
Scalable production of graphene with tunable and stable doping by electrochemical intercalation and exfoliation†
Ya-Ping
Hsieh
*a,
Wan-Yu
Chiang
a,
Sun-Lin
Tsai
a and
Mario
Hofmann
*b
aGraduate Institute of Opto-Mechatronics, National Chung Cheng University, Chiayi, 62102, Taiwan. E-mail: yphsieh@ccu.edu.tw
bDepartment of Material Science and Engineering, National Cheng Kung University, Tainan, 70101, Taiwan. E-mail: Mario@mail.ncku.edu.tw
First published on 16th November 2015
Abstract
Graphene's unique semimetallic band structure yields carriers with widely tunable energy levels that enable novel electronic devices and energy generators. To enhance the potential of this feature, a scalable synthesis method for graphene with adjustable Fermi levels is required. We here show that the electrochemical intercalation of FeCl3 and subsequent electrochemical exfoliation produces graphene whose energy levels can be finely tuned by the intercalation parameters. X-ray photoelectron spectroscopy reveals that a gradual transition in the bonding character of the intercalant is the source of this behavior. The intercalated graphene exhibits a significantly increased work function that can be varied between 4.8 eV and 5.2 eV by the intercalation potential. Transparent conducting electrodes produced by these graphene flakes exhibit a threefold improvement in performance and the doping effect was found to be stable for more than a year. These findings open up a new route for the scalable production of graphene with adjustable properties for future applications.
Introduction
Graphene is a two-dimensional carbon material that exhibits a wealth of unique properties which have attracted the attention of researchers in many fields ranging from electronic1 to energy storage devices.2–4 One particularly interesting property of graphene, brought about by its low carrier concentration and peculiar band structure, is its semimetallic nature. The ability to tune graphene's Fermi level over a wide range while retaining metallic conduction enables applications as tunable Schottky barrier devices,5 hole/electron conductors in solar cells,6 or ohmic contact materials.7While electrostatic control of the Fermi level using gate dielectrics8 or electrochemical gating9 enables proof-of-concept devices, a permanent method to adjust the Fermi level is required for many applications. Chemical doping is the most common approach to varying the carrier distribution by introducing charged ions in the vicinity of the graphene lattice.10 The stability of doping amount and characteristics over time, however, is a major issue of this approach.11 Many factors, such as the clustering of adsorbates and their reaction with oxygen, cause changes in the character and effectiveness of doping upon prolonged exposure to the environment.12
A solution to enhancing the stability of dopants could be through confinement effects: Intercalation between graphene layers has been shown to result in stable doping of graphene with FeCl3,13 Li,14,15 Al16 and other materials that are normally environmentally unstable. This robustness is not well understood and scaling of the confinement approach has been challenging. Intercalation of FeCl3 between layers of large-scale CVD-grown graphene showed a reduction in conductivity by 50% within 1 week17 in contrast to intercalation between multilayers of mechanically exfoliated graphene that was stable for over 1 year.18 The instability arises from larger inter-layer spacing of sequentially transferred graphene due to a limited interaction between the layers19 which increases contaminant mobility between layers. Furthermore, uniform intercalation of large-scale graphene sheets has been challenging because of the limited diffusion speed of dopants over the required length-scales.17
In order to overcome these issues, we investigate the doping effect in thin films of graphene flakes. Such films are composed of micrometer-sized grains of multilayer graphene which can be efficiently intercalated.13
This work combines electrochemical intercalation and exfoliation techniques because of their easy scalability and ability to finely tune the graphene's properties through manipulation of the process parameters.
Optimization of the intercalation process was shown to enable the formation of low-stage graphite intercalation compounds.13,20 The geometry, chemical composition, and production yield of graphene can then be adjusted by the electrochemical exfoliation parameters.21–24 We here demonstrate that the modification of the electrochemical intercalation process results in the change of graphene's carrier concentration. Spectroscopic analysis of the intercalated iron species revealed an increasing contribution of iron chloride with intercalation potential which results in an enhanced charge transfer to graphene. This mechanism permits fine adjustment of graphene's work function between 4.8 eV and 5.2 eV through control of the intercalation process. Thus produced graphene exhibits a threefold increase in performance over pristine graphene flake films. The doping effect was stable over one year due to a self-limiting oxidation effect.
