Solvent H-bond accepting ability induced conformational change and its influence towards fluorescence enhancement and dual fluorescence of hydroxy meta-GFP chromophore analogue†
Received
16th June 2016
, Accepted 5th August 2016
First published on 5th August 2016
Abstract
The effect of structural rigidity towards enhancement of fluorescence quantum yield of GFP chromophore analogues has been documented. In the present study, a new way of enhancing the fluorescence quantum yield of two ortho–meta GFP chromophore analogues meta-methoxy-ortho-hydroxy-benzylimidazolidinone (abbreviated as mOMe-HBDI) and meta-diethylamino-ortho-hydroxyl imidazolidinone (abbreviated as MOHIM) has been reported. This enhancement is controlled by the H-bond accepting ability (denoted as β value) of the solvent and happens only in the case of GFP chromophore analogues having ortho (hydroxyl)-meta (electron donating group) and not in the case of analogues having a para electron donating group. The ground state (solid) conformation of mOMe-HBDI has been obtained from single crystal X-ray analysis, exhibiting the existence of strong intramolecular H-bonding. However, in solution phase, as the solvent β value increases, the strength of intramolecular H-bonding decreases. This process has strong influence on the relative conformational orientation of phenyl and imidazolidinone rings. For mOMe-HBDI, fluorescence quantum yield increases with increase in β value of the solvents. However, the effect of solvent polarity cannot be completely ruled out. The lower wavelength emission band (∼480 nm) has been assigned to the normal charge-transferred (CT) species, whereas the highly Stokes shifted emission band (∼660 nm) has been assigned to the proton-transferred (PT) tautomer species for mOMe-HBDI. In solvents of low β value (say hexane) only the PT band and in solvents of high β value (say DMSO) only the CT band is observed. Quite interestingly, in solvents of intermediate β value both CT and PT bands, thus dual emission, are observed. For mOMe-HBDI when fluorescence decay is monitored at the normal CT emission band, it is observed to be biexponential in nature. The short component increases from ∼0.2 ns to 0.6 ns and the long component increases from 1 to 3.6 ns as the β value of the solvent increases. For a particular solvent, when fluorescence decay is monitored at the normal CT band, as the monitoring wavelength increases the amplitude of the long lifetime component increases and that of the short lifetime component decreases. Time-resolved area-normalised emission spectral (TRANES) analysis confirms the possible existence of two conformers having differential stabilisation by solvent polarity. When fluorescence decay is monitored at the PT band an instrument response limited (<60 ps) decay is noted. Strong support in favour of the above-mentioned structural, steady state and time-resolved optical observations and analyses has been obtained from the methoxy derivative mOMe-MBDI and MOMIM.
Introduction
The green fluorescent protein (GFP) chromophore (p-HBDI, Chart 1) residing inside a β-barrel exhibits very high fluorescence quantum yield (Φfl, ∼0.8) and high fluorescence lifetime (τfl, in the nanosecond range).1 However, the same p-HBDI exhibits very low Φfl (10−3 or lower) and τfl (several hundred femtoseconds to several picoseconds) in solution.1–15 Differential optical behaviour of p-HBDI in a β-barrel and in solution has been attributed to structural τ twist around the exocyclic double bond connecting benzene and imidazolidinone moieties.1–14 Thus, the quest of enhancing fluorescence behaviours of the GFP chromophore in normal organic solvents is very active. There are several reports regarding enhancement of Φfl (and τfl) due to structural restriction of τ twist.15–28 There are a few reports describing how enhancement of solvent polarity or hydrogen bond donating ability of solvents reduces Φfl.10,11,29 However, there has been no such detailed investigation till date regarding solvent-property-controlled enhancement of Φfl in the case of GFP chromophore analogues, especially enhancement of Φfl in high polarity solvents. Hydrogen bond donating and accepting abilities of solvents have been shown to have significant effect on the photophysics of molecules exhibiting excited state proton transfer.30–32 It has been shown that the relative intensity of charge transfer (CT) and proton transfer (PT) bands of these molecules is controlled by the relative dynamics of CT and PT. The latter is in turn controlled by polarity and hydrogen bond donating/accepting ability of the solvent.30 For several molecules it has been reported that an excited state intramolecular PT process could suppress the intramolecular CT process30,31 and vice versa.33 However, modulation of the extent of CT by excited state PT and its effect towards tuning Φfl and τfl for GFP chromophore analogues have not been investigated in detail.
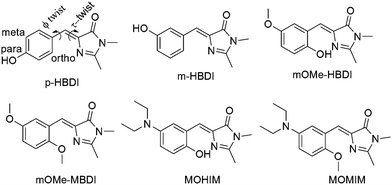 |
| | Chart 1 Chemical structure of GFP chromophore analogues. | |
It has been reported in the literature that, para and meta derivatives of GFP chromophore analogue (m-HBDI and p-HBDI, Chart 1) spectroscopically behave differently in solution.10,34–36 It has also been shown that an electron donating group like an amine group in the meta orientation can get involved in the charge transfer process.36–40 It has been reported in the literature that as solvent polarity increases fluorescence quantum yield and fluorescence lifetime decrease.10,29 Although there are several reports of observation of CT and PT emission bands and thus dual fluorescence of other molecules, there is only one report of dual fluorescence, i.e. that of CT and PT bands, of GFP chromophore analogues.41 It has been shown that by chemical structural control of the extent of CT, the relative intensity of CT and PT bands can be modulated.41 However, it has never been reported that H-bond accepting ability can enhance fluorescence quantum yield and fluorescence lifetime significantly. In order to shed light in the above direction four different molecules (mOMe-MBDI, m-OMe-HBDI, MOMIM and MOHIM, see Chart 1) have been synthesised. These molecules are not new and have been synthesised by different groups previously.10,42 Some photophysical data have also been reported. For example Tolbert et al. have synthesised mOMe-MBDI and measured the photophysical data in ethanol containing 1% DMSO solvent.42 A very high value of ϕfl (31%) is reported but no discussion about the same was given. Chou et al. have synthesised the compounds and measured the photophysical data in a few solvents.20 They observed proton transfer tautomer emission of mOMe-HBDI in nonpolar solvents like cyclohexane. However, complicated multiexponential decay for mOMe-HBDI in polar solvents was observed. Possible existence of E–Z isomerisation in the ground and excited states and deprotonation to the polar solvents were speculated.20 Recently mOMe-HBDI was studied by Shustova et al. in a solid scaffold-like MOF matrix.28 However, they observed green emission of mOMe-HBDI in MOF matrix in contrast to the red tautomer emission in solution.28 All these interesting results stimulated us to reinvestigate the photophysical properties of the compounds m-OMe-HBDI and m-OMe-MBDI in detail. Studies were extended to other similar o-hydroxy derivatives containing stronger electron donating diethylamino group in the meta position, viz. MOMIM and MOHIM. Comparison with data on previously reported analogues containing hydroxy groups at the ortho position has also been performed to support and validate the interpretation made in the present study. It could be successfully shown that fluorescence quantum yield and fluorescence lifetime were indeed enhanced as solvent H-bond accepting ability and solvent polarity increases.
