
Visible light mediated photoredox reactions catalyzed by recyclable PIB-bound ruthenium photoredox catalysts†
Yannan
Liang
 and
David E.
Bergbreiter
*
and
David E.
Bergbreiter
*
Department of Chemistry, Texas A&M University, P.O. Box 30012, College Station, Texas 77843-3012, USA. E-mail: bergbreiter@tamu.edu; Fax: +1 (979) 8454719; Tel: +1 (979) 8453437
First published on 8th October 2015
Abstract
This report describes the preparation of a PIB-bound Ru(II)-bipyridine complex [Ru(PIB-bpy)3Cl2], and its use as a recyclable photoredox catalyst to carry out both oxidative C–C bond cleavage of aldehydes and [2 + 2] cycloaddition of bis(enone)s. While this polymer-supported Ru catalyst could be successfully recovered and reused for 5 cycles with no loss of catalytic activity and with leaching levels of ca. 1% of the charged catalyst for these reactions, other reactions like photodimerization or alkyl halide synthesis that require a more polar solvent medium for successful reactions of the low molecular weight catalyst proceeded either with varying selectivity or in low yield because of solubility limitations of the PIB-supported catalyst in the more polar solvents that are often used for this chemistry.
1. Introduction
In recent years, the development of environmentally benign synthetic pathways has become increasingly important. Photochemistry and photocatalysis are examples of such chemistry. Catalytic reactions that use visible light as a sustainable, abundant, and environmentally benign energy source have recently received heightened attention with the development of new homogeneous organo- and metal photocatalysts for such tranformations.1,2 For example, tris(bipyridine)ruthenium(II) dichloride [Ru(bpy)3Cl2] and similar Ir complexes have received attention as new photoredox catalysts due to their stability, long excited state lifetime, and excellent photoredox properties.3,4 The reactivity of [Ru(bpy)3Cl2] and its application to a number of visible light induced reactions by groups including those of MacMillan,5 Stephenson,6,7 Yoon8–10 and others11–13 have shown the applicability of such catalysts to diverse chemistry including C–H functionalization, reductive dehalogenation, polymerizations, and cycloadditions.Our interest in this chemistry arose because of our interests in developing effective procedures for reuse of homogeneous catalysts, especially costly and potentially toxic transition metal catalysts. While such environmental concerns can often be addressed using catalysts immobilized on insoluble supports,14 catalyst homogeneity is especially important for photocatalysts like [Ru(bpy)3Cl2] in maximizing the effectiveness of light sources and where flow reactors might be used. Work by various groups including our own has shown that soluble polymer supports serve as useful tools to facilitate recovering catalysts and ligands.15–17 We also have shown that the separation/recovery procedures we have developed work with Ru photoredox polymerization catalysts.18 Here we explore the broader applicability and limitations of these soluble polymer-supported Ru photoredox catalysts in other catalytic reactions.
Our initial work showed that polyisobutylene (PIB)-ligated versions of the Ru(II) bipyridine dichloride complex could be prepared and used in light initiated polymerization reactions of alkyl acrylates.18 These soluble catalysts effect polymerizations like their low molecular weight homogeneous analogs and can be effectively separated from polymer products. With this [Ru(PIB-bpy)3Cl2] catalyst, this separation involved a liquid solid separation of a solution of the catalyst from the solid polymer product, a strategy like one we had previously used in styrene polymerization.19 While this solid/liquid separation strategy is unlikely to be useful in most reactions of this Ru photoredox catalyst where the products and catalysts are both soluble after a reaction or in flow reactors where the formation of solids can be problematic, we wanted to explore the potential of these separable and recyclable [Ru(PIB-bpy)3Cl2] catalyst in other reactions where a liquid/liquid separation of the catalyst could occur. The studies described below show both the potential and limitations associated with use of a PIB-supported alkane phase selectively soluble PIB supported photoredox catalyst in three examples of reactions that are typically effected in moderately polar solvents. Specifically we show how a [Ru(PIB-bpy)3Cl2] catalyst can be used successfully as recyclable catalyst in an oxidative C–C bond cleavage reaction, in a [2 + 2] cycloaddition reaction, and unsuccessfully in reactions that convert alcohols to bromides. The principle limitation in these cases is the issue of solvent polarity in that photoredox reactions that require solvents like acetonitrile do not necessarily work as well or at all when solvents compatible with a PIB polymer support are necessary.
2. Experimental
2.1 General information
Vinyl terminated PIB (Glissopal 2300) with an Mn of 2300 Da, was provided by BASF.20 Other reagents and solvents were purchased from commercial suppliers and used without further purification unless otherwise stated. All glassware was oven- dried or flame-dried before use. 1H NMR and 13C NMR spectra were obtained on an Inova 300 spectrometer operating at 299.91 MHz for proton and 75.41 MHz for carbon nuclei, and on an Inova 500 spectrometer operating at 499.95 MHz for proton and 125.72 MHz for carbon nuclei at ambient temperature. Chemical shifts are quoted in ppm and referenced to the residual proton in CDCl3. Spin multiplicities are indicated by the following symbols: s (singlet), d (doublet), t (triplet), dd (doublet of doublet) and m (multiplet). UV-vis spectra were recorded on a Shimadzu UV-2600 spectrometer. A NexION 300D inductively coupled plasma-mass spectrometer (ICP-MS) was used to determine Ru loading in the catalyst and crude products. PIB-OH 2,21 PIB-Br 3,21 2,3-diphenylpropanal 7,22 and bis(enone) 9 (ref. 23) were synthesized following the literature procedures.2.2 Synthesis of PIB-bound bipyridine ligand 5
A 100 mL round-bottomed flask was equipped with a magnetic stir bar and sealed with a rubber septum. Then 10 mL of distilled anhydrous THF was transferred into the flask by forced siphon. The THF was cooled to −78 °C. Diisopropylamine (0.46 mL, 3.3 mmol) was added by syringe, followed by the addition of 2.5 mL of a 1.6 M solution of n-butyllithium in hexane (4 mmol). After stirring for 45 min, a solution of 4,4′-dimethyl-2,2′-bipyridine 4 (0.61 g, 3.3 mmol) in 10 mL of anhydrous THF was added to the flask. The reaction mixture was allowed to stir at −78 °C for 2 h. At this point, a solution of PIB-Br 3 (5.1 g, 2.1 mmol) in 20 mL of anhydrous THF was transferred to the flask by forced siphon. This solution was allowed to stir at −78 °C and slowly warmed to ambient temperature for 20 h. Then the reaction was quenched by adding 5 mL of methanol and the solvent was removed under reduced pressure. The resulting residue was dissolved in 75 mL of hexane and this phase was extracted 3 times with 20 mL of 90% ethanol/water and 3 times with 20 mL of brine. Then the hexane layer was dried over anhydrous sodium sulfate and the solvent was removed under reduced pressure to obtain a crude product that was purified by silica gel column chromatography using hexane/ethyl acetate (9![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, v/v) as the eluent to obtain the product 5 (4.6 g, 74% yield) as light yellow viscous oil.