Experimental
Graphite flake electrodes were produced by mixing a binder [poly(vinylidene fluoride): 0.6 g and 1-methyl-2-pyrrolidone: 10 ml] with natural graphite flakes (Alfa Aesar, 10 mesh, 99% metal basis). Electrochemical intercalation was carried out in FeCl3 solution (FeCl3 (40%): 72 ml; HCl (36%): 18 ml; DI water: 10 ml) following previous reports.25,26 Electrochemical exfoliation was conducted in an aqueous H2SO4 electrolyte (0.47 M) as previously reported.27 A pulsed voltage of 30 V was applied using a computer-controlled Motech SP1200 power supply to exfoliate graphene. The obtained solution was deposited onto 2 × 2 cm2 quartz substrates without further purification using an airbrush with a 0.3 mm nozzle. Spectral transmittance of graphene films was measured using a KMAC Spectra Academy SV2100 and sheet resistance of these films was characterized by 4-Probe measurements in van-der-Pauw geometry.Results and discussion
To understand the effect of electrochemical intercalation on the graphite flakes, SEM images (FE-SEM ZEISS AURIGA) were taken before and after intercalation (Fig. 1(a) and (b)). An obvious expansion of the edges in the out-of-plane direction can be observed. This behavior might originate from the electrochemical etching of graphite edges by OH− ions which had been previously suggested as a required step to improve intercalation between graphite layers.28 | ||
| Fig. 1 SEM image of the graphite electrode (a) before and (b) after FeCl3 intercalation. | ||
Raman spectroscopy shows a negligible intensity of the defect related D-band peak at around 1350 cm−1 after intercalation (Fig. 2a) indicating that the quality of the graphite electrode is not significantly affected by this electrochemical etching process.
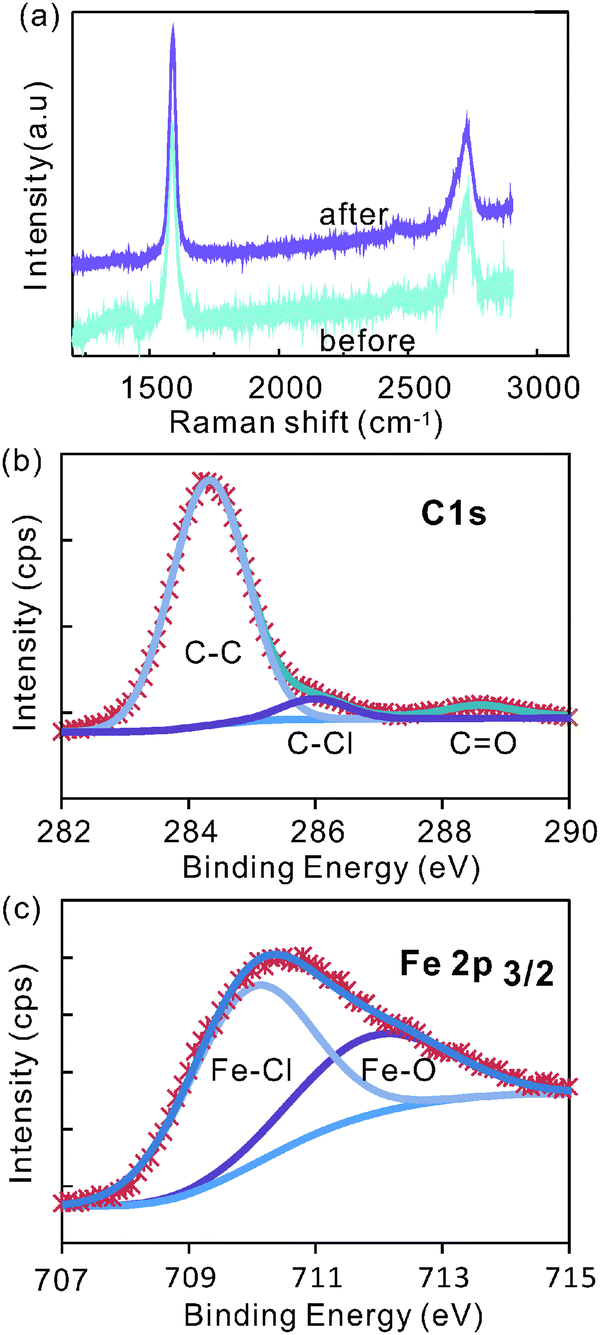 | ||
| Fig. 2 (a) Raman spectra before and after intercalation, and (b) C1s peak and (c) Fe 2p peak of XPS spectra after intercalation. | ||
This observation is supported by X-ray photoelectron spectroscopy (XPS) of graphite after intercalation (for more XPS-spectra see ESI,† Fig. S1). The main C1s peak can be deconvoluted into two sub-peaks with peak positions of 284.3 eV and 286 eV (Fig. 2b). The first peak was assigned to sp2-bonded carbon. The negligible amount of sp3-bonds (∼285 eV) in the graphite lattice corroborates the high quality of graphite after intercalation. The peak at 286 eV has been found to indicate bonding between chlorine and carbon atoms.29
The effect of chlorine interaction can also be seen in the Fe2p XPS peak. Two peaks were fitted into the Fe2p3/2 peak at around 709.9 and 712.3 eV (Fig. 2c). These peaks represent the bonds of iron to chlorine and to oxygen,30 respectively, and raise the question about the chemical composition of the intercalant.