Results and discussion
Ground state structure analysis from X-ray and NMR studies
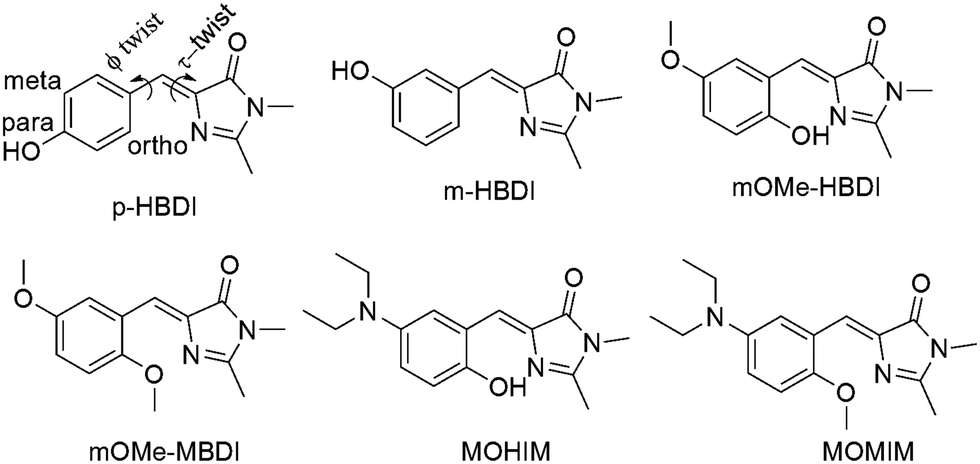
Solid state single crystal structures of mOMe-HBDI and mOMe-MBDI are shown in Fig. 1 ((a) and (b) respectively). The conformation of mOMe-HBDI exhibits nearly planar relative orientation of phenyl and imidazolidinone rings, with dihedral angle ∠N(5)–C(9)–C(7)–C(11) of −1.07°. A smaller distance of 2.597 Å between O(3)–N(5) and dihedral angle of 163° for ∠O(3)–H–N(5) confirm the formation of a strong intramolecular hydrogen bond between the hydroxyl group of the phenyl ring and the sp2 nitrogen of the imidazolidinone ring. In this conformation the proton attached to C12 of mOMe-HBDI is on the opposite side to the sp2 nitrogen of the imidazolidinone ring (shown by an arrow). The solid state conformation of mOMe-MBDI (Fig. 1b) adopts the almost planar Z-form in the solid state with a dihedral angle of −7.89° (∠C3–C4–C6–N1), with the methoxy group on the opposite side to the sp2 nitrogen of the imidazolidinone ring due to steric bulk. This conformation keeps the proton attached to C3 of mOMe-MBDI on the same side and spatially closer to the sp2 nitrogen of the imidazolidinone ring (shown with an arrow). The distance between the proton shown as “Ha” (Fig. 2b) and the sp2 nitrogen of imidazolidinone ring is ∼2.40 Å, with ∠C–Ha–N angle of ∼127°. Such a close spatial arrangement could cause the electronic environment around the proton (shown with an arrow) of mOMe-MBDI to be highly perturbed by the electronic environment of the imidazolidinone ring.43
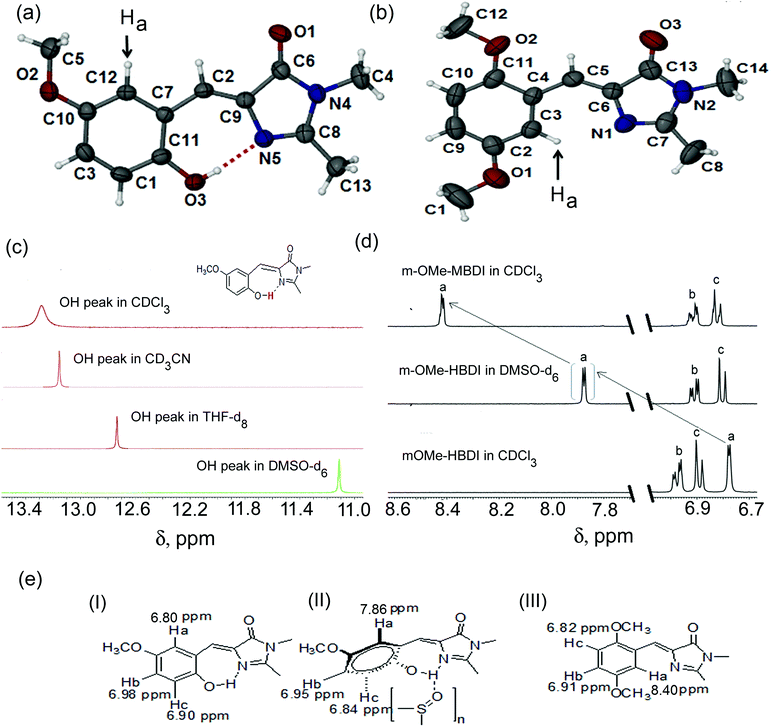 |
| | Fig. 1 Single crystal structure of (a) mOMe-HBDI and (b) mOMe-MBDI. Thermal ellipsoids are drawn at the 50% probability level. (c) 1H NMR spectra showing the variation in position of the hydroxyl proton in different solvents. (d) 1H NMR spectra showing the shift of the “Ha” proton, indicating solution state variation in conformation of mOMe-HBDI and mOMe-MBDI. (e) Solvent-dependent ground state conformation in solution ((I) mOMe-HBDI in CDCl3, (II) mOMe-HBDI in DMSO-d6, (III) mOMe-MBDI in CDCl3). The crystal structure of mOMe-HBDI was reported earlier by measuring the data at a different temperature (150 K). Data in the present study were recorded at room temperature (293 K). Changes in bond length and bond angle values have been observed.20 The crystal structure of MOMIM has already been reported.10 | |
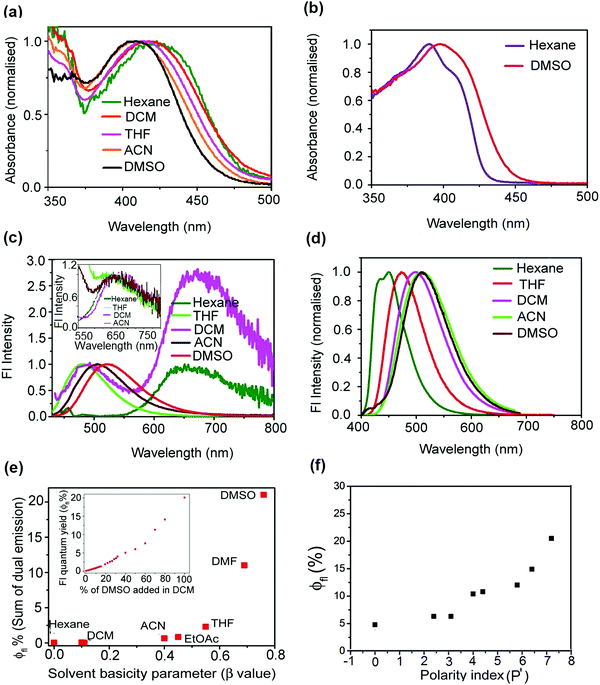 |
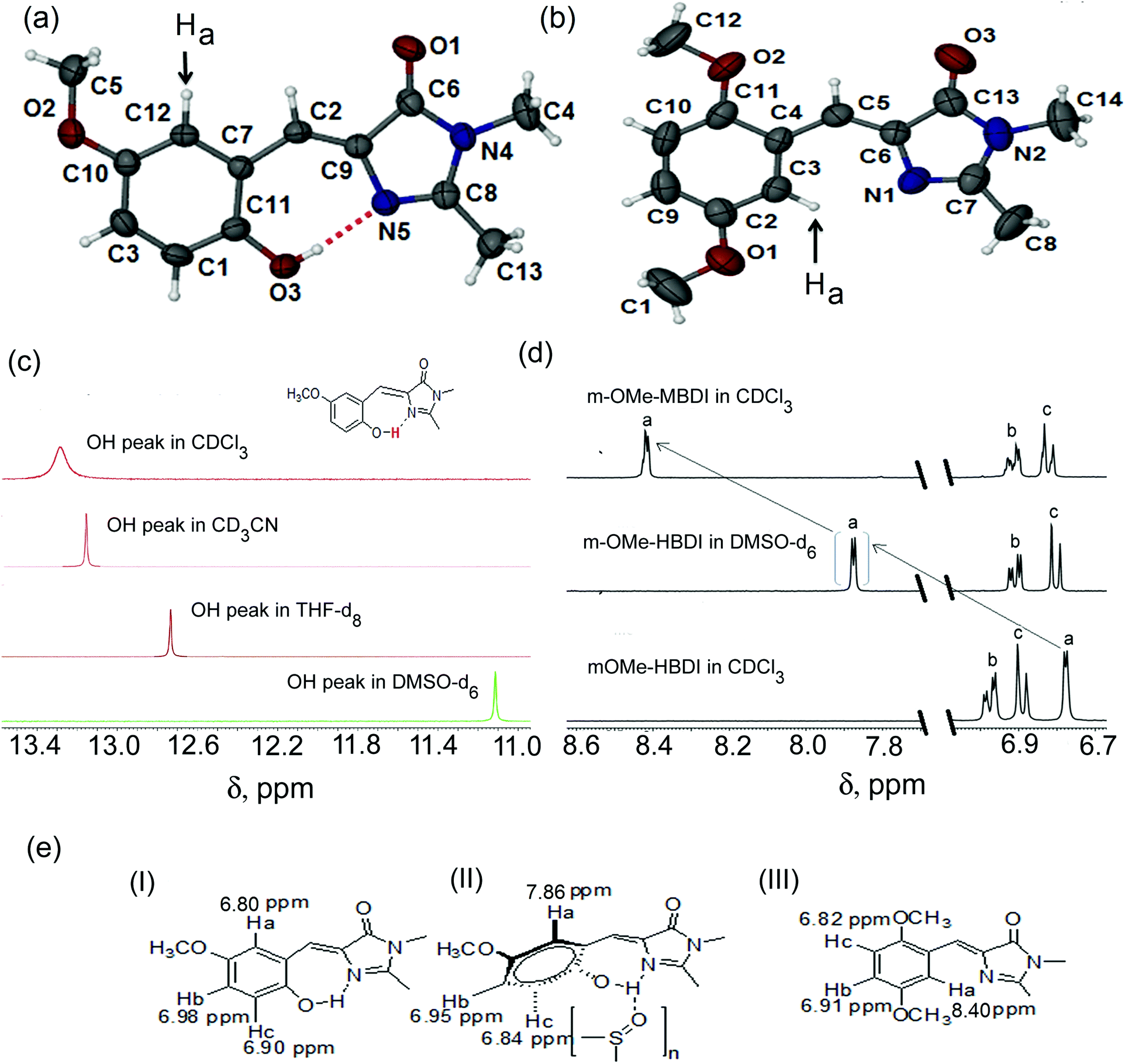
| | Fig. 2 Absorption spectra of mOMe-HBDI in different solvents (a). Absorption spectra of mOMe-MBDI in hexane and DMSO (b). Solvent dependent emission spectra of mOMe-HBDI (inset shows normalisation of proton transfer band of mOMe-HBDI in different solvents) (c) and mOMe-MBDI (d) (λex, corresponding absorption maxima of the lowest energy band, normalised to the charge transfer band). Solvent β value dependent enhancement of fluorescence quantum yield of mOMe-HBDI (inset shows enhancement of fluorescence quantum yield of mOMe-HBDI upon addition of DMSO in DCM) (e). Solvent polarity (P′)44 dependent enhancement of fluorescence quantum yield of mOMe-MBDI (f). | |
To arrive at the solution state conformation, 1H NMR has been performed for both mOMe-HBDI and mOMe-MBDI (for MOMIM and MOHIM see ESI,† S12). In mOMe-HBDI the presence of a strong intramolecular hydrogen bond between the o-hydroxyl group and the sp2 nitrogen of the imidazolidinone ring was confirmed from a large downfield shift of the hydroxyl proton obtained at 13.28 ppm in CDCl3.15,20 The corresponding peak was shifted to 13.08, 12.67, and 11.12 ppm in CD3CN, THF-d8, and DMSO-d6 respectively (Fig. 1c). Thus, as the H-bond accepting ability of the solvent increases the corresponding peak of hydroxyl proton shifts up-field indicating ‘partial rupture’ of the intramolecular H-bond.