:1, v/v) as the eluent to obtain the product 5 (4.6 g, 74% yield) as light yellow viscous oil.
PIB-bipyridine ligand (5): 1H NMR (300 MHz, CDCl3) δ: 8.58 (d, 2 H), 8.29 (s, 2 H), 7.19 (s, 2 H), 2.74 (m, 2.4 H), 2.48 (s, 2.4 H), 1.95–0.75 (m); 13C NMR (125 MHz, CDCl3) δ: 156.1, 153.2, 149.0, 148.9, 148.1, 124.6, 123.9, 122.0, 121.3, 59.5–22.7 (multiple peaks).
2.3 Synthesis of PIB-bound Ru(II)-bipyridine [Ru(PIB-bpy)3Cl2] complex 6
A 50 mL pressure vessel equipped with a magnetic stir bar was charged with PIB-bipyridine ligand 5 (1.53 g, 0.52 mmol), anhydrous RuCl3 (31.2 mg, 0.15 mmol), 5 mL of heptane, and 5 mL of ethanol and sealed with a rubber septum. N2 was bubbled through this solution for 15 min and then the rubber septum was replaced with the pressure vessel lid and closed tightly. This reaction mixture was allowed to stir at 90 °C for 24 h, at which point the reaction mixture was cooled to ambient temperature and 5 mL of water was added. The mixture was transferred to a separatory funnel, and another 50 mL of hexane was added to the funnel. The top non-polar phase was separated and extracted with 20 mL of 90% ethanol/water three times. Then the hexane layer was dried over anhydrous sodium sulfate and the hexane solvent was removed under reduced pressure. This crude product was purified by chromatography using neutral alumina column as a support and dichloromethane/methanol (9:1, v/v) as the eluent to afford the desired product (1.39 g, 87% yield) as red viscous oil with a metal loading of 9.47 × 10−3 g of Ru per g of 6 as determined by ICP-MS. If 6 were formed without any fractionation of the PIB2300 phase tag, it should have had a molecular weight of 8914 Da. The 10660 Da Mn for 6 calculated based on the ICP-MS analysis suggests that some fractionation of the PIB label occurred and that 6 is better described as having PIB2750 phase tags. The UV-visible spectroscopy of 6 had a λmax = 465 nm in hexane that was comparable to the previously reported data for a similar [Ru(PIB-bpy)3Cl2] complex in hexane that had a λmax = 463 nm (ε = 15500 M−1 cm−1).18
[Ru(PIB-bpy)3Cl2] complex (6): 1H NMR (300 MHz, CDCl3) δ: 8.46 (m, 2 H), 7.66 (m, 2 H), 7.36 (m, 2 H), 2.84 (m, 2.6 H), 2.64 (s, 2.8 H), 1.95–0.75 (m); 13C NMR (125 MHz, CDCl3) δ: 156.5, 156.4, 156.3, 156.2, 154.7, 151.2, 151.0, 150.7, 150.6, 150.1, 150.0, 129.3, 129.2, 129.1, 129.0, 128.1, 127.9, 125.0, 124.1 84.7–14.1 (multiple peaks).
2.4 General procedure for oxidative C–C bond cleavage catalyzed by 6
A 20 mL Schlenk tube equipped with a magnetic stir bar was charged with [Ru(PIB-bpy)3Cl2] 6 (178 mg, 16.6 μmol), 2,3-diphenylpropanal 7 (54 mg, 0.25 mmol), piperidine (75 μL, 0.75 mmol), 4.5 mL of dichloromethane, and 0.5 mL of acetonitrile. Then the Schlenk tube was connected to a balloon filled with oxygen, and the homogeneous reaction mixture was allowed to stir under the irradiation of a 30 W household fluorescent bulb at ambient temperature for 10 h. The reaction was followed by 1H NMR spectroscopy. After the reaction was completed, the solvent was removed under reduced pressure. Hexane was added to dissolve the residue. This hexane phase was then extracted three times with 5 mL portions of acetonitrile. The catalyst containing hexane phase was recovered, the hexane was removed, and the recovered catalyst was reused for the following cycle with fresh substrate in 4.5 mL of dichloromethane and 0.5 mL of acetonitrile. The product containing acetonitrile was concentrated under reduced pressure to afford crude product. The crude product was then purified by silica column chromatography using hexane/ethyl acetate (19:1, v/v) as the eluenting solvent to yield the desired product 8 (51.6 mg per cycle, 96% yield) as light yellow waxy solid, mp = 48–53 °C (lit. 50–55 °C).24
1,2-Diphenylethanone (8): 1H NMR (300 MHz, CDCl3) δ (ppm): 8.07 (d, 2H), 7.60 (t, 1H), 7.50 (t, 2H), 7.36 (m, 5H), 4.34 (s, 2H); 13C NMR (75 MHz, CDCl3) δ (ppm): 197.6, 136.6, 134.6, 133.2, 129.5, 128.7, 128.6, 126.9, 45.5.
2.5 General procedure for [2 + 2] cycloaddition of bis(enone) catalyzed by 6
A 20 mL Schlenk tube equipped with a magnetic stir bar was charged with [Ru(PIB-bpy)3Cl2] 6 (176 mg, 16.4 μmol), bis(enone) 9 (77.8 mg, 0.25 mmol), N,N-diisopropylethylamine (100 μL, 0.57 mmol), and LiBF4 (57.9 mg, 0.62 mmol). Addition of 4.5 mL of dichloromethane and 0.5 mL of acetonitrile formed a solution which was sealed with a rubber septum and degassed three times using the freeze pump thaw method. Then the reaction mixture was allowed to stir under the irradiation of a 30 W household fluorescent bulb at ambient temperature for 10 h. 1H NMR spectroscopy was used to follow the reaction. After the reaction was complete, the solvent was removed under reduced pressure. At this point, hexane and acetonitrile was added to dissolve the residue. The two phases were separated and the hexane layer was extracted 3 times with 5 mL of acetonitrile. The catalyst containing hexane phase was isolated, the hexane was removed, and the recovered catalyst was reused for the following cycle with fresh substrate in 4.5 mL of dichloromethane and 0.5 mL of acetonitrile. The solvent was removed from the combined product-containing acetonitrile phases under reduced pressure to afford the crude products which were purified by silica column chromatography using hexane/ethyl acetate (19:1, v/v) as the eluent to yield the reductive cyclization product 10b (30 mg per cycle, 39% yield), and hexane/ethyl acetate (9:1, v/v) as the eluent to yield the cycloaddition product 10a (32.6 mg per cycle, 42% yield) as a white solid, mp = 148–153 °C (lit. 155–157 °C).23
(1R,5S,6R,7S)-6,7-Dibenzoylbicyclo[3.2.0]heptane (10a): 1H NMR (300 MHz, CDCl3) δ: 7.78 (d, 4H), 7.47 (t, 2H) 7.38 (t, 4H), 3.89 (d, 2H), 3.23 (m, 2 H), 2.04 (m, 2H), 1.87 (m, 2H), 1.72 (m, 2H); 13C NMR (75 MHz, CDCl3) δ (ppm): 198.6, 136.3, 132.5, 128.5, 127.8, 48.3, 39.1, 32.5, 25.2.
