggregation may vary, depending on the physicochemical nature of the nanoparticles and the protein in question.18,19 Pervious studies showed that GO nanosheets exerted an inhibitory effect on the aggregation of amyloid-β.22 In this work, we used combined experimental and computational methods to show that GO nanosheets inhibited hIAPP aggregation. Our computational study indicates that the aggregation-inhibition effect of GO was due to its amphiphilic structure with abundant hydrogen bond donors and acceptors (i.e., hydroxyls and epoxide oxygen atoms on the surface, Fig. 2C and D) and the hydrophobic graphene moieties favoring aromatic interactions, which in turn rendered a strong binding affinity for amphiphilic proteins and peptides. Using computer simulations, we have recently shown that strong binding of peptides to nanoparticles resulted in aggregation inhibition, while weak binding could have a dual aggregation promoting and inhibiting effect depending on the relative protein–nanoparticle concentrations (Ding et al., unpublished data). Therefore, our studies and the studies by others22 suggest that the aggregation inhibition effect of GO nanosheets may be generic, not limited to one type of protein or peptide.In previous computational studies concerning the interactions between graphene (not GO) and hIAPP 22–28 (i.e. the amyloidogenic region), a strong binding governed by aromatic pi–pi stacking was observed.26,27 Due to the strong binding between peptides and graphene, the peptides adopted coil-like structures by maximizing interactions with the graphene sheet rather than with themselves. In our simulations of hIAPP binding with GO, we found that hydrogen bonding was the driving force while aromatic stacking also contributed significantly to the binding (Fig. S2 in the ESI†). The N-terminal helix of hIAPP was mostly retained and residues 22–29 bound less frequently to GO compared to the two termini of the peptide. This difference was due to the amphiphilic structural properties of GO. According to our simulation study with different hIAPP to GO concentration ratios, the average number of contacts between GO and the peptide featured a high-to-low transition near ∼3 hIAPP chains per GO nanosheet (Fig. S3, ESI†). These results suggest an optimal GO concentration for hIAPP aggregation inhibition, corresponding to a GO:hIAPP mass ratio of ∼0.75 or a GO surface area of 8.3 nm2 per hIAPP chain.
It is worth examining whether other anti-aggregation molecules have similar inhibition mechanisms. A set of small-molecule polyphenols exhibit anti-aggregation properties against several amyloidogenic proteins including hIAPP.6–8 Recently, we showed that polyphenolic curcumin and resveratrol inhibit hIAPP aggregation by stabilizing “off-pathway” hIAPP oligomers, where the small molecules formed a nanosized core and peptides bind to the surface.16 The stabilizing factors for the nano-assembly included inter-molecular hydrogen bonds and π–π stacking between aromatic residues such as Phe15 and Phe23 of hIAPP and the phenols, the same interactions important for hIAPP aggregation inhibition by GO. The strong binding of peptides with either the GO nanosheet or the polyphenol nanocluster resulted in reduced hIAPP self-association and subsequent inter-chain β-sheet formation (Fig. 3). As a result, the peptide oligomers in the molecular complex could not serve as the conformational template for the conformational conversion of incoming peptides and the growth of amyloid fibrils.34,46 The prevention of hIAPP self-association and aggregation by competitive binding has also been observed in protein-based inhibitors such as mutant hIAPPs,12 insulin11 and rIAPP.47
GO nanosheets have been shown in the literature to have great potential for drug delivery.28,29 The good water solubility, large surface area and biocompatibility may be further exploited for delivering small molecules such as polyphenols and other hIAPP aggregation inhibitors. The surface of GO may also be functionalized for cell and tissue targeting.48,49 Since our study has demonstrated the anti-aggregation properties of GO, these planar nanostructures with great flexibility in size and surface modification may have an added advantage both as a drug delivery vehicle with specific targeting and also an anti-aggregation medicine. Due to the complex architecture of pancreatic islets and cell signaling within the islets, however, there remain hurdles to be overcome by future studies towards the delivery of bare or functionalized GO ex vivo and in vivo. These efforts may be especially focused on tailoring the physicochemical properties of GO and GO derivatives for specific pancreatic targeting.
In summary, we investigated the inhibitory effects of GO nanosheets on hIAPP aggregation. Our simulation results showed that hIAPP strongly bound GO sheets due to the both hydrogen bonding and aromatic interactions. A reduction of the helical contents and an increase of the random coil structure of hIAPP were observed in the presence of GO, both in experiments (CD) and in DMD simulations. DMD simulations of multiple hIAPPs with and without GO nanosheets further vindicated that GO-binding prevented hIAPP aggregation. This binding between hIAPP and GO was confirmed by Raman spectroscopy. Our TEM study confirmed the reduced hIAPP aggregation in the presence of GO nanosheets. Furthermore, GO nanosheets protected insulin-producing NIT-1 cells from hIAPP-induced toxicity, likely through the disruption of hIAPP fibrillation. Therefore, our multiscale and multidisciplinary studies combining in silico and in vitro approaches point to the promise of GO as a nanotechnology for the treatment of T2D.