It would be interesting to know whether because of partial rupture of the H-bond there is any change in the relative conformation of benzyl and imidazolidinone rings. In mOMe-HBDI the proton “Ha” remains in the opposite direction with respect to the sp2 nitrogen of the imidazolidinone ring (Fig. 1a) and appears at δHa = 7.86 ppm in DMSO-d6 (Fig. 1d and e). In CDCl3 this proton “Ha” is far apart from the imidazolidinone ring and δHa in CDCl3 is 6.77 ppm. For mOMe-MBDI the δ value of the proton “Ha” appears at δHa = 8.42 ppm in CDCl3. From the structural aspect it is expected that because of steric bulk, the methoxy group (in mOMe-MBDI) will remain in the same conformation as in the solid phase. In that scenario the proton “Ha” of mOMe-MBDI will be spatially closer to the sp2 nitrogen of the imidazolidinone ring (see Fig. 1b). Deshielding of that proton by the imidazolidinone ring is evidenced by the large downfield shift of that proton (“Ha”) with respect to that of mOMe-HBDI in 1H NMR spectra. Thus, a Δδ value of ∼1.65 ppm between the “Ha” proton in mOMe-HBDI and the “Ha” proton in mOMe-MBDI (Fig. 1d and e, both in CDCl3) provides a clear indication of the spatial position with respect to the sp2 nitrogen of the imidazolidinone ring and hence the conformation in solution. So the position of the “Ha” proton (δ, ppm scale) in 1H NMR spectra of mOMe-MBDI and mOMe-HBDI could be correlated with its average distance from the sp2 nitrogen atom of the imidazolidinone ring. The δHa value of the “Ha” proton of mOMe-HBDI in DMSO-d6 is in between the farthest (mOMe-HBDI in CDCl3) and closest (mOMe-MBDI in CDCl3) positions of the “Ha” proton with respect to the imidazolidinone ring. Thus, in a strong H-bond accepting solvent like DMSO, not only does the H-bond get ruptured, but also the orientation of the benzyl ring with respect to the imidazolidinone ring is in between two oppositely oriented planar conformations. Thus, the solvent β value dependent conformation is noted (Fig. 1e).
Steady state spectral analysis
Absorption.
Absorption spectra of mOMe-HBDI and mOMe-MBDI in different solvents are depicted in Fig. 2a and b respectively (for MOMIM and MOHIM see ESI,† S13). Absorption maximum of mOMe-HBDI (425 nm) is 35 nm red shifted with respect to the absorption maximum of the mOMe-MBDI (390 nm) (both in hexane). A similar red shift (22 nm) of the long wavelength absorption band of MOHIM compared with MOMIM was observed in hexane (see ESI,† Table S3). This red shift indicates the formation of a seven-membered ring with intramolecular H-bond in mOMe-HBDI and MOHIM. Thus, the π electron gets more delocalised and hence the energy gap between the S0 and S1 electronic levels decreases. If that is true, then it could be anticipated that rupture of the intramolecular H-bond would cause a blue shift of absorption maxima. This is indeed what has been observed for mOMe-HBDI. As the H-bond accepting ability or β value of the solvent increases, the absorption maximum gets blue shifted from hexane (425 nm) to DMSO (408 nm) (Fig. 2a). The absorption maxima of mOMe-HBDI and mOMe-MBDI (408 nm and 399 nm respectively) as well as MOHIM and MOMIM (473 nm and 476 nm respectively) are quite close to each other in DMSO.
Emission.
The emission spectra of mOMe-HBDI and mOMe-MBDI in various fluid organic solvents are shown in Fig. 2c and d respectively (for MOMIM and MOHIM see ESI,† S13). Steady state photophysical data for mOMe-HBDI and mOMe-MBDI are depicted in Tables 1 and 2 respectively (for MOMIM and MOHIM see ESI,† Table S3). In the case of mOMe-HBDI, in solvents of low β value (say hexane) a highly Stokes shifted (>8000 cm−1) emission band (maximum ∼660 nm) is observed. The emission maximum of this band is independent of solvent polarity (see inset of Fig. 2c). However, as the solvent β value increases the intensity of this highly Stokes shifted band decreases and a new lower wavelength band appears. This lower wavelength band emission maximum changes from 485 nm to 525 nm. Similarly for MOHIM in solvents of low β value a highly Stokes shifted emission band with no solvatochromic shift was observed. However, in solvents of moderate or high β value a band with large solvatochromic shift was observed (see ESI,† Fig. S20 and Table S3). For mOMe-MBDI, as the solvent polarity increases the emission maximum increases (from 451 nm in hexane to 510 nm in DMSO), indicating the charge transfer nature of the emitting species. A similar observation was made for MOMIM (see ESI,† Fig. S19). Computational calculation shows that the electron gets transferred from the methoxy group or diethylamino group at the meta position to the imidazolidinone ring of mOMe-MBDI (from HOMO to LUMO, see ESI,† S10) or MOMIM.10 Comparing the emission behaviour with mOMe-MBDI, for mOMe-HBDI the lower wavelength band has been assigned to the charge transferred (CT) species and the longer wavelength band has been assigned to the proton transferred (PT) tautomer species. For mOMe-HBDI in hexane only the proton transferred tautomer band is observed. As the β value of the solvent increases, the intensity of this PT band decreases and dual fluorescence is observed (both CT and PT band). In solvents of very high β value only the CT band is observed. This kind of dual fluorescence of GFP chromophore analogues and effect of solvent polarity on relative magnitude of CT and PT bands has recently been reported in the literature.41 To the best of our knowledge this is only the second report of this kind. Absence of the PT band in solvents of high β value confirms that the H-bond is indeed ruptured and thus no PT band is observed. However, more interesting is the drastic variation in ϕfl of mOMe-HBDI from solvent to solvent (Fig. 2f). Solvent β value dependent photophysical data of mOMe-HBDI are depicted in Table 1. It can be noticed from Table 1 that there is almost 2000 times enhancement of fluorescence quantum yield from hexane to DMSO for mOMe-HBDI. Also, for MOHIM, with the much stronger electron donating diethylamino group at the meta position, enhancement of ϕfl occurs from hexane to DMSO (see ESI,† Table S3). But the extent of enhancement is much less (only 10–15 times in comparison with 2000 times in mOMe-HBDI). This difference is because the emission maximum of MOHIM is at 700 nm (red) and that of mOMe-HBDI is at 525 nm (green). Thus, another nonradiative decay