Characterization of the intensity of the Cl2p and Fe2p peaks permits extraction of the atomic concentration of iron and chlorine at varying intercalation voltages. We observe that the concentration of both elements is increasing with intercalation voltage (Fig. 3a) which is supported by the results from Energy-dispersive X-ray spectroscopy (EDS) (ESI,† Fig. S2).
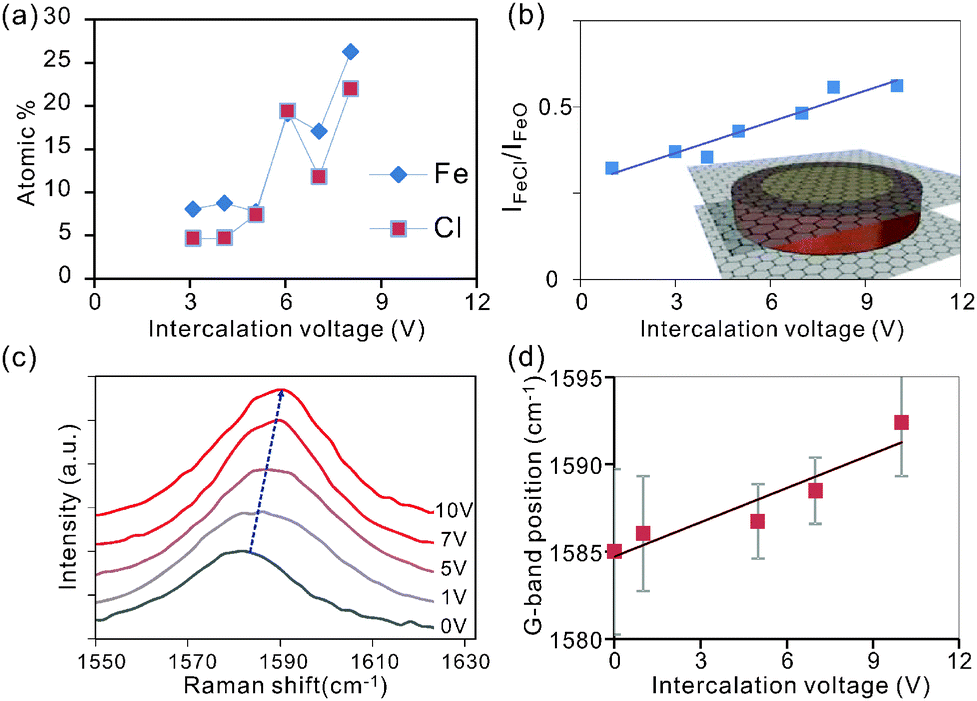 | ||
| Fig. 3 Influence of intercalation voltage on (a) elemental composition obtained by XPS, (b) intensities of the two peaks contributing to the Fe 2p peak, (inset) schematic of self-limiting oxidation process where surface iron oxides (red) protect a core of iron chloride (yellow), (c) Raman G-band of graphite intercalated at different voltages, and (d) Raman G-Band position vs. intercalation voltage. | ||
Surprisingly, the trend of an enhanced intercalation effect extends over a large voltage range between 0 and 10 V. This voltage window is significantly larger than in previous electrochemical intercalation cases.31 The observed difference is thought to originate from the parallel formation of gas bubbles at the large potentials needed for complete intercalation.31 This alternate reaction pathway will enhance the interfacial resistance and in turn decrease the efficiency of intercalation. Consequently, high voltages are required to simultaneously drive both reactions.