2,2′-((1S,2S)-Cyclopentane-1,2-diyl)bis(1-phenylethanone) (10b): 1H NMR (300 MHz, CDCl3) δ: 7.96 (dd, 4H), 7.56 (t, 2H), 7.47 (t, 4H), 3.22 (dd, 2H), 2.95 (dd, 2H), 2.20 (m, 2H), 1.99 (m, 2H), 1.64 (m, 2H), 1.29 (m, 2H); 13C NMR (75 MHz, CDCl3) δ: 200.3, 137.2, 132.9, 128.6, 128.1, 44.0, 41.6, 32.5, 23.7.
3. Results and discussion
The preparation of PIB-bound Ru(II)-bipyridine [Ru(PIB-bpy)3Cl2] complexes used here is shown in Scheme 1 and is similar to that that used previously.18 It began with commercially available alkene terminated PIB (Mn = 2300 Da), forming an alcohol 2via a hydroboration/oxidation reaction. This alcohol was then converted first into a mesylate and then into the bromide 3. In our prior work we showed that one PIB group per bipyridine is sufficient to produce a phase separable heptane soluble PIB-bound Ru complex. Attempts to convert both methyl groups of a 4,4′-dimethylbipyridine were always incomplete because they were frustrated by some unwanted E2 reactions of 3 with the lithiated bipyridine. Thus we modified our synthesis using 3 as the limiting reagent in reaction with the lithium derivative of 4,4′-dimethylbipyridine 4 formed using LDA at −78 °C. This led to a ca. 6:1 mixture of mono- and dialkylated PIB-bound bipyridine ligand 5 (1H NMR spectroscopic analysis was used to show that 15–20% of dialkylated product formed along with 5). The main by-product of this synthesis is unalkylated 4, which was easily separated from the mixture of PIB-containing products. Other by-products included alkene-terminated PIB that was also separated from 5 by chromatography. We had earlier shown the one PIB group/bipyridyl ligand was sufficient to make 5 or the Ru complex formed from 5 heptane phase selectively soluble. Following our prior procedure, PIB-bound bipyridine ligand 5 was then used to form the PIB-bound Ru(II)-bipyridine complex 6 by allowing PIB-bound bipyridine ligands 5 to react with anhydrous RuCl3 in a heptane/ethanol mixture at 90 °C. The product 6 was isolated as red viscous heptane soluble oil that was characterized by 1H NMR spectroscopy, 13C NMR spectroscopy, and UV-visible spectroscopy. Complex 6 had an absorbance at a λmax at 465 nm in hexane that was comparable to the previously reported data of [Ru(PIB-bpy)3Cl2] in hexane with λmax at 463 nm (ε = 15500 M−1 cm−1).18 The Ru complex 6 was as phase selectively soluble in heptane/CH3CN and heptane/DMF as the other analogs of 6 that were prepared form 100% dialkylated PIB-bound bipyridine or bipyridine ligands containing a 1:2 mixture of mono- and dialklyated bipyridine.
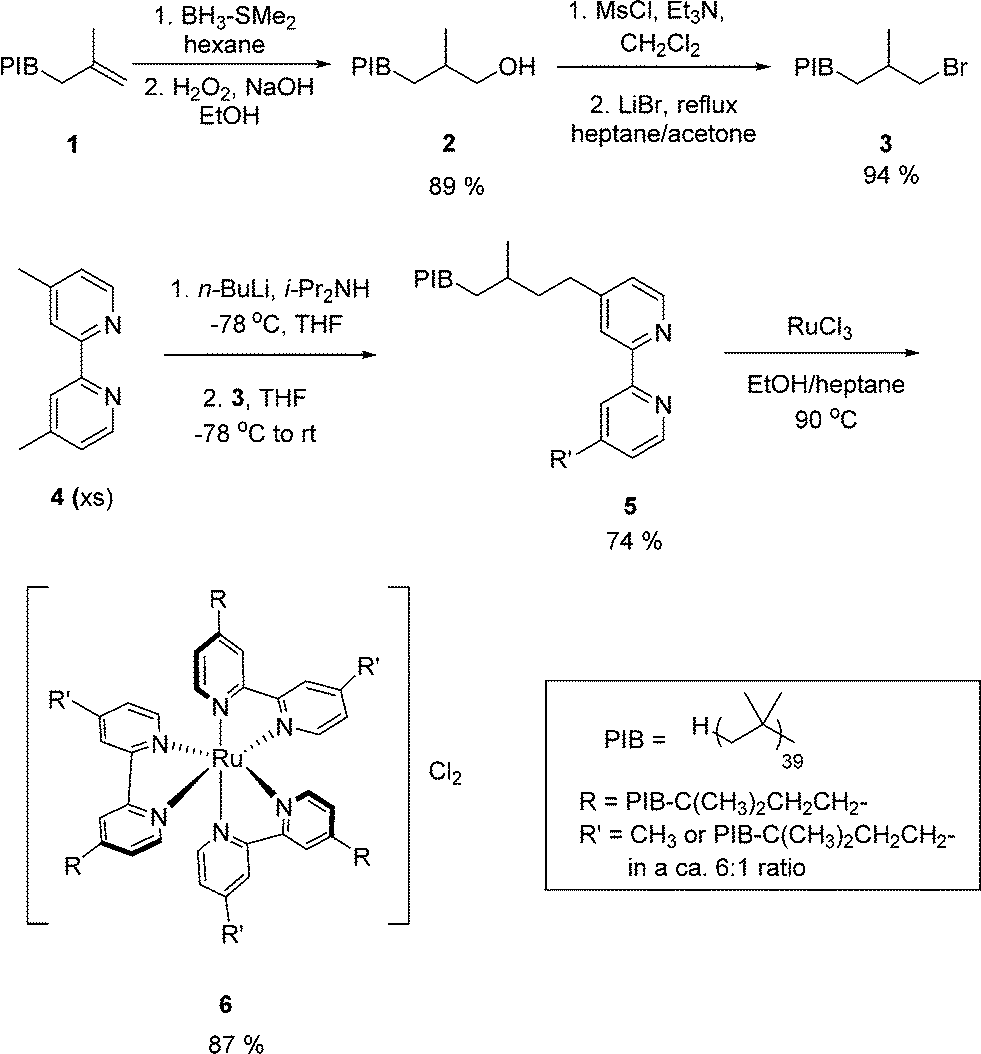 | ||
| Scheme 1 Synthesis of PIB-bound Ru(II)-bipyridine [Ru(PIB-bpy)3Cl2] complex 6. | ||
Having the PIB-supported ruthenium complex 6 in hand, we first investigated a visible-light mediated oxidative C–C bond cleavage reaction of aldehyde using this complex as a recyclable photoredox catalyst (Scheme 2). This reaction was first explored by Xia and his coworkers22 using low-molecular weight [Ru(bpy)3Cl2] as the catalyst. In our experiments, reactions were carried out using 2,3-diphenylpropanal 7 with 3 equiv. of piperidine in the presence of 6.6 mol% catalyst 6 in a 9/1 (vol/vol) CH2Cl2/CH3CN homogeneous system at ambient temperature using irradiation with a 30 W fluorescent bulb. After irradiation for 10 h, the starting material was fully converted to the desired oxidative C–C bond cleavage product 1,2-diphenylethanone 8. Reactions were followed by 1H NMR spectroscopy and the reactant aldehyde was fully converted to product (>98% conversion) after 10 h.