channel governed by the energy gap law becomes operative in DMSO.10,45,46 The presence of such nonradiative decay was previously investigated by our group in detail for MOMIM.10 The inset of Fig. 2e shows the enhancement of ϕfl with respect to addition of DMSO to a solution of mOMe-HBDI in DCM. In DMSO, mOMe-HBDI possesses a ϕfl of ∼21% which is almost the same value as obtained for mOMe-MBDI. Similarly, MOMIM and MOHIM possess almost the same ϕfl value (∼0.6%) in DMSO. The increase in ϕfl of mOMe-HBDI and MOHIM in different solvents could be easily correlated with increase in solvent H-bond accepting basicity parameter (β value) rather than solvent polarity. We can draw this inference because tetrahydrofuran (THF) is a solvent of lower polarity but higher H-bond accepting ability than acetonitrile (ACN), and ϕfl of mOMe-HBDI is almost four times higher in THF than in ACN (see Table 1). Quite interestingly, ϕfl of mOMe-MBDI is 4.8% in hexane and it gradually increases with gradual increase of polarity of non-hydroxylic solvents and reaches 15% in DMF and 21% in DMSO (see Fig. 2f and Table 2). In polar solvents with high hydrogen bond accepting ability e.g. DMSO, high fluorescence quantum yield (21%) for physically non-restricted GFP chromophore analogues has been obtained for mOMe-MBDI. However, for MOMIM ϕfl decreases from hexane (ϕfl ∼ 12%) to DMSO (ϕfl ∼ 0.6%). This was previously ascribed to the energy gap law governed nonradioactive decay in DMSO due to the much red shifted emission maximum (λem ∼ 690 nm for MOMIM in comparison with 510 nm for mOMe-MBDI).10 However, without the presence of electron donating groups like methoxy or diethylamino at the meta position of the benzene ring neither of the derivatives could show such large enhancement in fluorescence properties, at least in nonpolar solvents.36–40 This enhancement is due to the ortho–meta effect of electron donating groups in benzinoid compounds.36–40 Consequently OHIM, CHBDI and MHBDI, studied previously by our group, were unable to show such large enhancement in fluorescence properties in DMSO (ϕfl ∼ 10−4) although similar rupture of the intramolecular H-bond was observed.41 For the same reason, o-HBDI studied by different groups earlier was nonfluorescent in solvents of different polarity and H-bond accepting basicity.15 Here it is worth mentioning that only a single OMe or OH group as substituent at the meta position or at the ortho position of the benzene ring is not so efficient in enhancing the fluorescence quantum yield to such a large extent.15,34,35 Rather, combination of two electron donating OMe groups at both meta and ortho positions of the benzene ring could enhance the charge transfer character to such an extent that enhancement of photophysical properties (such as ϕfl and τfl) is of much larger magnitude in comparison with molecules containing a single OMe group. This is known as the “excited state ortho–meta effect” of the electron donating groups.39,47 In other words, mOMe-HBDI and mOMe-MBDI containing two electron donating groups at meta and ortho positions and having emission maxima in the blue-green region in a variety of polar solvents constitute an ideal choice of molecules to observe solvent polarity and H-bond accepting basicity dependent fluorescence enhancement. Interestingly, in a high polarity protic (i.e. H-bond donating) solvent like MeOH, ϕfl is 0.02, which can be attributed to solute–solvent H-bonding interaction.11 This kind of fluorescence enhancement that is controlled by polarity and hydrogen bond accepting ability of the solvent has not been reported earlier. It should also be mentioned here that the viscosity of DMSO (2.24 cP) is higher than that of hexane (0.31 cP) or DCM (0.44 cP). However, there are plenty of reports in the literature to show that this great increase in viscosity has negligible effect on τ-twist, and hence the enhancement of ϕfl of mOMe-MBDI and mOMe-HBDI in DMSO cannot be due to viscosity.3,8,48
Table 1 Steady state photophysical data of mOMe-HBDI in different solvents
| Solvents |
Solvent β-value |
λ
abs (nm) |
ε (M−1 cm−1) |
λ
em (nm) |
ϕ
fl
|
| Hexane |
0.00 |
425 |
NA |
656 |
∼10−4 |
| DCM |
0.10 |
419 |
1.0 × 104 |
485, 667 |
∼10−4 |
| ACN |
0.40 |
409 |
1.1 × 104 |
510, 658 |
0.006 |
| THF |
0.55 |
410 |
1.3 × 104 |
483, 658 |
0.022 |
| DMF |
0.69 |
409 |
1.3 × 104 |
517 |
0.112 |
| DMSO |
0.76 |
408 |
1.5 × 104 |
525 |
0.205 |
Table 2 Steady state photophysical data of mOMe-MBDI in different solvents
| Solvents |
Solvent polarity (P′)44 |
λ
abs (nm) |
ε (M−1 cm−1) |
λ
em (nm) |
ϕ
fl
|
| Hexane |
0.0 |
390 |
NA |
451 |
0.048 |
| DCM |
3.1 |
396 |
1.5 × 104 |
489 |
0.063 |
| THF |
4.0 |
390 |
1.2 × 104 |
478 |
0.104 |
| ACN |
5.8 |
393 |
1.6 × 104 |
510 |
0.104 |
| DMF |
6.4 |
393 |
1.7 × 104 |
503 |
0.149 |
| DMSO |
7.2 |
399 |
1.2 × 104 |
510 |
0.210 |
Fluorescence dynamics results
(a) For mOMe-MBDI.
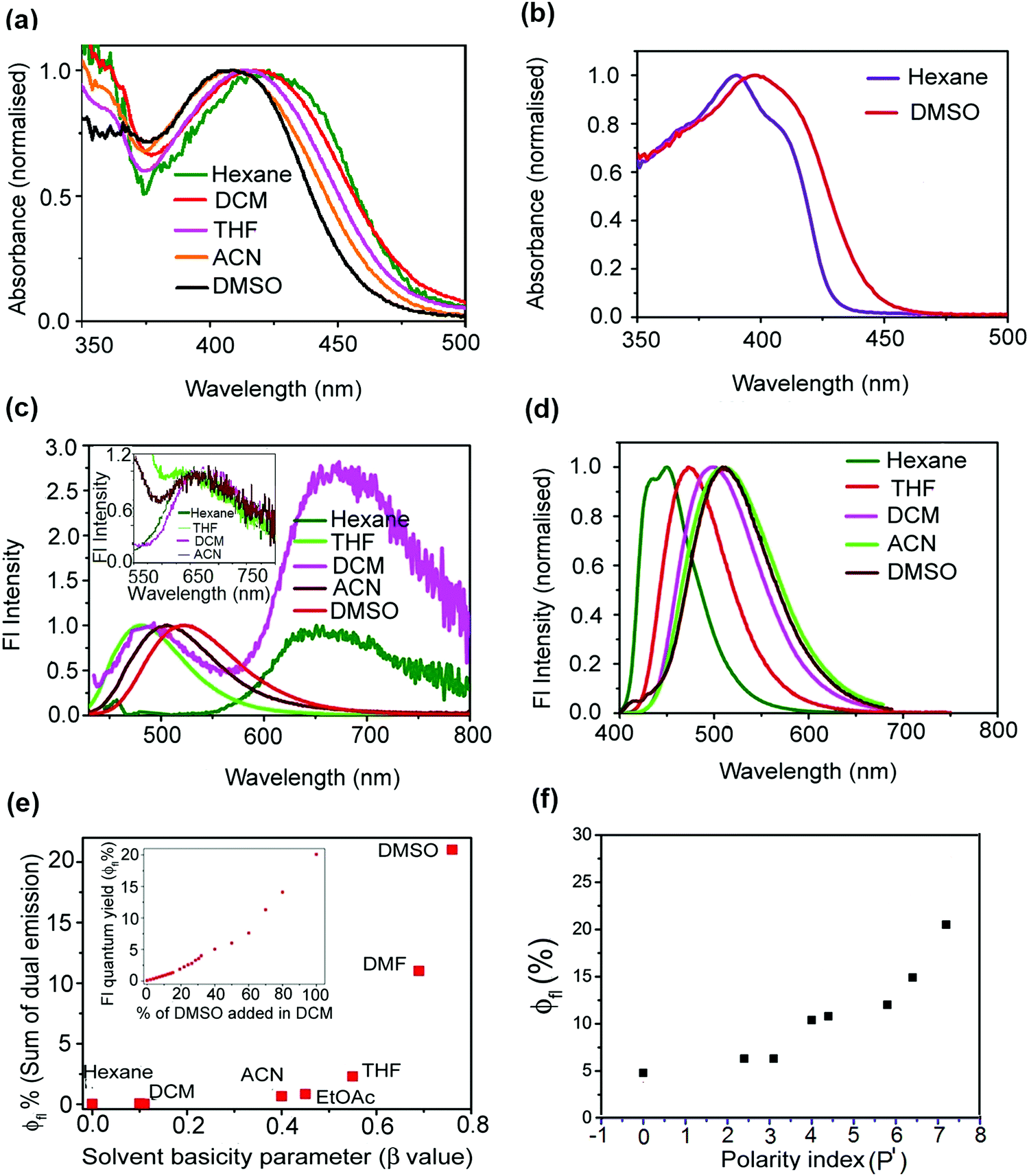
In order to understand the photophysics as well as fluorescence dynamics in a better way, time-resolved fluorescence spectroscopic measurements have been performed using femtosecond upconversion as well as picosecond–nanosecond based time-correlated single photon counting techniques. For mOMe-MBDI, single exponential decay behaviour was observed in hexane (Fig. 3a and Table 3) (for MOMIM see ESI,† S13 and Table S3). However, with increasing solvent polarity, excited state decay of mOMe-MBDI becomes biexponential as well as slower (Fig. 3a). The time constants are shown in Table 3. The magnitude of the short lifetime component (∼0.36 ns) increases from 0.35 ns to 0.60 ns whereas the magnitude of the long lifetime component increases from 1.2 ns to 3.2 ns as solvent polarity increases. The decay in hexane is independent of monitoring wavelength. However, in solvents of higher polarity fluorescence decay becomes monitoring wavelength dependent. For example, in acetonitrile as the monitoring wavelength increases, the amplitude of the longer lifetime component increases gradually and that of the short component decreases concomitantly (Fig. 3b and Table 3). The magnitude of both lifetimes, however, remains independent of monitoring wavelength for a particular solvent, say acetonitrile. Time-resolved area-normalised emission spectra (TRANES) show (Fig. 3c) an isoemissive point at 19![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 791 