The observed correlation between both Fe and Cl concentrations with intercalation voltage (Fig. 3(a)) demonstrates the higher attainable intercalation efficiency (lower intercalation stage) for higher electrochemical potentials.25
The ratio of Fe to Cl atoms was found to be 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1.5 in agreement with previous studies on gas-phase FeCl3 intercalation.17 The deviation of this ratio from the expected value of 1:3 indicates that a significant portion of FeCl3 undergoes oxidation.
:1.5 in agreement with previous studies on gas-phase FeCl3 intercalation.17 The deviation of this ratio from the expected value of 1:3 indicates that a significant portion of FeCl3 undergoes oxidation.
To reveal the difference in iron bonding, we investigate the relative peak intensities of the oxide and chloride-related bonds in Fe 2p3/2 (Fig. 2b). We observe that the iron chloride concentration is increasing with respect to iron oxide for increasing intercalation voltage (Fig. 3b).
The observed trend can be explained by a self-limiting oxidation process of iron intercalants.32 At low Fe concentrations associated with low intercalation voltages, small intercalant clusters form that can be completely oxidized upon exposure to the environment. Larger intercalation voltages will result in bigger agglomerates and oxidation is limited to the surface regions. Consequently, iron atoms in the center of the intercalant islands are protected from oxidation and maintain their chlorine bonds (inset Fig. 3b).
Raman analysis was employed to characterize the impact of the detected increasing FeCl3 concentration at higher intercalation voltages on charge transfer. An increasing blue shift of the 2D-peak (Fig. 3c) suggests an intercalation-induced hole doping of graphene.33–35 This behavior is expected since Fe3+ tends to reduce to metallic Fe by removing charges from graphene.36,37 The higher reduction potential of iron chloride compared to iron oxide can explain the enhanced doping effect at higher intercalation voltages which facilitate higher FeCl3 concentrations.
The adjustable charge transfer caused by the intercalation process was found to be retained even after electrochemical exfoliation. We observe that the work function of exfoliated graphene thin films is directly proportional to the intercalation voltages used in the intercalation step (Fig. 4a). The highest achievable work function was found to be 5.2 eV which is in agreement with previous results on gas-phase intercalation.38
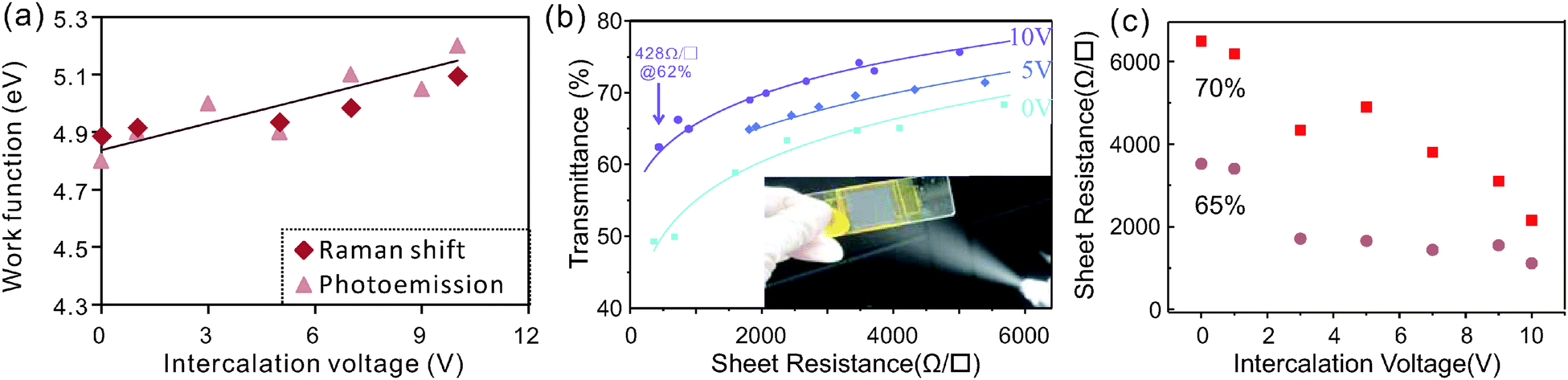 | ||
| Fig. 4 Control of graphene properties by the intercalation process. (a) Work function of sprayed graphene thin films from the photoelectric effect and Raman characterization, (b) TCF sheet resistance vs. transmittance for 3 intercalation voltages, (inset) photo of the spraying process, and (c) sheet resistance for different intercalation voltages at two fixed transmittances. | ||
To confirm that charge transfer is indeed the source of the work function change, Raman spectroscopy of the graphene thin films was carried out.
