
First efficient catalyst recycling for the iridium-catalysed hydroformylation of 1-octene
A.
Behr
*a,
A.
Kämper
a,
R.
Kuhlmann
a,
A. J.
Vorholt
a and
R.
Franke
bc
aTechnische Universität Dortmund, Lehrstuhl für Technische Chemie, Emil-Figge-Str. 66, 44227 Dortmund, Germany. E-mail: behr@bci.tu-dortmund.de; Fax: +49 231 755 2311; Tel: +49 231 755 2310
bEvonik Performance Materials GmbH, Paul-Baumann-Straße 1, 45772 Marl, Germany
cRuhr-Universität Bochum, 44780 Bochum, Germany
First published on 6th October 2015
Abstract
This paper describes the development of an efficient catalyst recycling concept for the iridium-catalysed hydroformylation of 1-octene through the investigation of biphasic systems, thermomorphic solvent systems and an ex situ extraction. Particularly high selectivities (>90%) towards the desired aldehydes as well as low rates of iridium leaching were observed using the monosulfonated triphenylphosphine ligand (TPPMS). In polar solvents such as propylene carbonate or N,N-dimethylformamide, low rates of catalyst leaching (0.2%) as well as high rates of product separation (nearly 80%) were achieved. High reaction rates and a long-term activity and stability of the catalyst were observed using the solvent N,N-dimethylformamide and the extraction with non-polar solvents.
Introduction
Hydroformylation, also known as “oxo-synthesis”, is one of the most important homogeneous-catalysed reactions in the chemical industry. In hydroformylation, olefins are converted to aldehydes in the presence of syngas (CO/H2) (see example in Fig. 1). Aldehydes are useful intermediates in the production of solvents, plasticizers, odorants and other fine chemicals.1–4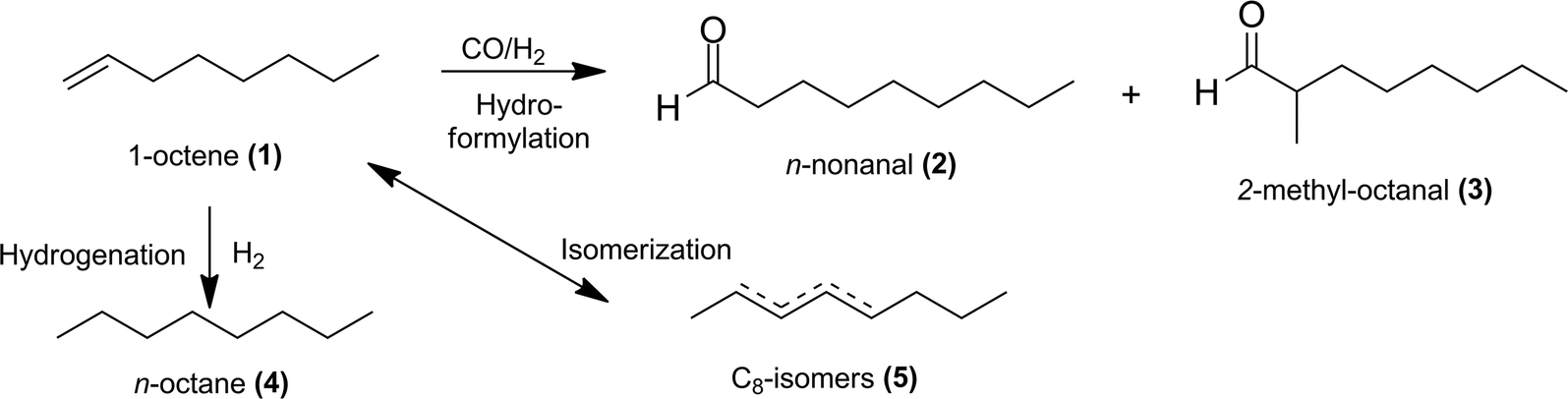 | ||
| Fig. 1 Hydroformylation of 1-octene and side reactions. | ||
Hydroformylation was first described by Otto Roelen in 1938. Its subsequent development for use in industrial processes led to the use of catalyst metals such as cobalt and, above all, rhodium. To this day, rhodium is the most active metal used in hydroformylation, yet it is also one of the rarest and, consequently, most expensive metals on earth. The dramatic fluctuations in rhodium prices can be ascribed to the high demand for this metal within the automotive industry (ca. 80%).3–6
Recent research by Beller et al. demonstrates that old assumptions from 1977 (Table 1) with regard to metal activities in hydroformylation should be re-examined. The values in Table 1 predict an activity ratio of rhodium to iridium as 10.000![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1.
:1.
| Metal | Rh | Co | Ir | Ru | Os, Tc | Mn | Fe |
|---|---|---|---|---|---|---|---|
| Activity | 1000 | 1 | 0.1 | 0.01 | 0.001 | 0.0001 | 0.000001 |
However, new catalyst systems provide alternatives for achieving satisfactory catalytic performances using iridium that are only 8 times lower than using rhodium.3,4,8,9
The use of noble metals such as rhodium or iridium in homogeneous catalysis raises the question as to how the catalyst can be recycled most effectively. Former developments in industrial hydroformylation have shown different ways of recycling cobalt and rhodium catalysts. In some cases the separation of the catalyst from the products is achieved through chemical transformation and reactivation of the catalytic complex or via distillation of the low-boiling products. An important hydroformylation process is the Ruhrchemie/Rhône-Poulenc process for converting short chain olefins such as propene or 1-butene. Here an efficient catalyst recycling is possible using an aqueous biphasic liquid–liquid system and the trisulfonated triphenylphosphine (TPPTS) ligand. Unfortunately, this recycling concept is not suitable for use with higher olefins such as 1-octene or 1-dodecene, necessitating the development of alternative recycling systems.3–5,10–13
Among the more promising recycling concepts for higher olefins are a biphasic liquid–liquid system involving either extraction or a thermomorphic solvent system such as the temperature-dependent multi-component solvent-system (TMS). Using an extraction or a TMS system has the great advantage of preventing potential mass transfer limitations as the reaction mixture occurs in one homogeneous liquid phase. The extraction method refers to a highly effective separation of catalyst and products using the appropriate extracting agent. However, this method is more expensive in terms of the equipment and process engineering involved. The use of TMS as described by Behr et al. presents an effective means for catalyst/product separation in a plant that is easy to operate.4,14 At the same time, finding the most effective and stable solvent system is somewhat laborious in terms of identifying the right composition of the TMS mixture. The solvents involved in these thermomorphic systems form a homogeneous one-phasic mixture in the tempered reactor. After the reaction, the mixture can be separated into two liquid phases using a separator at lower temperatures.13,15–21
This paper describes for the first time the development of a new catalyst recycling concept for the iridium-catalysed hydroformylation of 1-octene. First, opportunities of controlling and optimizing the reaction to achieve high yields and selectivities toward the desired aldehydes and to supress hydrogenation were to be identified. Aside from this, the experiments were intended to find an effective possibility of separating the catalyst from the product while avoiding catalyst and ligand leaching as much as possible in addition to preventing the deactivation of the catalyst. Achieving this predicated on identifying a suitable ligand in combination with an appropriate solvent system consisting of polar and non-polar solvents. The ideal system would provide for long-term catalyst stability using an effective recycling system suitable for use in a continuously operating miniplant.