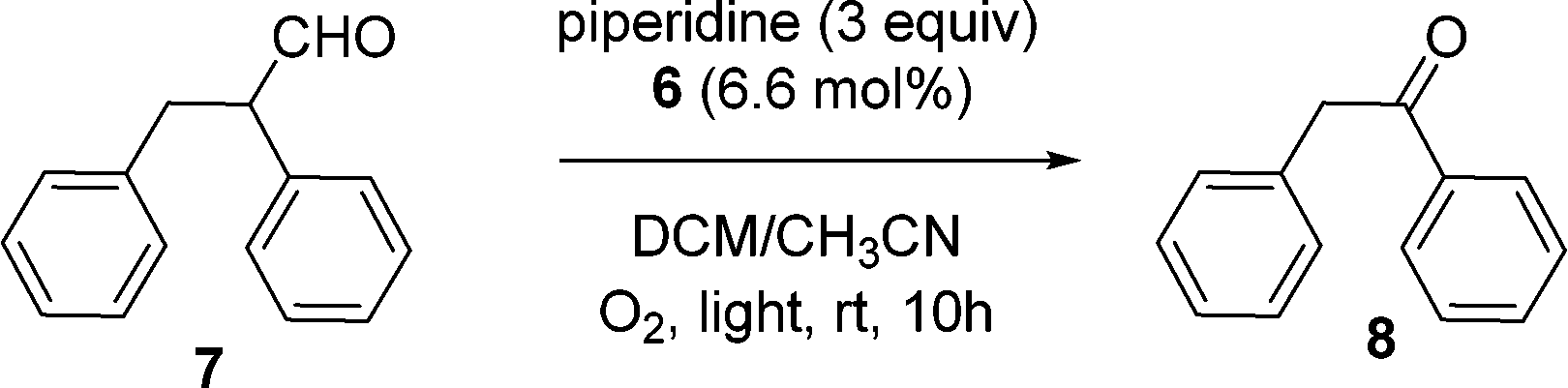 | ||
| Scheme 2 Oxidative C–C bond cleavage catalyzed by the PIB-bound Ru(II)-bipyridine complex 6. | ||
As shown in Table 1, the PIB-bound Ru(II)-bipyridine complex 6 showed excellent catalytic activity in this reaction, and can be successfully recovered and reused for 5 cycles with no significant loss of activity. The yield of the product was determined by 1H NMR spectroscopy using 1,1,2,2-tetrachloroethane as an internal standard showed that the yields were essentially quantitative in cycles 1–5. After each cycle, the solvent was removed under reduced pressure. The catalyst was then redissolved in hexane and the product was extracted from the catalyst solution in hexane using CH3CN. While the crude product was contaminated by excess amine, it could easily be purified by column chromatography. In our experiments, the crude products from all five cycles were combined and further purified by column chromatography. The isolated yield of the oxidative C–C cleavage product 8 in this combined material corresponded to 96% yield/cycle. These results are comparable to previous reported results using low-molecular weight [Ru(bpy)3Cl2] photoredox catalyst.22 Leaching of PIB-supported Ru catalyst into the polar phase during the recycling process was also tested using inductively coupled plasma mass spectroscopy (ICP-MS) to measure the Ru contamination in crude product from the third cycle of the reaction. The result of this test showed that the unpurified crude product contained 14 μg Ru which corresponds to ca. 0.9% of the initial amount of Ru used. This leaching was measured after the third cycle so that it measure leaching of catalysts. If the catalyst 6 were only ca. 99% pure, analysis of leaching in cycle 1 would not distinguish between catalyst leaching and an insignificant impurity in the starting catalyst.
| Cycle | Yield 8bc [%] |
|---|---|
| a Reactions were carried out with 0.25 mmol of aldehyde 7, 6.6 mol% of 6 and 3 equiv. of piperidine at ambient temperature using a 4.5 mL/0.5 mL mixture of CH2Cl2/CH3CN. b Yields were determined by 1H NMR spectroscopy using 1,1,2,2-tetrachloroethane as an internal standard. c The product of these five cycles was combined to facilitate isolation of 8. The yield of product 8 corresponded to an average isolated yield of 96% per cycle. | |
| 1 | 99 |
| 2 | 98 |
| 3 | 98 |
| 4 | 99 |
| 5 | 98 |
To try to avoid the extraction step in the strategy described above, a second strategy was also briefly studied. In this case we used a 1/4/1 (vol/vol/vol) heptane/THF/CH3CN solvent mixture. As can be seen in Fig. 1, the first cycle of the reaction was followed by 1H NMR spectroscopy, and the starting aldehyde 7 was fully converted to the desired product 8 after 10 h using visible light irradiation from a 30 W fluorescent bulb at room temperature at essentially the same rate as in the CH2Cl2/CH3CN solvent mixture. The presence of a significantly amount of heptane had no significant effect on the rate of the reaction with a similar yield at 10 h. There was a minor difference in activity with these solvent mixtures but the differences were not judged to be significant. This result was also not considered to be significantly different from the 94% yield of this product in 7 h reported previously using a low molecular weight catalyst in CH3CN.
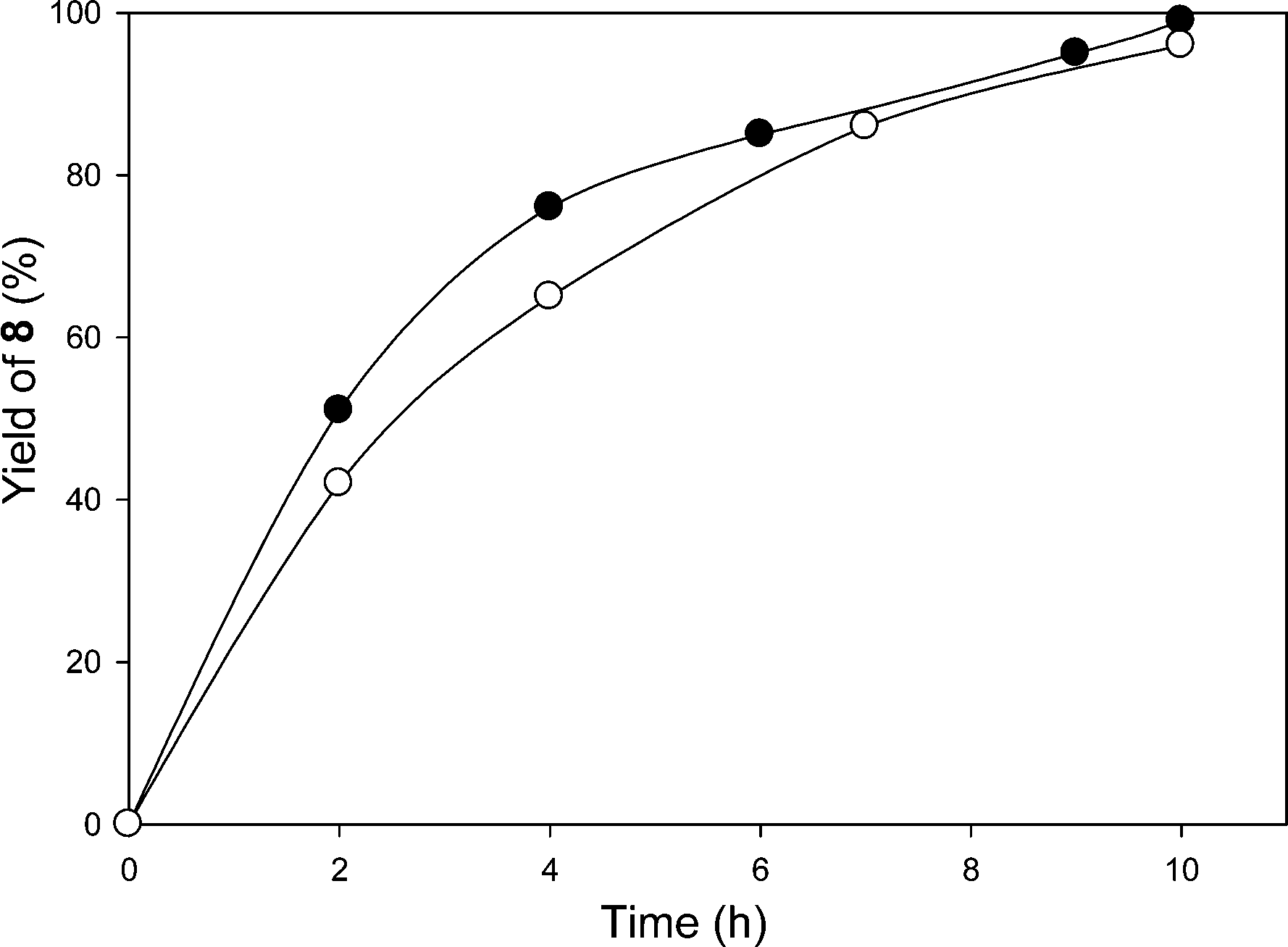 | ||
| Fig. 1 Plot of formation of product 8versus time in the first cycle of an oxidative C–C cleavage reaction of 7 in either a 9/1 (vol/vol) CH2Cl2/CH3CN (●) or a 1/4/1 (vol/vol/vol) heptane/THF/CH3CN (○) solvent mixture using complex 6 as the catalyst. | ||
The heptane/THF/CH3CN solvent mixture was examined because the PIB-bound Ru(II)-bipyridine complex can be easily recovered via a latent biphasic method using this solvent system by simply adding a 2 mL of water to the 6 mL reaction solution. This addition of water after the reaction causes this latent biphasic solvent mixture to separate into two phases producing an easily isolable less dense heptane-rich catalyst-containing phase that can be directly reused with fresh substrate for at least 4 cycles. However, while some product can be collected from the denser polar phase, the product 8 has only modest solubility in the water-containing polar phase. Thus, a good yield of 8 using this solvent system was only obtained by combining all portions of the reaction mixture at the end of the catalytic cycles. At this point, the catalyst could be separated from the product by solvent removal and extraction process (vide supra) or solvent could be removed from the combined phases and the product could be isolated by column chromatography. In either case, the average isolated yield of 4 cycles of 93% per cycle for the oxidative C–C cleavage product 8 was essentially unchanged from reactions using a CH2Cl2/CH3CN solvent mixture.
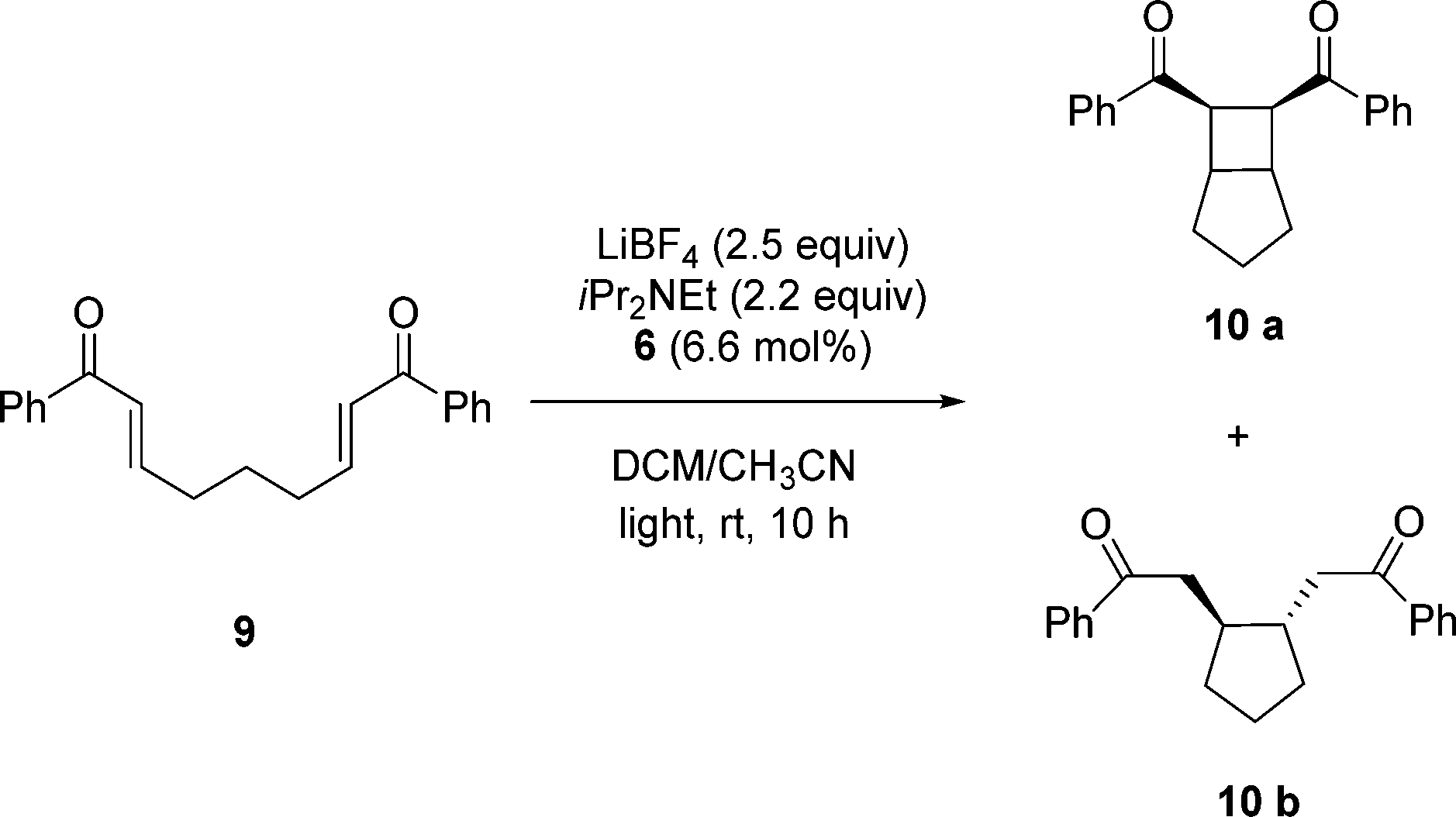
With success in an oxidative C–C bond cleavage using this recyclable PIB-bound Ru(II)-bipyridine complex, we next sought to apply this catalyst containing a ca. 6:1 mixture of bipyridyl ligands with one or two PIB groups to other reactions. Yoon and his coworkers8 have demonstrated that [2 + 2] cycloaddition of bis(enone)s is catalyzed by [Ru(bpy)3Cl2] under visible light in CH3CN as a solvent. This polar solvent is not suitable for 6 as PIB is completely insoluble in CH3CN.21 To use the PIB-bound Ru(II)-bipyridine complex 6 as a photoredox catalyst for [2 + 2] cycloaddition of bis(enone) 9 under homogeneous conditions, we used a 4.5 mL/0.5 mL mixture of CH2Cl2/CH3CN to dissolve 6.6 mol% of 6, bis(enone) 9, 2.5 equiv. of LiBF4, and 2.2 equiv. of iPr2NEt. The reaction mixture was then allowed to react at ambient temperature for 10 h using visible light irradiation from a 30 W fluorescent bulb in either CH2Cl2/CH3CN (Scheme 3). As shown in Fig. 2, the first cycle of the reaction was followed by 1H NMR spectroscopy, and the conversion of reactant bis(enone) 9 was over 97% after 10 h.
 | ||
| Scheme 3 [2 + 2] cycloaddition of bis(enone) catalyzed by the PIB-bound Ru(II)-bipyridine complex 6. | ||
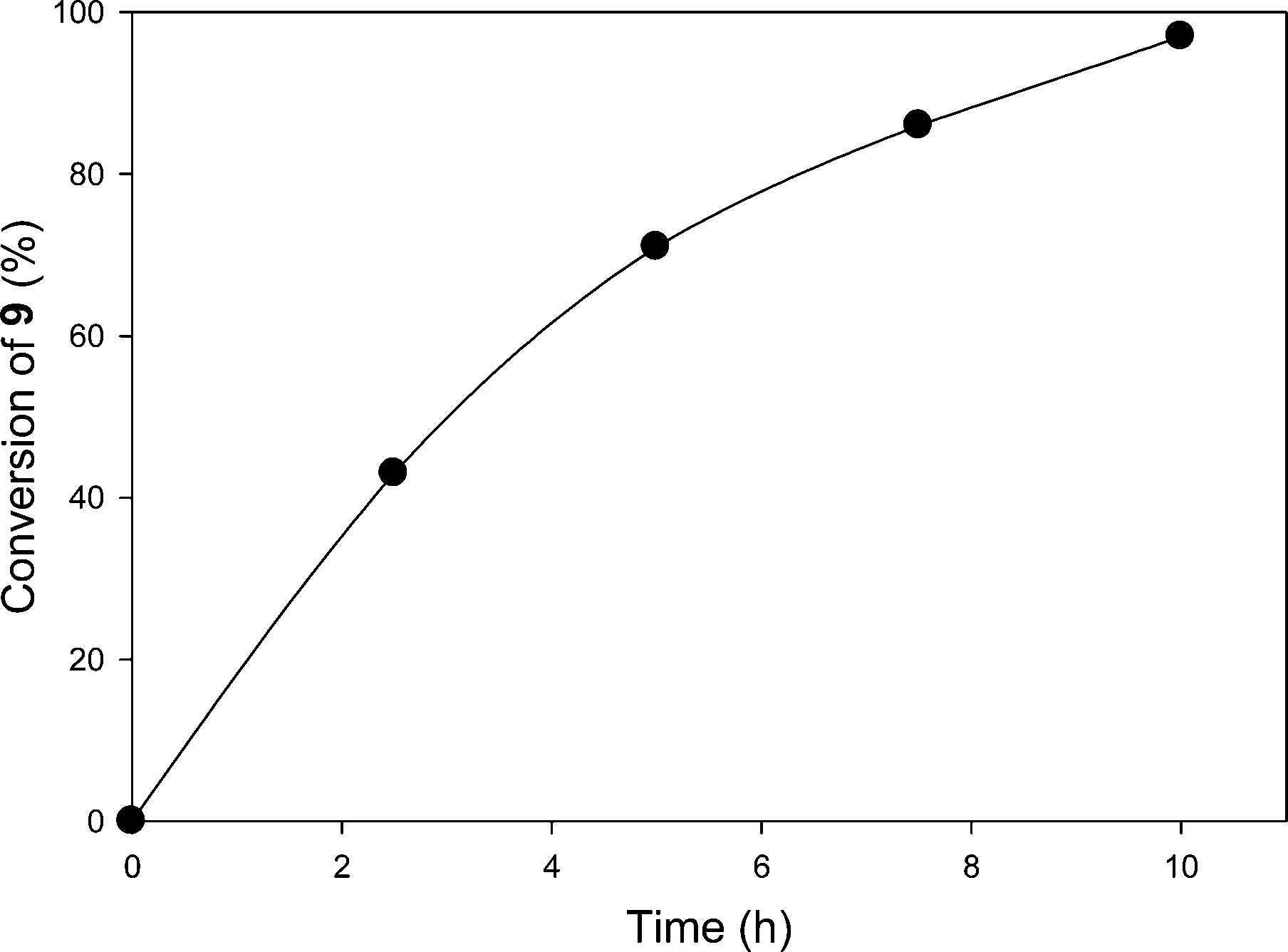 | ||
| Fig. 2 Plot of conversion of bis(enone) 9vs. time in the first cycle of [2 + 2] cycloaddition in a 9/1 (vol/vol) mixture of CH2Cl2/CH3CN using complex 6 as the catalyst. | ||
As can be seen in Table 2, the PIB-bound Ru(II)-bipyridine complex 6 showed good catalytic activity in this reaction, and the catalyst can be successfully recovered and reused for 5 cycles with no significant loss of activity. In this case, the product isolation, catalyst separation and catalyst recycling procedure followed the procedure described above for conversion of 7 to 8 in this same solvent mixture. However, while 6 could be recycled in this reaction and while the conversion of starting material to products was high, the selectivity for formation of the desired cycloaddition product 10a was modest under these solvent conditions. A significant amount of the reductive cyclization product 10b also formed as a side product in the reaction. When the crude products of these five cycles were combined, we were able to separate these two compounds by column chromatography. The average isolated yield of the cycloaddition product 10a was 210% (42% per cycle) and the reductive cyclization product 10b was 195% (39% per cycle). 1H NMR spectroscopic analysis of the crude product in cycles 1 and 2 showed that 10a and 10b were present in a roughly 1/1 ratio. The conversion in cycle 4 was anomalously lower than that in cycle 3 or 5. We did not further investigate this discrepancy. An ICP-MS analysis of the crude product from the third cycle of the reaction was analyzed by ICP-MS to measure the Ru contamination, and the result showed that 1.1% of catalyst 6 (18.6 μg Ru) leached into the CH3CN phase.
| Cycle | Yield of 10a and 10ab [%] | Yield 10bbcd [%] |
|---|---|---|