Variation in doping changes the bond-strength of neighboring carbon atoms which affects the Raman G-band position. In the limit of zero temperature and large doping the G-band shift can be related to the Fermi-level shift by a simple formula proposed by Pisana et al.39
| ℏΔωG = 4.39 × 10−3|EF| |
The thus extracted work function change agrees well with photoemission measurements (ESI,† Fig. S3).
The observed increase in work function due to the shifting Fermi level suggests a significant doping effect. The large initial value of the work function compared to the expected value of 4.3 eV for pristine graphene40 indicates that graphene is intrinsically p-doped due to environmental contaminants such as oxygen.41 The work function shift can be related to the dopant concentration by
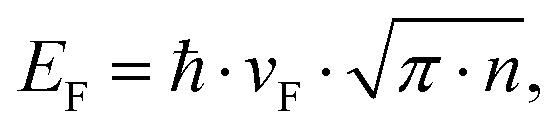
Due to the quadratic dependence of the density of states on energy, the intercalation-induced increase in work function from 4.8 eV to 5.2 eV will result in an increased doping level of 6 × 1013 cm2. Consequently, FeCl3 intercalation can increase the carrier concentration of exfoliated graphene by more than three times compared to graphene exfoliated from pristine graphite.
Such an increase in the carrier density is expected to significantly enhance the conductivity of graphene films which results in an increased performance of graphene-based transparent conducting films (TCFs). We therefore analyzed the sheet resistance of films composed of graphene that was intercalated at different voltages. Fig. 4(b) shows that more strongly intercalated graphene exhibits higher transmittances at similar sheet resistances.
The obtained values of sheet resistance and transmittances are comparable to graphene films grown on Nickel by chemical vapor deposition42 and were achieved without additional sample purification and processing. Further sample treatment processes such as centrifugation, acid treatment, and high temperature annealing have been proven to dramatically improve the performance of the exfoliated graphene.43
More importantly, the graphene sheet resistance at a given transmittance can be finely controlled by tuning the intercalation voltage (Fig. 4c), which proves that the higher doping of graphene increases the thin film performance. A threefold reduction in sheet resistance is found between pristine graphene and the material that was intercalated at 10 V. The value of the resistance decrease is similar to the estimated increase in carrier concentration indicating that the carrier mobility μ is not significantly affected by the addition of dopants. This robustness of the carrier transport in the presence of increased charge impurity scattering44 is caused by the relatively low concentration of iron impurities of 0.5 at% after expansion and due to additional limitations of carrier transport in the investigated thin flake films. Hall effect measurements of pristine films reveal relatively low carrier mobilities of μ = 1.5 cm2 V−1 s−1 which are similar to previously measured hopping mobilities in percolative networks.45 These low mobilities dominate carrier transport and only depend on the morphology and their stability is explained by our observation that the intercalation process has no significant influence on the flake morphology (ESI,† Fig. S4).
Our results show that the exfoliation from electrochemically intercalated graphite represents a route for enhancing the performance of graphene. An additional advantage of our method over currently used doping techniques is its environmental stability. Sprayed graphene flake samples were re-measured after one year and their sheet resistance remained virtually unchanged exhibiting unsystematic variations within 10% (see ESI,† Fig. S5). This result proves that FeCl3 can act as a stable source of doping for industrial applications.
Conclusions
In summary, using FeCl3 intercalated graphite for electrochemical exfoliation was shown to change the work function and performance of the resulting graphene. The doping process can be controlled by the intercalation voltage through an increase of the amount of reactive FeCl3. Exfoliated graphene thin films exhibit a threefold increased conductivity compared to pristine graphene. Our approach provides a solution for stable and heavy doping that is compatible with industrial scale production and enhances the performance of graphene TCFs for future applications.Acknowledgements
YP Hsieh and M. Hofmann acknowledge financial support from Applied Materials, Inc., the Ministry of Science and Technology, and Industrial Technology Research Institute of Taiwan.References
- A. K. Geim and K. S. Novoselov, Nat. Mater., 2007, 6, 183–191 CrossRef CAS PubMed
.
- Y. Yang, B. Qiao, X. Yang, L. Fang, C. Pan, W. Song, H. Hou and X. Ji, Adv. Funct. Mater., 2014, 24, 4349–4356 CrossRef CAS
- W. Song, X. Ji, W. Deng, Q. Chen, C. Shen and C. E. Banks, Phys. Chem. Chem. Phys., 2013, 15, 4799–4803 RSC
- Y.-X. Wang, S.-L. Chou, H.-K. Liu and S.-X. Dou, Carbon, 2013, 57, 202–208 CrossRef CAS
- L. Lancellotti, T. Polichetti, F. Ricciardella, O. Tari, S. Gnanapragasam, S. Daliento and G. Di Francia, Thin Solid Films, 2012, 522, 390–394 CrossRef CAS
- X. Li, H. Zhu, K. Wang, A. Cao, J. Wei, C. Li, Y. Jia, Z. Li, X. Li and D. Wu, Adv. Mater., 2010, 22, 2743–2748 CrossRef CAS PubMed
- K. E. Byun, H. J. Chung, J. Lee, H. Yang, H. J. Song, J. Heo, D. H. Seo, S. Park, S. W. Hwang, I. Yoo and K. Kim, Nano Lett., 2013, 13, 4001–4005 CrossRef CAS PubMed
- M. F. Craciun, S. Russo, M. Yamamoto and S. Tarucha, Nano Today, 2011, 6, 42–60 CrossRef CAS
- A. Das, S. Pisana, B. Chakraborty, S. Piscanec, S. K. Saha, U. V. Waghmare, K. S. Novoselov, H. R. Krishnamurthy, A. K. Geim, A. C. Ferrari and A. K. Sood, Nat. Nanotechnol., 2008, 3, 210–215 CrossRef CAS PubMed
- H. T. Liu, Y. Q. Liu and D. B. Zhu, J. Mater. Chem., 2011, 21, 3335–3345 RSC
- K. Ki Kang, R. Alfonso, S. Yumeng, P. Hyesung, L. Lain-Jong, L. Young Hee and K. Jing, Nanotechnology, 2010, 21, 285205 CrossRef PubMed
- C. W. Jang, J. M. Kim, J. H. Kim, D. H. Shin, S. Kim and S.-H. Choi, J. Alloys Compd., 2015, 621, 1–6 CrossRef CAS
- C.-Y. Su, A.-Y. Lu, Y. Xu, F.-R. Chen, A. N. Khlobystov and L.-J. Li, ACS Nano, 2011, 5, 2332–2339 CrossRef CAS PubMed
- A. Kumar, A. L. M. Reddy, A. Mukherjee, M. Dubey, X. Zhan, N. Singh, L. Ci, W. E. Billups, J. Nagurny, G. Mital and P. M. Ajayan, ACS Nano, 2011, 5, 4345–4349 CrossRef CAS PubMed
- W. Bao, J. Wan, X. Han, X. Cai, H. Zhu, D. Kim, D. Ma, Y. Xu, J. N. Munday, H. D. Drew, M. S. Fuhrer and L. Hu, Nat. Commun., 2014, 5, 4224 CAS
- M.-C. Lin, M. Gong, B. Lu, Y. Wu, D.-Y. Wang, M. Guan, M. Angell, C. Chen, J. Yang, B.-J. Hwang and H. Dai, Nature, 2015, 520, 324–328 CrossRef CAS PubMed
- S. Yi, F. Wenjing, L. H. Allen and K. Jing, Nanotechnology, 2014, 25, 395701 CrossRef PubMed
- I. Khrapach, F. Withers, T. H. Bointon, D. K. Polyushkin, W. L. Barnes, S. Russo and M. F. Craciun, Adv. Mater., 2012, 24, 2844–2849 CrossRef CAS PubMed
- M. Kalbac, H. Farhat, J. Kong, P. Janda, L. Kavan and M. S. Dresselhaus, Nano Lett., 2011, 11, 1957–1963 CrossRef CAS PubMed
- G. M. Morales, P. Schifani, G. Ellis, C. Ballesteros, G. Martínez, C. Barbero and H. J. Salavagione, Carbon, 2011, 49, 2809–2816 CrossRef CAS
- H. Mario, C. Wan-Yu, D. N. Tuân and H. Ya-Ping, Nanotechnology, 2015, 26, 335607 CrossRef PubMed
- K. Parvez, Z.-S. Wu, R. Li, X. Liu, R. Graf, X. Feng and K. Müllen, J. Am. Chem. Soc., 2014, 136, 6083–6091 CrossRef CAS PubMed
- S. Yang, S. Brüller, Z.-S. Wu, Z. Liu, K. Parvez, R. Dong, F. Richard, P. Samorì, X. Feng and K. Müllen, J. Am. Chem. Soc., 2015, 137, 13927–13932 CrossRef CAS PubMed
- A. Ambrosi and M. Pumera, Chem. – Eur. J., 2015 DOI:10.1002/chem.201503110
- Y. Geng, Q. Zheng and J. K. Kim, J. Nanosci. Nanotechnol., 2011, 11, 1084–1091 CrossRef CAS PubMed
- Z. Yan, Z. Zhuxia, L. Tianbao, L. Xuguang and X. Bingshe, Spectrochim. Acta, Part A, 2008, 70, 1060–1064 CrossRef PubMed
- M. Hofmann, W.-Y. Chiang, T. D. Nguyễn and Y.-P. Hsieh, Nanotechnology, 2015, 26, 335607 CrossRef PubMed
- W. W. Liu and J. N. Wang, Chem. Commun., 2011, 47, 6888–6890 RSC
-
D. Briggs, X-ray and Ultraviolet Photoelectron Spectroscopy, Peking University Press, 1984 Search PubMed
- A. P. Grosvenor, B. A. Kobe, M. C. Biesinger and N. S. McIntyre, Surf. Interface Anal., 2004, 36, 1564–1574 CrossRef CAS
- F. Kang, Y. Leng, T.-Y. Zhang and B. Li, Carbon, 1998, 36, 383–390 CrossRef CAS
- R. Nishitani, Y. Sasaki and Y. Nishina, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 3141–3144 CrossRef CAS
- A. Das, S. Pisana, B. Chakraborty, S. Piscanec, S. K. Saha, U. V. Waghmare, K. S. Novoselov, H. R. Krishnamurthy, A. K. Geim, A. C. Ferrari and A. K. Sood, Nat. Nanotechnol., 2008, 3, 210–215 CrossRef CAS PubMed
- C. Casiraghi, Phys. Rev. B: Condens. Matter Mater. Phys., 2009, 80, 233407 CrossRef
- D. Zhan, L. Sun, Z. H. Ni, L. Liu, X. F. Fan, Y. Wang, T. Yu, Y. M. Lam, W. Huang and Z. X. Shen, Adv. Funct. Mater., 2010, 20, 3504–3509 CrossRef CAS
- J. Nathaniel and X.-Q. Wang, Appl. Phys. Lett., 2012, 100, 213112 CrossRef
- T. Abe, M. Inaba, Z. Ogumi, Y. Yokota and Y. Mizutani, Phys. Rev. B: Condens. Matter Mater. Phys., 2000, 61, 11344–11347 CrossRef CAS
- W. Zhao, P. H. Tan, J. Liu and A. C. Ferrari, J. Am. Chem. Soc., 2011, 133, 5941–5946 CrossRef CAS PubMed
- S. Pisana, M. Lazzeri, C. Casiraghi, K. S. Novoselov, A. K. Geim, A. C. Ferrari and F. Mauri, Nat. Mater., 2007, 6, 198–201 CrossRef CAS PubMed
- W. Hu, Z. Li and J. Yang, J. Chem. Phys., 2013, 138, 124706 CrossRef PubMed
- A. Piazza, F. Giannazzo, G. Buscarino, G. Fisichella, A. L. Magna, F. Roccaforte, M. Cannas, F. M. Gelardi and S. Agnello, J. Phys. Chem. C, 2015, 119, 22718–22723 CAS
- W.-C. Yen, Y.-Z. Chen, C.-H. Yeh, J.-H. He, P.-W. Chiu and Y.-L. Chueh, Sci. Rep., 2014, 4, 4739 CAS
- C.-M. Gee, C.-C. Tseng, F.-Y. Wu, H.-P. Chang, L.-J. Li, Y.-P. Hsieh, C.-T. Lin and J.-C. Chen, Displays, 2013, 34, 315–319 CrossRef CAS
- C. K. Chua, A. Ambrosi, Z. Sofer, A. Macková, V. Havránek, I. Tomandl and M. Pumera, Chem. – Eur. J., 2014, 20, 15760–15767 CrossRef CAS PubMed
- G. Eda and M. Chhowalla, Nano Lett., 2009, 9, 814–818 CrossRef CAS PubMed
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5cp06395g |
| This journal is © the Owner Societies 2016 |