Materials and methods
The reactions were carried out in 25 ml autoclaves and in a one pot 300 mL Parr autoclave. The following example is given for preparing the reaction mixture for use in a 25 ml autoclave. The precursor Ir(cod)(acac) (0.0713 mmol, 0.8 mol% of 1-octene), ligand (0.18 mmol, P/Ir = 2.5/1) and LiCl (0.14 mmol, Li/Ir = 2/1) were introduced into the autoclave. Then 1-octene (8.91 mmol) and solvents (polar or respectively non-polar) were added. The autoclaves were closed and pressurized using a 2:1 CO/H2 mixture of syngas (30 bar). The reaction was started at a temperature of 100 °C. The autoclaves were emptied of the remaining gas and stripped with argon at the end of the reaction. The quantitative analysis was performed via gas chromatography (GC) (HP 6890 Agilent Technologies) using an HP-INNOWAX column. This procedure was repeated for the reaction in the Parr autoclave, though the substances weighed 10 times more in these cases. Additionally, a semi-continuous stream of syngas (CO/H2 = 1/1) at 30 bar was introduced to ensure constant pressure.
For ex situ extraction, the reaction mixture was weighed and analysed before the non-polar solvent was added. Usually a DMF:non-polar solvent ratio of 1:1 was applied. Additionally, the amount of non-polar solvent was investigated and optimized, which is shown in Fig. 6. The total mixture was stirred (700 rpm) for 15 minutes and separated in a cooled separation funnel for 30 minutes at 0 °C. After separation both phases were weighed and analysed via GC for product distribution and via ICP-OES for iridium and phosphorus leaching. The results of ICP-OES (ppm) were multiplied with the amount of non-polar respectively polar phase to get the amount of iridium and phosphorus in both phases. Finally, the results of the non-polar phase were correlated to the total amount of iridium and phosphorus added to the reaction mixture.
For application of thermomorphic solvent systems, suitable solvent mixtures were investigated and determined in previous solubility tests at laboratory scale. Therefore, polar solvent (DMF, PC) and 1-octene or nonanal were mixed up in specific compositions and heated up to reaction temperature. If necessary, certain amounts of non-polar solvent (heptane, 2,2,4-trimethylpentane, …) were added and the solution was examined for miscibility. Then the mixture was cooled, separated and analysed via GC for identifying the appropriate miscibility gap and for promising solvent mixtures. These mixtures were performed in hydroformylation reactions and separated for 30 min at 0 °C in a separation funnel. The analysis were similar to the extraction procedure described above.
Results and discussion
Generally, the hydroformylation of 1-octene (1) leads to linear (2) and branched (3) aldehydes, which are summarized as yields (Y) of C9-aldehydes (2 + 3) in this paper. Consequently, the described chemoselectivity S refers to the total amount of aldehydes (2 + 3) and shows the reaction efficiency of the unwanted isomerization (5) and hydrogenation products (4). It is essential to keep the formation of the by-product n-octane (4) to a minimum by adding LiCl and the proper ratio of CO/H2. A separate paper will discuss the influences of these additives in achieving higher chemoselectivities. The optimized experiments lead to high selectivities (S), yields (Y) and conversions (X) with homogeneous solvent systems in the iridium-catalysed hydroformylation of 1-octene using an Ir(cod)(acac)/PPh3 catalyst system with N-methyl-2-pyrrolidone (NMP) as a solvent (Table 2, entry 1). However, previous studies have demonstrated that NMP is not an appropriate solvent for catalyst recycling in liquid–liquid systems. This required a search for