cm−1. Above the isoemissive point (in cm−1 scale) emission intensity decreases and below the isoemissive point the emission intensity increases with increase in time. However, the possible existence of two different states, viz. LE state (unsolvated) and solvent stabilised ICT state, is excluded owing to the observed nanosecond timescale of relaxation. (Solvent relaxation occurs in hundreds of femtoseconds or within a few picoseconds.) Thus a simpler model consisting of two different conformers could be considered. However, no rise time was noticed in 60 ps or longer timescale. Following a recent report for para derivatives of modified p-HBDI by Meech et al., we ascribe planar or nearly planar conformers to State II (see Fig. 3d), which have slightly red shifted absorption and emission bands compared with the ϕ-twisted conformers ascribed to State I (see Fig. 3d).3,11 It is also expected that charge transfer efficiency for the planar conformation (State II) would be more than for the ϕ-twisted conformer (State I). Thus, two conformers are expected to be differently stabilised by polar solvents. In hexane both the conformers have similar excited state lifetimes as charge transfer stabilisation by the solvent is negligible. However, in polar solvents the planar conformer (State II) will have more solvent stabilisation than the twisted one (State I). Hence the excited state lifetime of the planar conformer (State II) will be longer (1.2 to 3.1 ns) than that of the twisted conformer (State I) (0.35 to 0.57 ns). This is further supported by the fact that as the monitoring wavelength increases the amplitude of the long component increases. In comparison with para derivatives the extent of charge transfer (for both nearly twisted conformation (State I) and nearly planar conformation (State II)) is higher for meta analogues. Hence a longer excited state lifetime is expected for meta analogues and that is what has been observed (∼0.35–0.57 ns and 1.2–3.1 ns for meta analogues in comparison with 0.28 ps and 0.85 ps for para derivative ABDI11). It is necessary to understand why ϕfl and τfl of mOMe-MBDI increase with increase in polarity of the solvent. Following the Michl and Bona
791 cm−1. Above the isoemissive point (in cm−1 scale) emission intensity decreases and below the isoemissive point the emission intensity increases with increase in time. However, the possible existence of two different states, viz. LE state (unsolvated) and solvent stabilised ICT state, is excluded owing to the observed nanosecond timescale of relaxation. (Solvent relaxation occurs in hundreds of femtoseconds or within a few picoseconds.) Thus a simpler model consisting of two different conformers could be considered. However, no rise time was noticed in 60 ps or longer timescale. Following a recent report for para derivatives of modified p-HBDI by Meech et al., we ascribe planar or nearly planar conformers to State II (see Fig. 3d), which have slightly red shifted absorption and emission bands compared with the ϕ-twisted conformers ascribed to State I (see Fig. 3d).3,11 It is also expected that charge transfer efficiency for the planar conformation (State II) would be more than for the ϕ-twisted conformer (State I). Thus, two conformers are expected to be differently stabilised by polar solvents. In hexane both the conformers have similar excited state lifetimes as charge transfer stabilisation by the solvent is negligible. However, in polar solvents the planar conformer (State II) will have more solvent stabilisation than the twisted one (State I). Hence the excited state lifetime of the planar conformer (State II) will be longer (1.2 to 3.1 ns) than that of the twisted conformer (State I) (0.35 to 0.57 ns). This is further supported by the fact that as the monitoring wavelength increases the amplitude of the long component increases. In comparison with para derivatives the extent of charge transfer (for both nearly twisted conformation (State I) and nearly planar conformation (State II)) is higher for meta analogues. Hence a longer excited state lifetime is expected for meta analogues and that is what has been observed (∼0.35–0.57 ns and 1.2–3.1 ns for meta analogues in comparison with 0.28 ps and 0.85 ps for para derivative ABDI11). It is necessary to understand why ϕfl and τfl of mOMe-MBDI increase with increase in polarity of the solvent. Following the Michl and Bona![[c with combining breve]](https://www.rsc.org/images/entities/char_0063_0306.gif) ić-Koutecký model for a meta derivative (say mOMe-MBDI), the planar singlet state (1S*) will be more stabilised than the τ-twisted geometry (1P*) in the excited state.10,37,49–52 It is also reported that nonradiative decay occurs from the twisted state, thereby reducing the ϕfl.37 Thus, as the energy barrier from solvent stabilised CT state (1S*, see Fig. 3d) to τ-twisted state (1P*, see Fig. 3d) increases there will be less chance of twisted state formation; hence there is less chance of Z–E isomerisation. Z–E isomerisation has been shown to be a major nonradiative decay channel for GFP chromophore analogues.1–14,36 Thus, as the chance of Z–E isomerisation decreases there will be enhanced ϕfl. For mOMe-MBDI the magnitude of ϕZE decreases from CDCl3 (54%) to DMSO-d6 (41%) (see ESI,† S11). This is because of solvent polarity mediated stabilisation of mOMe-MBDI and thereby concomitant increase of the Z–E isomerisation energy barrier (Fig. 3d).
ić-Koutecký model for a meta derivative (say mOMe-MBDI), the planar singlet state (1S*) will be more stabilised than the τ-twisted geometry (1P*) in the excited state.10,37,49–52 It is also reported that nonradiative decay occurs from the twisted state, thereby reducing the ϕfl.37 Thus, as the energy barrier from solvent stabilised CT state (1S*, see Fig. 3d) to τ-twisted state (1P*, see Fig. 3d) increases there will be less chance of twisted state formation; hence there is less chance of Z–E isomerisation. Z–E isomerisation has been shown to be a major nonradiative decay channel for GFP chromophore analogues.1–14,36 Thus, as the chance of Z–E isomerisation decreases there will be enhanced ϕfl. For mOMe-MBDI the magnitude of ϕZE decreases from CDCl3 (54%) to DMSO-d6 (41%) (see ESI,† S11). This is because of solvent polarity mediated stabilisation of mOMe-MBDI and thereby concomitant increase of the Z–E isomerisation energy barrier (Fig. 3d).