Acknowledgements
The work was supported by ARC Project No. CE140100036 (Davis), NIH R15ES022766-01A1 (Ding), NSF CBET-1232724 (Ding & Ke), and NHMRC Project Grant APP1071350 (Gurzov). Davis is thankful for the award of an Australian Laureate Fellowship from the ARC. Gurzov is supported by a Juvenile Diabetes Research Foundation (JDRF) fellowship. The St Vincent's Institute receives support from the Operational Infrastructure Support Scheme of the Government of Victoria. The authors acknowledge Dr Eric Hanssen for assistance with the TEM imaging. All simulations were performed on the Palmetto high performance cluster at Clemson University CCIT.References
- C. G. Glabe, Neurobiol. Aging, 2006, 27, 570–575 CrossRef CAS PubMed.
- P. Westermark, A. Andersson and G. T. Westermark, Physiol. Rev., 2011, 91, 795–826 CrossRef CAS PubMed.
- L. Haataja, T. Gurlo, C. J. Huang and P. C. Butler, Endocr. Rev., 2008, 29, 303–316 CrossRef CAS PubMed.
- A. Lorenzo, B. Razzaboni, G. C. Weir and B. A. Yankner, Nature, 1994, 368, 756–760 CrossRef CAS PubMed.
- E. T. A. S. Jaikaran and A. Clark, Biochim. Biophys. Acta, Mol. Basis Dis., 2001, 1537, 179–203 CrossRef CAS.
- D. E. Ehrnhoefer, J. Bieschke, A. Boeddrich, M. Herbst, L. Masino, R. Lurz, S. Engemann, A. Pastore and E. E. Wanker, Nat. Struct. Mol. Biol., 2008, 15, 558–566 CAS.
- S. Sparks, G. Liu, K. J. Robbins and N. D. Lazo, Biochem. Biophys. Res. Commun., 2012, 422, 551–555 CrossRef CAS PubMed.
- R. Mishra, D. Sellin, D. Radovan, A. Gohlke and R. Winter, ChemBioChem, 2009, 10, 445–449 CrossRef CAS PubMed.
- J. R. Brender, K. Hartman, R. P. R. Nanga, N. Popovych, R. de la Salud Bea, S. Vivekanandan, E. N. G. Marsh and A. Ramamoorthy, J. Am. Chem. Soc., 2010, 132, 8973–8983 CrossRef CAS PubMed.
- R. Jayawardena, P. Ranasinghe, P. Galappatthy, R. Malkanthi, G. R. Constantine and P. Katulanda, Diabetol. Metab. Syndr., 2012, 4, 13 CrossRef CAS PubMed.
- J. L. Larson and A. D. Miranker, J. Mol. Biol., 2004, 335, 221–231 CrossRef CAS PubMed.
- A. Abedini, F. Meng and D. P. Raleigh, J. Am. Chem. Soc., 2007, 129, 11300–11301 CrossRef CAS PubMed.
- A. Kapurniotu, A. Schmauder and K. Tenidis, J. Mol. Biol., 2002, 315, 339–350 CrossRef CAS PubMed.
- M. Tatarek-Nossol, L.-M. Yan, A. Schmauder, K. Tenidis, G. Westermark and A. Kapurniotu, Chem. Biol., 2005, 12, 797–809 CrossRef CAS PubMed.
- P. Nedumpully-Govindan and F. Ding, Sci. Rep., 2015, 5, 8240 CrossRef CAS PubMed.
- P. Nedumpully-Govindan, A. Kakinen, E. H. Pilkington, T. P. Davis, P. C. Ke and F. Ding, submitted.
- P. Nedumpully-Govindan, Y. Yang, R. Andorfer, W. Cao and F. Ding, Biochemistry, 2015 DOI:10.1021/acs.biochem.5b0089.
- C. Li and R. Mezzenga, Nanoscale, 2013, 5, 6207 RSC.
- M. Zaman, E. Ahmad, A. Qadeer, G. Rabbani and R. H. Khan, Int. J. Nanomed., 2014, 9, 899–912 Search PubMed.
- S. Linse, C. Cabaleiro-Lago, W.-F. Xue, I. Lynch, S. Lindman, E. Thulin, S. E. Radford and K. A. Dawson, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 8691–8696 CrossRef CAS PubMed.
- C. Chung, Y.-K. Kim, D. Shin, S.-R. Ryoo, B. H. Hong and D.-H. Min, Acc. Chem. Res., 2013, 46, 2211–2224 CrossRef CAS PubMed.
- M. Mahmoudi, O. Akhavan, M. Ghavami, F. Rezaee and S. M. A. Ghiasi, Nanoscale, 2012, 4, 7322 RSC.
- J. E. Kim and M. Lee, Biochem. Biophys. Res. Commun., 2003, 303, 576–579 CrossRef CAS PubMed.
- A. K. Jana and N. Sengupta, Biophys. J., 2012, 102, 1889–1896 CrossRef CAS PubMed.
- H. Li, Y. Luo, P. Derreumaux and G. Wei, Biophys. J., 2011, 101, 2267–2276 CrossRef CAS PubMed.
- J. Guo, J. Li, Y. Zhang, X. Jin, H. Liu and X. Yao, PLoS One, 2013, 8, e65579 CAS.
- J. Liu, Z. Yang, H. Li, Z. Gu, J. A. Garate and R. Zhou, J. Chem. Phys., 2014, 141, 22D520 CrossRef PubMed.
- H. Shen, L. Zhang, M. Liu and Z. Zhang, Theranostics, 2012, 2, 283–294 CrossRef CAS PubMed.
- J. Liu, L. Cui and D. Losic, Acta Biomater., 2013, 9, 9243–9257 CrossRef CAS PubMed.
- R. Tuma, J. Raman Spectrosc., 2005, 36, 307–319 CrossRef CAS.
- K. N. Kudin, B. Ozbas, H. C. Schniepp, R. K. Prud'homme, I. A. Aksay and R. Car, Nano Lett., 2008, 8, 36–41 CrossRef CAS PubMed.
- Z. Yan, J. Wang, Y. Zhang, M. Qin and W. Wang, Phys. Rev. E: Stat., Nonlinear, Soft Matter Phys., 2010, 81, 021910 CrossRef PubMed.
- Z. Yan, J. Wang, J. Zhang, M. Qin and W. Wang, Phys. Rev. E: Stat., Nonlinear, Soft Matter Phys., 2010, 82, 031917 CrossRef PubMed.
- F. Ding, J. J. LaRocque and N. V. Dokholyan, J. Biol. Chem., 2005, 280, 40235–40240 CrossRef CAS PubMed.
- S. Barton, R. Jacak, S. D. Khare, F. Ding and N. V. Dokholyan, J. Biol. Chem., 2007, 282, 25487–25492 CrossRef CAS PubMed.
- D. C. Latshaw and C. K. Hall, Biophys. J., 2015, 109, 124–134 CrossRef CAS PubMed.
- R. Laghaei, N. Mousseau and G. Wei, J. Phys. Chem. B, 2011, 115, 3146–3154 CrossRef CAS PubMed.
- J. J. W. Wiltzius, S. A. Sievers, M. R. Sawaya and D. Eisenberg, Protein Sci., 2009, 18, 1521–1530 CrossRef CAS PubMed.
- A. S. Reddy, L. Wang, S. Singh, Y. L. Ling, L. Buchanan, M. T. Zanni, J. L. Skinner and J. J. de Pablo, Biophys. J., 2010, 99, 2208–2216 CrossRef CAS PubMed.
- P. Chen, S. A. Seabrook, V. C. Epa, K. Kurabayashi, A. S. Barnard, D. A. Winkler, J. K. Kirby and P. C. Ke, J. Phys. Chem. C, 2014, 118, 22069–22078 CAS.
- J. A. Williamson, J. P. Loria and A. D. Miranker, J. Mol. Biol., 2009, 393, 383–396 CrossRef CAS PubMed.
- D. F. Moriarty and D. P. Raleigh, Biochemistry, 1999, 38, 1811–1818 CrossRef CAS PubMed.
- D. Zanuy and R. Nussinov, J. Mol. Biol., 2003, 329, 565–584 CrossRef CAS PubMed.
- K. Hamaguchi, H. R. Gaskins and E. H. Leiter, Diabetes, 1991, 40, 842–849 CrossRef CAS PubMed.
- E. N. Gurzov, F. Ortis, D. A. Cunha, G. Gosset, M. Li, A. K. Cardozo and D. L. Eizirik, Cell Death Differ., 2009, 16, 1539–1550 CrossRef CAS PubMed.
- W. Xi, W. Li and W. Wang, J. Phys. Chem. B, 2012, 116, 7398–7405 CrossRef CAS PubMed.
- C. T. Middleton, P. Marek, P. Cao, C. Chiu, S. Singh, A. M. Woys, J. J. de Pablo, D. P. Raleigh and M. T. Zanni, Nat. Chem., 2012, 4, 355–360 CrossRef CAS PubMed.
- S. Shi, K. Yang, H. Hong, H. F. Valdovinos, T. R. Nayak, Y. Zhang, C. P. Theuer, T. E. Barnhart, Z. Liu and W. Cai, Biomaterials, 2013, 34, 3002–3009 CrossRef CAS PubMed.
- X. Yang, Y. Wang, X. Huang, Y. Ma, Y. Huang, R. Yang, H. Duan and Y. Chen, J. Mater. Chem., 2011, 21, 3448–3454 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental and computational methods; Fig. S1 – CD spectra; Fig. S2 – residue-wise contact frequencies of a single hIAPP with a GO nanosheet; and Fig. S3 – GO–hIAPP contact number per chain as a function of the number of hIAPP chains. See DOI: 10.1039/c5cp05924k |
| This journal is © the Owner Societies 2016 |