suitable solvents able to form a specific polar phase with the catalyst complex with an effective means of separating products through a non-polar phase (Fig. 2). For example, propylene carbonate (PC) and N,N-dimethylformamide (DMF) were identified as potential (semi)-polar solvents for a liquid–liquid catalyst recycling via biphasic systems, TMS systems or ex situ extraction. For biphasic systems a non-polar product phase can be formed with and without a non-polar solvent, as described in Fig. 2. In this case often a second liquid phase in the reactor leads to high mass transfer limitations and a decrease of the reaction rate. For improving the catalyst performance in the reactor the use of TMS systems is an alternative method, which result in a homogeneous mixture at reaction temperature and a separation of the two liquid phases in a cooled separator. The addition of a non-polar solvent after the reaction describes the ex situ extraction, which has the advantage of a total homogeneous reaction mixture and an improved space-time yield.| Entry | Ligand | Solvent | X [%] (1) | Y [%] | S [%] (2) + (3) |
l:b |
||
|---|---|---|---|---|---|---|---|---|
| (2) + (3) | (4) | (5) | ||||||
| Reaction conditions: 8.91 mmol 1-octene, 4 g solvent, 0.0713 mmol Ir(cod)(acac), P/Ir = 2.5/1, LiCl/Ir = 2/1, 30 bar (CO/H2 = 2/1), 100 °C, 16 h, 700 rpm.a Precipitation of the catalyst complex (Ir, P). | ||||||||
| 1 | PPh3 | NMP | 95 | 90 | 4 | 1 | 94 | 76:24 |
| 2 | PPh3a | PC | 65 | 59 | 2 | 4 | 91 | 79:21 |
| 3 | PPh3a | DMF | 54 | 49 | 2 | 6 | 91 | 77:23 |
| 4 | TPPMS | NMP | 89 | 83 | 4 | 2 | 93 | 73:27 |
| 5 | TPPMS | PC | 70 | 64 | 2 | 4 | 91 | 77:23 |
| 6 | TPPMS | DMF | 68 | 61 | 4 | 3 | 90 | 75:25 |
| 7 | TPPMSa | Water | 87 | 33 | 37 | 16 | 38 | 77:23 |
| 8 | TPPTSa | NMP | 87 | 76 | 5 | 6 | 87 | 74:26 |
| 9 | TPPTS | Water | 62 | 32 | 23 | 7 | 52 | 73:27 |
| 10 | Biphephos | NMP | 74 | 47 | 19 | 8 | 64 | 73:27 |
| 11 | Xantphos | NMP | 11 | 4 | 2 | 5 | 36 | 76:24 |
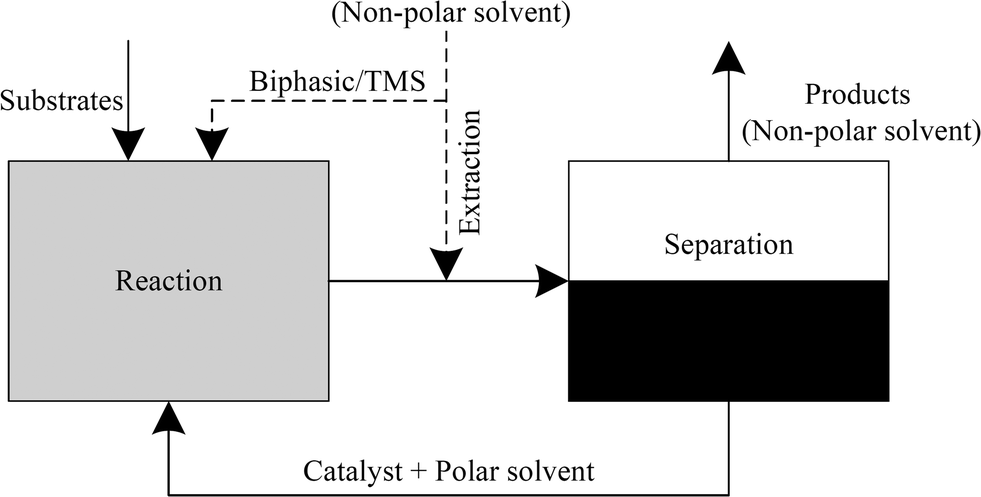 | ||
| Fig. 2 Flow-scheme for catalyst recycling in biphasic fluid systems. | ||
Initial experiments using DMF and PC in combination with triphenylphosphine (PPh3) resulted in high chemoselectivities towards the desired aldehydes (91%). However, the low catalytic activities or conversions were due to the low solubility of the catalyst complex, which was found as a solid after the reaction (Table 2, entries 2 and 3).
It was therefore necessary to identify a more polar ligand for use in this recycling concept. Sulfonated ligands such as monosulfonated triphenylphosphine (TPPMS) and trisulfonated triphenylphosphine (TPPTS) also performed well in homogeneous NMP solutions (Table 2, entries 4 and 8). For this reason, the sulfonated phosphine ligands were also investigated in both DMF and PC. With TPPMS in both PC and DMF, desirable catalytic performances were observed (Table 2, entries 5 and 6), whereas TPPTS in water resulted in the excessive formation of undesired hydrogenation products (Table 2, entry 9).
As described by Beller et al., the use of phosphorous ligands and of bidentate ligands such as biphephos or xantphos in particular results in poor performances and selectivities (Table 2, entries 10 and 11). Compared to rhodium-catalysed hydroformylation, the addition of xantphos or biphephos does not result in an improvement of the regioselectivity towards the more desired terminal aldehydes.8,9,22,23
The monosulfonated TPPMS ligand performed well in DMF and PC with a chemoselectivity of about 90% and a regioselectivity of about 75:25. Furthermore, when varying different reaction parameters in the iridium-catalysed system, no considerable influences on the regioselectivity were identified.
Propylene carbonate based recycling systems
The use of PC as a polar solvent proved promising for iridium catalyst recycling (Table 2, entry 5). Table 3 shows the results of using different recycling concepts.| Entry | PC [wt%] | 1-Octene [wt%] | Non-polar solvent [wt%] | X [%] (1) | Y [%] | S [%] | Leaching [%] | |||
|---|---|---|---|---|---|---|---|---|---|---|
| (2) + (3) | (4) | (5) | (2) + (3) | Ir | P | |||||
| Conditions: Ir(cod)(acac) = 0.8 mol% (substrate), TPPMS/Ir = 2.5/1, LiCl/Ir = 2/1, 30 bar (CO/H2 = 2/1), 100 °C, 16 h, 700 rpm, tSeparation = 30 min, TSeparation = 0 °C.a Thermomorphic solvent system.b Biphasic system. | ||||||||||
| 1a | 80 | 20 | — | 70 | 64 | 2 | 4 | 91 | 0.3 | 0.4 |
| 2a | 78 + (2% H2O) | 20 | — | 74 | 69 | 2 | 3 | 93 | 0.3 | 0.4 |
| 3a | 67 | 33 | — | 60 | 52 | 3 | 5 | 86 | 2 | 0.9 |
| 4b | 60 | 20 | n-Dodecane (20%) | 63 | 50 | 7 | 6 | 80 | 0.3 | 0.2 |
| 5b | 58 + (2% H2O) | 20 | n-Heptane (20%) | 63 | 54 | 3 | 4 | 86 | 0.3 | 0.4 |
Due to its high polarity, the liquid–liquid separation from non-polar substances was predicted to be good. In this case, separating the non-polar phase consisting of a high amount of 1-octene, octane and aldehydes from the polar propylene carbonate/catalyst phase was possible. As demonstrated by the experiments shown in entries 1 and 2 in Table 3, good catalytic performances were observed with chemoselectivities of up to 93% and at a maximum conversion of 74% if no additional non-polar solvent was used. The addition of small amounts of water (Table 3, entry 2) improved hydroformylation performance and results in better product separation. The catalyst and ligand losses shown in Table 3 as Ir and P leaching at rates of 0.3% and 0.4% are relatively low, indicating that catalyst recycling in the polar phase works well. Further studies indicated that the mixtures in entries 1–3 are quasi temperature dependent mixtures. This means that the components 1-octene and PC form a biphasic mixture at the beginning of batch reactions. As the reaction progresses, the formation of aldehydes increases the polarity of the reaction mixture. After a certain yield for the phase mediating product, a homogeneous mixture is formed that can be separated into two liquid phases after cooling down to room temperature and cooling temperature. In Table 3 entry 3, the chemoselectivity (86%) and conversion (60%) is lower than in entries 1 and 2, which is attributed to the higher amount of substrate and consequently to higher mass transfer limitations. Furthermore, the rate of iridium and phosphorous leaching is higher (2%/0.9%) due to higher concentrations of substrate and products which result in increasing mediation into the non-polar phase.
The addition of non-polar solvents such as n-dodecane or n-heptane to build a biphasic system (Table 3, entries 4 and 5) provided yet another means of designing a PC-based recycling system. The assumption was that this would improve separation of the polar catalyst and non-polar product phase. The mass transfer limitations are much higher than in the entries 1 and 2, which leads to lower conversions and chemoselectivities whereas the Ir leaching is low at a rate of approximately 0.3%.
The application of a temperature dependent system with PC was therefore deemed to be far more suitable (Table 3, entries 1–3).
Considering that further operation in continuous processes should be done under steady state conditions, a specific point within the batch reaction must be referenced. As described above, this point was given with the further reaction rate and yield for nonanal. The kinetic behaviour of the hydroformylation of 1-octene in propylene carbonate was subsequently investigated (Fig. 3).
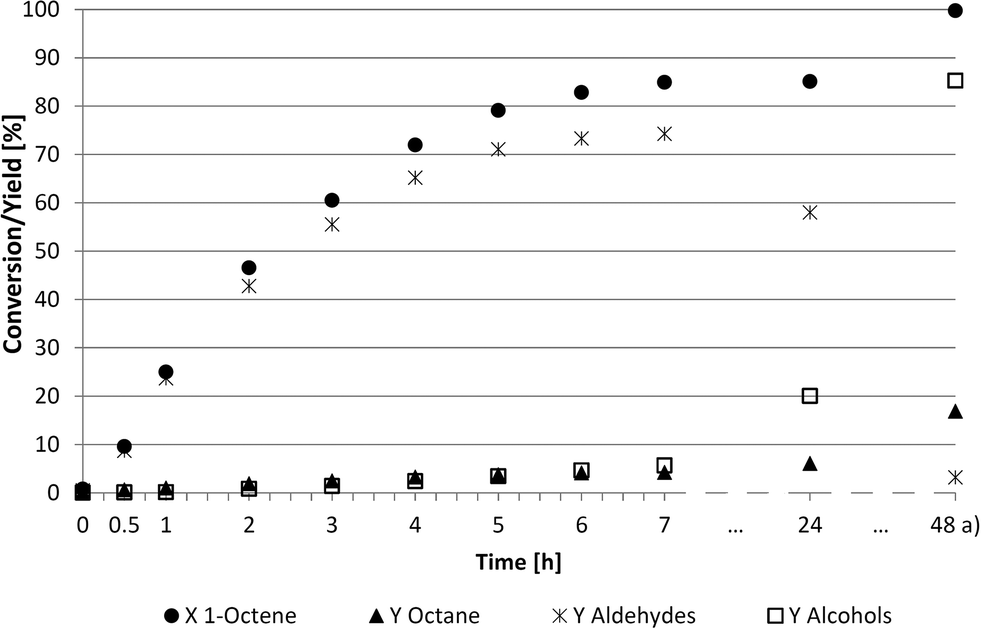 | ||
| Fig. 3 Kinetic investigations of the hydroformylation of 1-octene in propylene carbonate. Reaction conditions: 89.1 mmol 1-octene, 39 g PC, 1 g H2O, 0.713 mmol Ir(cod)(acac), TPPMS/Ir = 2.5/1, LiCl/Ir = 2/1, 30 bar (CO/H2 = 2/1, const. 1/1), 100 °C, 700 rpm, a) pure hydrogen from 24 h to 48 h. | ||
This experiment under semi-continuous conditions resulted in good catalytic performance and high chemoselectivities (90%) towards the desired aldehydes. After 7 hours, the conversion reached a preliminary maximum of approximately 85%. Considering the kinetic trend of Fig. 3, an operation point respectively residence time in the reactor was determined to be between 3 and 6 h.
Unexpectedly, some hydrogenation to the corresponding alcohol, nonanol, was observed after 6 h. The reaction was subsequently stopped after 24 h and the Parr autoclave was emptied and flushed with pure hydrogen to convert most of the remaining aldehydes to alcohols. This tandem reaction was not observed in previous experiments using other solvents in iridium-catalysed hydroformylation.
In the chemical industry, long chain alcohols are often desired products. However, a homogeneous reaction mixture occurs after cooling and an efficient catalyst recycling in PC becomes more difficult due to the much higher polarity of nonanol. Consequently, other solvents with higher polarity such as water or the addition of non-polar solvents became necessary. But mentioned previously, the catalytic performance and chemoselectivity using water-based solvent systems and biphasic systems do not provide satisfactory results (Tables 2 and 3). Alternative recycling concepts should be examined more closely in order to identify an efficient recycling concept for iridium-catalysed hydroformylation of 1-octene.