Table 3 Time-resolved photophysical data of mOMe-MBDI and mOMe-HBDI in different solvents. Amplitudes of fluorescence decay components are shown in parentheses
| Solvent |
Solvent polarity index (P′) |
λ
mon (nm) |
Lifetime (ns) |
λ
mon (nm) |
Lifetime |
| mOMe-MBDI |
mOMe-HBDI |
| Hexane |
0.0 |
451 |
τ
1 = 0.35 |
656 |
τ
1 = <60 ps |
| DCM |
3.1 |
489 |
τ
1 = 0.35 (29.3), τ2 = 1.2 (70.7) |
485 |
τ
1 = 0.17 ns (30.6), τ2 = 1.03 ns (69.4) |
| 667 |
τ
1 = 0.75 ps (34), τ2 = 8.05 ps (66) |
| THF |
4.0 |
478 |
τ
1 = 0.58 (30.1), τ2 = 2.0 (69.9) |
483 |
τ
1 = 0.38 ns (32.2), τ2 = 1.86 ns (67.8) |
| 658 |
τ
1 = <60 ps |
| ACN |
5.8 |
510 |
τ
1 = 0.60 (30.7), τ2 = 2.72 (69.3) |
510 |
τ
1 = 0.51 ns (28.9), τ2 = 2.91 ns (71.1) |
| 658 |
τ
1 = 0.36 ps (35), τ2 = 6.8 ps (65) |
| DMF |
6.4 |
503 |
τ
1 = 0.58 (23.4), τ2 = 2.71 (76.6) |
517 |
τ
1 = 0.55 ns (28), τ2 = 3.37 ns (72) |
| DMSO |
7.2 |
510 |
τ
1 = 0.57 (22.7), τ2 = 3.1 (77.3) |
525 |
τ
1 = 0.60 ns (21), τ2 = 3.61 ns (79) |
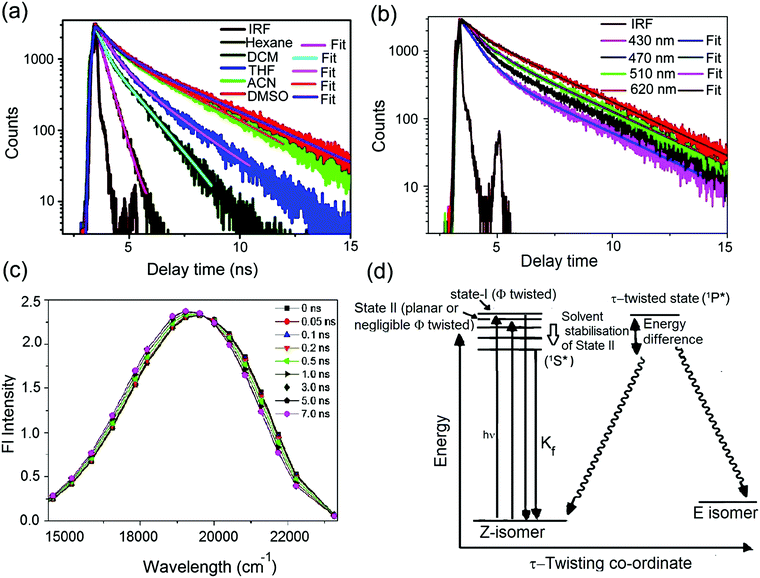 |
| | Fig. 3 (a) Variation in fluorescence decay (TCSPC) of mOMe-MBDI in different fluid organic solvents of different polarity index. (b) Fluorescence decay of mOMe-MBDI in acetonitrile monitored at different wavelengths of steady state emission spectra. (c) Construction of time-resolved area-normalised emission spectra (TRANES) in acetonitrile. (d) Excited state relaxation dynamics of mOMe-MBDI in acetonitrile. State I (twisted) and State II (planar or nearly planar) are excited states of two conformers that differ in the extent of Φ twist. | |
Thus, τfl will be increased and hence ϕfl will be increased as polarity of the solvent increases. An energy level diagram is provided for mOMe-MBDI, and also the steady state property ϕfl can be correlated to the time-resolved property τfl (see Tables 2 and 3). For mOMe-MBDI, the CT stabilisation induced emission maximum is in the blue region (∼510 nm in ACN). However, for MOMIM and similar compounds, the CT stabilisation induced emission maximum is in the red region (690 nm for MOMIM in ACN). As the emission is in the red region a nonradiative decay channel which is governed by the energy gap law becomes effective.10,29 Hence both ϕfl and τfl decrease with increase of solvent polarity (see ESI,† Table S3). Hence, it could be shown that chemical structural modification induced control of electronic energy levels plays a crucial role in controlling ϕfl and τfl.
(b) For mOMe-HBDI.
No Z–E isomerisation was observed for mOMe-HBDI or MOHIM. This result is consistent with the previously observed results on similar o-hydroxy derivatives of p-HBDI.53–55 This negligible Z–E isomerisation for derivatives bearing the o-hydroxy group was ascribed to the structure of the molecule at the conical intersection due to the presence of the intramolecular H-bond, which favours the return of the molecule to the Z-form after S1 → S0 internal conversion.53,55 In fact, in contrast to the τ-twisting co-ordinate for the methoxy derivatives,11,36 the ϕ-twisting co-ordinate is reported to be a dominant nonradiative relaxation pathway for corresponding hydroxy derivatives.54,55 The decay behaviour of mOMe-HBDI and MOHIM is much more complicated. Monitoring at the proton transfer band of mOMe-HBDI, a system response limited rise (<280 fs) and biexponential decay with time constant of 360 fs and 6.8 ps were observed (see Table 3, ESI,† S8 and Fig. S14). The faster decay component of 360 fs was also observed for similar excited state intramolecular proton transfer (ESIPT) systems reported previously.54 But its origin is not fully understood. Intramolecular vibrational redistribution, vibronic relaxation, and internal conversion etc. have been proposed as possible origin.54 The longer decay component (6.8 ps) is associated with the population decay of the proton transferred tautomer species. Similar ultrafast decay of proton transfer tautomer emission associated with rotation around the exocyclic bond in the excited state has been reported earlier for o-HBDI and similar intramolecularly H-bonded analogues of o-HBDI.15,54 Monitoring at the normal CT band, a slower biexponential excited state population decay has been observed (Fig. 4). Similar to the methoxy derivative mOMe-MBDI, as solvent β value and polarity increase the decay become slower. Unlike mOMe-MBDI, for mOMe-HBDI the magnitude of both the short and long lifetime components increases (from 0.17 ns to 0.60 ns and from 1 ns to 3.6 ns respectively) (see Table 3). For a particular solvent (say acetonitrile), when decay was monitored at the CT band, as monitoring wavelength increases the amplitude of the longer lifetime component increases. Thus, for a solvent with significantly high β value and high polarity, the fluorescence decay of mOMe-HBDI appears to be similar to that of mOMe-MBDI. The current result also indicates that the normal band originates due to the ground state weakening or partial rupture of the H-bond. This assertion is based on the lack of correlation between the decay of normal emission and evolution of proton transfer tautomer emission.15 Similar to mOMe-MBDI, TRANES with an isoemissive point indicates that the emission is occurring from two different conformers. Thus, similar to mOMe-MBDI, it can be concluded for mOMe-HBDI that the normal emission is taking place from two different conformers having differential stabilisation by solvent polarity and H-bond accepting ability. In order to check whether in solvents of high β value anion formation takes place or not, steady state optical studies in alkaline ACN and alkaline DMF have been carried out. In alkaline ACN solution, the emission maximum of the anionic form of mOMe-HBDI is at 639 nm whereas in normal ACN the emission maximum is at 507 nm (ESI,† S6 and Fig. S11). A similar observation has been obtained for DMF as well. Thus, involvement of the anionic form in the observed photophysics and fluorescence decay of mOMe-HBDI was excluded. For MOHIM, monitoring at the emission band maxima in different solvents (see ESI,† Fig. S20 and Table S3), multiexponential decay is observed. In hexane, an ultrafast decay component (<60 ps, system response limited) is observed along with two other components in the nanosecond time region. From previous literature reports54 and from the current study on mOMe-HBDI, the system response limited faster decay possibly originates from the tautomer depopulation. In contrast, the longer decay component in the nanosecond time region is probably coming from the emission of the normal species. Thus, even in hexane we could expect the possible existence of both the PT tautomer and normal CT species in the excited state. However, in DMSO no such ultrafast decay component (<64 ps) is observed for MOHIM. Thus, in DMSO the normal CT species exclusively dominates the excited state decay of MOHIM. Detailed spectroscopic investigations show that intramolecular proton transfer can take place for mOMe-HBDI and MOHIM in hexane, and DCM. In these solvents of lower β value, mOMe-HBDI and MOHIM possess ϕfl of ∼0.01% (sum of dual emission). It was also observed that the proton transfer band emits in the sub-picosecond time scale (8.05 ps in DCM for mOMe-HBDI). This fact indicates that the proton transfer state of mOMe-HBDI and MOHIM acts as a weakly fluorescent state and causes ultrafast nonradiative decay of the excited state population. In contrast, in DMSO, owing to partial rupture of the intramolecularly H-bonded planar conformation, proton transfer does not take place in the excited state of mOMe-HBDI and MOHIM. As a result emission takes place exclusively from the normal CT state. Hence, owing to the barrier to twisting induced by the same ortho–meta effect (as observed for mOMe-MBDI), mOMe-HBDI possesses very high values of ϕfl (21%) and τfl (3.2 ns) in DMSO. However, for MOHIM ϕfl does not increase much in DMSO mainly due to the initiating of energy gap law governed nonradiative decay as the emission maximum (700 nm) is in the red region.10,45,46
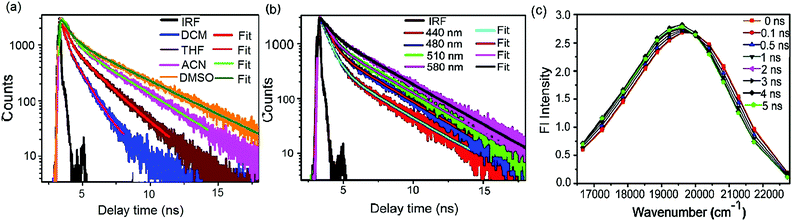 |