| a Reactions were carried out with 0.25 mmol of bisenone 9, 6.6 mol% of 6, 2.5 equiv. of LiBF4, and 2.2 equiv. of iPr2NEt at ambient temperature using a 4.5 mL/0.5 mL mixture of CH2Cl2/CH3CN. b Yields were determined by 1H NMR spectroscopy using 1,1,2,2-tetrachloroethane as an internal standard. c The average isolated yield of product 10a for 5 cycles is 42% per cycle. d The average isolated yield of product 10b for 5 cycles is 39% per cycle. | ||
| 1 | 98 | 51 |
| 2 | 99 | 41 |
| 3 | 94 | 39 |
| 4 | 65 | 29 |
| 5 | 98 | 60 |
While the yield of the recovered catalyst 6 was not measured, we did observe that there was no visual leaching of the highly chromogenic catalyst 6 into the product phase. Moreover, in recycling 6, we used recycled 6 in the same volume of solvent and observed that a visually identical intensity orange-red solution of 6 was reformed in cycles 2 and 3.
We postulated that the formation of a mixture of two products in this reaction is due to the solvent change. Hence, we carried out an experiment using the conventional low molecular weight [Ru(bpy)3Cl2] catalyst for the [2 + 2] cycloaddition of bis(enone) 9 in 4.5 mL/0.5 mL of CH2Cl2/CH3CN under the same conditions used with 6. While the bis(enone) 9 in this experiment was fully consumed, it again formed a mixture of products 10a and 10b after 1 h. In this case, the two products were isolated by column chromatography to yield 31% of product 10a and 61% of product 10b.
We found that the use of an alternative heptane/THF/CH3CN solvent system for the [2 + 2] cycloaddition was less successful. The conversion of the starting bis(enone) 9 under visible light irradiation was slower and required over 24 h to before it was above 90%. Again, while we were able to separate and reuse 6 for 4 cycles using the latent biphasic strategy, both cycloaddition product 10a and reductive cyclization product 10b again formed. In this case, the average isolated yield of the cycloaddition product 10a was 16% per cycle and the reductive cyclization product 10b was 60% per cycle.
The solubility limitations of 6 that required the presence of a less polar solvent and that led to a mixture of products in the [2 + 2] chemistry were even more problematic in attempts to use 6 to convert alcohols to bromides (Scheme 4).25 In this case, we were able to use a low molecular weight catalyst [Ru(bpy)3Cl2] in DMF to form 1-bromo-3-phenylpropane (Scheme 4). However, when this same reaction was carried out using 3-phenyl-1-propanol with 2 equiv. of CBr4, and 2 equiv. of NaBr in the presence of 1.3 mol% catalyst 6 in a 4/1 (vol/vol) mixture of CH2Cl2 and DMF at ambient temperature for 20 h under irradiation of blue LED no bromide formed. Based on our experience with the [2 + 2] cycloadditions, we thought that the solvent change could be the cause of failure for this reaction. A control experiment using [Ru(bpy)3Cl2] as catalyst to carry out this reaction in the same 4/1 (vol/vol) mixture of CH2Cl2/DMF also produced no product, suggesting that the lack of solubility of 6 in polar solvents would preclude its use in this chemistry.
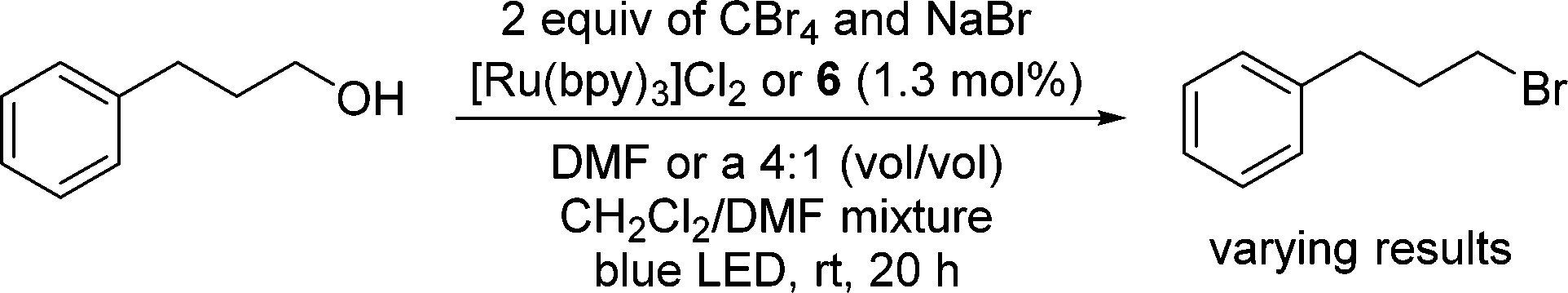 | ||
| Scheme 4 Attempted conversion of alcohol to halide by the PIB-bound Ru(II)-bipyridine complex 6. | ||
4. Conclusions
In conclusion, a PIB-bound Ru(II)-bipyridine complex was prepared, and we demonstrated that this PIB-supported Ru complex can be used as a recyclable photoredox catalyst to carry out oxidative C–C bond cleavage of aldehyde in CH2Cl2/CH3CN homogeneous system efficiently. In addition, a visible-light induced [2 + 2] cycloaddition of bis(enone) can also occur with high conversions of the bis(enone) using this PIB-bound Ru(II)-bipyridine complex. However, using solvents that dissolve the PIB catalyst lead to formation of both cycloaddition and cyclization products. In a third case, the substitution of solvents other that CH3CN was completely unsuccessful. The results indicated that this PIB-supported Ru catalyst can be successfully recycled and reused for at least 5 cycles though successful use of this soluble catalyst requires a solvent tolerant reaction. This illustrates a general limitation of these PIB supported catalysts (and other phase selectively soluble polymer supported catalysts) in that the different solvent systems needed for a specific support can lead to longer reaction times, altered selectivity, or, in the case of the alcohol to halide reaction, no reaction at all. These PIB-supported catalysts and a biphasic liquid/liquid separation based catalyst recycling strategy can be applied to other photoredox reactions if the reaction is tolerates less polar solvent. If this type of catalyst recycling were to be effective in photoredox [2 + 2] cycloadditions or in alcohol to halide reactions, a polar solvent soluble polymer support would be required so that a more polar medium could be used for the photoredox catalysis chemistry.Acknowledgements
Financial support of this research by the National Science Foundation (Grant CHE-1362735) and the Robert A. Welch Foundation (Grant A-0639) and the gift samples of Glissopal (PIB) from BASF are gratefully acknowledged. We also thank Thomas Malinski for the ICP-MS analysis.References
- D. A. Nicewicz and T. M. Nguyen, ACS Catal., 2014, 4, 355–360 CrossRef CAS.
- T. Koike and M. Akita, Inorg. Chem. Front., 2014, 1, 562–576 RSC.
- C. K. Prier, D. A. Rankic and D. W. C. MacMillan, Chem. Rev., 2013, 113, 5322–5363 CrossRef CAS PubMed.
- J. M. R. Narayanam and C. R. J. Stephenson, Chem. Soc. Rev., 2011, 40, 102–113 RSC.
- D. A. Nicewicz and D. W. C. MacMillan, Science, 2008, 322, 77–78 CrossRef CAS PubMed.
- J. M. R. Narayanam, J. W. Tucker and C. R. J. Stephenson, J. Am. Chem. Soc., 2009, 131, 8756–8757 CrossRef CAS PubMed.
- J. W. Beatty and C. R. J. Stephenson, Acc. Chem. Res., 2015, 48, 1474–1484 CrossRef CAS PubMed.
- M. A. Ischay, M. E. Anzovino, J. Du and T. P. Yoon, J. Am. Chem. Soc., 2008, 130, 12886–12887 CrossRef CAS PubMed.
- T. P. Yoon, M. A. Ischay and J. Du, Nat. Chem., 2010, 2, 527 CrossRef CAS PubMed.
- T. P. Yoon, ACS Catal., 2013, 3, 895–902 CrossRef CAS PubMed.
- M.-H. Larraufie, R. Pellet, L. Fensterbank, J.-P. Goddard, E. Lacôte, M. Malacria and C. Ollivier, Angew. Chem., Int. Ed., 2011, 50, 4463–4466 CrossRef CAS PubMed.
- G. Zhang, I. Y. Song, K. H. Ahn, T. Park and W. Choi, Macromolecules, 2011, 44, 7594–7599 CrossRef CAS.
- Y.-Q. Zou, J.-R. Chen, X.-P. Liu, L.-Q. Lu, R. L. Davis, K. A. Jørgensen and W.-J. Xiao, Angew. Chem., Int. Ed., 2012, 51, 784–788 CrossRef CAS PubMed.
- G. J. Barbante, T. D. Ashton, E. H. Doeven, F. M. Pfeffer, D. J. D. Wilson, L. C. Henderson and P. S. Francis, ChemCatChem, 2015, 7, 1655–1658 CrossRef CAS.
- J. L. Pan, B. J. Zhang, X. W. Jiang, L. F. Zhang, Z. P. Cheng and X. L. Zhu, Macromol. Rapid Commun., 2014, 35, 1615–1621 CrossRef CAS PubMed.
- P. W. Liu, Z. B. Ye, W.-J. Wang and B.-G. Li, Macromolecules, 2013, 46, 72–82 CrossRef CAS.
- D. E. Bergbreiter, ACS Macro Lett., 2014, 3, 260–265 CrossRef CAS.
- N. Priyadarshani, Y. Liang, J. Suriboot, H. S. Bazzi and D. E. Bergbreiter, ACS Macro Lett., 2013, 2, 571–574 CrossRef CAS.
- D. E. Bergbreiter, P. N. Hamilton and N. M. Koshti, J. Am. Chem. Soc., 2007, 129, 10666–10667 CrossRef CAS PubMed.
- BASF, http://www.performancechemicals.basf.com/ev/internet/polyisobutene/en/content/EV3/polyisobutene/glissopal (accessed August 4, 2015).
- J. Li, S. Sung, J. Tian and D. E. Bergbreiter, Tetrahedron, 2005, 61, 12081–12092 CrossRef CAS.
- H. Sun, C. Yang, F. Gao, Z. Li and W. Xia, Org. Lett., 2013, 15, 624–627 CrossRef CAS PubMed.
- T.-G. Baik, A. L. Luiz, L.-C. Wang and M. J. Krische, J. Am. Chem. Soc., 2001, 123, 6716–6717 CrossRef CAS PubMed.
- D. Taber, E. I. Becker and P. E. Spoerri, J. Am. Chem. Soc., 1954, 76, 776–781 CrossRef CAS.
- C. Dai, J. M. R. Narayanam and C. R. J. Stephenson, Nat. Chem., 2011, 3, 140–145 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Figures of 1H and 13C NMR spectra of the catalyst and products. See DOI: 10.1039/c5cy01287b |
| This journal is © The Royal Society of Chemistry 2016 |