DMF based recycling systems
Due to its lower polarity and smaller miscibility gap with 1-octene in comparison to PC, DMF is more suitable as a solvent for recycling systems including thermomorphic mixtures or an ex situ extraction. A homogeneous mixture was formed during the hydroformylation of 1-octene with Ir(cod)(acac)/TPPMS complex and DMF without limiting mass transfer even if no mediating aldehyde is used. Table 4 presents the results for DMF-based recycling systems. Entries 1–3 provide results for reactions in DMF without commixing other solvents. The homogeneous product phase required subsequent extraction using a non-polar solvent to separate the catalyst phase (see Fig. 6). The chemoselectivity towards the desired aldehydes is at a rate of approximately 90%. The addition of small amounts of water increases the conversion at a rate of up to 74%. Additionally, water improves the polarity of the mixture, which has the advantage of providing a more effective extraction. Entries 4 to 7 provide the results for thermomorphic solvents systems consisting of DMF and alkanes such as n-heptane, n-dodecane or 2,2,4-trimethylpentane. With a composition of approximately 40 wt% DMF and 40 wt% alkane, aldehyde selectivities of approximately 90% were detected (entries 4–7). The use of 2,2,4-trimethylpentane in particular led to a higher conversion rate of about 74% and a low rate of leaching at approximately 0.2% (Ir) and 0.3% (P) (entry 6).| Entry | DMF [wt%] | 1-Octene [wt%] | Non-polar solvent [wt%] | X [%] (1) | Y [%] | S [%] (2) + (3) | Leaching [%] | |||
|---|---|---|---|---|---|---|---|---|---|---|
| (2) + (3) | (4) | (5) | Ir | P | ||||||
| Conditions: Ir(cod)(acac) = 0.8 mol% (substrate), TPPMS/Ir = 2.5/1, LiCl/Ir = 2/1, 30 bar (CO/H2 = 2/1), 100 °C, 16 h, 700 rpm, tSeparation = 30 min, TSeparation = 0 °C.a Extraction with non-polar solvent necessary.b Thermomorphic solvent systems. | ||||||||||
| 1a | 60 | 40 | — | 65 | 58 | 3 | 4 | 89 | n.a. | n.a. |
| 2a | 80 | 20 | — | 68 | 61 | 4 | 3 | 90 | n.a. | n.a. |
| 3a | 78 + (2% H2O) | 20 | — | 74 | 67 | 2 | 5 | 91 | (see Fig. 6) | |
| 4b | 40 | 20 | n-Dodecane (40%) | 60 | 52 | 4 | 4 | 87 | 0.2 | 0.6 |
| 5b | 40 | 20 | n-Heptane (40%) | 70 | 63 | 4 | 3 | 90 | 0.3 | 0.8 |
| 6b | 40 | 20 | 2,2,4-Trimethyl-pentane (40%) | 74 | 66 | 4 | 4 | 89 | 0.2 | 0.3 |
| 7b | 39 + (1% H2O) | 20 | 2,2,4-Trimethyl-pentane (40%) | 63 | 55 | 4 | 4 | 90 | 0.2 | 0.1 |
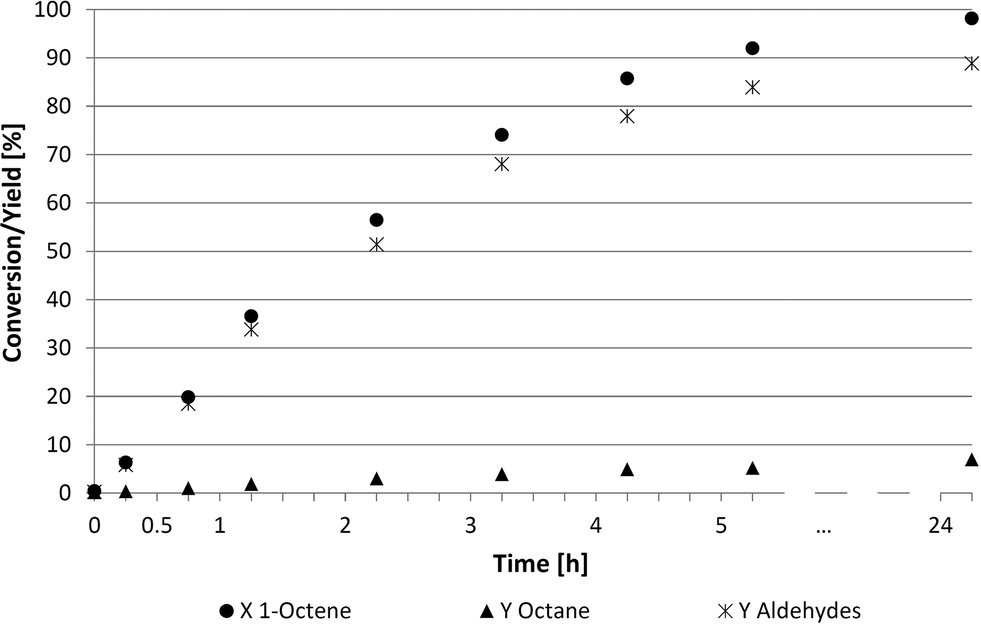
A more detailed look into the kinetics (Fig. 4 and 5) was required to better understand these two suitable recycling systems (Table 4, entries 3 and 6). The reaction rate in the thermomorphic system consisting of DMF and 2,2,4-trimethylpentane was surprisingly slow. The full conversion of 1-octene was reached only after 24 h, while only 60% of 1-octene was converted into aldehydes (52%) and octane (4%) after 7 h. Further investigations of this thermomorphic system showed that the iridium catalyst formed a specific small phase with the ligand TPPMS at higher temperatures. These emulsion drops lead to high mass transfer limitations and lower reaction rates. The chemoselectivity towards the desired aldehydes was at 91% whereas the regioselectivity was steady at 75:25.
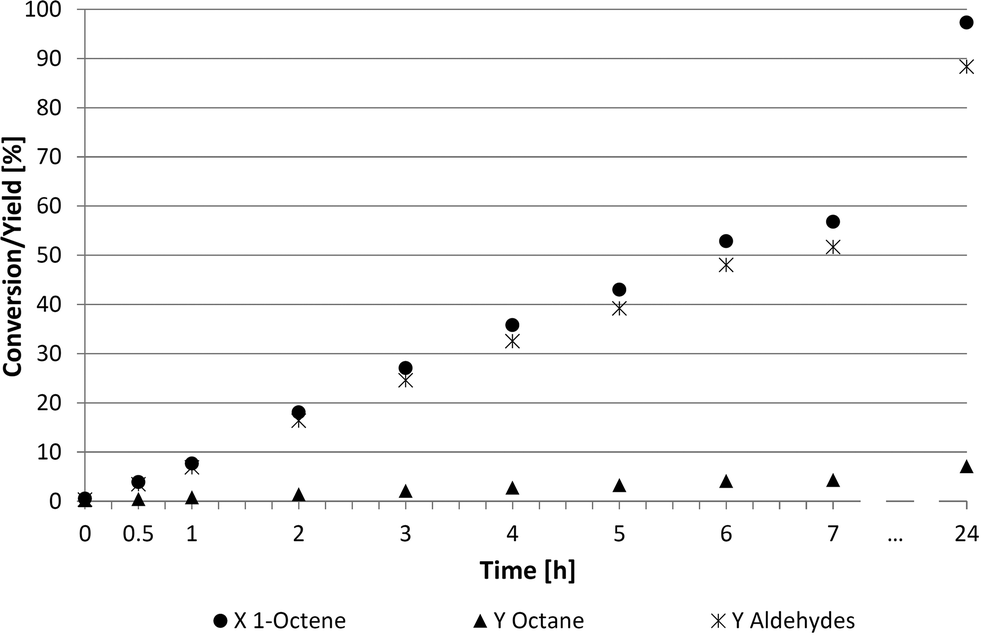 | ||
| Fig. 4 Kinetics of the hydroformylation in thermomorphic DMF-2,2,4-trimethylpentane system. Reaction conditions: 89.1 mmol 1-octene, 20 g DMF, 20 g 2,2,4-trimethylpentane, 0.713 mmol Ir(cod)(acac), TPPMS/Ir = 2.5/1, LiCl/Ir = 2/1, 30 bar (CO/H2 = 2/1, const. 1/1), 100 °C, 700 rpm. | ||
 | ||
| Fig. 5 Kinetics of the hydroformylation in DMF. Reaction conditions: 89.1 mmol 1-octene, 39 g DMF, 1 g H2O, 0.713 mmol Ir(cod)(acac), P/Ir = 2.5/1, LiCl/Ir = 2/1, 30 bar (CO/H2 = 2/1, const. 1/1), 100 °C, 700 rpm. | ||
Examining the kinetics of the homogeneous DMF system (Fig. 5) shows a higher reaction rate than described in Fig. 4. The reaction was also faster compared to NMP and PC (Fig. 3) based systems. This system led to a conversion of 90% and a chemoselectivity of 91% after 6 h. No consecutive reaction to undesired alcohols was observed.
An effective means of extraction with a non-polar solvent such as 2,2,4-trimethylpentane was investigated to separate the product from the catalyst. The plots for the product separation and the iridium and phosphorus leaching in the extraction experiments using 2,2,4-trimethylpentane are shown in Fig. 6.
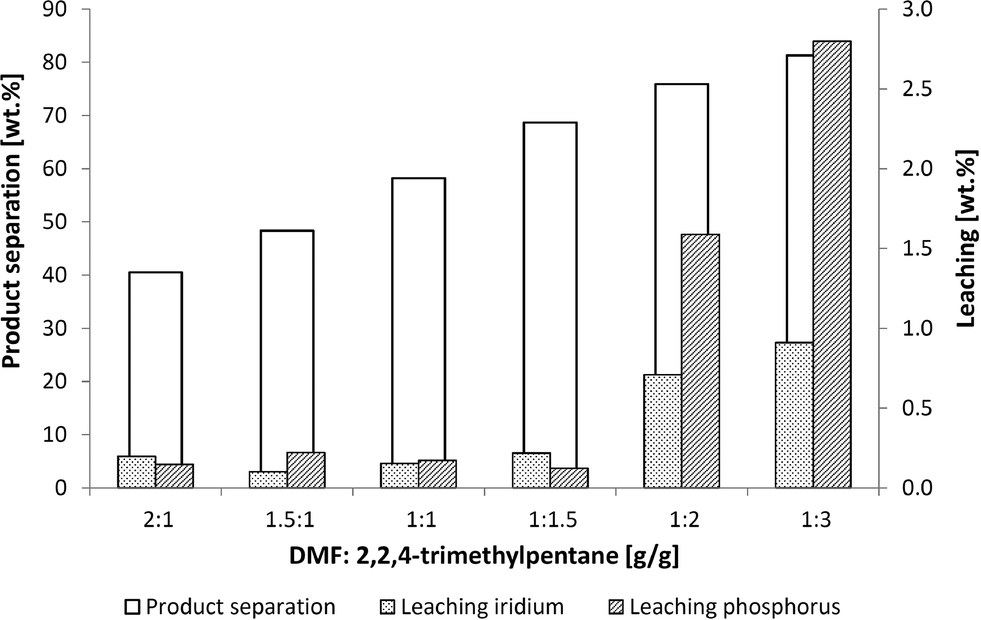 | ||
| Fig. 6 Influence of DMF:2,2,4-trimethylpentane ratio on leaching and product separation. Conditions: 2 g reaction mixture (see Fig. 5), mixing time 15 min, separation time 30min at 0 °C. | ||
The product separation increased to 90% using a higher amount of non-polar extracting agent. Running the experiment with a DMF:2,2,4-trimethylpentane ratio of 1:2 and 1:3, the catalyst and ligand leaching increased to rates of up to 3% whereas the product separation was observed at a maximum of 80%. It was therefore necessary to find a compromise between leaching and product separation that led to a ratio of 1:1.5. Low rates of leaching at 0.2% and a product separation of 70% are possible at this ratio.
The DMF-based system with downstream extraction was selected for use in further examinations due to its favourable results as compared to the PC recycling concept previously described (Table 5).
| Recycling concept | Selectivity/activity | Leaching/separation |
|---|---|---|
| − Not suitable, o medium, + well suitable. | ||
| PC–Biphasic | − | o |
| PC–TMS | o | + |
| DMF–TMS | o | + |
| DMF–Extraction | + | + |
Both a selective and efficient separation of product and catalyst is possible using the homogeneous DMF-based recycling system. Further information with regard to long-term catalyst system stability was needed to determine whether further applications at a miniplant scale or at an industrial scale would be viable. As such, the catalyst phase was separated via extraction using 2,2,4-trimethylpentane following the reaction and was then returned to the autoclave. New 1-octene was then added and the reaction was restarted. The long-term activity of the iridium complex was verified in five recycle runs with the same catalyst mixture (Fig. 7). A selectivity of about 90% and a conversion of about 80% were observed. The leaching of iridium and phosphorous leaching at rates of up to 0.3% and product separation at rates of up to 80% indicated that this recycling system is both stable and suitable in long-term applications.
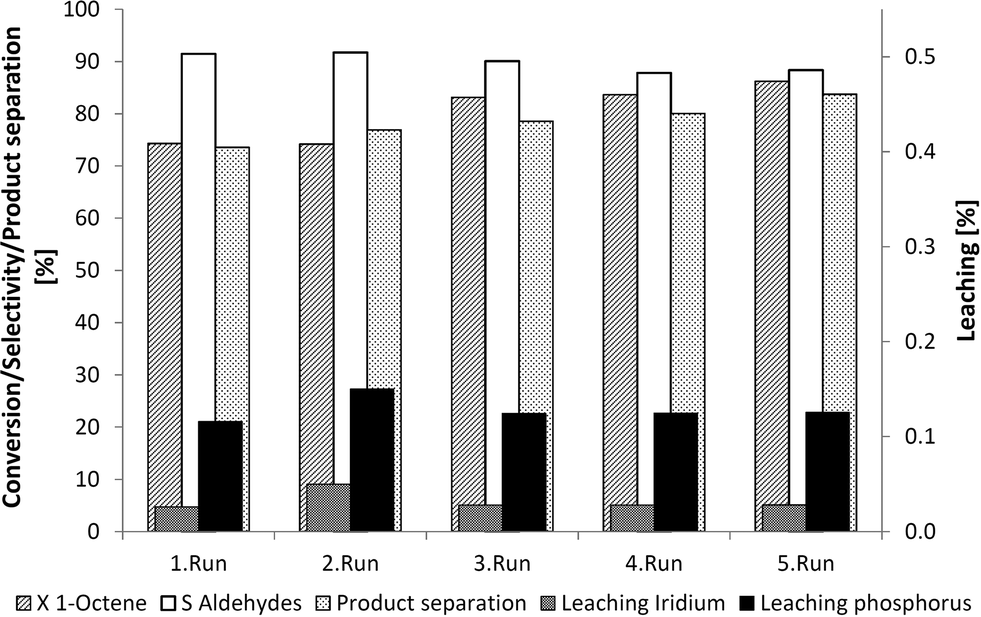 | ||
| Fig. 7 Recycling of the Ir/TPPMS-catalyst system in DMF with following 2,2,4-trimethylpentane. Reaction conditions: 89.1 mmol 1-octene, 39 g DMF, 1 g H2O, 0.713 mmol Ir(cod)(acac), TPPMS/Ir = 2.5/1, Li/Ir = 2/1, 30 bar (CO/H2 = 2/1, const. 1/1), 5 h, 100 °C, 700 rpm, extraction with 60 g 2,2,4-trimethylpentane for 10 min, separation of the biphasic mixture for 1 h at 0 °C. | ||
Finally, we performed an experiment using Rh(CO)2(acac) as metal precursor for comparing iridium- and rhodium-catalyzed hydroformylation in DMF. The results are presented in Table 6 and were determined with the application of nearly same reaction and separation conditions. Obviously and as expected, the rhodium catalyst is more active than the iridium catalyst, which is shown with higher TON and TOF of 423 and 228 h−1. The chemoselectivity with 86% is lower, which is due to the formation of undesired octenes. Comparable to iridium experiments, the rhodium and phosphorus leaching is three times respectively nine times higher after extraction with 2,2,4-trimethylpentane. Hence, despite the lower activity, we developed a suitable iridium system with higher selectivities and much lower catalyst leaching, which is competitive to experiments using a rhodium catalyst.
| Catalyst (concentration) | Time [h] | X [%] (1) | Y [%] | S [%] (2) + (3) |
l![[thin space (1/6-em)]](https://www.rsc.org/images/entities/i_char_2009.gif) :b :b
|
TON [−] | TOF [h−1] | Leaching [%] | |||
|---|---|---|---|---|---|---|---|---|---|---|---|
| (2) + (3) | (4) | (5) | Ir/Rh | P | |||||||
| Conditions: 89.1 mmol 1-octene, 39 g DMF, 1 g H2O, TPPMS/metal-precursor = 2.5/1, LiCl/metal-precursor = 2/1, 30 bar (CO/H2 = 2/1), 100 °C, 700 rpm, extraction with 2,2,4-trimethylpentane DMF/iso-octane = 1/1.5, tSeparation = 30 min, TSeparation = 0 °C. | |||||||||||
| Ir(cod)(acac) (0.8 mol%) | 4.25 | 86 | 78 | 5 | 3 | 91 | 75:25 |
109 | 27 | 0.2 | 0.2 |
| Rh(CO)2(acac) (0.2 mol%) | 1.5 | 88 | 76 | <1 | 12 | 86 | 71:29 |
423 | 282 | 0.6 | 1.8 |
Conclusions
In summary, selective recycling systems for the iridium-catalysed hydroformylation of 1-octene based on dimethylformamide (DMF) and propylene carbonate (PC) as polar solvents were investigated for the first time. High yields and chemoselectivities towards the desired oxo-products at rates of approximately 91% were observed. The results demonstrated good activity for the catalyst complex formed by the precursor Ir(cod)(acac) and the monosulfonated triphenylphosphine ligand (TPPMS) at conversion rates up to 90% after a reaction time of 6 h. The catalyst complex and oxo-products were separated selectively and effectively using the TPPMS ligand at iridium leaching rates of only 0.2%. The combination of this catalyst system with the appropriate polar solvent PC or DMF and a non-polar solvent such as 2,2,4-trimethylpentane allowed for different recycling systems: a thermomorphic solvent system and a homogeneous reaction followed by ex situ extraction. Ultimately, a DMF–H2O system with a downstream extraction allowed for both a highly selective hydroformylation of 1-octene and an efficient separation of catalyst from the products. The stability and activity of the iridium catalyst was confirmed over the course of 5 recycle runs. Because of higher chemoselectivities and lower catalyst leaching the iridium system seems to be very competitive to rhodium-catalysed hydroformylation, which was performed during our investigations, too. Additionally, the results obtained with this recycling concept enable a further investigation of this reaction in a continuously operated miniplant. The results of those subsequent experiments will soon be published in a separate paper.Acknowledgements
The described experiments were part of the PROFORMING project (no. 03X3559). The authors would like to thank BMBF (German Federal Ministry of Education and Research), Evonik Industries, LIKAT (Leibniz Institute for Catalysis) and SUPREN (Sustainable Process Engineering) for their cooperation and helpful input. The authors also thank Umicore AG & Co. KG for donation of transition metal compounds.References
- H. Bahrmann, H. Bach and G. D. Frey, Oxo Synthesis, in Ullmann's Encyclopedia of Industrial Chemistry, Wiley-VCH, Weinheim, 2000, pp. 610–616 Search PubMed.
- C. D. Frohning, C. W. Kohlpaintner and H.-W. Bohnen, Carbon Monoxide and Synthesis Gas Chemistry, in Applied Homogeneous Catalysis with Organometallic Compounds, Wiley-VCH, Weinheim, 2002, pp. 29–194 Search PubMed.
- R. Franke, D. Selent and A. Börner, Chem. Rev., 2012, 112, 5675–5732 CrossRef CAS PubMed.
- A. Behr and P. Neubert, Applied homogeneous catalysis, Wiley-VCH, Weinheim, 2012 Search PubMed.
- G. D. Frey, J. Org. Chem., 2014, 754, 5–7 CrossRef CAS.
- J. Matthey, Platinum 2013, Johnson Matthey Public Limited Company, 2013 Search PubMed.
- F. P. Pruchnik, Organometallic chemistry of the transition elements, Springer-Verlag, 1990 Search PubMed.
- I. Piras, R. Jennerjahn, R. Jackstell, A. Spannenberg, R. Franke and M. Beller, Angew. Chem., Int. Ed., 2011, 50, 280–284 CrossRef CAS PubMed.
- J. Pospech, I. Fleischer, R. Franke, S. Buchholz and M. Beller, Angew. Chem., 2013, 125, 2922–2944 CrossRef.
- C. De, R. Saha, S. K. Ghosh, A. Ghosh, K. Mukherjee, S. S. Bhattacharyya and B. Saha, Res. Chem. Intermed., 2013, 39, 3463–3474 CrossRef CAS.
- C. W. Kohlpaintner, R. W. Fischer and B. Cornils, Appl. Catal., A, 2001, 221, 219–225 CrossRef CAS.
- G. Frey and G. Dämbkes, 75 Jahre Oxo-Synthese, Klartext, Essen, 2013 Search PubMed.
- B. Cornils and W. A. Herrmann, Aqueous-Phase Organometallic Catalysis, Wiley-VCH, Weinheim, 2004 Search PubMed.
- A. Behr, B. Turkowski, R. Roll, R. Schöbel and G. Henze, Multiphase Catalysis in Temperature-Dependent Multi-Component Solvent (TMS) Systems, in Regulated Systems for Multiphase Catalysis, ed. W. Leitner and M. Hölscher, Springer-Verlag, Berlin/Heidelberg, 2008, pp. 19–52 Search PubMed.
- E. Schäfer, Y. Brunsch, G. Sadowski and A. Behr, Ind. Eng. Chem. Res., 2012, 51, 10296–10306 CrossRef.
- K. Kunna, C. Müller, J. Loos and D. Vogt, Angew. Chem., Int. Ed., 2006, 45, 7289–7292 CrossRef CAS PubMed.
- M. Jakuttis, A. Schönweiz, S. Werner, R. Franke, K.-D. Wiese, M. Haumann and P. Wasserscheid, Angew. Chem., Int. Ed., 2011, 50, 4492–4495 CrossRef CAS PubMed.
- M. Kraume, Chem. Ing. Tech., 2013, 85, 1–13 CrossRef.
- R. Schomäcker, M. Schwarze, H. Nowothnick, A. Rost and T. Hamerla, Chem. Ing. Tech., 2011, 83, 1343–1355 CrossRef.
- M. Müller, Y. Kasaka, D. Müller, R. Schomäcker and G. Wozny, Ind. Eng. Chem. Res., 2013, 52, 7259–7264 CrossRef.
- S. L. Desset, U. Hintermair, Z. Gong, C. C. Santini and D. J. Cole-Hamilton, Top. Catal., 2010, 53, 963–968 CrossRef CAS.
- G. Kiedorf, D. M. Hoang, A. Müller, A. Jörke, J. Markert, H. Arellano-Garcia, A. Seidel-Morgenstern and C. Hamel, Chem. Eng. Sci., 2014, 115, 31–48 CrossRef CAS.
- J. Markert, Y. Brunsch, T. Munkelt, G. Kiedorf, A. Behr, C. Hamel and A. Seidel-Morgenstern, Appl. Catal., A, 2013, 462–463, 287–295 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2016 |