| | Fig. 4 (a) Variation in fluorescence decay (time-correlated single-photon counting, TCSPC) of mOMe-HBDI in different fluid organic solvents of different polarity. (b) Fluorescence decay of mOMe-HBDI in acetonitrile monitored at different wavelengths of steady state emission spectra (normal charge transfer band). (c) Construction of time-resolved area-normalised emission spectra (TRANES) for mOMe-HBDI in acetonitrile monitored at the charge transfer normal emission band. | |
The larger the population of the proton transferred tautomer state (proportional to the ground state intramolecular hydrogen bonding strength in different solvents), the lower the overall ϕfl. Thus, the lower the chance of proton transfer or higher the chance of charge transfer, the higher are ϕfl and τfl. Hence it is necessary to know the nature of the proton transferred state. Steady state emission spectra of mOMe-HBDI show that the proton transfer band has negligible solvatochromic shift of emission maxima in solvents of varying polarity (hexane to ACN). Similar results have been reported in the literature.31 This means that in the proton transferred species there is negligible CT character. This suppression of CT character is mainly due to relocation of the proton between donor and acceptor centre in the excited state of mOMe-HBDI.31,33,56 Suppression of charge transfer by the proton transfer process makes the charge transfer induced (ortho–meta effect) enhancement of ϕfl and τfl inefficient for mOMe-HBDI. The highly Stokes shifted proton transfer band also results in ultrafast deactivation of the excited state proton transferred tautomer species. In accordance with literature reports, this kind of nonradiative decay could be assigned to overtone vibration associated with the motion of the hydrogen bond.20,54 In hexane, formation of the intramolecularly H-bonded planar conformation favours excited state proton transfer and consequently mOMe-HBDI is very weakly fluorescent (ϕfl ∼ 0.01%) because of the more nonradiative nature of fluorescence decay in hexane. However, in DMSO, owing to partial rupture of the intramolecular H-bond (evidenced from the NMR spectral analysis and steady state optical analyses), emission occurs exclusively from the normal charge transfer state. This results in not-so-large Stokes shifted emission and thus a smaller proportion of non-radiative decay, thereby causing a higher fluorescence quantum yield (ϕfl = 21%) of mOMe-HBDI in DMSO. It would be interesting to investigate the relative dynamics of CT and PT for mOMe-HBDI and MOHIM in different solvents by means of femtosecond pump–probe technique using ultrashort time resolution (less than 100 fs). Thus, more detailed information about the dynamics of CT and PT and its influence on relative intensity of CT and PT bands could be understood. These experiments will be performed in the near future.
Conclusions
The effect of H-bond accepting ability and polarity of the solvent towards enhancement of fluorescence quantum yield and fluorescence lifetime of meta GFP chromophore analogues has been reported in detail for the first time. From single crystal X-ray analysis it could be shown that strong intramolecular H-bonding exists in the hydroxy derivative (mOMe-HBDI). However, as the solvent H-bond accepting ability increases, the strength of intramolecular H-bonding decreases and this process has strong influence on the conformational structure or more precisely relative orientation of phenyl and imidazolidinone rings. For the hydroxy derivative, mOMe-HBDI fluorescence quantum yield increases with increase in H-bond accepting ability or β value of the solvents. However, an effect of solvent polarity cannot be completely ruled out. Quite interestingly dual fluorescence has been observed in the case of mOMe-HBDI. The lower wavelength emission band (∼480 nm) has been assigned to normal charge transferred species, whereas the highly Stokes shifted emission band (∼660 nm) has been assigned to proton transferred tautomer species. In solvents of low β value only the PT band and in solvents of high β value only the CT band is observed, whereas in solvents of intermediate β value both CT and PT bands, and hence dual emission, are observed. For the methoxy derivative mOMe-MBDI, fluorescence quantum yield increases as solvent polarity increases. This observation is in stark contrast with existing literature reports where fluorescence quantum yield of several GFP chromophore analogues has been reported to decrease with increase in solvent polarity. For the methoxy derivative mOMe-MBDI, fluorescence decay has been observed to be biexponential. The short component changes from ∼0.4 ns to 0.6 ns whereas the long component changes from 1 ns to 3 ns as solvent polarity increases. Amplitudes of both short and long lifetimes are almost independent of solvent polarity. However, for a particular solvent (say acetonitrile), as monitoring wavelength increases the amplitude of the longer component increases. This observation hints at the possible existence of two conformers which have differential stabilisation by solvent. For mOMe-HBDI, when fluorescence decay is monitored at the normal emission band it is observed to be biexponential in nature. For mOMe-HBDI, when fluorescence decay is monitored at the normal charge transfer band the short component increases from ∼0.2 ns to 0.6 ns and the long component increases from 1 ns to 3.6 ns. For a particular solvent, when fluorescence decay is monitored at the normal charge transfer band, as the monitoring wavelength increases the amplitude of the long lifetime component increases and that of the short lifetime component decreases. As for mOMe-MBDI, TRANES analysis of mOMe-HBDI also reveals the existence of two conformers having differential charge transfer stabilisation by solvent. When the fluorescence decay is monitored at the tautomer band an instrument response limited (<280 fs) rise followed by a decay (with two time constants, one of 0.4 to 0.8 ps and another of 6.8 to 8.1 ps) were observed. For both mOMe-MBDI and mOMe-HBDI, as solvent polarity increases, the ICT state (1S*) is more stabilised and the energy barrier from the ICT state (1S*) to the twisted state (1P*) increases. Thus, there will be less chance of twisted state formation; hence there is less chance of Z–E isomerisation. Thus, there will be enhanced ϕfl and τfl.
Conflict of interest
The authors declare no competing financial interests.
Acknowledgements
P. K. M. thanks IISER-Kolkata for financial help and instrumental facilities. Support from the Fast-Track Project (SR/FT/CS-52/2011) of DST-India is gratefully acknowledged. P. K. M. acknowledges Prof. P. Ramamurthy (NCUFP) for his help in femtosecond decay measurements. T. C. and M. M. thank CSIR India for their respective Fellowships.
References
- H. Niwa, S. Inouye, T. Hirano, T. Matsuno, S. Kojima, M. Kubota, M. Ohashi and F. I. Tsuji, Proc. Natl. Acad. Sci. U. S. A., 1996, 93, 13617–13622 CrossRef CAS.
- P. Altoe, F. Bernardi, M. Garavelli, G. Orlandi and F. Negri, J. Am. Chem. Soc., 2005, 127, 3952–3963 CrossRef CAS PubMed.
- J. Conyard, M. Kondo, I. A. Heisler, G. Jones, A. Baldridge, L. M. Tolbert, K. M. Solntsev and S. R. Meech, J. Phys. Chem. B, 2011, 115, 1571–1577 CrossRef CAS PubMed.
- M. S. Baranov, K. A. Lukyanov, A. O. Borissova, J. Shamir, D. Kosenkov, L. V. Slipchenko, L. M. Tolbert, I. V. Yampolsky and K. M. Solntsev, J. Am. Chem. Soc., 2012, 134, 6025–6032 CrossRef CAS PubMed.
- M. Zimmer, Chem. Rev., 2002, 102, 759–781 CrossRef CAS PubMed.
- K. L. Litvinenko, N. M. Webber and S. R. Meech, J. Phys. Chem. A, 2003, 107, 2616–2623 CrossRef CAS.
- A. Follenius-Wund, M. Bourotte, M. Schmitt, F. Iyice, H. Lami, J. J. Bourguignon, J. Haiech and C. Pigault, Biophys. J., 2003, 85, 1839–1850 CrossRef CAS PubMed.
- S. Rafiq, B. K. Rajbongshi, N. N. Nair, P. Sen and G. Ramanathan, J. Phys. Chem. A, 2011, 115, 13733–13742 CrossRef CAS PubMed.
- I. Petkova, G. Dobrikov, N. Banerji, G. Duvanel, R. Perez, V. Dimitrov, P. Nikolov and E. Vauthey, J. Phys. Chem. A, 2010, 114, 10–20 CrossRef CAS PubMed.
- T. Chatterjee, M. Mandal, V. Gude, P. P. Bag and P. K. Mandal, Phys. Chem. Chem. Phys., 2015, 17, 20515–20521 RSC.
- G. J. Huang, C. W. Cheng, H. Y. Hsu, C. Prabhakar, Y. P. Lee, E. W. G. Diau and J. S. Yang, J. Phys. Chem. B, 2013, 117, 2695–2704 CrossRef CAS PubMed.
- K. L. Litvinenko, N. M. Webber and S. R. Meech, J. Phys. Chem. A, 2003, 107, 2616–2623 CrossRef CAS.
- D. Mandal, T. Tahara and S. R. Meech, J. Phys. Chem. B, 2004, 108, 1102–1108 CrossRef CAS.
- M. E. Martin, F. Negri and M. Olivucci, J. Am. Chem. Soc., 2004, 126, 5452–5464 CrossRef CAS PubMed.
- K. Y. Chen, Y. M. Cheng, C. H. Lai, C. C. Hsu, M. L. Ho, G. H. Lee and P. T. Chou, J. Am. Chem. Soc., 2007, 129, 4534–4535 CrossRef CAS PubMed.
- A. Baldridge, K. M. Solntsev, C. Song, T. Tanioka, J. Kowalik, K. Hardcastle and L. M. Tolbert, Chem. Commun., 2010, 46, 5686–5688 RSC.
- A. Baldridge, S. Feng, Y. T. Chang and L. M. Tolbert, ACS Comb. Sci., 2011, 13, 214–217 CrossRef CAS PubMed.
- M. S. Baranov, K. A. Lukyanov, A. O. Borissova, J. Shamir, D. Kosenkov, L. V. Slipchenko, L. M. Tolbert, I. V. Yampolsky and K. M. Solntsev, J. Am. Chem. Soc., 2012, 134, 6025–6032 CrossRef CAS PubMed.
- T. Chatterjee, D. Roy, A. Das, A. Ghosh, P. P. Bag and P. K. Mandal, RSC Adv., 2013, 3, 24021–24024 RSC.
- W. T. Chuang, C. C. Hsieh, C. H. Lai, C. H. Lai, C. W. Shih, K. W. Chen, W. Y. Hung, Y. H. Hsu and P. T. Chou, J. Org. Chem., 2011, 76, 8189–8202 CrossRef CAS PubMed.
- L. Wu and K. Burgess, J. Am. Chem. Soc., 2008, 130, 4089–4096 CrossRef CAS PubMed.
- Y. H. Hsu, Y. A. Chen, H. W. Tseng, Z. Zhang, J. Y. Shen, W. T. Chuang, T. C. Lin, C. S. Lee, W. Y. Hung, B. C. Hong, S. H. Liu and P. T. Chou, J. Am. Chem. Soc., 2014, 136, 11805–11812 CrossRef CAS PubMed.
- A. Baldridge, S. R. Samanta, N. Jayaraj, V. Ramamurthy and L. M. Tolbert, J. Am. Chem. Soc., 2010, 132, 1498–1499 CrossRef CAS PubMed.
- J. S. Paige, K. Y. Wu and S. R. Jaffrey, Science, 2011, 333, 642–646 CrossRef CAS PubMed.
- K. P. Kent, L. M. Oltrogge and S. G. Boxer, J. Am. Chem. Soc., 2009, 131, 15988–15989 CrossRef CAS PubMed.
- A. Baldridge, A. Amador and L. M. Tolbert, Langmuir, 2011, 27, 3271–3274 CrossRef CAS PubMed.
- K. P. Kent and S. G. Boxer, J. Am. Chem. Soc., 2011, 133, 4046–4052 CrossRef CAS PubMed.
- E. A. Dolgopolova, T. M. Moore, W. B. Fellows, M. D. Smith and N. B. Shustova, Dalton Trans., 2016, 45, 9884–9891 RSC.
- G. J. Huang, J. H. Ho, C. Prabhakar, Y. H. Liu, S. M. Peng and J. S. Yang, Org. Lett., 2012, 14, 5034–5037 CrossRef CAS PubMed.
- P. T. Chou, M. L. Martinez and J. H. Clements, J. Phys. Chem., 1993, 97, 2618–2622 CrossRef CAS.
- C. C. Hsieh, C. M. Jiang and P. T. Chou, Acc. Chem. Res., 2010, 43, 1364–1374 CrossRef CAS PubMed.
- P. K. Mandal and A. Samanta, J. Phys. Chem. A, 2003, 107, 6334–6339 CrossRef CAS.
- S. Jana, S. Dalapati and N. Guchhait, J. Phys. Chem. A, 2013, 117, 4367–4376 CrossRef CAS PubMed.
- J. Dong, K. M. Solntsev, O. Poizat and L. M. Tolbert, J. Am. Chem. Soc., 2007, 129, 10084–10085 CrossRef CAS PubMed.
- K. M. Solntsev, O. Poizat, J. Dong, J. Rehault, Y. Lou, C. Burda and L. M. Tolbert, J. Phys. Chem. B, 2008, 112, 2700–2711 CrossRef CAS PubMed.
- J. S. Yang, G. J. Huang, Y. H. Liu and S. M. Peng, Chem. Commun., 2008, 1344–1346 RSC.
- F. D. Lewis, S. R. Kalgutkar and J. S. Yang, J. Am. Chem. Soc., 1999, 121, 12045–12053 CrossRef CAS.
- H. K. Sinha and K. Yates, J. Am. Chem. Soc., 1991, 113, 6062–6067 CrossRef CAS.
- H. E. Zimmerman, J. Am. Chem. Soc., 1995, 117, 8988–8991 CrossRef CAS.
- H. E. Zimmerman, J. Phys. Chem. A, 1998, 102, 5616 CrossRef CAS.
- T. Chatterjee, M. Mandal, A. Das, K. Bhattacharyya, A. Dutta and P. K. Mandal, J. Phys. Chem. B, 2016, 120, 3503–3510 CrossRef CAS PubMed.
- J. S. Lee, A. Baldridge, S. Feng, Y. SiQiang, Y. K. Kim, L. M. Tolbert and Y. T. Chang, ACS Comb. Sci., 2011, 13, 32–38 CrossRef CAS PubMed.
- P. J. Langley, J. Hulliger, R. Thaimattamb and G. R. Desiraju, New J. Chem., 1998, 1307–1309 RSC.
- L. R. Snyder, J. Chromatogr., 1974, 92, 223–230 CrossRef CAS.
-
N. J. Turro, V. Ramamurthy and J. C. Scaiano, Modern molecular photochemistry of organic molecules, University Science Books, Sausalito, CA, 2010 Search PubMed.
- J. V. Caspar and T. J. Meyer, J. Phys. Chem., 1983, 87, 952–957 CrossRef CAS.
- H. E. Zimmerman, J. Phys. Chem. A, 1998, 102, 5616–5621 CrossRef CAS.
- J. Dong, K. M. Solntsev and L. M. Tolbert, J. Am. Chem. Soc., 2006, 128, 12038–12039 CrossRef CAS PubMed.
- V. Bonaić-Koutecký, J. Köhler and J. Michl, Chem. Phys. Lett., 1984, 104, 440–443 CrossRef.
- V. Bonaić-Koutecký and J. Michl, J. Am. Chem. Soc., 1985, 107, 1765–1766 CrossRef.
- C. W. Cheng, G. J. Huang, H. Y. Hsu, C. Prabhakar, Y. P. Lee, E. W. G. Diau and J. S. Yang, J. Phys. Chem. B, 2013, 117, 2705–2716 CrossRef CAS PubMed.
- E. M. Crompton and F. D. Lewis, Photochem. Photobiol. Sci., 2004, 3, 660–668 CAS.
- G. Cui, Z. Lan and W. Thiel, J. Am. Chem. Soc., 2012, 134, 1662–1672 CrossRef CAS PubMed.
- C. C. Hsieh, P. T. Chou, C. W. Shih, W. T. Chuang, M. W. Chung, J. Lee and T. Joo, J. Am. Chem. Soc., 2011, 133, 2932–2943 CrossRef CAS PubMed.
- Y. J. Ai, R. Z. Liao, W. H. Fang and Y. Luo, Phys. Chem. Chem. Phys., 2012, 14, 13409–13414 RSC.
- A. P. Demchenko, K. C. Tang and P. T. Chou, Chem. Soc. Rev., 2013, 42, 1379–1408 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details, synthesis and characterisation of GFP chromophore analogues, additional NMR data and stack plot, crystallographic table, additional steady state and time-resolved spectroscopic data, etc. CCDC 1417289 and 1417290. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6cp04219h |
|
| This journal is © the Owner Societies 2016 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence