
Electrochemical technologies for wastewater treatment and resource reclamation
Yujie
Feng
*a,
Lisha
Yang
a,
Junfeng
Liu
a and
Bruce E.
Logan
b
aState Key Laboratory of Urban Water Resource & Environment, Harbin Institute of Technology, Harbin 150090, PR China. E-mail: yujief@hit.edu.cn; Fax: +86 451 86287017; Tel: +86 451 86287017 Tel: +86 451 86283068
bDepartment of Civil and Environmental Engineering, The Pennsylvania State University, 212 Sackett Building, University Park, PA 16802, USA
First published on 5th May 2016
Abstract
Research developments in environmental electrochemistry and their potential to contribute to a cleaner environment are reviewed here for wastewater treatment applications. Most environmental pollutants can be successfully eliminated or converted to non-toxic materials by one or more processes, including electrochemical oxidation, electrochemical reduction, electrocoagulation and electrocoagulation/flotation, electrodialysis, and electrochemical advanced oxidation processes. Specific examples of applications for pollutant removal and reclamation of wastewater are given for the different processes, along with research needs and improvements for commercial application of these electrochemical processes.
 Yujie Feng | Prof. Yujie Feng obtained her Ph.D. and M. Phil. from Harbin Institute of Technology and Bachelor's degree from Tianjin University. She has been working at Harbin Institute of Technology as a Lecturer (1994–1998), Associate Professor (1998–2002) and Professor (since 2002–present). She is currently the Deputy Director of the State Key Laboratory of Urban Water Resource and Environment (HIT) of the National Ministry of Science & Technology. She is also a Visiting Professor at Penn State University, USA and Liaoning University, China and a Fellow of International Water Association (IWA). Her research is focused on wastewater treatment and energy recovery, and risk evaluation of toxic compounds or nano-materials in engineering systems. |
 Lisha Yang | Lisha Yang is a Ph.D. student at Harbin Institute of Technology (2013–present). She received her Bachelor's degree and Master's degree in Environmental Science and Engineering at Heilongjiang University. Her research interest is in the development of new routes for preparation of nano-sized coating electrode materials for applications in electrochemical technologies for wastewater treatment and resource reclamation. |
 Junfeng Liu | Dr. Junfeng Liu obtained his Ph.D. and M. Phil. from Harbin Institute of Technology. Dr. Liu is currently a lecturer at the School of Municipal and Environmental Engineering, Harbin Institute of Technology. His research interests are in the fields of environmental electrocatalytic materials, photocatalytic materials and wastewater refractory organic pollutant removal technology. He has published more than 30 papers, including 14 papers indexed by SCI. |
 Bruce E. Logan | Dr. Logan earned his B.S. and M.S. in Chemical Engineering and Environmental Engineering, respectively, at Rensselaer Polytechnic Institute and his Ph.D. in Environmental Engineering at University of California, Berkeley. He started his career as an Assistant Professor at the University of Arizona, and became an Associate Professor, then a Professor before moving to The Pennsylvania State University. Dr. Logan is currently an Evan Pugh Professor and the Stan and Flora Kappe Professor of Environmental Engineering in the Department of Civil and Environmental Engineering at The Pennsylvania State University. |
Water impactElectrochemical treatment of pollutants in water can be accomplished in many different ways, for example, by direct oxidation and reduction reactions, through the production of reactive chemical species, or by releasing chemicals that achieve physical removal. These different electrochemical processes are critically reviewed here, noting specific challenges to advance existing and new technologies for cost-effective water treatment. |
1. Introduction
As a consequence of industrial development activities, technological progress and profit have sometimes prevailed over environmental concerns, resulting in an increasing number of detrimental pollutants that have found their way into the environment. Thus, the removal of environmental pollutants has become a major issue and a crucial factor for the sustainable development of modern industrial processes, which must comply with regulations to ensure clean environments.Industrial electrochemistry has undergone development towards cleaner processes and more environmentally friendly products, which is one of the strategies for environmental protection. As a consequence, a special research field of environmental electrochemistry has been developed, which is based on using electrochemical techniques to remove impurities from gases, liquids, or soils, to prevent or minimize environmental pollution. The first book devoted to the potential of electrochemistry for environmental protection appeared four decades ago by Bockris.1 Since then, several books and reviews have been devoted to this topic,2–20 which have indicated the inherent advantages of several different electrochemical technologies based on their environmental compatibility, due to the fact that the main reagent, the electron, is a ‘clean reagent’. Other attractive advantages are related to versatility, high energy efficiency, amenability to automation, and cost-effectiveness.
From the viewpoint of high efficiency and low resource consumption, electrochemical technologies can be used either as a pretreatment step to increase the biodegradability of a pollutant or as an advanced treatment method further to reduce COD or color in the water to achieve relevant effluent standards.
Based on analyzing the characteristics of the wastewater quality, two combined electrochemical processes have been proposed by the research group of Feng and co-workers,21,22 as shown in Fig. 1.
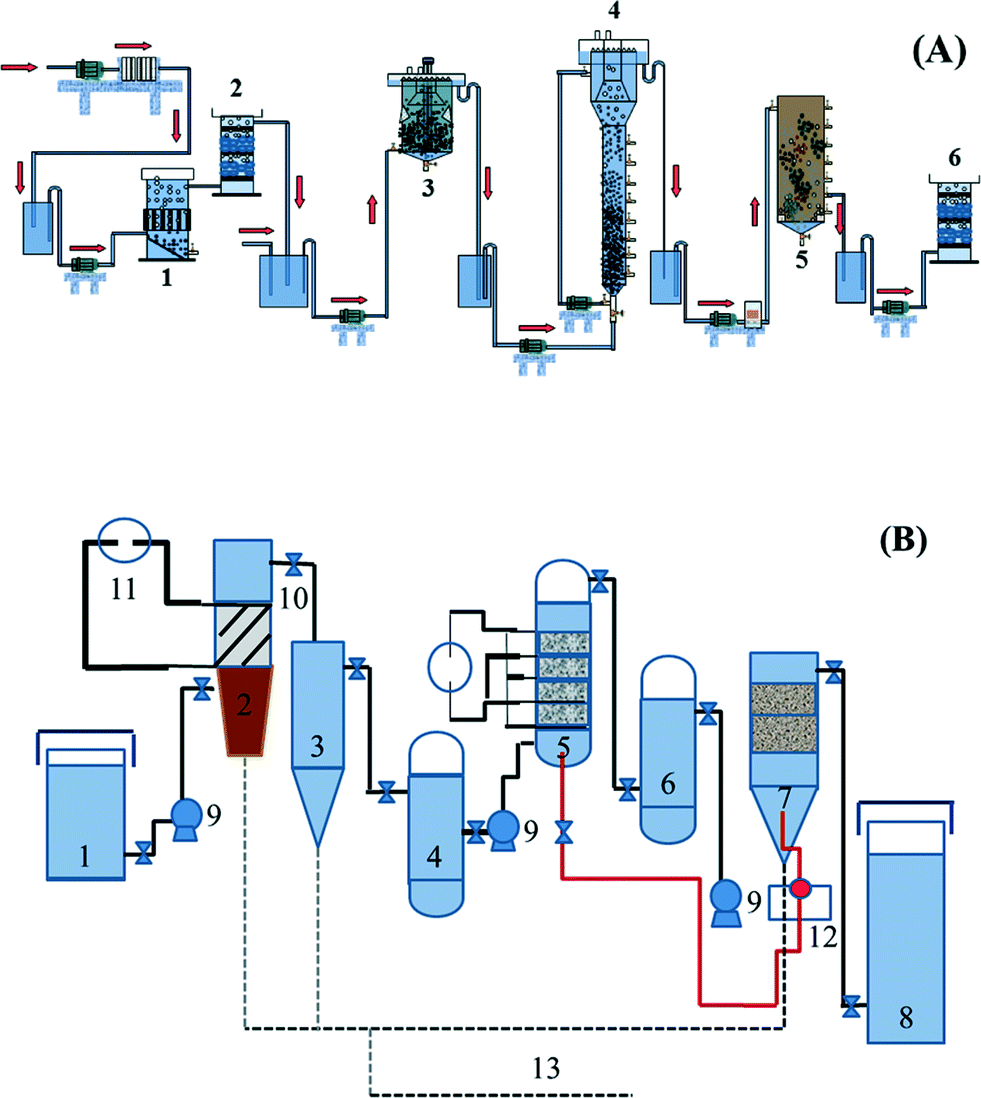 | ||
| Fig. 1 Flow diagram of combined electrochemical processes: (A) two-phase anaerobic (CSTR + EGSB)–aerobic (SBR)–electrocatalytic oxidation (1. electroflocculation reactor, 2. electrochemical reactor, 3. CSTR, 4. EGSB, 5. SBR, 6. electrochemical reactor); (B) coagulation–electrocatalytic oxidization–biological contact oxidation combined process (1. adjustable water tank, 2. electrocoagulation, 3. sedimentation tank, 4. regulating tank, 5. three-dimensional fixed bed electrochemical device system, 6. regulating tank, 7. biological contact oxidization device system, 8. effluent trough, 9. pump, 10. valve, 11. DC power supply, 12. aeration system, 13. sludge collection system).21,22 | ||
In this paper, we reviewed the electrochemistry-based approaches for removal of pollutants in wastewaters, such as electrochemical oxidation, electrochemical reduction, electrocoagulation and electrocoagulation/flotation, and electrodialysis processes. Emerging technologies such as electro-Fenton and photoelectro-Fenton processes, photoelectrocatalysis, and sonoelectrocatalysis are also discussed along with the relative advancements and recent achievements. The fundamentals of each technology are briefly discussed in order to better understand their advantages and limitations for practical applications in the removal and treatment of environmental pollutants in water and wastewater.
2. Electrochemical oxidation
Electrochemical oxidation is considered to be a very powerful tool for breaking up even the most resistant organic compounds.23 Anodic oxidation of organic contaminants can be performed in several different ways, including both direct and indirect oxidation.2.1 Direct anodic oxidation
In direct anodic oxidation (or direct electron transfer to the anode), the pollutants are destroyed after adsorption on the anode surface, without the involvement of any substances other than the electron. Such oxidation is theoretically possible at more negative potentials than those needed for water splitting and oxygen evolution. However, this process usually results in electrode fouling due to the formation of polymeric layers on its surface and consequently leads to very poor chemical decontamination.24,25 Gattrell and Kirk investigated the electro-oxidation of phenol with platinum and peroxidized platinum anodes using cyclic voltammetry and chronoamperometry. Their studies demonstrated that phenol can be irreversibly adsorbed on metallic platinum, quickly passivating the electrode.262.2 Indirect anodic oxidation via intermediates of oxygen evolution
To avoid the drawbacks of direct oxidation, the indirect oxidation method based on the oxygen evolution region can be used, which has an advantage over direct electrolysis in that it does not need addition of oxidation catalysts to the solution, and it does not produce by-products.In this process, the electrochemical reaction leads to partial or total decontamination of the electrogenerated species at the anode due to physically adsorbed “active oxygen” (adsorbed hydroxyl radicals ˙OH) or chemisorbed “active oxygen” (oxygen in the lattice of a metal oxide (MO) anode).27,28 This physisorbed ˙OH is the second strongest oxidant known after fluorine, with a high standard potential (E0 = 2.80 V vs. SHE) that ensures the complete combustion of organic compounds, and the chemisorbed “active oxygen” participates in the formation of selective oxidation products.
As has been established in many studies,29,30 the nature of the anode material influences not only the efficiency of the process, but also the electrode selectivity. For example, “active anodes” with low oxygen evolution overpotentials such as IrO2, RuO2 or Pt favor the partial and selective oxidation of pollutants, while “non-active anodes” with high oxygen evolution overpotentials such as SnO2, PbO2 or boron-doped diamond (BDD) can facilitate complete combustion, and thus they are regarded as ideal electrodes for the complete oxidation of organics to CO2 in wastewater treatment (Fig. 2).
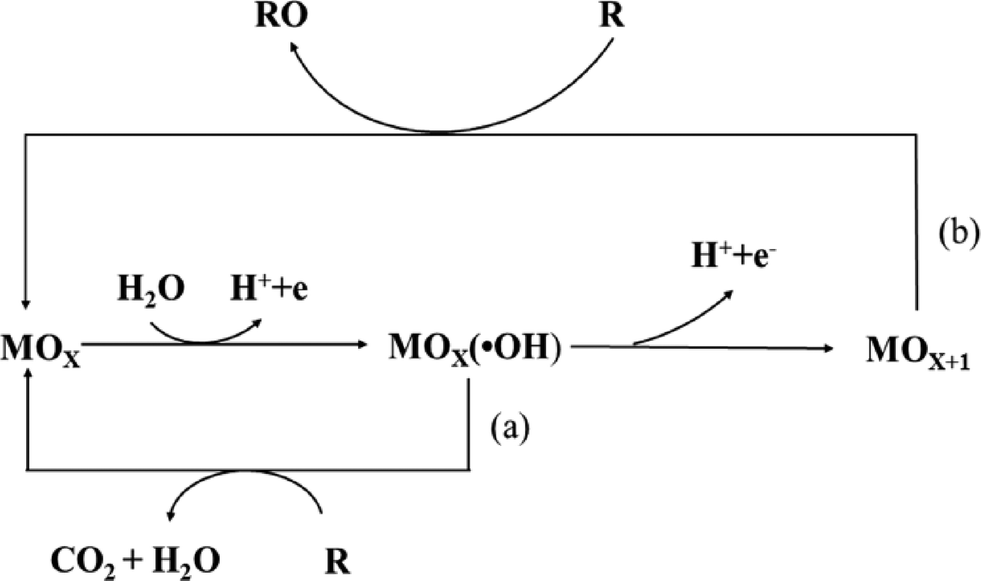 | ||
| Fig. 2 Scheme of the electrochemical oxidation of organic compounds on (a) “active” and (b) “non-active” anodes. | ||
The oxidation of a wide range of biorefractory organic compounds on platinum anodes has been reported in many studies.31–38 Platinum electrodes have a relatively low oxygen evolution overpotential (i.e., 1.6 V vs. SHE in 0.5 M H2SO4) which can enable selective conversion of pollutants at a low current efficiency. In a study by Feng et al.30 on the electro-oxidation of phenol using platinum electrodes, it was found that the concentration of phenol rapidly decreased to zero, but residual TOC concentrations suggested that the overall degradation reactions significantly slowed down due to the formation of intermediate products.
The electro-oxidation of phenol on platinum anodes was investigated in depth by Comninellis and Pulgarin.29,33 Their experimental results indicated that aromatic intermediates (hydroquinone, catechol, benzoquinone) initially were formed during electrolysis, and subsequently the aromatic ring opened with the formation of aliphatic acids (e.g., maleic, fumaric, and oxalic acid) which resisted further electro-oxidation. Thus, complete TOC removal could not be achieved, and the current efficiency decreased during the electrolysis process (Fig. 3).
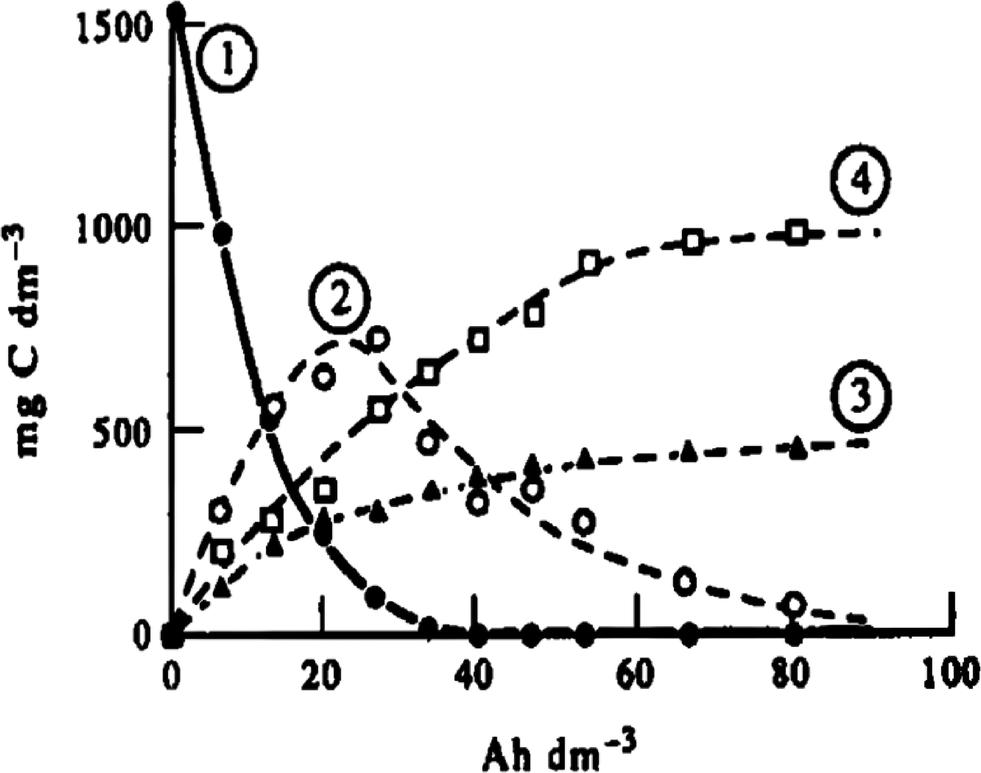 | ||
| Fig. 3 Evolution of (1) phenol, (2) aromatic intermediates, (3) aliphatic acids, and (4) CO2 during oxidation of phenol at the Pt anode: i = 50 mA cm−2, T = 70 °C.29,33 | ||
During the past two decades, DSA-type anodes coated with a layer of RuO2 and IrO2 have begun to be extensively employed in the field of wastewater treatment.30,40–47
However, when these electrodes are used at high current densities, organic oxidation can yield low current efficiencies for complete combustion since they favor the secondary reaction of oxygen evolution. Electrochemical destruction of 4-chlorophenol in an aqueous medium using a platinum anode coated with a RuO2 film has been studied by Johnson et al.27 They found that this type of electrode was stable and active when used in a cell with a solid Nafion membrane without the addition of a soluble supporting electrolyte. However, the time required for complete COD and TOC removal was too long and the current efficiency was low.
Titanium is also used as an inexpensive coating on electrodes. The electro-oxidation of 1,4-benzoquinone in water, in which the benzoquinone concentration and intermediate products during oxidation with Ti/IrO2 and Ti/SnO2 anodes were monitored, has been investigated by Pulgarin et al.48 It was found that the most important factor was the composition of the anode. With the Ti/IrO2 anode, the primary oxidation step was easily achieved (benzene ring rupture), resulting in the accumulation of carboxylic acids formed as the final non-toxic products. With the Ti/SnO2 anode, carboxylic acids were formed at a much faster reaction rate and then oxidized, producing only CO2 as the final product.
Electro-oxidation of Reactive Blue 19 solutions in a three-electrode quartz cell equipped with a Ti/Ru0.3Ti0.7O2 anode was investigated by Pelegrini et al. They obtained 35% decolorization efficiency and 9.6% TOC removal after 2 h of electrolysis.49
Electrochemical degradation of phenol with five different types of anodes (three RuO2-based electrodes, Ti/PbO2 and Pt electrodes) was evaluated by Feng et al.30
As shown in Fig. 4, the relative performance for phenol degradation of the three RuO2 electrodes decreased in the order: Ti/Sb–Sn–RuO2–Gd > Ti/Sb–Sn–RuO2 > Ti/RuO2. However, these electrodes were less efficient than Pt or Ti/PbO2 electrodes. Aromatic ring opening occurred using all these electrodes, but with the three RuO2-based electrodes, phenol was decomposed into aromatic intermediates, such as benzoquinone and hydroquinone, or several carboxylic acids, such as maleic acid, succinic acid, and oxalic acid. Full mineralization to CO2, or complete TOC removal, only was obtained for the Ti/PbO2 anode (Fig. 4).
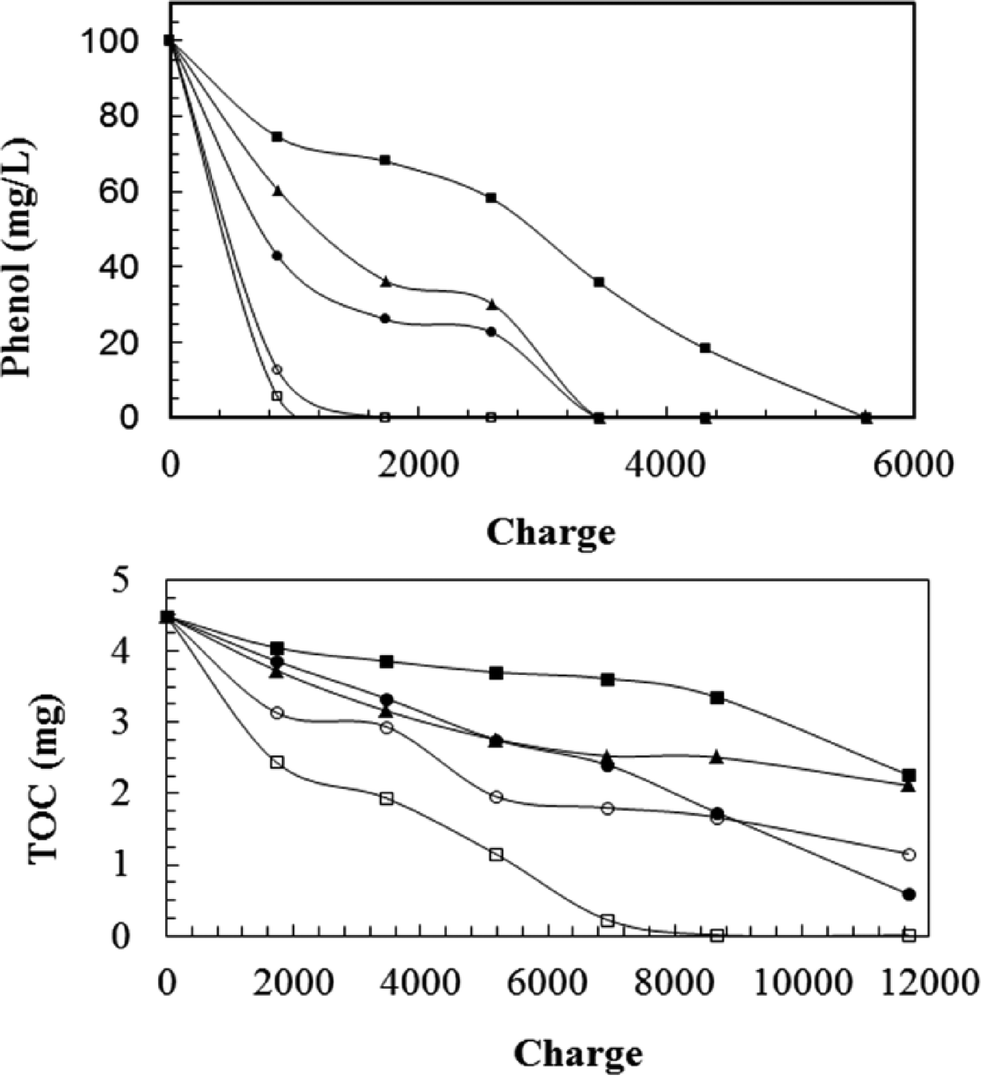 | ||
| Fig. 4 Electrochemical degradation of 100 ppm phenol in 60 ml of electrolyte as a function of charge passed for different electrode materials, i = 10 mA cm−2 (–■–) Ti/RuO2; (–▲–) Ti/Sb–Sn–RuO2; (–●–) Ti/Sb–Sn–RuO2–Gd; (–□–) Ti–PbO2; and (–○–) Pt.30 | ||
DSA-type anodes coated with a layer of RuO2 or IrO2 can be used efficiently for organics degradation by indirect electrolysis by in situ generation of active chlorine through the oxidation of chloride ions present in the solution.45,50–54 For example, Kraft55 showed that DSA-type electrodes (IrO2 and IrO2–RuO2) gave higher current efficiencies during electrochemical chlorine production than boron-doped diamond (BDD) and platinum (Pt) anodes (Fig. 5).
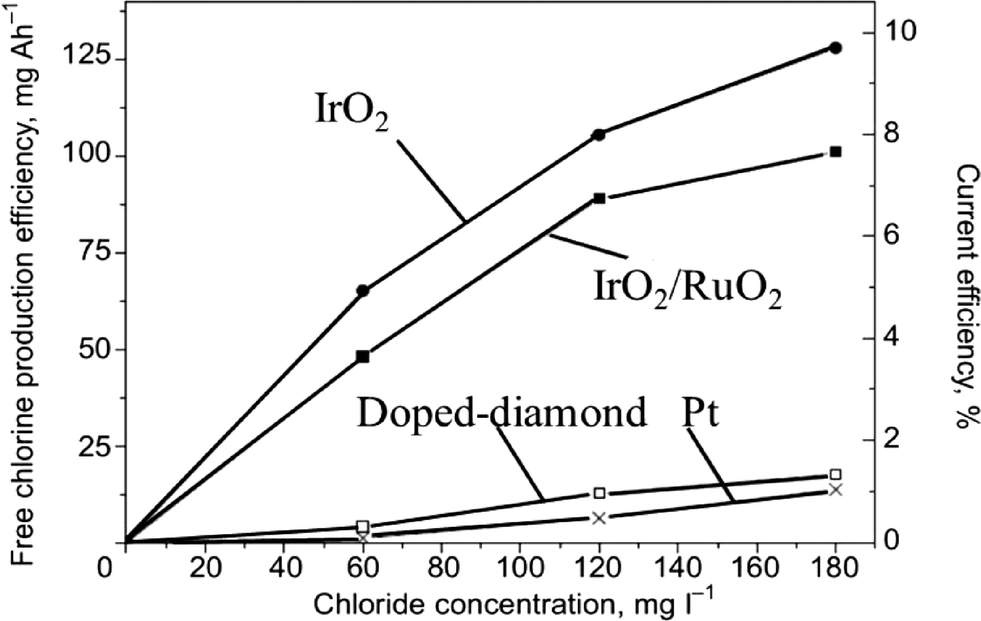 | ||
| Fig. 5 Dependence of the electrochemical free chlorine production efficiency on the chloride content of electrolyzed water under standard conditions using four different anode materials (iridium oxide, mixed iridium/ruthenium oxides, platinum, doped diamond).55 | ||
Scialdone et al. investigated the electrochemical oxidation of organics on IrO2–Ta2O5 anodes in the presence and absence of NaCl, in a continuous batch recirculation reaction system equipped with a parallel plate undivided electrochemical cell. Their results showed that in the presence of NaCl, high current efficiency (CE) was generally obtained using DSA anodes at high current densities and low flow rates.56
An electrochemical oxidation approach for the treatment of a high-salinity reverse osmosis (RO) concentrate was investigated using boron-doped diamond (BDD) and titanium-based dimensionally stable anodes (Ti/IrO2–RuO2). The results showed that both direct oxidation and indirect oxidation by active chlorines played a role in the treatment of the RO concentrate, but the contribution was different for the anodes. The highest COD removal was observed using the BDD electrode at the same current density as that of the other electrode, but the least energy consumption was obtained using the Ti/IrO2–RuO2 electrode.57
In order to improve the stability of Pb electrodes, one approach that is being investigated is the incorporation of metallic or nonmetallic species such as Fe2+,66–70 Bi3+,71–75 Co2+,76,77 and F− (ref. 69, 76, 78–80) into the PbO2 crystalline matrix.
Andrade et al. showed that incorporation of F, Fe, and Co ions into the PbO2 film enhanced its chemical stability compared to that of pure PbO2 in the oxidation of simulated wastewaters containing Blue Reactive 19 dye or phenol.69,76
Another approach for stabilizing lead electrodes is based on introducing a transition layer between the coating and the substrate. Antimony-doped tin oxide has been widely investigated as a transition layer for PbO2 elecrodes.81–87 The lattice size of SnO2 is between β-PbO2 and TiO2, and therefore, the Sb-doped SnO2 transition layer can enhance the solid solubility and hence reduce the internal stress of the Ti/SnO2/β-PbO2 electrode and improve the binding force between the PbO2 coating and Ti substrate, and it also inhibits the formation of a TiO2 layer.
Bi et al. explored the electro-deposition of PbO2 on the Ti substrate with an Sb-doped SnO2 undercoating, using a traditional acidic nitrate solution. They investigated the morphology and microstructure of the PbO2 coatings by varying the electro-deposition temperature and time. Their results indicated that the electrochemical performance of the deposited PbO2 was largely a function of the resulting morphologies and microstructures on the electrodes.82
In order to reveal the mechanism of the enhanced electrochemical performance of the TiO2-NTs/SnO2–Sb/PbO2 electrode, the interlayer of Sb-doped SnO2 (SnO2–Sb) and TiO2 nanotubes (TiO2-NTs) on Ti were introduced into the PbO2 electrode system.83 This electrode with nanotubes had a more regular and compact morphology than previous Ti/SnO2–Sb/PbO2 electrodes, as well as better oriented crystals with smaller sizes (Table 1). Kinetic analyses indicated that the electrochemical oxidation of nitrobenzene on the PbO2 electrodes followed a pseudo-first-order reaction, and mass transport was enhanced at the constructed electrode.
| Electrode | Unit cell parameter | ||
|---|---|---|---|
| a (Å) | c (Å) | V (Å3) | |
| a The lattice parameters of SnO2 (JCPDF 41-1445). b The lattice parameters of PnO2 (JCPDF 65-2826). | |||
| (Standard SnO2)a | 4.738 | 3.187 | 71.54 |
| SnO2–Sb in Ti/SnO2–Sb interlayer | 4.727 | 3.185 | 71.17 |
| SnO2–Sb in TiO2-NTs/SnO2–Sb interlayer | 4.704 | 3.173 | 70.21 |
| (Standard PbO2)b | 4.955 | 3.383 | 83.06 |
| PbO2 in Ti/SnO2–Sb/PbO2 electrode | 4.952 | 3.380 | 82.88 |
| PbO2 in TiO2-NTs/SnO2–Sb/PbO2 electrode | 4.950 | 3.379 | 82.79 |
An alternative approach for stabilizing Pb electrodes was to position an interlayer of TiO2 nanotubes (NTs) between a fluorine resin (FR)-doped PbO2 coating and the Ti substrate.88 The improvement in surface properties and microstructure was investigated by comparison to traditional PbO2 electrodes. This treatment improved the electrochemical resistance of the electrode in a Na2SO4 solution to 12.2 Ω with PbO2/TiO2-NTs/Ti, compared to 147 Ω with PbO2/Ti. The service life of PbO2/TiO2-NTs/Ti was increased to about 335 h, which was 7.1 times that of the PbO2/Ti electrodes. Pb4+ was detected in the electrolyte after a 50 h electrochemical degradation test using PbO2/Ti (1.1 × 10−5 M) and PbO2/TiO2-NTs/Ti (3.4 × 10−6 M) electrodes, but none was detected with the FR-PbO2/TiO2-NTs/Ti electrodes. This suggests that firm bonding between PbO2 and the substrate was achieved, with the PbO2 stably deposited onto TiO2-NTs, and this association was improved with FR doping. Contact between SO42− and Pb4+ was likely blocked by the FR, inhibiting the anodic dissolution of the PbO2 coating.
To gain better knowledge on the ability of these Pb anodes to eliminate pollutants, many researchers have undertaken comparative studies on the performance of electrochemical oxidation with boron-doped diamond (BDD) and PbO2 anodes. A comparative study between PbO2 and BDD anodes for electrooxidation of cresols (o-, m- and p-cresol) showed that complete electrochemical incineration was achieved at the same time as the initial pollutant was removed, since BDD(˙OH) simultaneously destroys all oxidation by-products formed. The mineralization process of the m-cresol effluent on PbO2 under comparable conditions is much less efficient due to the lower oxidation ability of PbO2(˙OH). Despite this fact, a shorter electrolysis time is needed for the total disappearance of m-cresol with PbO2 than with BDD.89 Electrochemical oxidation of ibuprofen (Ibu) using a Ti/Pt/PbO2 electrode and a boron-doped diamond (BDD) electrode was investigated by Ciríaco et al., showing a much higher degradation efficiency for the BDD anode of 20 mA cm−2 than that for the PbO2-based electrode.90
In contrast, work by Zhao et al.88 showed that a FR-PbO2/TiO2-NTs/Ti anode had similar morphology and surface wetting properties, a higher OEP, and better electrochemical performance than a boron-doped diamond film (BDD) electrode (Table 2). The improved surface hydrophilic properties of PbO2 electrodes could produce better conditions leading to the increased chemical adsorption ability on the surface and subsequently lower utilization of ˙OH than BDD anodes. The physical resistance of the PbO2 electrode was much lower than that of BDD, and therefore it had higher conductivity. Hydroxyl radical utilization is significantly enhanced on a PbO2 electrode, which has been shown to produce a higher oxidation rate and higher removal efficiency for 2,4-dichlorophenoxyacetic acid than a BDD electrode.91,92
| Electrode | Loading capacity (g m−2) | R OCPct (Ω) | Extrapolated OEP0a (V vs. SCE) | Service lifetime (h) |
|---|---|---|---|---|
| a The extrapolated OEP0 at zero current is obtained by using an extrapolation technique from the anodic polarization (j–E) curve tested in 0.5 M H2SO4 aqueous solution, which is equivalent to the minimum decomposition potential for water. | ||||
| FR-PbO2/TiO2-NTs/Ti | 971.6 | 12.2 | 2.50 | 335 |
| PbO2/TiO2-NTs/Ti | 950.1 | 34.8 | 1.90 | 170 |
| PbO2/Ti | 705.9 | 147 | 1.80 | 47 |
| BDD | — | 34![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 500 500 |
2.40 | — |
A comparative study of the electrochemical mineralization of environmentally persistent long-chain perfluorinated carboxylic acids (PFCAs) with Ti/SnO2–Sb/Ce–PbO2 and Ti/BDD anodes was carried out using galvanostatic control at room temperature.93 The results showed that the performance of the PbO2 electrode was comparable with that of a BDD electrode. After 180 min of electrolysis, the PFNA removal efficiencies on the BDD and PbO2 electrodes were 98.7 ± 0.4% and 97.1 ± 1.0%, respectively, while the corresponding PFDA removal efficiencies were 96.0 ± 1.4% and 92.2 ± 1.9%.
Other authors have reported similar performance using PbO2 and BDD anodes during oxalic acid incineration.52,94 There is a strong interaction between this compound and Pb(IV) sites which promotes anodic oxidation, and the rate of oxidation was only limited by mass transfer to the electrode surface at high current densities and low substrate concentrations.
Titanium anodes coated with Sb-doped tin oxide have been considered as one of the most suitable electrodes in the electrochemical oxidation of refractory organics because of their large overpotential for oxygen evolution (1.9 V vs. SHE) and high yield of hydroxyl radicals.30,96,97
In general, two different mechanisms can be distinguished for the oxidation of organic pollutants on SnO2 anodes. One is direct oxidation, and the other is indirect oxidation. Direct oxidation can only take place on the surface of SnO2 anodes. Indirect oxidation can occur via hydroxyl radicals, which are generated by oxygen vacancies on the SnO2 anodes.98 Electrochemical oxidation of organic pollutants on SnO2 anodes mainly depends on indirect oxidation.
Research has shown that the electrochemical characteristics and service life of SnO2 anodes are influenced by the preparation method as well as other factors. Ti metal is often used as a base material for SnO2 electrodes because of its low cost and stability. The key aspect of SnO2 anode preparation is achieving a stable catalytic coating on the Ti base, while also ensuring that the catalytic coating is stable and well adhered to the base material.
The particle size of the SnO2 crystal has a great influence on the electrochemical characteristics of SnO2 anodes. Smaller particle sizes means larger surface areas, which will improve the overall electrocatalytic reaction rates. The particle size of SnO2 crystals prepared by electrodeposition and sol–gel methods is on the nanometer scale, which provides a very high specific surface area. Of all preparation methods, the sol–gel method is a relatively simple, effective, and convenient way to produce effective SnO2 nanocoatings.97
The performance of Sb-doped SnO2 anodes has been previously investigated using phenol. It has been shown in the work of Stucki and co-workers that doped SnO2 anodes oxidized a wide range of organic compounds with an efficiency about five times higher than that of platinum anodes.34,99 Similar results were also obtained by Comninellis and Pulgarin,96 in their studies on the electrochemical oxidation of phenol on doped-SnO2 and platinum anodes. They found that aliphatic acids were rapidly oxidized, with only very small amounts of aromatic intermediates, using a SnO2 anode, but the Pt anode had low degradation rates and produced a large amount of intermediates. Comninellis measured a CE of 0.58 using an SnO2–Sb2O5 electrode, and obtained 71% degradation of phenol, compared to lower CEs of 0.18 (PbO2), 0.17 (IrO2), 0.14 (RuO2) and 0.13 (Pt) at a current density of 500 A m−2 (pH = 12.5, initial phenol concentration = 10 mm, and reaction temperature of 70 °C).100
Electrochemical degradation of phenol was investigated by Feng et al. using three different types of anodes: Ti/SnO2–Sb, Ti/RuO2, and Pt. Although phenol was oxidised by all of the anodes at a current density of 20 mA cm−2 or a cell voltage of 4.6 V, the intermediate products of phenol degradation, including benzoquinone and organic acids, were subsequently rapidly oxidized by the Ti/SnO2–Sb anode but accumulated in solution using Ti/RuO2 and Pt. The degradation rates of the Ti/RuO2 and Pt anodes were considerably slower, as the SnO2–Sb coating improved the catalytic reaction and allowed rapid organic oxidation driven by hydroxyl radicals generated from anodic water electrolysis (Fig. 6).101
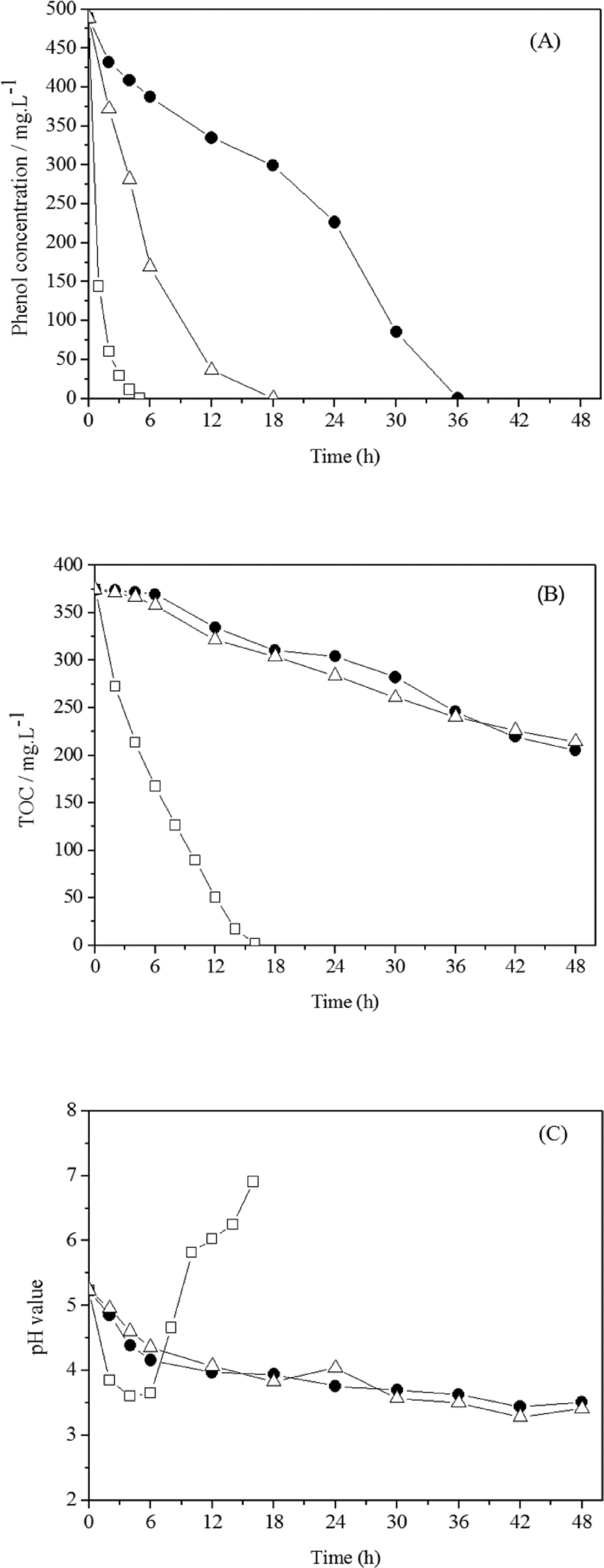 | ||
| Fig. 6 Electrochemical degradation of phenol (490 mg L−1) on Ti/SnO2–Sb, Ti/RuO2 and Pt anodes at a current density of 20 mA cm−2: (A) phenol degradation, (B) TOC removal, and (C) pH variation.101 | ||
The great potential of SnO2 for electrochemical treatment of many different organic chemicals has been shown, including: phenol,96,102–108 aliphatic acids,106 dyes,109,110 drugs,111,112 Bisphenol A (BPA),113 nitrophenol,114 2,4-dichlorophenol,115 4-chlorophenol,116 pentachlorophenol,117,118 perfluorooctanoic acid,109,119 perfluorinated carboxylic acids (PFCAs),93 industrial wastewater120 and naphthylamine.121 Nevertheless, the major drawback that has prevented the practical applications of Ti/SnO2–Sb electrodes is their relatively short service lifetime. The short lifetime has been ascribed to the formation of a resistive layer between the substrate and coating, a passivation layer formed on the outer surface of the coating, and selective loss of the catalyst into the electrolyte solution.
Many methods have been proposed to further improve SnO2 electrodes. For example, doping SnO2 electrodes with rare earth metals can improve the electro-catalytic decomposition of organics such as phenol, as shown in several examples summarized in Table 3.122
| Electrode | The optimum molar ratio of rare earth metals to Sn | The time of degradation of phenol from 100 mg L−1 to below 10 mg L−1 (h) | Oxygen evolution potential (V (vs. Ag/AgCl)) | The sequence of ˙OH-producing capacity | Oxidation mechanisms |
|---|---|---|---|---|---|
| Ti–Ce–Sb–SnO2 | 1:50 |
3.0 | 2.16 | The redox couple of Ce4+/Ce3+ | |
| Ti–Eu–Sb–SnO2 | 1:50 |
2.5 | 2.21 | Adsorbed˙OH species | |
| Ti–Gd–Sb–SnO2 | 1:50 |
2.5 | 2.23 | ||
| Ti–Dy–Sb–SnO2 | 1:200 |
1.6 | 2.24 | ||
| Ti–Nd–Sb–SnO2 | 1:200 |
<1.5 | 2.28 |
The addition of Gd can improve the morphology and performance of Ti/SnO2–Sb electrodes. Both phenol and intermediate products (e.g., benzoquinone) were shown to decompose more rapidly at 2% doping of Gd for the electrodes, over a doping range of 0–10% (Fig. 7).103
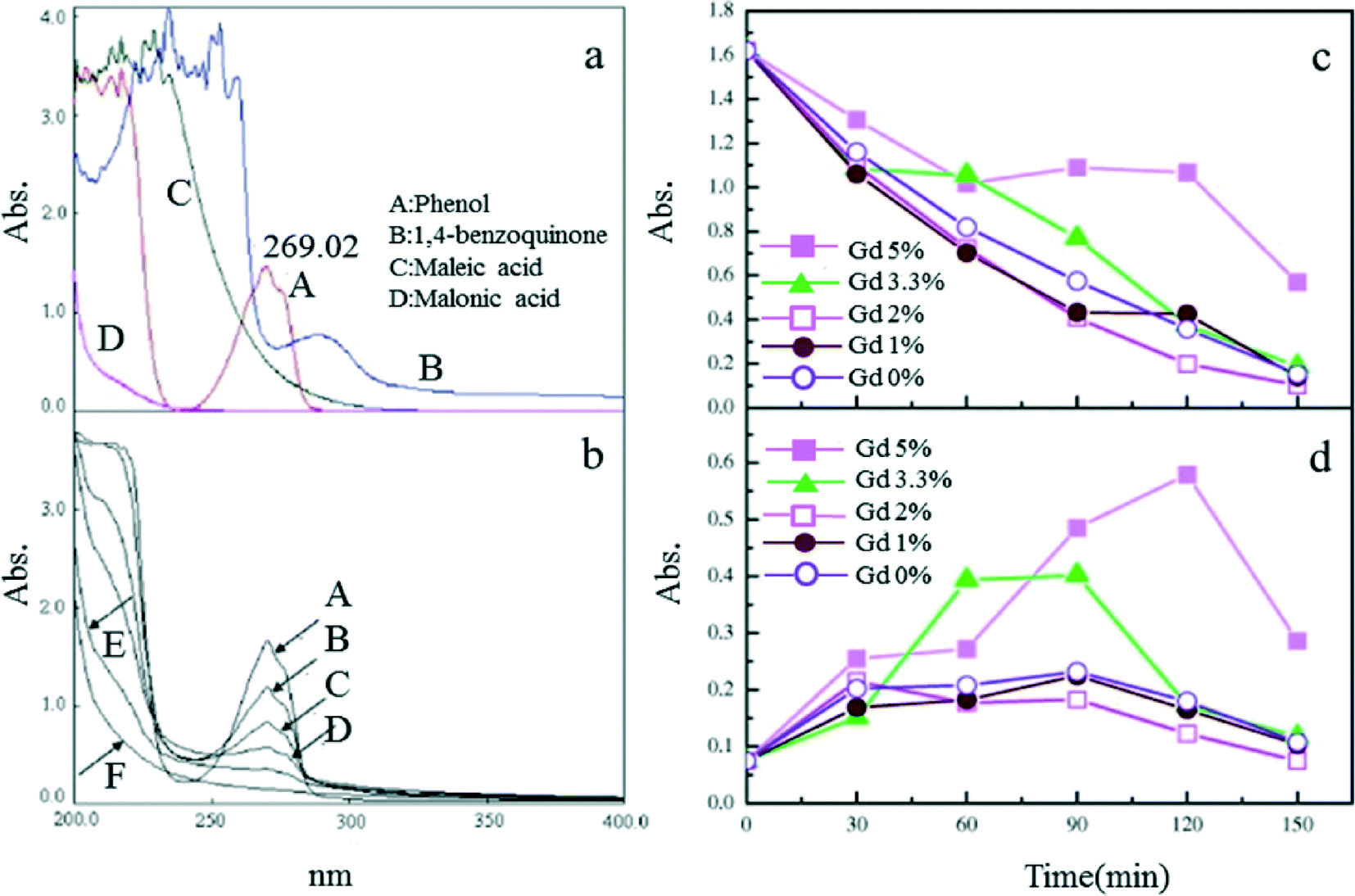 | ||
| Fig. 7 (a) UV scan curves of electrolytes for phenol and some possible intermediates; (b) UV scan of electrolytes of the 2% Gd-doped Ti/SnO2–Sb anode; (c) UV absorbance at 269 nm and (d) at 290 nm of electrolytes as a function of time for different compositions of Gd-doped Ti/SnO2–Sb electrodes, i = mA cm−2.103 | ||
The enhanced performance of Gd-doped Ti/Sb–SnO2 electrodes is due to the increased adsorption capacity for hydroxyl radicals on the electrode surface, and the lower mobility of oxygen atoms in the SnO2 lattice.
Other new methods to improve the lifetime of SnO2 electrodes include using TiO2 NTs or CNTs serving as carriers for further loading of the nanocrystal catalyst both to enhance the performance of the Ti/SnO2 electrode and to improve its service lifetime, without reducing its O2 evolution potential.
Another approach to improve the stability based on changing the electrode microstructure was proposed by Zhao et al., in which a TiO2-NTs/SnO2 electrode that had high oxygen evolution potential, excellent electrocatalytic performance, and relatively long-term stability was constructed by implanting Sb-doped SnO2 into highly ordered TiO2 nanotubes (TiO2-NTs) grown in situ on a Ti substrate under controlled conditions. The service lifetime of this electrode (TiO2-NTs/SnO2) was 2.4 times higher than that of a traditional Sb-doped SnO2 (SnO2) electrode. Based on TOC removal rates, the TiO2-NTs/SnO2 electrode also completely mineralized benzoic acid (BA).123
The substrate architecture is a main factor in the improved performance of TiO2-NTs/SnO2–Sb anodes for organic pollutant degradation. The pore diameter and length of the TiO2-NTs substrates of the TiO2-NTs/SnO2–Sb electrode were shown to be critical factors in enhanced pollutant degradation, with TiO2-NTs electrodes that had an 85 nm pore diameter and a 51 m length having the best performance.107
A SnO2–Sb2O4-based anode modified with Cr3C2 and CNTs was examined for phenol oxidation.102 The service lifetimes of the Ti/SnO2–Sb2O4–Cr3C2 and Ti/SnO2–Sb2O4–CNT–Cr3C2 electrodes were 7.4 times and 5.6 times longer than that of the Ti/SnO2–Sb2O4 electrode, respectively. The Ti/SnO2–Sb2O4–CNT–Cr3C2 electrode showed the highest oxygen evolution potential, COD removal and current efficiency (CE).
Carbon nanotubes (CNTs) can be incorporated into Ti/SnO2–Sb electrodes using a pulse electrodeposition method to improve performance.124 An electrode modified with CNTs had a higher specific surface area and smaller lattice size, which provided more active sites for hydroxyl radical generation and contaminant oxidation compared to electrodes without CNTs. The oxygen evolution potential of Ti/SnO2–Sb–CNT is 2.23 V, which is 0.07 V higher than that of an electrode lacking CNTs. As a result, the competitive reaction was weakened, the current efficiency was improved, and an accelerated lifetime test indicated that the service life would be 1816 h at a current density of 50 mA cm−2, which is 4.8 times longer than that of the Ti/SnO2–Sb electrode without CNTs. Ti/SnO2–Sb–CNT electrodes have been demonstrated to have a superior electrochemical oxidation and degradation ability using Acid Red 73 as a model organic pollutant. The CNT-modified electrode had a 1.9× higher kinetic rate constant, 1.3× greater chemical oxygen demand (COD) and total organic carbon (TOC) removal efficiencies, 1.4× improved mineralization current efficiency, and a similar (0.98×) specific energy consumption compared to Ti/SnO2–Sb electrodes (Fig. 8).
 | ||
| Fig. 8 (a) TOC removal efficiency and (b) mineralization current efficiency (MCE) in 1.0 g L−1 Acid Red 73 and 0.1 M Na2SO4 solution at a current density of 50 mA cm−2.124 | ||
Among the several materials which have been proposed as anodes, synthetic BDD exhibits several technologically important properties that distinguish it from conventional electrodes, such as an inert surface with low adsorption properties and a strong tendency to resist deactivation, remarkable corrosion stability even in strongly acidic media, and an extremely high O2 evolution overvoltage.125,126 Therefore, it has received great attention in the past decades as a new electrode material.
The behavior of BDD anodes for the electrochemical oxidation of different organic compounds has been investigated in depth by Comninellis and co-workers160–168 and Cañizares and co-workers.128,130–133,141 In a research study on Si/BDD anodes for a wide range of pollutants by Comninellis et al.,168 it was found that independent of the organic pollutant nature, the current efficiency and the amount of intermediates were affected by local concentrations of ˙OH relative to organics concentration on the anode surface. Based on this observation, they proposed a comprehensive kinetic model to relate COD concentrations and current efficiencies for the electrochemical oxidation of a wide range of pollutants with BDD electrodes. The energy consumption during the process was predicted based on experimental conditions including the applied current intensity, organic concentration, and mass-transfer coefficient.
Based on the studies of oxidation of different phenolic compounds (phenol, chlorophenols, and nitrophenols) and carboxylic acids on BDD anodes, Cañizares et al. found that the organic compounds were completely mineralized regardless of the characteristics of the wastewater (initial concentration, pH, and supporting media) and operating conditions (temperature and current density) used. They also found that the phenolic compounds could be oxidized by the hydroxyl radicals on the electrode surface, and also by inorganic oxidants electrogenerated on the BDD anodes in the bulk of the solution, depending on the electrolyte composition, such as peroxodisulfuric acid from sulfuric acid oxidation.132
The rate of oxidation of azo dyes at BDD/Si electrodes can be tuned indirectly by changing the boron doping level. Bogdanowicz et al. first investigated the influence of the level of [B]/[C] ratio on the degradation and mineralization of aromatic pollutants like azo-dyes.144 They found that the mechanical and chemical stability of the electrodes resulted from a microcrystalline layer with a relatively high sp3/sp2 band ratio. The influence of commonly used electrolytes, NaCl and Na2SO4, on the dye removal efficiency was also investigated. They found that the efficiency of the BDD process depended on the electrode's doping level. Higher amounts of dopant on the surface of the BDD electrode resulted in the higher efficiency of dye removal in both electrolytes (Fig. 9).
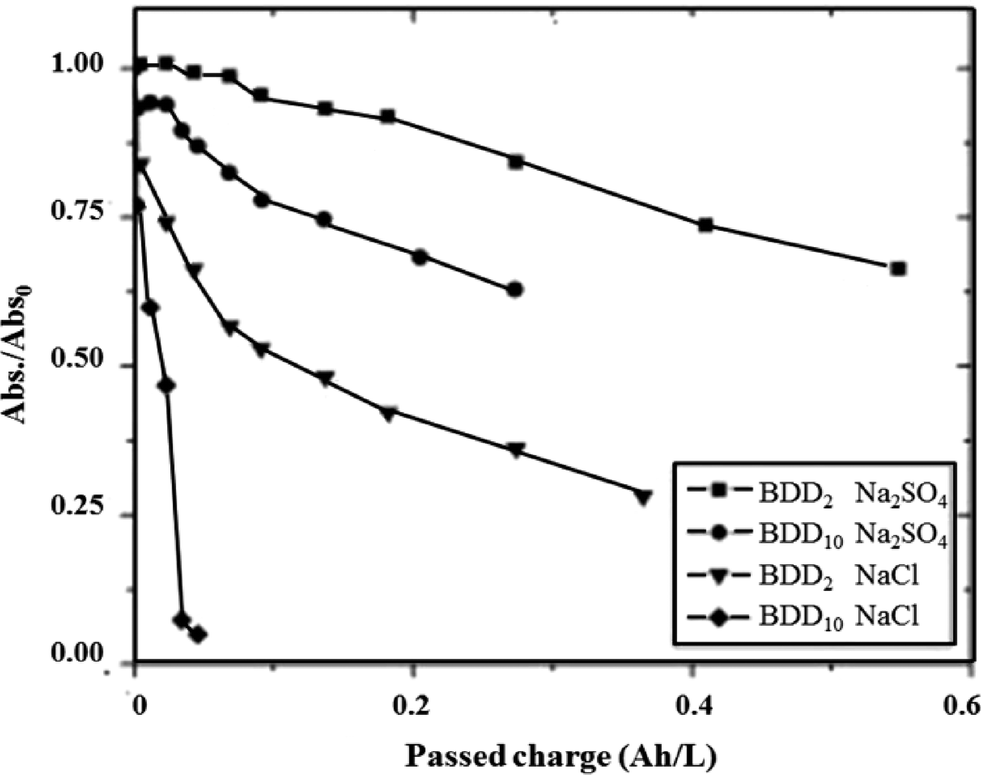 | ||
| Fig. 9 Rubin F-2B concentration as a function of charge passed Q at BDD2 ([B]/[C] = 2000) and BDD10 ([B]/[C] = 10000) in NaCl and Na2SO4 electrolytes. Experimental conditions: [Rubin F-2B]initial = 20 mg L−1, pHinitial = 6.2, [NaCl] = 0.12 M, [Na2SO4] = 0.05 M, T = 20 ± 2 °C.144 | ||
BDD electrodes show good performance for the electrochemical oxidation of pesticides and drugs. The degradation of 2,4-D herbicide in a recirculation flow plant has been studied with Pt/air-diffusion and BDD/BDD electrodes by electrochemical oxidation and electro-Fenton processes. In both treatments, the use of a single BDD/BDD cell always achieved a higher degree of degradation, with 59% mineralization and 0.42 kW h g−1 TOC specific energy after 300 min of electrolysis for the electro-Fenton process at 25 mA cm−2.151
The use of BDD anodes has also been widely investigated for the removal of surfactants from wastewater. The degradation rates of seven perfluorinated compounds (PFCs) with different carbon chain lengths and head groups were compared by Zhuo et al. using BDD electrodes.155 Intermediates of PFCs were detected, with degradation rates showing pseudo-first-order kinetics of perfluoroalkyl carboxylates and sulfonates that increased with carbon chain length. They also proposed electrochemical oxidation mechanisms of PFCs on a BDD anode, where PFC decomposition began with a direct, one-electron transfer from a carboxyl or sulfonate group to the BDD, with the formed PFC radicals decarboxylated or desulfonated to yield a perfluoroalkyl radical which permitted a defluorination reaction between a perfluoroalkyl radical and a hydroxyl radical (Fig. 10).
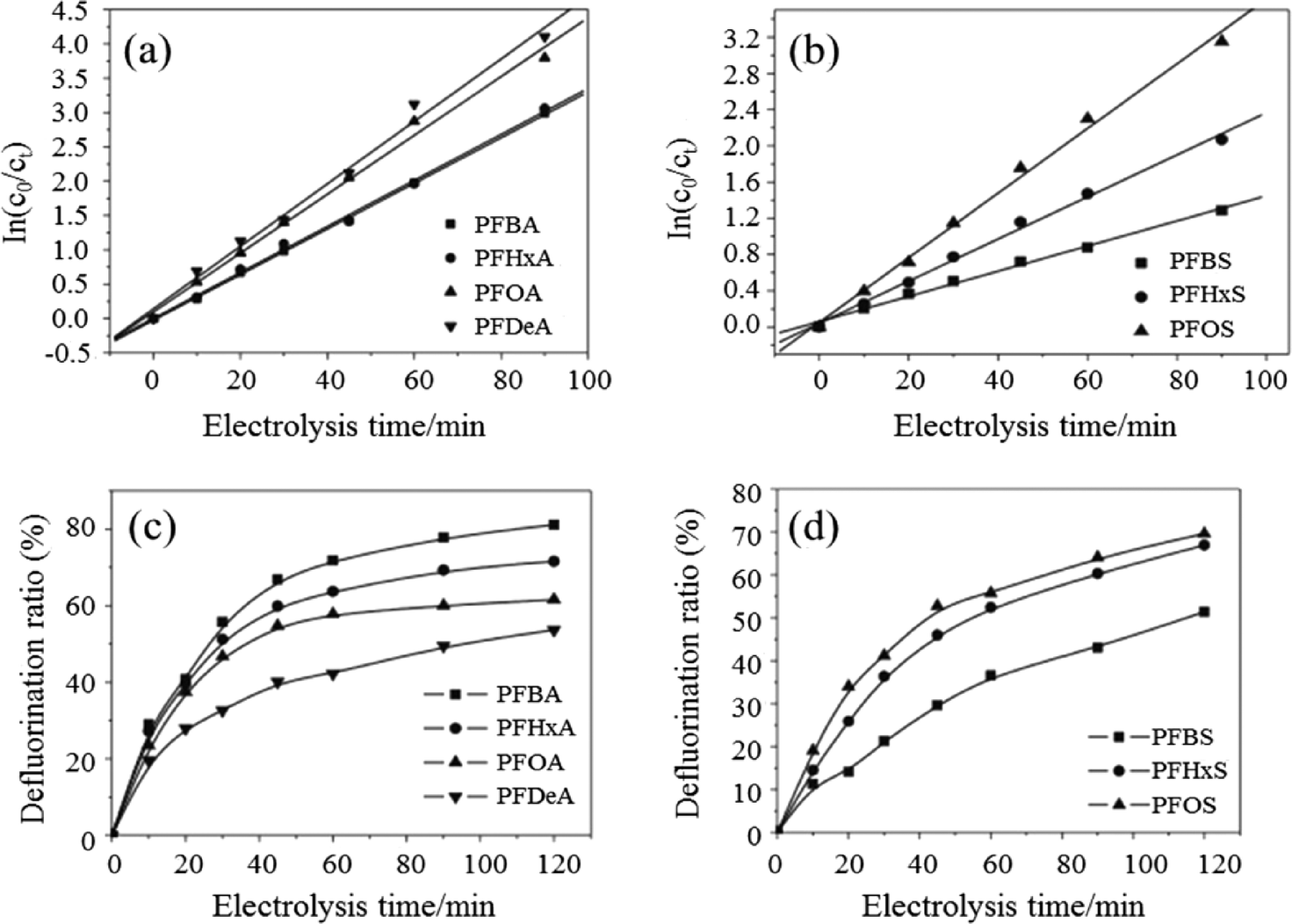 | ||
| Fig. 10 Pseudo-first-order kinetic reactions for (a) PFXA and (b) PFXS decomposition; the defluorination ratios for (c) PFXA and (d) PFXS on a BDD electrode (reaction conditions: [PFXA]0 = [PFXS]0 = 0.114 mM; i = 23.24 mA cm−2; T = 32 °C; electrolyte = 1.4 g L−1 NaClO4).155 | ||
Electrochemical oxidation of a pesticide residue 2,6-dichlorobenzamide (BAM) with Si/BDD and Ti/Pt–Ir anodes was studied to compare non-active with active anodes.149 The results showed that BDD, as a non-active anode, was more efficient than the Pt–Ir electrode, and it produced a lower amount of degradation intermediates due to the non-selective nature of the hydroxyl radicals formed on the anode. The initial degradation pathway was found to be different for the two cells, where the BDD electrode gave rise to both a cathodic and an anodic pathway, compared to the Pt–Ir cell which only had an anodic pathway.
Electrochemical degradation of bisphenol A (BPA) was examined on four different anode materials: Ti/BDD, Ti/Sb–SnO2, Ti/RuO2 and Pt. BPA was readily destroyed on the Ti/Sb–SnO2 and Ti/BDD anodes, while the Pt anode had a moderate ability to remove BPA and the Ti/RuO2 anode did not effectively oxidise BPA. Compared to the Pt and Ti/RuO2 anodes, the Ti/Sb–SnO2 and Ti/BDD anodes were found to have higher oxygen evolution potentials and higher anodic potentials for BPA electrolysis at the same current densities. In comparison to the Ti/Sb–SnO2 anode, the Ti/BDD anode with high durability and good reactivity for organic oxidation appeared to be the most promising for the effective EC treatment of BPA and similar endocrine disrupting chemical (EDC) pollutants.113
Electrochemical oxidation of ibuprofen (Ibu) was examined using Ti/Pt/PbO2 and BDD electrodes in a batch cell at different current densities (10, 20 and 30 mA cm−2) in a Na2SO4 electrolyte. Very good degradation of Ibu was achieved, with COD removal between 60 and 95%, and TOC removal from 48 to 92%, in 6 h experiments, with higher rates obtained with the BDD electrode. The combustion efficiency (ηC), which can be estimated from the rate of decrease of TOC compared to that of COD, indicated slightly higher removal with the BDD at lower current densities, with 100% removal for both types of anodes at 30 mA cm−2.90
BDD and Ti/IrO2–RuO2 electrodes were compared to test their effectiveness for electrochemical oxidation of an azo dye (Reactive Red 120) in acidic media (1 M HClO4).150 Ti/IrO2–RuO2 exhibited a low oxidation power with high selectivity to organic intermediates and low TOC removal (10% at 25 °C and 40% at 80 °C), while the use of the BDD electrodes induced total mineralization to CO2. In both cases, the decoloration of the solution was rapid, but very rapid, nearly 100% removal was achieved with the BDD (2 A h L−1) compared to a slower rate with Ti/IrO2–RuO2 (25 A h L−1). The effectiveness of these materials was examined based on the instantaneous current efficiency (ICE) (%), which was determined from COD measurements using the following equation:
| ICE =(CODt −CODt+Δt)FV/8IΔt | (1) |
485 C mol−1), V is the electrolyte volume (dm3), and 8 is the oxygen equivalent mass (geq.−1). The ICE was up to 0.13 in the case of Ti/IrO2–RuO2, and up to 0.45 for the BDD.
The electrochemical mineralization of environmentally persistent long-chain perfluorinated carboxylic acids (PFCAs), perfluorononanoic acid (C8F17COOH, PFNA) and perfluorodecanoic acid (C9F19COOH, PFDA). was investigated over Ti/SnO2–Sb–Ce (SnO2), Ti/SnO2–Sb/Ce–PbO2 (PbO2), and Ti/BDD (BDD) anodes. The energy consumption was calculated based on the electrical efficiency per log order reduction (EE/O) in the electrochemical oxidation process, as follows:
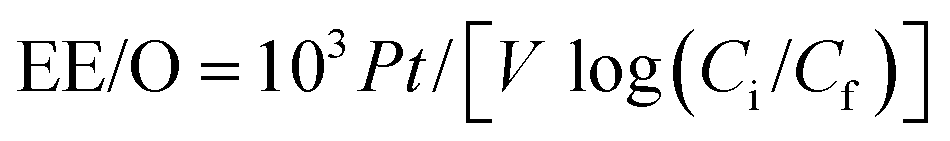 | (2) |
For PFNA, EE/O was 54 W h L−1 for SnO2 and 72 W h L−1 for the PbO2 electrodes. The lowest EE/O value of 42 W h L−1 was achieved with the BDD electrode. The EE/O values for PFDA were EE/O(SnO2) = 1.4 × EE/O(PbO2) and 1.9 × EE/O(BDD).93
The anodic oxidation of methamidophos (MMD), a highly toxic pesticide used worldwide, was studied in a sodium sulfate aqueous solution using Pb/PbO2, Ti/SnO2, or Si/BDD electrodes at 30 °C. Under galvanostatic conditions, it was observed that the performance of the electrode material was influenced by pH and current density, and the MMD degradation using Pb/PbO2 in acidic media (pH 2.0 and 5.6) generated formaldehyde as the main product. Under the same conditions, Ti/SnO2 had low formaldehyde production compared to the Pb/PbO2 electrode, while the Si/BDD electrodes did not show any formaldehyde production. The ATR-FTIR characteristics of MMD in crystalline form and in aqueous solution were established, which showed the formation of phosphate as the reaction progressed, suggesting complete MMD mineralization using the Si/BDD electrode.169
Diamond films are usually deposited on a titanium or silicon substrate. A diamond layer was deposited onto a 3-D porous Pt nano-sheet perpendicular to the BDD hybrid film using a simple and facile double template method. Physical and electrochemical results indicated that the 3-D porous Pt/BDD/Si electrode had a high catalytic ability and was resistant to poisoning for methanol electro-oxidation, because of the larger electrochemically active area and porous structure and the activity of the BDD substrate.170
BDD electrodes have been fabricated using numerous other materials. Although high-quality BDD films can be deposited on silicon, tantalum, niobium and tungsten, they are unsuitable for application in wastewater treatment because of their poor mechanical strength, the low conductivity of Si, and the high cost of Ta, Nb and W. Considering the tradeoffs in performance and cost, BDD films synthesized on a titanium substrate are preferable due to their good conductivity, high strength, low price, high anticorrosion, and quick repassivation behavior. However, the poor adhesion strength of diamond with Ti has been a problem in realizing high-performance BDD/Ti electrodes. For example, it was observed that an extreme difference in temperature during substrate cooling (from 850 °C to ambient temperatures) resulted in a large thermal residual stress on the formation of a TiC interlayer, reducing diamond film adhesion to the substrate, and a short service life of the Ti/BDD electrode.171 For this reason, various methods have been developed to improve adhesion, such as fabrication by the microwave plasma-enhanced chemical vapor deposition (MPCVD) method. A sand-blasted treaded substrate and the introduced buffer layer were favorable for producing electrodes with improved adhesion and good electrochemical properties.172
The electrochemical degradation of amaranth, a type of azo dye, using an activated carbon fiber (ACF) electrode was investigated under potentiostatic or galvanostatic conditions.173,174 With either approach, three different decolorization processes occurred: adsorption, electroreduction, and electrooxidation. The adsorption was insignificant for the removal of color, COD and TOC. The electrooxidation and electroreduction benefitted color, COD and TOC removal, and electroreduction was more effective than electrooxidation.
Another azo dye, alizarin red S (ARS), was electrochemically oxidized using activated carbon fiber (ACF) felt as an anode. The initial pH, current density and the type of supporting electrolyte all played an important role in ARS degradation. The large specific surface area and higher mesopore percentage of ACF anodes provided effective electrochemical degradation of the dye, as shown by an increase in color removal efficiency from 54 to 84% as the specific surface area of the ACF anodes was increased from 894 to 1682 m2 g−1.175
ACF electrodes have been tested with other materials. For example, they were used with TiO2 (TiO2/ACF), with the electrode prepared using a simple and inexpensive sol–gel-adsorption method.176 Tests using phenol and other organic pollutants showed that intermediates were always produced, demonstrating that the adsorbed ˙OH generated on the TiO2/ACF–graphite anode was the most active species in the electrochemical oxidation system.
The use of an electrospun ACF electrode modified with CNTs (e-CNT/ACF) was examined for the electrochemical degradation of the dye, methyl orange (MO).178 Results showed that the CNTs in the web-like e-CNT/ACF composites helped to improve the pore distribution and conductivity of the composite electrode, resulting in ∼90% degradation of MO in 60 min, which was much higher than that obtained using only the commercial woven ACF under similar conditions.
Two pieces of ACF with CNT packed evenly between them were used as the anode and cathode to treat wastewater contaminated with a dye (X-3B) in a system called a seepage carbon nanotube (SCNT) reactor.179 This reactor was designed to facilitate contaminant mass transfer from bulk solution to the electrode surface, in order to improve electrochemical wastewater treatment rates. Comparison of color and COD removals showed that the SCNT electrode reactor removed total color by 94.4% and COD by 57.6% in 90 min levels of treatment were much higher than 32.8% (color) and 28.0% (COD) obtained using conventional electrochemical reactors (with ACF-CNT electrodes positioned vertically at the center of the same reactor). The research also showed that the current efficiency of the SCNT reactor was 340% higher than that of conventional reactors, and the energy consumption to mineralize the same amount of organics was only 16.5% of that for conventional reactors.
A novel poly(aniline-co-o-aminophenol) (PAOA)-modified carbon felt electrode reactor was designed and investigated for fluoride removal from aqueous solutions. This reactor design was innovative because it operated under a wider pH range due to the coating of the electrode with a copolymer PAOA ion exchange film. Contaminant mass transfer from bulk solution to the electrode surface was enhanced by the use of porous carbon felt as an electron-conducting carrier material compared to other reactors.180 The electro-oxidation of Ce(III) on a carbon felt anode that proceeded with a high current efficiency was studied which showed that at a current of 2 A, oxidation of cerium had a current efficiency of 92% with the majority of Ce (>80%) oxidized to Ce(IV) within 40 min.181
Carbon nanotubes (CNTs) have been used as carbon-based electrodes for chemical degradation or pollutant removal. The use of a carbon nanotube filter for electrochemical water treatment was investigated by Vecitis and co-workers.182–185 They found enhanced performance of the three-dimensional electrodes due to the high electrode surface area and porosity, and therefore an increased number of electrochemically active surface sites.
The primary passivation mechanisms and electrode regeneration methodologies of electrochemical filtration with three-dimensional carbon nanotube (CNT) networks were investigated using phenol. Polymerization of phenol on the CNT surface resulted in a reduction of current and electrochemical performance. Polymerization therefore needs to be prevented to stabilize the performance of these electrodes, or the electrodes must be cleaned. Calcination and redispersion in HCl (pH = 1.7), toluene, and hexanes are effective for removal (>97%) of the passivating electropolymer coating. However, prevention is better than post-treatment for dealing with passivation of CNT, and conducting electrochemical filtration at a higher potential could be useful in avoiding the generation of polymers rather than trying to regenerate the electrodes after passivation.
In all cases with these sp2-hybridized carbonaceous anode materials, however, electro-oxidation is generally accompanied by surface corrosion. Thus, the application of three-dimensional electrodes will require further investigation into the electrode passivation mechanisms, electrode regeneration techniques, and passivation prevention methods.185
2.3 Indirect electro-oxidation via in situ generated chemical oxidants
To avoid the deactivation of the anode during direct oxidation of chemicals, an alternative approach is indirect oxidation by destroying pollutants through the electrochemical generation of chemical reactants such as active chlorine, ozone, persulphate, and hydrogen peroxide.| 2Cl → Cl2 + 2e− | (3) |
| Cl2 + H2O → HClO + H+ + Cl− | (4) |
| HClO ↔ ClO− + H+ | (5) |
In the presence of active chlorine, oxygen transfer can be carried out by the adsorbed oxychloro species, which are considered intermediates of the chlorine evolution reaction as shown in Fig. 11.188
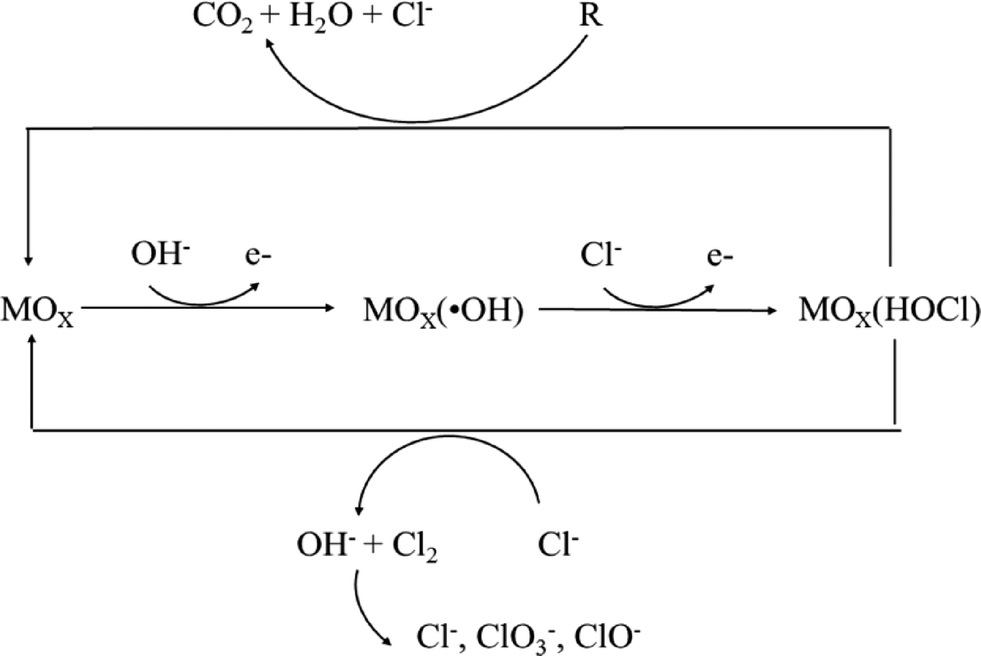 | ||
| Fig. 11 Reaction scheme of chlorine-mediated electrochemical oxidation of organics. | ||
The predominance of different chemical species in active chlorine is well known to be a function of pH. Cl2 is the predominant stable species at pH <3; when the pH is between 5 and 6, active chlorine exists as hypochlorous acid (HClO) and hypochlorous anions (OCl−) are present at pH ≥6; at higher pH values (>7.5), hypochlorous anions (OCl−) are the predominant species. HClO is the most powerful oxidant among the active chlorine species for oxidation of organics, and thus reactions are best conducted in acidic rather than alkaline media.
The choice of the anode material utilized is important for in situ generation of active chlorine, and DSA-type anodes coated with a layer of RuO2 or IrO2 are particularly effective due to their good electrocatalytic properties for chlorine evolution, as well as their long-term mechanical and chemical stability.194–197
Other non-active electrodes, including BDD, SnO2 and PbO2, are not useful for this approach under some conditions because the high anodic potential typical of these electrodes can lead to oxidation of Cl− to form chlorate (ClO3−) and perchlorate (ClO4−), which do not have any oxidizing capacity.198 For example, Palmas et al. showed that chlorates and perchlorates could easily be formed from further oxidation of hypochlorite on BDD anodes.199 Similar results were reported by Lacasa et al. who also showed that the anode material significantly influenced the speciation of chlorine, with the formation of perchlorate obtained using BDD electrodes.200 Thus, these electrodes are not useful for treatment of drinking water due to the hazardous risks associated with the formation of these chemical species. Chlorate and perchlorate formation is minimal on DSA-type and Pt electrodes.201,202
Active chlorine production is suitable for the treatment of some types of wastewaters, such as olive oil, textile, and tannery effluents.203–229 Due to an abundance of chloride in these wastewaters, there is usually no need for addition of chloride salts for effective treatment. For example, Turro et al. investigated the behavior of a Ti/IrO2–RuO2 anode for the electrochemical oxidation of landfill leachate under different concentrations of the supporting electrolyte. They found that the addition of 20 mM NaCl gave results similar to those with no salt addition. In this case, the leachate contained 175 mM chlorides, and therefore increasing the Cl− concentration by ∼10% had a marginal effect on indirect oxidation.230
Under active chlorine mediation, the risk of formation of chlorinated organic compounds during electrolysis results has increased wastewater toxicity, which ultimately could limit the wide application of this approach to wastewater treatment. For example, analysis of the reaction products during the oxidation of phenol in the presence of NaCl with DSA-type Ti/SnO2 and Ti/IrO2 anodes showed that organochlorinated intermediates were formed. Although these compounds were then mineralized to CO2 or oxidized to volatile chlorinated compounds (i.e., chloroform), the toxicity of the solution remained above desirable limits.187 In another study, increasing the concentration of NaCl from 20 mM to 100 mM resulted in higher COD removal, but the enhanced production of organochlorinated compounds resulted in a solution with high ecotoxicity.229 Future work using saline solutions must therefore focus on developing both effective electrodes and experimental conditions which do not result in the formation of organochlorinated intermediates that will limit the overall reduction in toxic chemical species in water.
| Fe2+ + H2O2 → Fe3+ + ˙OH + OH− | (6) |
Electro-Fenton methods have become very attractive because of the much higher degradation rates of organic pollutants than those using traditional Fenton approaches due to the continuous regeneration of Fe2+ at the cathode via:
| Fe3+ + e− → Fe2+ | (7) |
This electrochemical approach thus avoids Fe3+ accumulation in the medium, thereby eliminating the production of iron sludge. The electro-Fenton process has been successfully used to treat non-biodegradable or refractory organic compounds such as phenolic compounds,240–246 dyes,247–249 pesticides and herbicides,250–254 leachates,255–257 drugs,258–260 and reverse osmosis concentrates.261
BDD electrodes are particularly effective for the electro-Fenton process. The mineralization process and decay kinetics of atrazine and cyanuric acid were examined by means of an electro-Fenton process with Pt or BDD anodes using an undivided cell with a carbon-felt cathode under galvanostatic conditions. The electro-Fenton process was more effective with the BDD for the degradation of both compounds. There was nearly total mineralization of atrazine based on 97% total organic carbon (TOC) removal. Efficient removal was due to rapid oxidation by ˙OH formed at the BDD compared to those in the bulk solution in a conventional Fenton reaction. Cyanuric acid was more slowly mineralized, mainly via ˙OH produced at the BDD surface, but it was not degraded using a Pt anode. These results highlight that electrochemical processes using a BDD anode are more powerful than the classical electro-Fenton process with Pt or PbO2 anodes.262
The main limitation of Fenton-based approaches is the requirement of low pH conditions. Fe3+ precipitation from solution at pH ≥3.5 can lead to the termination of this reaction. The electro-Fenton system also requires a low solution pH, typically within the pH range 2–4, and consequently, it is impractical if large amounts of chemicals are needed to acidify the water prior to treatment. Therefore, the inherent drawback of the need for a low pH limits the wide application of Fenton-based approaches, although they are useful when the solutions are initially quite acidic.
One approach proposed to overcome this low pH requirement was an electro-Fenton-like (EFL) system that used a Keggin-type iron-substituted heteropolytungstate anion PW11O39Fe(III)(H2O)4− to substitute for Fe3+ in the conventional electro-Fenton system. Experimental results using 0.1 M dimethyl phthalate (DMP) showed complete degradation in <80 min at pH = 6.86 at a potential of −0.5 V, with aeration using an O2 flow rate of 60 mL min−1. A TOC removal of 56% was achieved within 120 min. A comparison with a conventional electro-Fenton oxidation treatment of DMB showed that this approach had a higher efficiency of chemical degradation, suggesting its potential use in treatment of water and wastewater with a more neutral pH.263
Further development of electro-Fenton methods is proceeding based on coupling of the process with other AOPs with the aim of obtaining a synergetic effect for water and wastewater treatment. The most developed integrated process is the photoelectro-Fenton approach, where the contaminated solution treated under electro-Fenton conditions is exposed to UV illumination favouring the generation of homogeneous ˙OH and the photodegradation of complexes of Fe(III) with organics:238
| Fe(OH)2 + Fe2+ + ˙OH | (8) |
The mineralization of flumequine, an antimicrobial agent belonging to the first generation of synthetic fluoroquinolones which is detected in natural waters, was studied by using the electro-Fenton and photoelectro-Fenton approaches with UVA light. The photoelectro-Fenton process was more powerful, resulting in almost total mineralization with 94–96% total organic carbon removal.264
Solar photoelectro-Fenton processes (SPEF) are also being investigated, where the electrical energy needed by the electro-Fenton reactor is produced from solar energy.
The degradation of the industrial textile dye Disperse Blue 3, examined using the solar photoelectro-Fenton process, showed a positive effect of sunlight and Fe2+, with complete decolorization and mineralization of solutions in relatively short time periods.265 Decolorization and mineralization of another dye, Sunset Yellow FCF (SY) azo dye, were examined by using several processes: anodic oxidation coupled with electrogenerated H2O2 (AO-H2O2), electro-Fenton, UVA-illuminated photoelectro-Fenton, and SPEF.266 The results showed that the most powerful method was solar photoelectro-Fenton, achieving almost total mineralization and higher degradation compared to UVA-illuminated photoelectro-Fenton due to the higher UV intensity of sunlight, which quickly photolyzed Fe(III)–carboxylate complexes that could not be destroyed by ˙OH in traditional electro-Fenton processes.
The mineralization of the antibiotic chloramphenicol in a synthetic sulfate solution at pH = 3 was studied by anodic oxidation with electrogenerated H2O2 (AO-H2O2), electro-Fenton, UVA photoelectro-Fenton, and SPEF processes. The results demonstrated that SPEF with a BDD anode was the best method among these approaches examined for chemical mineralization.267
Another integrated process is the sonoelectro-Fenton approach, where the solution in the electrochemical reactor is simultaneously irradiated with ultrasound.268 Compared to the conventional electro-Fenton approach, a significant synergetic effect was obtained by this coupled reaction due to the additional effect of enhancement of the mass transfer rate to the electrode. For example, during the sonoelectrolytic process, despite the existence of some degassing, the high yield of hydrogen peroxide produced at the anode significantly enhanced the rate of mass transfer of oxygen toward the cathode due to the sonication.268
Another electro-Fenton-based approach, called the peroxi-electrocoagulation method, was successfully used to degrade chlorophenoxy and chlorobenzoic herbicides, and 2,4,5-trichlorophenoxyacetic acid. In this process, the soluble Fe2+ ions that react with cathodically generated H2O2 are continuously supplied to the solution from the sacrificial oxidation of an iron anode. Fenton's reaction occurs with this iron and produces Fe3+ ions, inducing coagulation. Thus, the pollutants are removed by the combined action of degradation by ˙OH generated by Fenton's reaction and their coagulation with Fe(OH)3 precipitates formed from the anodic corrosion.269 Treatment of sodium dodecyl sulfate (SDS) surfactant wastewater by the peroxi-electrocoagulation process showed that SDS in the aqueous phase was effectively removed with a mean energy consumption of 1.63 kW h kgSDS−1, with the SDS removal efficiency reaching 82%.270 The removal of phenol from water using this peroxi-electrocoagulation method was examined using a mild steel anode and a graphite cathode in a pilot-scale reactor. Ferric hydroxide and hydroxyl radicals generated in the cell removed phenol from the water, making it drinkable, demonstrating the feasibility of the process.271
3. Electrochemical reduction
3.1 Electrodeposition
Aqueous effluents containing metal contaminants from some electrochemical industries, such as the metallurgical and electroplating industries, printed circuit boards, and battery manufacturing require special treatment to remove toxic metal ions or recycling of valuable materials. The electrochemical recovery of metals from water has been practiced in the form of electrometallurgy for a long time.272 The first recorded example was in the mid-17th century in Europe which involved the electrochemical recovery of copper from cupriferous mine waters.273The electrochemical mechanism for metal recovery is based on cathodic deposition, which provides an efficient way to remove heavy/toxic metals or recover precious metals from water and wastewater without leaving any residues during metal separation.274
One example of the selective electrochemical removal of metals is the recovery of gold-rich alloys using a filter-press-type electrochemical flow reactor with highly polished vitreous carbon (VC) and titanium (Ti) flat cathodes.275 Vitreous carbon and titanium were determined to be outstanding cathode materials for gold recovery from gold plating wastewater. Both materials are easy to polish and chemically resistant to a wide range of chemicals. Cathodic efficiencies were higher on the Ti cathode in which 23% was observed for a gold-rich alloy recovered at 1.0 V, and 15% when only gold was considered (Table 4).
| Cathode | VC | Ti | ||
|---|---|---|---|---|
| E vs. SCE (V) | Alloy/% | Au/% | Alloy/% | Au/% |
| −1.0 | — | — | 23 | 15 |
| −1.1 | 11 | 5 | 19 | 11 |
| −1.2 | 11 | 5 | 17 | 8 |
| −1.4 | 18 | 9 | — | — |
| −1.5 | 21 | 7 | — | — |
| −1.6 | 16 | 5 | — | — |
Metals can also be recovered from aqueous solutions containing chelating agents such as EDTA, nitrilotriacetic acid and citrate. Using a two-chamber cell containing a commercial cation exchange membrane, a minimum of 40% of the metal was recovered with the greatest amount of 90% obtained for copper.276 Electrodeposition can be integrated with ultrasound to increase metal recovery. It was found that copper removal increased to 95.6%, and EDTA was also removed (84% COD removal) from the wastewater.277
The fluid mechanical environment is also important in reactor efficiency. A hydrodynamic study of a bench-scale electrochemical reactor using parallel plates with an inert fluidized bed (glass beads) showed the importance of fluid motion on the removal of cadmium and lead ions from an aqueous synthetic wastewater (Fig. 12).278
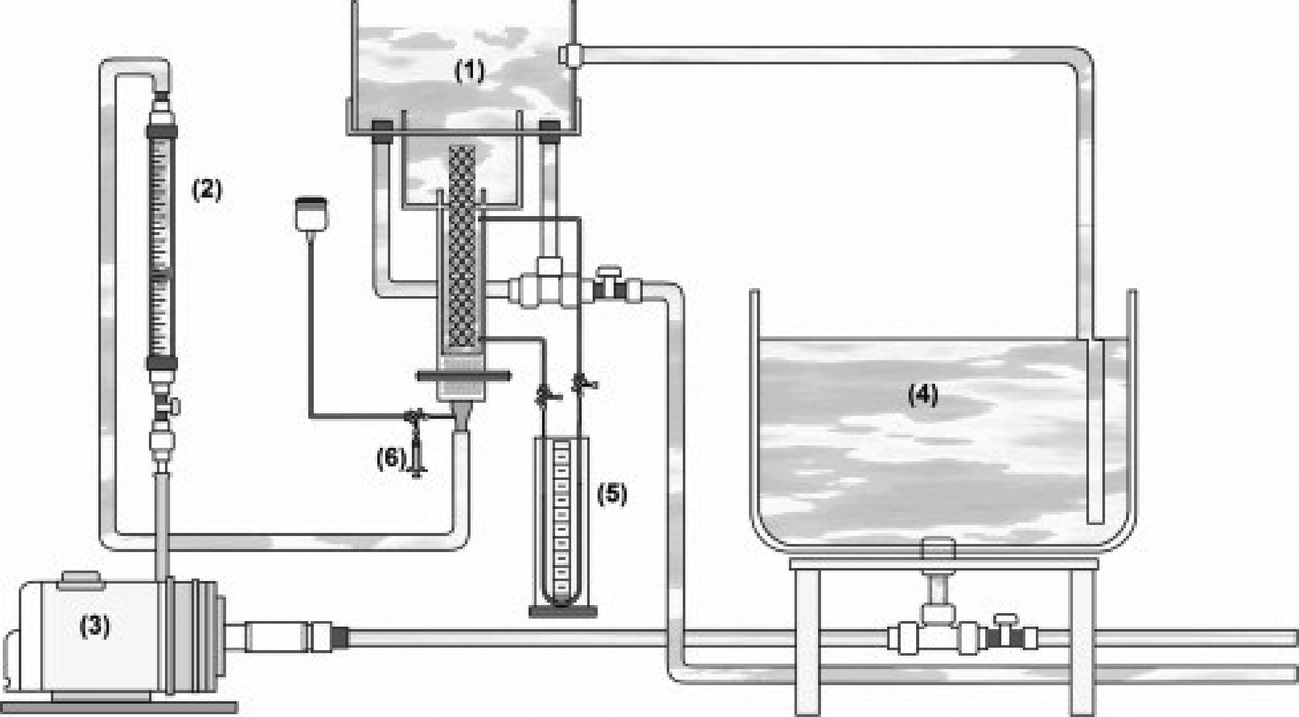 | ||
| Fig. 12 Schematic representation of the complete apparatus developed and employed in the experimental procedure: (1) electrochemical reactor; (2) hydraulic system; (3) centrifuge pump; (4) Łrecirculating batch flow system; (5) pressure gauge; (6) a dispositive to facilitate the injection of a tracer substance.278 | ||
An electrochemical reactor with a rotating cylinder electrode (RCE) and a pH controller were utilized to optimize the electrochemical recovery of nickel from a synthetic nickel plating wastewater. Control of the pH to ∼4 was crucial for recovering high-purity nickel, while preventing the precipitation of hydroxides and oxides (Fig. 13).279
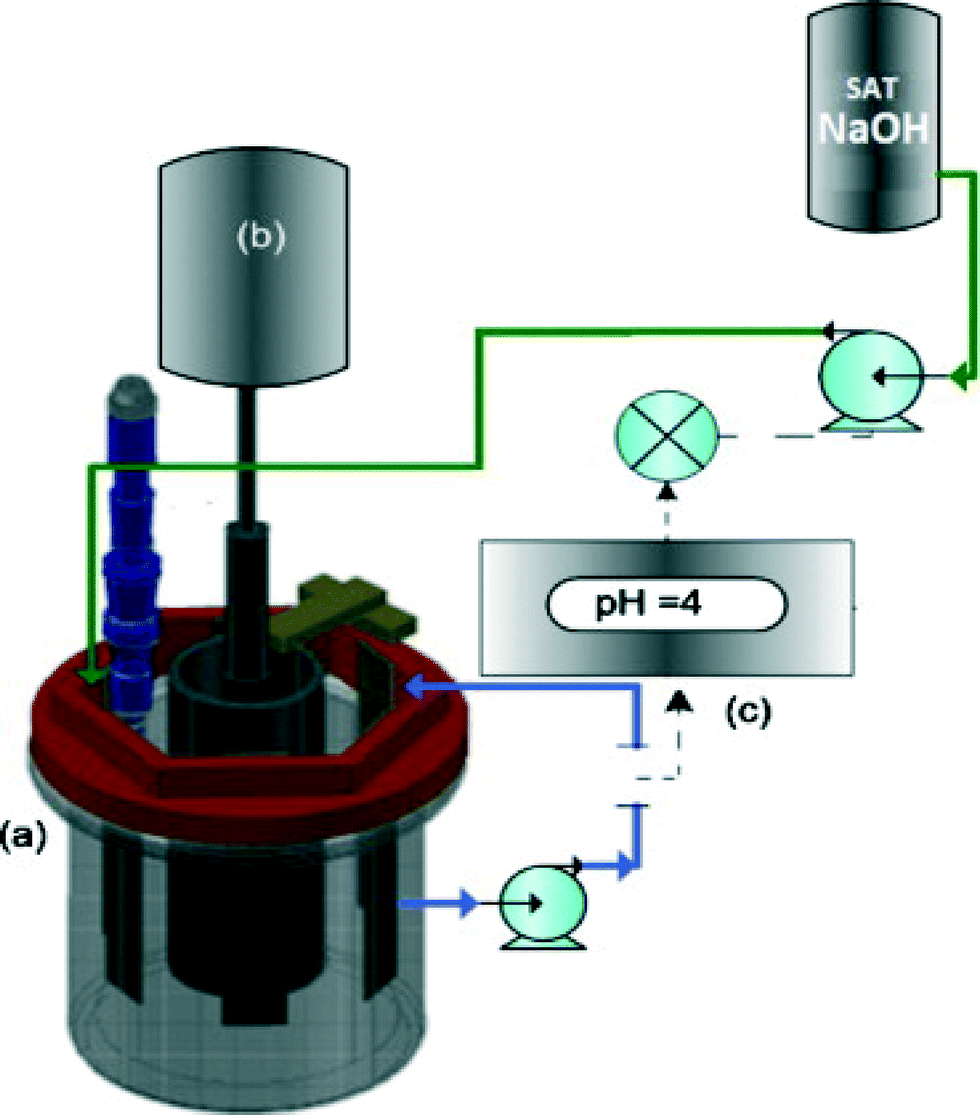 | ||
| Fig. 13 Sketch of the electrochemical reactor with a rotating cylinder electrode utilized to perform the electrochemical recovery of nickel: (a) rotating cylinder electrode, (b) rotating motor, (c) pH control system.279 | ||
3.2 Cathodic electrochemical dechlorination
Chlorinated organic compounds (COCs) are prevalent in many wastewaters, particularly those from industries that use solvents, as well as chemical industries that produce herbicides, fungicides, and pesticides. The presence of these COCs in the environment poses threats to human health due to their toxicity and high stability. Many methods have been used for cleaning wastewaters containing COCs. Compared to conventional physico-chemical, biological and chemical dehalogenation techniques, electrochemical reductive dechlorination has emerged as an attractive technique to destroy COCs due to its mild reaction conditions and the avoidance of possible secondary pollutants.280–285Another electrode widely used for the dechlorination of COCs is a palladium-based electrode.289–304 The use of a Pd cathode leads to the preferential production of totally saturated products, as a consequence of their catalytic activity for hydrogen evolution and hydrogenation reactions. The ability of palladium to adsorb hydrogen helps to promote indirect hydrodehalogenation.305
Nanostructured Pd thin films prepared by a cyclic voltammetric deposition method were shown to be effective in the electrochemical reductive dechlorination of carbon tetrachloride (CT). Electrochemical characterization and CT removal using gas chromatography showed that adsorbed hydrogen was important for removing CT from acidic solutions through a surface reaction with chemisorbed CT molecules, providing a good mechanistic reason for the efficiency of electrochemical dechlorination.312
Electrochemical reduction of several polychloroethanes has been investigated using electrodes containing different transition metals, including PCEs (1,1-dichloroethane (1,1-DCA), 1,2-dichloroethane (1,2-DCA), 1,1,1-trichloroethane (1,1,1-TCA), 1,1,2-trichloroethane (1,1,2-TCA), 1,1,1,2-tetrachloroethane (1,1,1,2-TeCA), 1,1,2,2-tetrachloroethane (1,1,2,2-TeCA), hexachloroethane (HCA), 1,1-dichloroethylene (1,1-DCE), 1,2-dichloroethylene (1,2-DCE), tetrachloroethylene (TCE) and chloroethylene (CE)). The electrocatalytic activity of PCE was found to be affected by the specific structure, with the efficiency of the electrocatalytic degradation increasing with the number of Cl atoms bound to the same carbon center. The number of Cl atoms bound to a second C atom had different effects: there was an activation enhancement due to polar effects. For the most active electrodes examined, the order of increasing electrocatalytic reactivity was: 1,1-DCA < 1,1,1-TCA < 1,2-DCA < HCA < 1,1,2,2-TeCA < 1,1,2-TCA < 1,1,1,2-TeCA. Two distinct reduction mechanisms for the reductive cleavage of PCEs were observed. Geminal PCEs showed sequential hydrodechlorination where one chlorine atom was lost in each reduction step, until completely dechlorinated ethane was obtained. Alternatively, reduction of vicinal PCEs involved removal of two chlorine atoms in an overall 2e− process resulting in the formation of the corresponding (chloro)ethylene, which could be further reduced by using more negative potentials.301
The size of the metal particles on the electrode is important. Electrospun polyacrylic acid (PAA)/polyvinyl alcohol (PVA) polymer nanofibers were used to immobilize Fe/Pd bimetallic nanoparticles to treat trichloroethylene (TCE)-contaminated groundwater. This bi-metal nanostructure that was immobilized within the polymer nanofibers was much more effective in remediation of TCE, especially at high initial concentrations, than colloidal-sized Fe/Pd nanoparticles.313
Other materials have been used for successful electrochemical dechlorination of chemicals. TCE degradation was investigated in a recirculated solution of an electrolysis system containing a cast iron anode and a copper foam cathode. The cast iron anode generated a reducing electrolyte that prevented the electron and proton combination with dissolved oxygen, thus the reduction of TCE on the copper foam cathode was enhanced. The conductivity of the electrolyte was an important factor for both the final elimination efficiency (FEE) of TCE and specific energy consumption. Under optimal conditions, FEE reached up to 98%, at an energy consumption of 6.49 kW h kg−1. This electrolysis system was proposed to remediate groundwater contaminated by chlorinated organic solvents or wastewater contaminated with chlorinated compounds.314
Several types of electrodes have been examined for degradation of 2,4-dichlorophenoxyacetic acid (2,4-D). Palladium/nickel foam (Pd/Ni foam) electrodes resulted in 87% removal of 2,4-D within 4 h, with generation of phenoxyacetic acid (PA), o-chlorophenoxyacetic acid (o-CPA) and p-chlorophenoxyacetic acid (p-CPA) as intermediates. The palladium loading and the NaCl concentration impacted the dechlorination kinetics of 2,4-D.300 With Pd/PPy–SLS/foam–Ni electrodes, 2,4-D was completely dechlorinated. The stability of the electrode was good as the dechlorination efficiency was maintained at 100% even after being reused 8 times.321
Roughened silver–palladium cathodes (Pd/Ag(r)) have also been tested for electrocatalytic hydrogenolysis (ECH) dechlorination of 2,4-D in an aqueous solution.321 The mechanism of ECH dechlorination of 2,4-D on the Pd/Ag(r) cathode can be described as occurring in several steps. First, 2,4-D is adsorbed to Ag followed by the generation of chemisorbed hydrogen on Pd. Hydrogenolysis of 2,4-D then proceeds as in catalytic hydrogenation by the reaction of the adsorbed 2,4-D with chemisorbed hydrogen, followed by desorption of the product. The effects of OH−, Cl−, and ethanol were discussed, and the results showed that Cl− was detrimental to ECH dechlorination, whereas OH− was able to promote the dechlorination of 2,4-D. Ethanol deactivated the Pd/Ag(r) cathode if an aqueous alkaline solution was used for the reaction. Under optimal conditions, 25 mM 2,4-D was selectively dechlorinated to phenoxyacetic acid with 85% removal at a CE of 66% at 298 K after 6 h. The only products generated besides phenoxyacetic acid were 2-chlorophenoxyacetic acid and 4-chlorophenoxyacetic acid.
A multifunctional Pd/C gas-diffusion cathode was examined for phenol degradation in successive environments of hydrogen gas and then air. Hydrogen gas was initially fed for 5 min at a rate of 25 mL s−1 to reach saturation. Hydrogen gas was then fed into the gas compartment for the first 60 min (electrolysis time) of the experiment, followed by air. The electrochemical reduction and oxidation was examined using three different chlorinated phenols [4-chlorophenol (4-CP), 2,4-dichlorophenol (2,4-DCP) and pentachlorophenol (PCP)].322 The Pd/C gas-diffusion cathode reductively dechlorinated the phenols when hydrogen gas was present, and then it accelerated the two-electron reduction of O2 to H2O2 by feeding air for oxidation of the chemicals. The combined process of reduction and oxidation improved the chlorinated phenol degradation efficiency with the removal efficiency of chlorinated phenols reaching nearly 100%. The degradation sequence was the best for 4-CP, followed by 2,4-DCP, and then PCP.
1,2,3-Trichlorobenzene (1,2,3-TCB) was used as a model for the degradation of a persistent organic pollutant (POP) under an inert gas atmosphere, as summarized in Table 5.323 Trace amounts of dichlorobenzene were observed with different amounts depending on the electrode material. Electrodes with Ru and Pd were selective mainly for meta-position dechlorination, while those with Pt groups were selective mainly for ortho-position (o-position) dechlorination. A PdO sintered electrode had an especially high selectivity for meta-position (m-position) dechlorination.
| Cathode electrode | Dechlorination rate (%) |
|---|---|
| Sintered RuO2 (major)/Pt/PdO | 91 |
| Pt (major)/IrO2/RuO2 | 81 |
| Sintered RuO2 | 59 |
| Sintered PdO | 96 |
| Sintered Pt | 53 |
| Sintered PdO/Pt | 97 |
| Sintered Pd/Pt | 82 |
| Plain Pd plate | 70 |
The electrochemical dechlorination of chlorobenzenes was studied in the presence of various arene mediators such as naphthalene, biphenyl, phenanthrene, anthracene, and pyrene.324 As the amount of naphthalene was reduced to 0.01 equivalents, there was complete dechlorination of monochlorobenzene with 77 × 104 C mol−2 of electric quantity. This same amount of total charge was then used in the presence of four different types of arene mediators: biphenyl, phenanthrene, anthracene, and pyrene. Complete dechlorination was achieved with all mediators except anthracene. Similar results were also obtained when this reaction was applied to 1,3-di- and 1,2,4-trichlorobenzene, with phenanthrene appearing to be the most effective mediator among those examined.
3.3 Cathodic electrochemical denitrification
Electrochemical reduction of nitrate and nitrite ions has gained more attention in the past several years,325–329 particularly for the treatment of nitrate-containing ground waters. As for many other chemicals, nitrate reduction products depend on the nature of the electrode surface,330–334 making this reaction very interesting from a mechanistic point of view.Electrochemical nitrate reduction was investigated on coinage (copper, silver and gold) and transition-metal electrodes (platinum, palladium, rhodium, ruthenium, iridium) at 0.1 M nitrate ions in acid solutions.333 The conclusion was that the rate-determining step on most of these electrodes was the reduction of nitrate to nitrite, based on the values of the Tafel slope, the kinetic order, and the effect of co-adsorbing anions. Cyclic voltammetry showed that current densities at given applied voltages for nitrate reduction depended strongly on the nature of the electrode. The activities for the electroreduction of nitrate decreased in the order Rh > Ru > Ir > Pd and Pt for the transition-metal electrodes, and in the order Cu > Ag > Au for the coinage metals. On-line mass spectrometry measurements for Pt and Rh showed no formation of gaseous products such as nitric oxide (NO), nitrous oxide (N2O) or nitrogen (N2), suggesting that ammonia and hydroxylamine were the main products using transition-metal electrodes. This conclusion was in agreement with the known mechanism for NO reduction, which forms N2O or N2 only if NO is in solution. For Cu, measurements showed the production of gaseous NO, which could be explained by the weaker binding of NO to Cu as compared to transition metals.
Different cathode materials were examined for nitrate reduction in an electrocatalytic reactor consisting of a solid polymer electrolyte/Pt electrode assembly. The reactivity and selectivity of electrocatalytic reduction of NO3−/NO2− in the membrane electrode assembly (MEA) reactors were largely dependent on the metallic composition of the cathode. Pt alone was relatively inactive, but rates were significantly improved by the deposition of Ni, Cu, Ag and/or Rh onto the Pt electrode. Although the activity was enhanced mostly by Cu or Ag, their reaction mechanisms were quite different. Catalytic hydrogenation of nitrate (H2–NO3− reaction) occurred on the Cu–Pt cathode, compared to electrochemical nitrate reduction using a Ag–Pt cathode.334
These results suggest that synergistic positive electrochemical impacts can be obtained when using two or more metallic species on the surface of the working electrodes.335–338 Anastasopoulos et al. synthesized compositional gradient thin films of PdCu alloys and found that a low concentration of Pd in copper resulted in the normally irreversible copper surface redox becoming reversible. This change led to a sharp increase in the rate of both nitrate and nitrite reduction. In both cases, these effects were thought to be associated with the ability of Pd to activate water.339
Sn modification of polycrystalline palladium or platinum films on gold electrodes enhanced nitrate reduction. Modification with Sn was the key to these rates as pure Pd did not have a measurable impact on nitrate reduction. The dominant volatile products identified were N2O and smaller amounts of N2, while NH2OH was the dominant non-volatile product.340
The activity for electrocatalytic reduction of nitrate on the amorphous Pd33Ni60P7 electrode was tested with cyclic voltammetric in a neutral 0.1 mol L−1 KClO4 solution with or without KNO3. The Pd33Ni60P7 alloy enhanced nitrate reduction in comparison with the electrodeposited films of Pd, Ni and Pd–Ni, likely attributed to its amorphous structure. The reduction reaction of NO3− on the electrode was found to be a totally irreversible process.341
The use of carbon as a substrate material for the preparation of the working electrode has major advantages over other materials, due to its good conductivity, high surface area, good fluid permeability, relatively high overpotential for hydrogen evolution, and high chemical stability over a wide range of pH values. Hybrid electrode materials, with metals dispersed on activated carbon fiber (ACF) and surface-functionalized carbon nanotubes (CNTs), have therefore been pursued for nitrate reduction. Pd/Sn-modified activated carbon fiber (ACF) electrodes were fabricated by the impregnation of Pd2+ and Sn2+ ions onto ACF by Wang et al. Electrocatalytic reduction of nitrate was shown, by this Pd/Sn–ACF electrode, with the Pd/Sn(4/1)-modified ACF electrode producing the highest rates of nitrate reduction over a pH range of 5.3–7.6.342
Other approaches to enhance nitrate reduction include deposition onto traditional glassy carbon electrodes to study the reaction kinetics. A GC/MWCNT–Rh electrode was prepared by electrodepositing multi-walled carbon nanotubes modified with rhodium particles on a glassy carbon electrode. It is interesting to observe that during prolonged electrochemical reduction of nitrite, the catalytic activity of this hybrid electrode remained almost unchanged demonstrating good temporal stability. This suggests the substantial absence of poisoning due to irreversible and strong adsorption of reactants, intermediates, and/or reaction products on the active catalytic sites.343
Nitrate concentrations can also impact rates, showing that they do not proceed according to zero-order kinetics.344–349
Katsounaros et al. showed that the impact of nitrate concentration on the rate of nitrate reduction could be adequately described by a simple Langmuir–Hinshelwood kinetic model. The selectivity to nitrogen increased from 70 to 83% as the concentration of nitrate increased from 100 to 1500 mg L−1, and then it remained almost constant at higher nitrate concentrations. Ammonia exhibited the opposite trend, with a decrease from 25 to 11%. Faradaic efficiency (% FE) increased with the increase of nitrate concentration from 25% at 0.1 M to 78% at 1 M, with 95% of the nitrate reduced in both cases. At high concentrations of nitrate, hyponitrite and hydroxylamine were detected as intermediates of the reduction.350
To develop a theoretical model of the electrochemical reduction of nitrate ions, a powerful analytical method, called the Homotopy Analysis Method (HAM), was used. This approach, which provides a convenient way to control and adjust the convergence region and rate of approximation series when necessary, was used to obtain approximate analytical solutions to a nonlinear ordinary differential equation. The obtained analytical expressions of concentrations and current were found to provide satisfactory agreement with numerical solutions.351
4. Electrocoagulation/electrocoagulation–flotation
Electrocoagulation (EC) is a process which causes the in situ electrochemical production of coagulated species and metal hydroxides that destabilize and aggregate particles or precipitate and adsorb the dissolved contaminants.352–354Compared to the conventional coagulation process, the electrocoagulation process has been proven to be very effective for contaminant removal from water with two outstanding characteristics. On the one hand, it provides better removal capabilities for the same species than chemical coagulation without addition of chemicals. On the other hand, it produces less sludge, thus lowering the sludge disposal cost.355–359
In practice, an electrocoagulation (EC) process will be often followed by an electroflotation (EF) process that is a simple process where pollutants can float to the surface of a water body via tiny bubbles of hydrogen and oxygen gases generated from water electrolysis. This combined system can be considered as an electrocoagulation–flotation (ECF) process.360,361
4.1 Effects of various operating parameters on EC
It can be noticed from the literature that the efficiency of EC processes significantly depends on the operating parameters. Solution pH and current density are the variables that have been studied widely.362–365A study of the influence of the pH of waste in the coagulation with aluminum by conventional and electrochemical dosing showed that a simple change in the pH of the waste could result in a significant change in the efficiency of the coagulation process, and that if the same pH conditions were found at the end of the treatment, the efficiencies of the solution dosing and electrochemical dosing technologies were very similar.366
The effect of initial pH on the batch removal of synthetically prepared wastewater having high concentration of humic substances has been investigated by the electrocoagulation method using plate electrodes. The effects of initial pH on an electrocoagulation system may be twofold. These are distribution of the aluminum hydrolysis product, transformation of the humic substance related to the initial pH and finally the effects of the gel layer especially at high humic substance concentrations and high initial pH formed on the anode surface. They observed that the initial humic substance concentration and initial pH have great effects on the removal rate and efficiency. For example, in the range of 200–500 mg L−1, a decrease in the removal efficiency was observed due to gel layer formation on the surface of the anode. In order to prevent this gel formation, the initial pH of the wastewater was adjusted to 5.0 and high removal efficiencies were observed.367
The removal of hexavalent chromium from synthetic solutions with different pH values using the electrocoagulation method was studied. The results showed that the pH of the solution has a significant effect on the Cr(VI) removal efficiency and the maximum chromium removal efficiency was obtained at pH = 4. They further reported that the pH of the synthetic solution after the EC process increased with an increase in the electrolysis time due to the generation of OH˙ in the EC process.368
It should be noted that the selection should be fixed with respect to other operating parameters like solution pH, temperature and flow rate.
The effect of the current density and flow rate of a continuous-flow electrocoagulation–flotation reactor on the removal efficiency for direct red 81 dye was investigated by Salim Zodi et al. The results indicated that the current efficiency (Faradaic yield) was strongly dependent on flow rates and current densities. For example, the efficiency of this specific reactor produced a considerable DR 81 removal from 71.5% at 100 A m−2 to 90.2% at 200 A m−2 for the same flow rate of 10 l h−1. When the flow rate was increased to 28 l h−1, the dye removal efficiency increased from 61.5% at 100 A m−2 to 76.8% at 200 A m−2. Hence, to achieve higher DR 81 removal efficiency, the current density needs to be higher when the flow rate is increased.370
It is a known fact that the operating current density in ECF processes directly determines the coagulant dosage and the rate of bubble generation, which influence both mixing and mass transfer in the reactor.
For example, the work by Holt et al. has reported that at low operating currents in which settling dominates, the pollutant fraction that is removed by flotation increases as the current increases because at higher operating current densities, bubble densities increase, resulting in a greater upward momentum flux and thus faster removal of pollutants and coagulants by flotation from the active reactor volume to the surface.371
On the contrary, the obtained results from the studies by Mohora show that the increase in operating current density caused a decrease in reactor DOC removal efficiency. They concluded that for higher operating current densities, more aluminum was available per unit of time in the ECF reactor volume but its residence time in the active reactor volume was shorter, which caused the decline in NOM removal efficiency.372
4.2 Application of EC and ECF processes
Electrocoagulation was first proposed in the nineteenth century and the first treatment plant was erected in 1889 in London for the treatment of sewage wastewater. EC or ECF processes are now used widely in the treatment of many types of wastewaters including dye and textile wastewater,373–377 refractory oily wastewater,378–380 municipal wastewater,381–385 manufacturing wastewater,386–388 and wastewaters with phenol,389 toxic metals390–393 and inorganic metals.394–398 Bench and pilot-scale research studies on using EC and ECF technologies to remove pollutants from many types of water and wastewaters were recently reviewed by Emamjomeha et al.,399 while Khandegar et al. provided a more focused review on removal of dyes from textile effluents.400The performance of EC/ECF technologies can be enhanced by integrating the process into a process train with other technologies. For example, Farhadi et al. compared electrocoagulation with photoelectrocoagulation, peroxi-electrocoagulation, and peroxi-photoelectrocoagulation for the removal of COD from pharmaceutical wastewater. Under optimum operating conditions for each process, the COD removal efficiency was in the order of peroxi-electrocoagulation > peroxi-photoelectrocoagulation > photoelectrocoagulation > electrocoagulation.401 Cotillas et al. looked into coupling of electrocoagulation with iron electrodes and UV irradiation (photoelectrocoagulation) for the simultaneous removal of turbidity and bacteria (Escherichia coli) from treated municipal wastewaters. Coupling UV irradiation to electrocoagulation was shown to improve the process performance compared to electrocoagulation alone. An examination of the effect of current density on process performance showed that there was a synergistic interaction of both technologies at low current densities (1.44 A m−2), but an antagonistic effect at high current densities (7.20 A m−2). This antagonistic effect was caused by the less efficient transmission of UV irradiation to the bulk solution due to the increase in the concentration of solids.402
Grey water treatment using electrocoagulation (EC) was examined in an integrated process with a submerged membrane bioreactor (SMBR). The combined EC–SMBR process resulted in a 13% reduction in membrane fouling. COD and color removal increased from 86% and 91.2%, respectively, using only the submersed membrane bioreactor, to 88.6% and 93.7% with EC–SMBR.403
Treatment using EC was enhanced when combined with ozone treatment or hydrogen peroxide treatment. Song et al. showed that the color removal efficiencies of the azo dye (C.I. Reactive Black 5), reached 10% by treatment with only ozone and 83% with only EC, but the combined process achieved 94% color removal.404 A combined electrocoagulation process followed by electrogenerated hydrogen peroxide treatment was evaluated for copper ion removal from an industrial wastewater.405 The maximum COD removal was 80%, which was about 20% greater in terms of COD removal than that of copper electrocoagulation alone. This additional organics reduction was due to a Fenton-like reaction between Cu(II) ions that remained in solution and the peroxide, generating OH˙ which oxidized the organics that were not adsorbed by the electrocoagulation treatment.
5. Electrodialysis
An electrodialysis (ED) process is an electrochemical separation process where ions are moved across polymeric anion- and cation-exchange membranes in a potential field (voltage). When an electrical potential difference is applied across an alternating series of cation- and anion-exchange membranes between two electrodes, positive ions migrate to the cathode (negative electrode) and negative ions migrate towards the anode. The presence of the ion exchange membranes traps them in alternating compartments, resulting in streams of dilute and concentrated ions.406Development of ED processes was initiated in the 1950s for production of table salt, when polymeric IEMs were manufactured on a commercial scale. Since then, ED has been a useful process for separation of ions and salts using IEMs. The development of a bipolar membrane, which is a composite membrane consisting of a cation-exchange layer and an anion-exchange layer pressed together, realized the splitting of solvents into H+ and OH−/CH3O− at the interface under a reverse potential bias. The use of this bipolar membrane has resulted in new technologies categorized as bipolar membrane electrodialysis (BMED).407 This solvent splitting technique has been used in more applications than other conventional ED techniques (CED) in chemical or biochemical synthesis, food processing, and pollution control (Fig. 14).
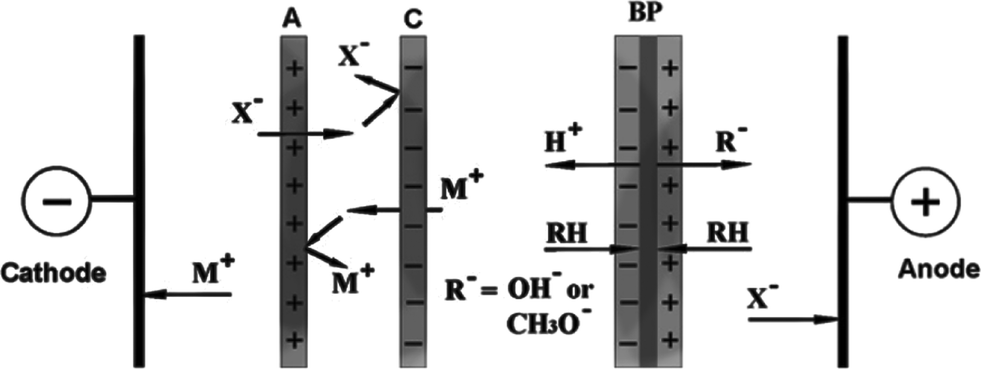 | ||
| Fig. 14 Schematic of BMED (BP, bipolar membrane; A: anionic membrane; C: cationic membrane; M+: cation; X−: anion; H+: hydrogen ion; OH−: hydroxide ion; CH3O−: methoxide ion).407 | ||
BMED was combined with an organic extraction process and an ion exchange process to develop an improved method for Cu2+ recovery (Fig. 15).408 This integrated process required three steps: first, extraction of copper ions from the mixture using an organic extractant HR (a complexing agent, where R = OH− or CH3O−) at a slightly alkaline pH, which was maintained by the OH− ions supplied by a bipolar membrane; second, movement of the bound Cu2+ ions across the cation-exchange resins to decrease the ohmic resistance of the compartment filled with HR to the other compartment along with the organic solution; third, release of the bound Cu2+ ions by substitution with the H+ ions from water splitting, which regenerates the HR. Tests showed that this approach, along with the use of ion exchange resins as a chamber spacer, improved current densities and substantially increased performance. An average current efficiency of 90% was achieved at a current density below 5 mA cm−2 with a gel ion-exchange resin, compared to a porous cation resin.409
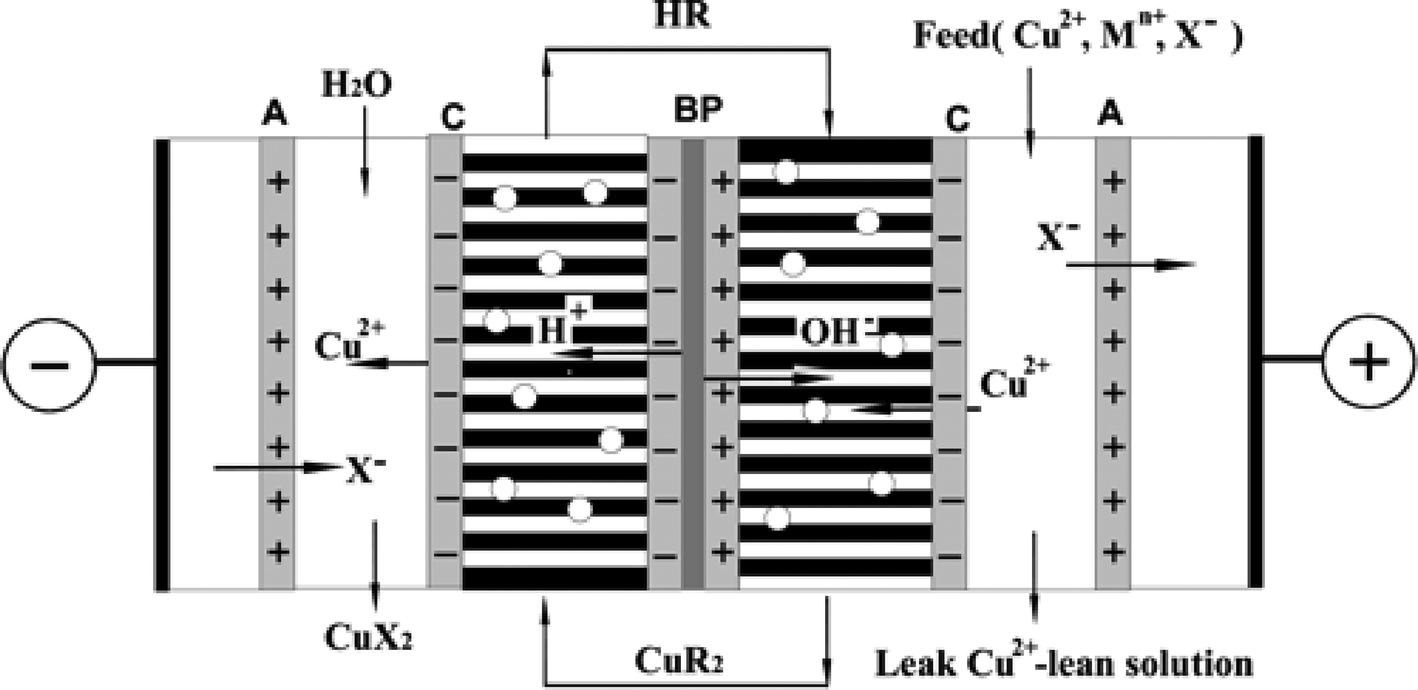 | ||
| Fig. 15 Concentration of copper from mixture solutions.408 | ||
A BMED process as a tool for the reclamation of NaOH from glyphosate neutralization liquor was investigated as well as its subsequent use as an absorbent for CO2 capture.410 A bench-scale BMED process was also used to examine the influence of operating conditions on the recovery of HCl and NaOH from seawater RO concentrates. The results showed that this technology was a technically feasible option for the production of 1.0 M or higher acid and base solutions that could potentially be used within the treatment plant, with current efficiencies in the 60–90% range (Fig. 16).411
 | ||
| Fig. 16 Bipolar membrane electrodialysis scheme to obtain acid and base (BP: bipolar membrane; C: cationic membrane; A: anionic membrane).411 | ||
ED has also been integrated with processes used in water treatment and reuse, including pressure-driven membrane processes, such as microfiltration (MF), ultrafiltration (UF), nanofiltration (NF), and reverse osmosis (RO). A pilot-scale wastewater treatment and reuse system was developed using ED and MF, which indicated that the treated water using such integration could meet the standards of water reuse and its quality remained stable for more than 6 months.412
UF was used as a pre-treatment process prior to ED (UF–ED) to treat river water and use it to add to surface waters to balance the depletion of surface water caused by excessive exploitation.413 The results showed acceptable reductions in concentrations for the following ions: Cl−, SO42−, NO3−, HCO3−, Na+, Mg2+, K+ and Ca2+. The use of NF, rather than UF, resulted in insufficient removal of these ions, especially the monovalent ions.
ED was also tested in conjunction with a membrane bioreactor (MBR).414,415 A CED process was used to concentrate the feed before it was pumped into MBRs to increase the solution conductivity, and thus provide a more stable voltage in the ED stack. Water with high nitrate content was purified by using this integrated process. Denitrification by the MBR of the concentrate produced by CED resulted in the treated water with a nitrate concentration below the acceptable value of 50 mg L−1.
6. Photoelectrochemical and sonoelectrochemical approaches
In recent years, the integration of electrochemistry with photocatalysis and sonochemistry methods has led to a new and interesting possibility for treatment of pollutants from wastewater.6.1 Photo-assisted electrochemical methods
In the recent years, there has been growing interest in the integration of photocatalysis and electrocatalysis for the treatment of toxic and/or recalcitrant organic compounds. Consequently, a number of studies have shown that DSA-type oxide electrodes can be utilized in photo-assisted electrochemical degradation processes in which the limitation of generation of highly reactive oxidants can be overcome through UV light irradiation. In addition, these anodes can generate chloro oxidant species (Cl2, HOCl, and OCl−), when electrolysis is carried out using a solution with a high chloride concentration under certain pH conditions. Thus, the combination of the chloro oxidant species generation and UV irradiation can result in the formation of highly reactive species.416–424The photo-assisted electrochemical (PAE) removal of dimethyl phthalate ester (DMP) was examined using a one-compartment filter press flow cell and a commercial DSA anode.424 Removal rates were similar to those reported using electrooxidation and a BDD anode. The highest rates of DMP and TOC removal were attained at high NaCl concentrations and current densities, due to the generation of high concentrations of reactive oxidants, such as ˙OH radicals, as well as other oxidants (h+, O2˙−, and Cl˙), in the PAE method. Thus, the PAE method improved the production of these chemicals and the rate of pollutant transport and degradation.
6.2 Sonoelectrolysis (SECT)
In an electrochemical oxidation process for the treatment of pollutants, the current efficiency usually gradually decreases during treatment. Thus, the pollutants and their intermediates are often adsorbed onto the electrode surface during oxidation and reduce the active sites on the electrode surface, resulting in partial or complete poisoning of the electrode. Treatment efficiency requires that consistently higher oxidation efficiency be maintained. In addition, an electrochemical process is often limited by the mass transport in the system, which also decreases the current efficiency.Recently, sonochemical technologies have been proposed in order to activate the electrode surface and enhance mass transfer efficiency. This hybrid process has been applied to degradation of several organic pollutants, including textile dyes,425,426 aromatics,427,428 nitro compounds,429 and chlorinated compounds.430,431 The efficient conductive diamond electrochemical oxidation (CDEO) coupled with ultrasound (US) was recently used for the degradation of progesterone in wastewater. Synergistic effects were clearly observed on the oxidation rates due to the improvement of mass transfer to the conductive-diamond surface of the electrode.432
7. Combination of electrochemical processes for resource reclamation from wastewaters
In recent years, various electrochemical approaches have been used to degrade organic matter in aqueous solutions, but wastewater treatment facilities consume a large amount of energy. Therefore, it is useful to develop novel hybrid technologies that can purify water and save or even generate electrical power.The possibility for both electrochemical degradation of wastewater organics and simultaneous production of hydrogen fuel has been recently explored. The electrochemical oxidation process with an option of microfiltration for real municipal wastewater treatment was investigated in terms of organic matter removal and hydrogen generation.433 Hydrogen production was enhanced with municipal wastewater due to the presence of specific organics and by adding NaCl to increase conductivity. The average current efficiency and the energy efficiency increased by approximately 10% when wastewater organics were present, whereas they increased by >20% when 50 mM NaCl was added to wastewater.
Other research studies have shown that it is possible to carry out wastewater treatment, hydrogen production, and waste heat recovery in a single electrochemical processing device. A theoretical analysis showed that converting hydrogen energy to electricity and utilizing waste heat recovery for adsorption chiller or heat pump applications resulted in good energy savings.434
Hydrogen gas production with simultaneous COD removal was achieved by electrochemical treatment of landfill leachate. The rates and yields of hydrogen gas were investigated using different applied voltages with aluminum electrodes and a DC power supply. Hydrogen gas was concluded to be mainly produced by ED of the leachate organics based on negligible H2 gas production using water in control experiments. The highest COD removal (77%) was obtained with an applied voltage of 4 V.435
The recovery of heavy metals from industrial aqueous solutions has received great attention in recent years, mainly due to more stringent laws for the protection of the environment. Conventional techniques for metal ion abatement, such as hydroxide precipitation or direct electroreduction, do not result in sufficient removal. One alternative technique is electrodialysis with the advantage that the low concentration of heavy metals can be concentrated, and the remaining effluent water can be diluted for reuse. However, the disadvantage of this approach is that it does not work well at high metal concentrations due to membrane fouling. Therefore, a future research direction could be some hybrid method to enhance electrodialysis processes through methods that reduce membrane fouling. For example, for copper recovery and water reuse from copper electroplating wastewater, a laboratory-scale process was developed that combined electrolysis (EI) and ED. The results showed that this combined process could achieve high recovery of both copper and water using wastewater with high or low concentrations of copper. Almost 99.5% of copper and 100% of water could be recovered.436
Compared to copper, the recovery of nickel is more challenging because the electrodeposition of nickel on the cathode is difficult due to the hydrogen evolution reaction, and therefore the recovery efficiency is low.437 The feasibility of nickel recovery and water reuse was investigated using three electrochemical processes (EI, ED and electrodeionization (EDI)) for both high and low nickel concentrations in wastewater. Almost 99.8% of nickel could be recovered, with a purity of 93.9%, and nearly 100% of water could be reused.438
ED has been combined with membrane processes, such as membrane filtration (MF), nanofiltration (NF), and reverse osmosis (RO) processes along with precipitation–neutralization processes to treat recycled water and sulfuric acid rinses from lead battery production lines.439 On average, 88 wt% sulfuric acid and 25 wt% rinse water were recovered. This treatment resulted in savings due to both water and acid recovery, along with additional savings due to a reduction in the chemicals needed for neutralization and the costs of sludge disposal.
An integrated ED–electrochlorination process was also used to treat wastewater. The process reduced the wastewater conductivity and TOC concentrations, and produced a valuable hypochlorite solution in the electrode rinse compartment (Fig. 17).440
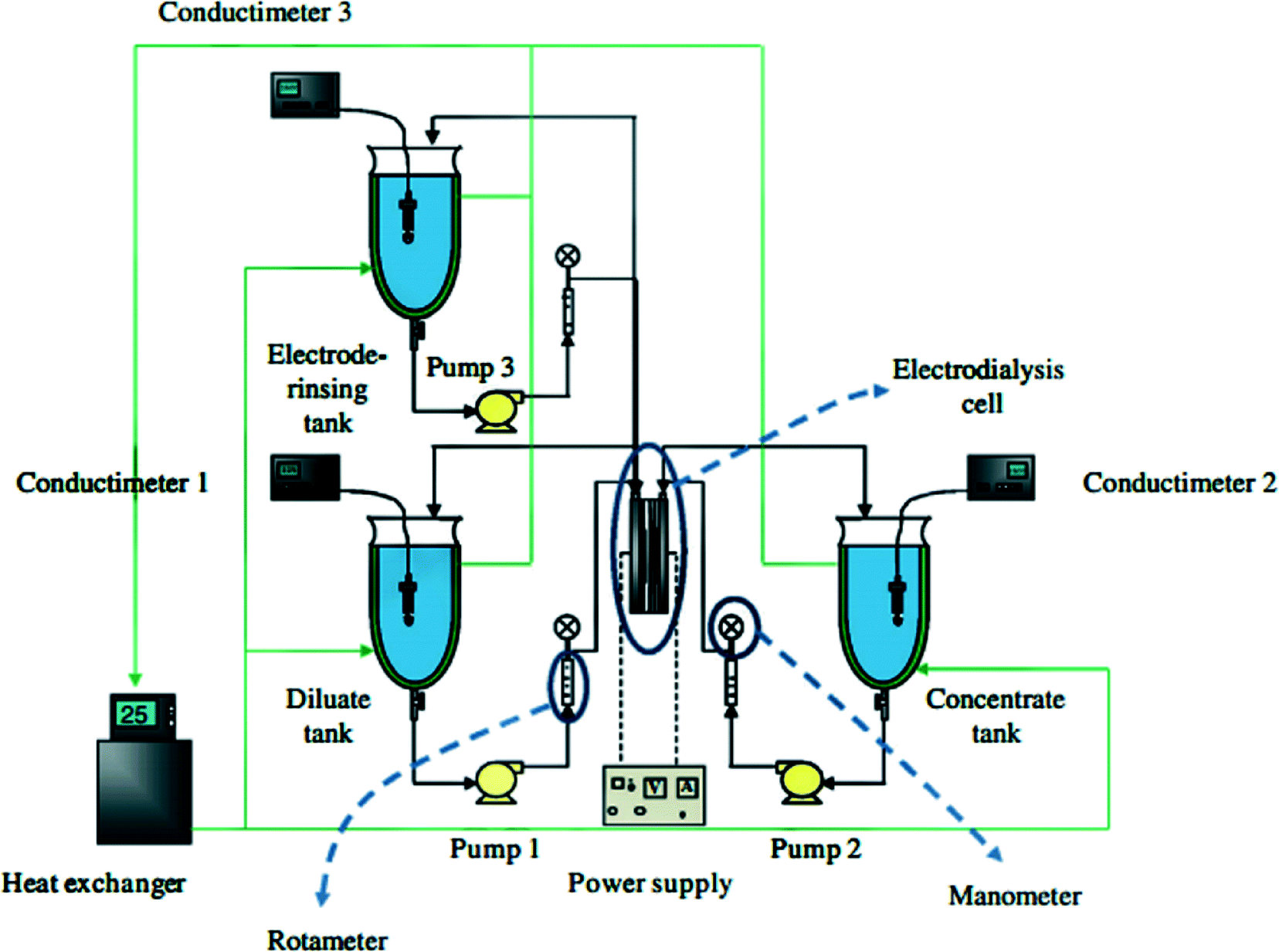 | ||
| Fig. 17 Experimental set-up.440 | ||
8. Conclusions
Environmental electrochemistry can be used to achieve a cleaner environment, as evidenced by the increasing number of new technologies and successes in treatment of polluted waters that were provided here in this review. Some of the most promising aspects are based on the use of different oxidation strategies and combinations of different technologies.Electrochemical oxidation is considered to be a very powerful tool able to mineralize completely non-biodegradable organic matter. Anodic oxidation of organic contaminants can be performed in several different ways including direct and indirect oxidation. Compared with direct anodic oxidation that leads to very poor decontamination, indirect anodic oxidation via intermediates of oxygen evolution can avoid electrode fouling, in which the nature of the electrode material strongly affects both process selectivity and efficiency.
Other strategies of indirect oxidation pathways for organic oxidation, such as active chlorine mediation and E-Fenton processes, are powerful and effective approaches for wastewater treatment. For the interaction of active chlorine with organics, further research directions should focus on developing novel electrode materials that can suppress the side formation of organochlorinated intermediates. Results from the literature cited here show that the electro-Fenton process with a BDD anode was the more powerful treatment for organic pollutants with a higher amount of reactive ˙OH than is expected to be formed on a BDD alone. Further development of the electro-Fenton process seems to be towards integrated processes, such as photoelectro-Fenton, sonoelectro-Fenton and peroxi-electrocoagulation methods, with the aim of obtaining a synergetic effect for water and wastewater treatment.
Although laboratory and pilot tests have been successful, industrial applications of these electrochemical oxidation methods are still limited, due to the relatively high energy consumption needed to treat low concentrations of chemicals in wastewater.
Future development of electrochemical oxidation techniques will require development of anode materials with specific characteristics that can make the process economically competitive with other conventional technologies. Energy consumption could be reduced using so-called “advanced electrochemical oxidation processes”, based on the combination of anodic and cathodic electrogeneration of highly oxidizing hydroxyl radicals.
Three electrochemical reduction processes, electrodeposition, cathodic electrochemical dechlorination, and electrochemical denitrification, were presented as the proposed technologies for the removal and reclamation of pollutants from wastewater.
For electrodeposition, the main advantage is that the deposited metals can be easily recycled by electrometallurgical processes. However, the surface of the cathode is modified during electrodeposition and it may need additional treatment. The electrodeposition of some heavy metals, like nickel, on the cathode is difficult due to the hydrogen evolution reaction which can greatly reduce the recovery efficiency of the metal.
For cathodic electrochemical dechlorination, the choice of the cathode materials has been found to have a major effect on the efficiency of this process, as it governs the reaction pathway and the selectivity. One of the main issues in the electrochemical reductive dechlorination of organic chlorides is the energy consumption associated with the process. The cost of the electricity needed for driving the electrolysis is too high to enable commercial success of this approach, and therefore suitable electrocatalysts are needed to lower the cell potential and thus reduce energy demands.
Electrochemical denitrification is receiving increased attention because of its advantages as an environmentally friendly, safe, selective, and cost-effective technique. From an environmental viewpoint, it is advantageous in that no chemicals are needed for the process, and nitrogen compounds formed by the electrocatalytic reduction of nitrate can have value. The efficiency of electrode reactions depends strictly upon the chemical and physical nature of the working electrodes. Synergistic electrochemical effects, in terms of catalytic activities, can be obtained when two or more metallic species are mixed together on the surface of the working electrodes. Although the reaction mechanisms on binary or ternary electrodes have been extensively examined, many subjects including the specific role of the foreign metal, the optimal surface composition, the surface morphology, and other factors are still under investigation and evaluation.
Electrocoagulation (EC) and electrocoagulation/flotation (ECF) processes have become effective technologies to remove pollutants from many types of water and wastewaters, and the performance of EC/ECF technologies can be enhanced by integrating the process into a process train with other technologies. The amount of chemicals required is small, and the amount of sludge produced is less than that required compared to conventional coagulation. However, this method has disadvantages such as anode passivation and sludge deposition on the electrodes that can inhibit continuous operation of the process. In addition, high concentrations of iron and aluminum ions can be released into the effluent that would have to be removed. Among these variables in the electrocoagulation (EC) and electrocoagulation/flotation (ECF) processes, solution pH and current density are two key operating parameters which significantly affect the efficiency of the electrocoagulation (EC) processes.
ED is one of the most recent technologies that has been used to separate plating chemicals from rinse water. The advantages of ED are that a low concentration of heavy metals can be concentrated, and that the treated effluent water can be diluted for reuse. However, ED does not work well for high concentrations of metals due to membrane fouling. The integration of ED with other pressure-driven membrane processes, such as microfiltration (MF), ultrafiltration (UF), nanofiltration (NF), and reverse osmosis (RO), will help further development of this electrochemical technology to obtain better performance of these systems for water treatment and reuse.
The integration of electrochemistry with photocatalysis and sonochemistry methods has led to a new and interesting possibility for the treatment of pollutants in wastewater. Using a photo-assisted process, it is possible to promote the direct generation of highly reactive species, such as ˙OH radicals, and also a series of other oxidants (h+, O2˙−, and Cl˙) in the bulk solution that would be absent in a purely electrochemical approach. Sonochemical technology shows promise as a method to improve the electrochemical process by activating the electrode surface and enhancing mass transfer efficiency. Presently, greater effort is needed to fully understand the degradation process that occurs with hybrid electrochemical technologies.
The electrochemical processing of wastewaters can have high energy demands, and therefore developing novel hybrid technologies that can purify water and generate or save energy is urgently needed to advance the applications of these technologies. The possibility for both electrochemical degradation of wastewater organics and simultaneous resource reclamation of materials in wastewater during treatment could make many of these processes useful compared to existing technologies.
Acknowledgements
This work was supported by the National Natural Science Fund for Distinguished Young Scholars (Grant No. 51125033) and the State Key Laboratory of Urban Water Resource and Environment (Harbin Institute of Technology) (No. 2015DX05).References
- J. O'M. Bockris, Electrochemistry of Cleaner Environments, Plenum Press, New York, 1972 Search PubMed.
- G. Kreysa and K. Jüttner, in Electrochemical Engineering and Energy, ed. F. Lapique, A. Storck and A. A. Wragg, Plenum Press, New York, 1994, p. 225 Search PubMed.
- G. C. Cushnie, Electroplating Wastewater Pollution Technology, Noyes Publications, New Jersey, USA, 1985 Search PubMed.
- K. Rajeshwar and J. G. Ibanez, Environmental Electrochemistry: Fundamentals and Applications in Pollution Abatement, Academic Press, London, 1997 Search PubMed.
- J. D. Genders and N. L. Weinberg, Electrochemistry for a Cleaner Environment, Electrosynthesis Co, New York, 1992 Search PubMed.
- D. Pletcher and F. C. Walsh, Industrial Electrochemistry, 2nd edn, Blackie, London, 1993 Search PubMed.
- C. A. C. Sequeira, Environmentally Oriented Electrochemistry, Elsevier, Amsterdam, 1994 Search PubMed.
- K. Rajeshwar, J. Ibanez and G. M. Swain, J. Appl. Electrochem., 1994, 24, 1077 CrossRef CAS.
- P. Tatapudi and J. M. Fenton, in Advances in electrochemical sciences and engineering, ed. H. Gerischer and C. W. Tobias, VCH-Veragsgesellschaft, Weinheim, 1995, ch. 4, p. 363 Search PubMed.
- K. Rajeshwar and J. Ibanez, Environmental Electrochemistry, Academic Press, San Diego, CA, 1997 Search PubMed.
- D. Simonson, Chem. Soc. Rev., 1997, 26, 181 RSC.
- K. Jüttner, U. Galla and H. Schmieder, Electrochim. Acta, 2000, 45, 3575 CrossRef.
- S. Trasatti, Int. J. Hydrogen Energy, 1995, 20, 835 CrossRef CAS.
- L. J. J. Janssen and L. Koene, Chem. Eng. J., 2002, 85, 137 CrossRef CAS.
- S. Vasudevan and M. A. Oturan, Environ. Chem. Lett., 2014, 12, 97 CrossRef CAS.
- G. H. Chen, Sep. Purif. Technol., 2004, 38, 11 CrossRef CAS.
- W. T. Mook, M. H. Chakrabarti, M. K. Aroua, G. M. A. Khan, B. S. Ali, M. S. Islam and M. A. Abu Hassan, Desalination, 2012, 285, 1 CrossRef CAS.
- C. A. Martínez-Huitle and E. Brillas, Appl. Catal., B, 2009, 87, 105 CrossRef.
- M. Panizza and G. Cerisola, Chem. Rev., 2009, 109, 6541 CrossRef CAS PubMed.
- J. Bersier, P. M. Bersier, L. Carlsson, T. Fuchigami, E. Steckhan and J. Yoshida, Electrochemistry V, Springer-Verlag, Berlin Heidelberg, 1994 Search PubMed.
- W. Zhao, PhD thesis, Harbin Institute of Technology, 2012 Search PubMed.
- L. M. Zhu, PhD thesis, Harbin Institute of Technology, 2011 Search PubMed.
- C. A. Martinez-Huitle and S. Ferro, Chem. Soc. Rev., 2006, 35, 1324 RSC.
- M. A. Rodrigo, P. A. Michaud, I. Duo, M. Panizza, G. Cerisola and C. Comninellis, J. Electrochem. Soc., 2001, 148, 60 CrossRef.
- E. Chatzisymeon, A. Dimou, D. Mantzavinos and A. Katsaounis, J. Hazard. Mater., 2009, 167, 268 CrossRef CAS PubMed.
- M. Gattrell and D. J. Kirk, J. Electrochem. Soc., 1993, 140, 1534 CrossRef CAS.
- S. K. Johnson, L. L. Houk, J. Feng, R. S. Houk and D. C. Johnson, Environ. Sci. Technol., 1999, 33, 2638 CrossRef CAS.
- H. Chang and D. C. Johnson, J. Electrochem. Soc., 1990, 137, 2452 CrossRef CAS.
- Ch. Comninellis, Electrochim. Acta, 1994, 39, 1857 CrossRef CAS.
- Y. J. Feng and X. Y. Li, Water Res., 2003, 37, 2399 CrossRef CAS PubMed.
- J. D. Rodgers, W. Jedral and N. Bunce, Environ. Sci. Technol., 1999, 33, 1453 CrossRef CAS.
- F. Bonfatti, S. Ferro, F. Lavezzo, M. Malacarne, G. Lodi and A. Debattisti, J. Electrochem. Soc., 1999, 146, 2175 CrossRef CAS.
- Ch. Comninellis and C. Pulgarin, J. Appl. Electrochem., 1991, 21, 703 CrossRef CAS.
- R. Kotz, S. Stucki and B. Carcer, J. Appl. Electrochem., 1991, 21, 14 CrossRef.
- A. Socha, E. Chrzescijanska and E. Kusmierek, Dyes Pigm., 2005, 67, 71 CrossRef CAS.
- R. A. Torres, W. Torres, P. Peringer and C. Pulgarin, Chemosphere, 2003, 50, 97 CrossRef CAS PubMed.
- F. Vigier, C. Coutanceau, J. M. Léger and J. L. Dubois, J. Power Sources, 2008, 175, 82 CrossRef CAS.
- J. H. Bezerra Rocha, M. M. Soares Gomes, E. Vieira dos Santos, E. C. Martins de Moura, D. Ribeiro da Silva, M. A. Quiroz and C. A. Martínez-Huitle, Electrochim. Acta, 2014, 140, 419 CrossRef.
- H. B. Beer, US Pat., 3632498, 1972 Search PubMed.
- M. Panizza and G. Cerisola, Electrochim. Acta, 2003, 48, 3491 CrossRef CAS.
- M. Panizza and G. Cerisola, Electrochim. Acta, 2004, 49, 3221 CrossRef CAS.
- G. R. P. Malpass, R. S. Neves and A. J. Motheo, Electrochim. Acta, 2006, 52, 936 CrossRef CAS.
- A. M. Z. Ramalho, C. A. Martínez-Huitle and D. R. da Silva, Fuel, 2010, 89, 531 CrossRef CAS.
- H. Zhang, F. Liu, X. G. Wu, J. H. Zhang and D. B. Zhang, Chem. Eng. J., 2009, 4, 568 CAS.
- E. Chatzisymeon, A. Dimou, D. Mantzavinos and A. Katsaounis, J. Hazard. Mater., 2009, 167, 268 CrossRef CAS PubMed.
- E. Turro, A. Giannis, R. Cossu, E. Gidarakos, D. Mantzavinos and A. Katsaounis, J. Hazard. Mater., 2011, 190, 460 CrossRef CAS PubMed.
- D. Valero, V. García-García, E. Expósito, A. Aldaz and V. Montiel, Sep. Purif. Technol., 2014, 123, 15 CrossRef CAS.
- C. Pulgarin, N. Adler, P. Peringer and Ch. Comninellis, Water Res., 1994, 28, 887 CrossRef CAS.
- R. Pelegrini, P. Peralta-Zamora, A. R. de Andrade, J. Reyes and N. Durán, Appl. Catal., B, 1999, 22, 83 CrossRef CAS.
- M. E. H. Bergmann and A. S. Koparal, J. Appl. Electrochem., 2005, 35, 1321 CrossRef CAS.
- E. Chatzisymeon, S. Fierro, I. Karafyllis, D. Mantzavinos, N. Kalogerakis and A. Katsaounis, Catal. Today, 2010, 151, 185 CrossRef CAS.
- C. A. Martínez-Huitle, M. A. Quiroz, Ch. Comninellis, S. Ferro and A. de Battisti, Electrochim. Acta, 2004, 50, 949 CrossRef.
- D. Rajkumar and J. G. Kim, J. Hazard. Mater., 2006, 136, 203 CrossRef CAS PubMed.
- O. Scialdone, A. Galia and S. Randazzo, Chem. Eng. J., 2011, 174, 266 CrossRef CAS.
- A. Kraft, Platinum Met. Rev., 2008, 52, 177 CrossRef CAS.
- O. Scialdone, S. Randazzo, A. Galia and G. Silvestri, Water Res., 2009, 43, 2260 CrossRef CAS PubMed.
- M. H. Zhou, L. Liu, Y. L. Jiao, Q. Wang and Q. Q. Tan, Desalination, 2011, 277, 201 CrossRef CAS.
- N. B. Tahar and A. Savall, J. Electrochem. Soc., 1998, 145, 3427 CrossRef CAS.
- N. B. Tahar and A. Savall, J. New Mater. Electrochem. Syst., 1999, 2, 19 CAS.
- D. C. Johnson, J. Feng and L. L. Houk, Electrochim. Acta, 2000, 46, 323 CrossRef CAS.
- J. Iniesta, E. Expósito, J. Gonzalez-García, V. Montiel and A. Aldaz, J. Electrochem. Soc., 2002, 149, 57 CrossRef.
- R. Amadelli and A. B. Velichenko, J. Serb. Chem. Soc., 2001, 66, 835 CAS.
- S. Abaci, U. Tamer, K. Pekmez and A. Yildiz, Appl. Surf. Sci., 2005, 240, 112 CrossRef CAS.
- L. S. Andrade, T. T. Tasso, D. L. Silva, R. C. Rocha-Filho, N. Bocchi and S. R. Biaggio, Electrochim. Acta, 2009, 54, 2024 CrossRef CAS.
- X. Y. Duan, F. Ma, Z. X. Yuan, L. M. Chang and X. T. Jin, J. Taiwan Inst. Chem. Eng., 2013, 44, 95 CrossRef CAS.
- J. Feng, D. C. Johnson, S. N. Lowery and J. J. Carey, J. Electrochem. Soc., 1994, 141, 2708 CrossRef CAS.
- A. B. Velichenko, R. Amadelli, G. L. Zucchini, D. V. Girenko and F. I. Danilov, Electrochim. Acta, 2000, 45, 4341 CrossRef CAS.
- S. E. Treimer, J. R. Feng, M. D. Scholten, D. C. Johnson and A. J. Davenport, J. Electrochem. Soc., 2001, 148, 459 CrossRef.
- L. S. Andrade, L. A. M. Ruotolo, R. C. Rocha-Filho, N. Bocchi, S. R. Biaggio, J. Iniesta, V. García-García and V. Montiel, Chemosphere, 2007, 66, 2035 CrossRef CAS PubMed.
- Y. H. Jiang, Z. X. Hu, M. H. Zhou, L. Zhou and B. D. Xi, Sep. Purif. Technol., 2014, 128, 67 CrossRef CAS.
- J. Iniesta, J. Gonzales-García, E. Expósito, V. Montiel and A. Aldaz, Water Res., 2001, 35, 3291 CrossRef CAS PubMed.
- K. T. Kawagoe and D. C. Johnson, J. Electrochem. Soc., 1994, 141, 3404 CrossRef CAS.
- O. Shmychkova, T. Luk'yanenko, A. Velichenko, L. Meda and R. Amadelli, Electrochim. Acta, 2013, 111, 332 CrossRef CAS.
- L. M. Chang, Y. Zhou, X. Y. Duan, W. Liu and D. D. Xu, J. Taiwan Inst. Chem. Eng., 2014, 45, 1338 CrossRef CAS.
- O. Shmychkova, T. Luk'yanenko, A. Yakubenko, R. Amadelli and A. Velichenko, Appl. Catal., B, 2015, 162, 346 CrossRef CAS.
- L. S. Andrade, R. C. Rocha-Filho, N. Bocchi, S. R. Biaggio, J. Iniesta, V. García-García and V. Montiel, J. Hazard. Mater., 2008, 153, 252 CrossRef CAS PubMed.
- A. B. Velichenko, R. Amadelli, E. A. Baranova, D. V. Girenko and F. I. Danilov, J. Electroanal. Chem., 2002, 527, 56 CrossRef CAS.
- F. L. Souza, J. M. Aquino, K. Irikura, D. W. Miwa, M. A. Rodrigo and A. J. Motheo, Chemosphere, 2014, 109, 187 CrossRef CAS PubMed.
- R. Amadelli, L. Armelao, A. B. Velichenko, N. V. Nikolenko, D. V. Girenko, S. V. Kovalyov and F. I. Danilov, Electrochim. Acta, 1999, 45, 713 CrossRef CAS.
- A. B. Velichenko, D. V. Girenko, S. V. Kovalyov, A. N. Gnatenko, R. Amadelli and F. I. Danilov, J. Electroanal. Chem., 1998, 454, 203 CrossRef CAS.
- S. Song, J. Q. Fan, Z. Q. He, L. Y. Zhan, Z. W. Liu, J. M. Chen and X. H. Xu, Electrochim. Acta, 2010, 55, 3606 CrossRef CAS.
- H. L. Bi, C. Z. Yu, W. Gao and P. Cao, Electrochim. Acta, 2013, 113, 446 CrossRef CAS.
- Y. Chen, H. Y. Li, W. J. Liu, Y. Tu, Y. H. Zhang, W. Q. Han and L. J. Wang, Chemosphere, 2014, 113, 48 CrossRef CAS PubMed.
- Q. Z. Dai, H. Shen, Y. J. Xia, F. Chen, J. D. Wang and J. M. Chen, Sep. Purif. Technol., 2013, 104, 9 CrossRef CAS.
- Y. H. Zheng, W. Q. Su, S. Y. Chen, X. Z. Wu and X. M. Chen, Chem. Eng. J., 2011, 174, 304 CrossRef CAS.
- X. P. Yang, R. Y. Zou, F. Huo, D. C. Cai and D. Xiao, J. Hazard. Mater., 2009, 164, 367 CrossRef CAS PubMed.
- H. An, H. Cui, W. Y. Zhang, J. P. Zhai, Y. Qian, X. C. Xie and Q. Li, Chem. Eng. J., 2012, 209, 86 CrossRef CAS.
- G. H. Zhao, Y. H. Zhang, Y. A. Lei, B. Y. Lv, J. X. Gao, Y. N. Zhang and D. M. Li, Environ. Sci. Technol., 2010, 44, 1754 CrossRef CAS PubMed.
- C. Flox, C. Arias, E. Brillas, A. Savall and K. Groenen-Serrano, Chemosphere, 2009, 74, 1340 CrossRef CAS PubMed.
- L. Ciríaco, C. Anjo, J. Correia, M. J. Pacheco and A. Lopes, Electrochim. Acta, 2009, 54, 1464 CrossRef.
- X. Zhu, M. Tong, S. Shi, H. Zhao and J. Ni, Environ. Sci. Technol., 2008, 42, 4914 CrossRef CAS PubMed.
- L. Gherardini, P. A. Michaud, M. Panizza, Ch. Comninellis and N. Vatistas, J. Electrochem. Soc., 2001, 148, 78 CrossRef.
- H. Lin, J. F. Niu, J. L. Xu, H. O. Huang, D. Li, Z. H. Yue and C. H. Feng, Environ. Sci. Technol., 2013, 47, 13039 CrossRef CAS PubMed.
- C. A. Martinez-Huitle, S. Ferro and A. de Battisti, J. Appl. Electrochem., 2005, 35, 1087 CrossRef CAS.
- S. Supothina and M. R. D. Guire, Thin Solid Films, 2000, 371, 1 CrossRef CAS.
- Ch. Comninellis and C. Pulgarin, J. Appl. Electrochem., 1993, 23, 108 CrossRef CAS.
- J. F. Liu, Y. J. Feng, L. X. Sun and Z. G. Qian, Mater. Sci. Technol., 2006, 14, 200 CAS.
- Y. H. Cui and Y. J. Feng, J. Mater. Sci., 2005, 40, 4695 CrossRef CAS.
- S. Stucki, R. Kotz, B. Career and W. Suter, J. Appl. Electrochem., 1991, 21, 99 CrossRef CAS.
- Ch. Comninellis, Trans. I. Chem. E., 1992, 70, 219 CAS.
- X. Y. Li, Y. H. Cui, Y. J. Feng, Z. M. Xie and J. D. Gu, Water Res., 2005, 39, 1972 CrossRef CAS PubMed.
- F. P. Hu, Z. Q. Dong, X. W. Cui and W. X. Chen, Electrochim. Acta, 2011, 56, 1576 CrossRef CAS.
- Y. J. Feng, Y. H. Cui, B. E. Logan and Z. Q. Liu, Chemosphere, 2008, 70, 1629 CrossRef CAS PubMed.
- J. W. Lv, Y. J. Feng, J. F. Liu, Y. P. Qu and F. Y. Cui, Appl. Surf. Sci., 2013, 28, 900 CrossRef.
- Y. J. Feng, Y. H. Cui, J. F. Liu and B. E. Logan, J. Hazard. Mater., 2010, 178, 29 CrossRef CAS PubMed.
- Y. H. Cui, Y. J. Feng, J. F. Liu and N. Q. Ren, J. Hazard. Mater., 2012, 239, 225 CrossRef PubMed.
- Q. Wang, T. Jin, Z. X. Hu, L. Zhou and M. H. Zhou, Sep. Purif. Technol., 2013, 102, 180 CrossRef CAS.
- S. Y. Yang, Y. S. Choo, S. Kim, J. Lee and H. Parkd, Appl. Catal., B, 2012, 111, 317 CrossRef.
- A. I. del Río, J. Fernández, J. Molina, J. Bonastre and F. Cases, Electrochim. Acta, 2010, 55, 7282 CrossRef.
- F. N. Chianeh and J. B. Parsa, Chem. Eng. Res. Des., 2014, 92, 2470 CrossRef.
- J. Q. Fan, G. H. Zhao, H. Y. Zhao, S. N. Chai and T. C. Cao, Electrochim. Acta, 2013, 94, 21 CrossRef CAS.
- J. Radjenovic, B. I. Esche and K. Rabaey, Water Res., 2011, 45, 3205 CrossRef CAS PubMed.
- Y. H. Cui, X. Y. Li and G. H. Chen, Water Res., 2009, 43, 1968 CrossRef CAS PubMed.
- Y. Liu, H. L. Liu, J. Ma and J. J. Li, J. Hazard. Mater., 2012, 213, 222 CrossRef PubMed.
- J. F. Niu, D. Maharana, J. L. Xu, Z. Chai and Y. P. Bao, J. Electrochem. Soc., 2013, 25, 1424 CAS.
- J. T. Kong, S. Y. Shi, X. P. Zhu and J. R. Ni, J. Electrochem. Soc., 2007, 19, 1380 CAS.
- M. Quiroz, S. Reyna and J. Sanchez, J. Solid State Electrochem., 2003, 7, 277 CrossRef CAS.
- J. F. Niu, Y. P. Bao, Y. Li and Z. Chai, Chemosphere, 2013, 92, 1571 CrossRef CAS PubMed.
- J. F. Niu, Y. P. Bao, Y. Li and Z. Chai, Water Res., 2012, 46, 2281 CrossRef PubMed.
- B. Wang, W. P. Kong and H. Z. Ma, J. Hazard. Mater., 2007, 146, 295 CrossRef CAS PubMed.
- F. T. Chen, S. C. Yu, X. P. Dong and S. S. Zhang, J. Hazard. Mater., 2012, 227, 474 CrossRef PubMed.
- Y. H. Cui, Y. J. Feng and Z. Q. Liu, Electrochim. Acta, 2009, 54, 4903 CrossRef CAS.
- G. H. Zhao, X. Cui, M. Liu, P. Q. Li, Y. G. Zhang, T. C. Cao, H. X. Li, Y. Z. Lei, L. Liu and D. M. Li, Environ. Sci. Technol., 2009, 43, 1480 CrossRef CAS PubMed.
- L. C. Zhang, L. Xu, J. He and J. J. Zhang, Electrochim. Acta, 2014, 117, 192 CrossRef CAS.
- M. A. Q. Alfaro, S. Ferro, C. A. Martinez-Huitle and Y. M. Vong, J. Braz. Chem. Soc., 2006, 17, 227 CrossRef CAS.
- E. Brillas and C. A. Martínez-Huitle, Synthetic diamond films preparation, electrochemistry, characterization and applications, John Wiley & Sons Inc., Hoboken, New Jersey-USA, 2011 Search PubMed.
- J. J. Carey, C. S. Christ and S. N. Lowery, US Pat., 5399247, 1995 Search PubMed.
- B. Nasr, G. Abdellatif, P. Cañizares, C. Saez, J. Lobato and M. A. Rodrigo, Environ. Sci. Technol., 2005, 39, 7234 CrossRef CAS PubMed.
- A. Morao, A. Lopes, M. T. P. de Amorim and I. C. Goncalves, Electrochim. Acta, 2004, 49, 15875 CrossRef.
- P. Cañizares, C. Saez, J. Lobato and M. A. Rodrigo, Ind. Eng. Chem. Res., 2004, 43, 1944 CrossRef.
- P. Cañizares, C. Saez, J. Lobato and M. A. Rodrigo, Electrochim. Acta, 2004, 49, 4641 CrossRef.
- P. Cañizares, J. Lobato, R. Paz, M. A. Rodrigo and C. Saez, Water Res., 2005, 39, 2687 CrossRef PubMed.
- P. Cañizares, L. Martinez, R. Paz, C. Saez, J. Lobato and M. A. Rodrigo, J. Chem. Technol. Biotechnol., 2006, 81, 1331 CrossRef.
- J. Y. Jiang, M. Chang and P. Pan, Environ. Sci. Technol., 2008, 42, 3059 CrossRef CAS PubMed.
- M. Mascia, A. Vacca, A. M. Polcaro, S. Palmas, J. R. Ruiz and A. D. Pozzo, J. Hazard. Mater., 2010, 174, 314 CrossRef CAS PubMed.
- N. Rabaaoui, K. M. Saada, Y. Moussaoui, M. S. Allagui, A. Bedoui and E. Elaloui, J. Hazard. Mater., 2013, 250, 447 CrossRef PubMed.
- R. B. A. Souza and L. A. M. Ruotolo, J. Environ. Chem. Eng., 2013, 1, 544 CrossRef CAS.
- S. C. Elaoud, M. Panizza, G. Cerisola and T. Mhiri, Desalination, 2011, 272, 148 CrossRef CAS.
- X. P. Zhu, J. R. Ni, J. J. Wei, X. Xing, H. N. Li and Y. Jiang, J. Hazard. Mater., 2010, 184, 493 CrossRef CAS PubMed.
- J. Hastie, D. Bejan, M. Teutli-Leon and N. J. Bunce, Ind. Eng. Chem. Res., 2006, 45, 4898 CrossRef CAS.
- P. Cañizares, A. Gadri, J. Lobato, B. Nasr, R. Paz, M. A. Rodrigo and C. Saez, Ind. Eng. Chem. Res., 2006, 45, 3468 CrossRef.
- C. Flox, S. Ammar, C. Arias, E. Brillas, A. V. Vargas-Zavala and R. Abdelhedi, Appl. Catal., B, 2006, 67, 93 CrossRef CAS.
- R. E. Palma-Goyes, F. L. Guzmán-Duque, G. Peñuela, I. González, J. L. Navad and R. A. Torres-Palma, Chemosphere, 2010, 81, 26 CrossRef CAS PubMed.
- R. Bogdanowicz, A. Fabiańska, L. Golunski, M. Sobaszek, M. Gnyba, J. Ryl, K. Darowickic, T. Ossowski, S. D. Janssens, K. Haenen and E. M. Siedleck, Diamond Relat. Mater., 2013, 39, 82 CrossRef CAS.
- M. H. Zhou, H. Särkkä and M. Sillanpää, Sep. Purif. Technol., 2011, 78, 290 CrossRef CAS.
- F. Guenfoud, M. Mokhtaria and H. Akrout, Diamond Relat. Mater., 2014, 46, 8 CrossRef CAS.
- I. Sires, P. L. Cabot, F. Centellas, J. A. Garrido, R. M. Rodriguez, C. Arias and E. Brillas, Electrochim. Acta, 2006, 52, 75 CrossRef CAS.
- A. M. Polcaro, A. Vacca, M. Mascia and S. Palmas, Electrochim. Acta, 2005, 50, 1841 CrossRef CAS.
- H. T. Madsen, E. G. Søgaard and J. Muff, Chemosphere, 2014, 109, 84 CrossRef CAS PubMed.
- M. Haidar, A. Dirany, I. Sirés, N. Oturan and M. A. Oturan, Chemosphere, 2013, 91, 1304 CrossRef CAS PubMed.
- O. García, E. Isarain-Chávez, A. Ei-Ghenymy, E. Brillas and J. M. Peralta-Hernández, J. Electrochem. Soc., 2014, 728, 1 Search PubMed.
- M. Panizza, M. Delucchi and G. Cerisola, J. Appl. Electrochem., 2005, 35, 357 CrossRef CAS.
- G. Lissens, J. Pieters, M. Verhaege, L. Pinoy and W. Verstraete, Electrochim. Acta, 2003, 48, 1655 CrossRef CAS.
- B. Louhichi, M. F. Ahmadi, N. Bensalah, A. Gadri and M. A. Rodrigo, J. Hazard. Mater., 2008, 158, 430 CrossRef CAS PubMed.
- Q. F. Zhuo, S. B. Deng, B. Yang, J. Huang, B. Wang, T. T. Zhang and G. Yu, Electrochim. Acta, 2012, 77, 17 CrossRef CAS.
- A. Katsoni, D. Mantzavinos and E. Diamadopoulos, Water Res., 2014, 57, 76 CrossRef CAS PubMed.
- E. Chatzisymeon, N. P. Xekoukoulotakis, E. Diamadopoulos, A. Katsaounis and D. Mantzavinos, Water Res., 2000, 43, 3999 CrossRef PubMed.
- A. J. C. da Silva, E. V. dos Santos, C. C. de Oliveira Morais, C. A. Martínez-Huitle and S. S. L. Castro, Chem. Eng. J., 2013, 233, 47 CrossRef.
- A. M. S. Solano, C. K. C. de Araújo, J. V. de Melo, J. M. Peralta-Hernandez, D. R. da Silva and C. A. Martínez-Huitle, Appl. Catal., B, 2013, 130, 112 CrossRef.
- M. Panizza, P. A. Michaud, G. Cerisola and Ch. Comninellis, J. Electroanal. Chem., 2001, 507, 206 CrossRef CAS.
- J. Iniesta, P. A. Michaud, M. Panizza, G. Cerisola, A. Aldaz and Ch. Comninellis, Electrochim. Acta, 2001, 46, 3573 CrossRef CAS.
- J. Iniesta, P. A. Michaud, M. Panizza and Ch. Comninellis, Electrochem. Commun., 2001, 3, 346 CrossRef CAS.
- L. Gherardini, P. A. Michaud, M. Panizza, C. Comninellis and N. Vatistas, J. Electrochem. Soc., 2001, 148, 78 CrossRef.
- D. Gandini, E. Mahe, P. A. Michaud, W. Haenni, A. Perret and Ch. Comninellis, J. Appl. Electrochem., 2000, 30, 1345 CrossRef CAS.
- F. Montilla, P. A. Michaud, E. Morallon, J. L. Vazquez and Ch. Comninellis, Electrochim. Acta, 2002, 47, 3509 CrossRef CAS.
- A. Perret, W. Haenni, N. Skinner, X. M. Tang, D. Gandini, Ch. Comninellis, B. Correa and G. Foti, Diamond Relat. Mater., 1999, 8, 820 CrossRef CAS.
- R. Bellagamba, P. A. Michaud, Ch. Comninellis and N. Vatistas, Electrochem. Commun., 2002, 4, 171 CrossRef CAS.
- Ch. Comninellis and G. Chen, Electrochemistry for the environment, Springer, New York, NY-USA, 2010 Search PubMed.
- C. A. Martinez-Huttle, A. De Battisti, S. Ferro, S. Reyna, M. Cerro-López and M. A. Quiro, Environ. Sci. Technol., 2008, 42, 6929 CrossRef.
- X. L. Tong, G. H. Zhao, M. C. Liu, T. C. Cao, L. Liu and P. Q. Li, J. Phys. Chem. C, 2009, 113, 13787 CAS.
- L. Guo and G. Chen, Diamond Relat. Mater., 2007, 16, 1530 CrossRef CAS.
- L. L. Du, J. R. Sun, H. Cui, H. D. Li, T. Cui and H. B. Lin, Chem. Res. Chin. Univ., 2012, 28, 507 CAS.
- L. Fan, Y. Zhou, W. Yang, G. Chen and F. Yang, Dyes Pigm., 2008, 76, 440 CrossRef CAS.
- L. Fan, Y. Zhou, W. Yang, G. Chen and F. Yang, J. Hazard. Mater., 2006, 137, 1182 CrossRef CAS PubMed.
- F. Y. Yi, S. X. Chen and C. Yuan, J. Hazard. Mater., 2008, 157, 79 CrossRef CAS PubMed.
- P. P. Jin, R. Chang, D. Q. Liu, K. Zhao, L. Zhang and Y. J. Ouyang, J. Environ. Chem. Eng., 2014, 2, 1040 CrossRef CAS.
- Y. H. Han, X. Quan, X. Ruan and W. Zhang, Sep. Purif. Technol., 2008, 59, 43 CrossRef CAS.
- Y. Y. Sun, G. Wang, Q. Dong, B. Q. Qian, Y. L. Meng and J. H. Qiu, Chem. Eng. J., 2014, 253, 73 CrossRef CAS.
- J. Yang, J. Wang and J. P. Jia, Environ. Sci. Technol., 2009, 43, 3796 CrossRef CAS PubMed.
- H. Cui, Y. Qian, H. An, C. C. Sun, J. P. Zhai and Q. Li, Water Res., 2012, 46, 3943 CrossRef CAS PubMed.
- J. Ludek, W. Y. Zhou and M. Kumagai, J. Rare Earths, 2006, 24, 257 CrossRef.
- G. Gao and C. D. Vecitis, Environ. Sci. Technol., 2011, 45, 9726 CrossRef CAS PubMed.
- C. D. Vecitis, M. H. Schnoor, M. S. Rahaman, J. D. Schiffman and M. Elimelech, Environ. Sci. Technol., 2011, 45, 3672 CrossRef CAS PubMed.
- C. D. Vecitis, G. D. Gao and H. Liu, J. Phys. Chem. C, 2011, 115, 3621 CAS.
- G. D. Gao and D. Chad, Electrochim. Acta, 2013, 98, 131 CrossRef CAS.
- J. Mieluch, A. Sadkowski, J. Wild and P. Zoltowski, Przem. Chem., 1975, 54, 513 CAS.
- Ch. Comninellis and A. Nerini, J. Appl. Electrochem., 1995, 25, 23 CrossRef CAS.
- F. Bonfatti, S. Ferro, F. Lavezzo, M. Malacarne, G. Lodi and A. De Battisti, J. Electrochem. Soc., 2000, 147, 92 CrossRef.
- B. J. Hernlem, Water Res., 2005, 39, 2245 CrossRef CAS PubMed.
- H. X. Shi, J. H. Qu, A. M. Wang and J. T. Ge, Chemosphere, 2005, 60, 326 CrossRef CAS PubMed.
- D. Ž. Mijin, M. L. Avramov Ivic, A. E. Onjiac and B. N. Grgur, Chem. Eng. J., 2012, 204, 151 CrossRef.
- M. G. Tavares, L. V. A. da Silva, A. M. Sales Solano, J. Tonholo, C. A. Martínez-Huitle and C. L. P. S. Zanta, Chem. Eng. J., 2012, 204, 141 CrossRef.
- A. Kapałka, A. Katsaounisb, N. L. Michelsa, A. Leonidova, S. Souentie, Ch. Comninellis and K. M. Udert, Electrochem. Commun., 2010, 12, 1203 CrossRef.
- T. L. Luu, J. Kim and J. Yoon, J. Ind. Eng. Chem., 2015, 21, 400 CrossRef.
- E. Chatzisymeon, S. Fierro, I. Karafyllis, D. Mantzavinos, N. Kalogerakis and A. Katsaounis, Catal. Today, 2010, 151, 185 CrossRef CAS.
- N. Papastefanakis, D. Mantzavinos and A. Katsaounis, J. Appl. Electrochem., 2010, 40, 729 CrossRef CAS.
- M. Gotsi, N. Kalogerakis, E. Psillakis, P. Samaras and D. Mantzavinos, Water Res., 2005, 39, 4177 CrossRef CAS PubMed.
- H. Bergmann, T. Iourtchouk, K. Schops and K. Bouzek, Chem. Eng. J., 2002, 85, 111 CrossRef CAS.
- S. Palmas, A. M. Polcaro, A. Vacca, M. Mascia and F. Ferrara, J. Appl. Electrochem., 2007, 37, 63 CrossRef CAS.
- E. Lacasa, J. Llanos, P. Cañizares and A. Manuel, Chem. Eng. J., 2012, 184, 66 CrossRef CAS.
- A. Kraft, M. Stadelmann, M. Blaschke, D. Kreysig, B. Sandt, F. Schröder and J. Rennau, J. Appl. Electrochem., 1999, 29, 861 CAS.
- A. Kraft, M. Wuensche, M. Stadelmann and M. Blaschke, Recent Res. Dev. Electrochem., 2003, 6, 27 CAS.
- M. Panizza, C. Bocca and G. Cerisola, Water Res., 2000, 34, 2601 CrossRef CAS.
- B. Wang, W. Kong and H. Ma, J. Hazard. Mater., 2007, 146, 295 CrossRef CAS PubMed.
- M. Panizza and G. Cerisola, Environ. Sci. Technol., 2004, 38, 5470 CrossRef CAS PubMed.
- P. B. Moraes and R. Bertazzoli, Chemosphere, 2005, 58, 41 CrossRef CAS PubMed.
- A. Vlyssides, P. Karlis, M. Loizidou, A. Zorpas and D. Arapoglou, Environ. Technol., 2001, 22, 1467 CrossRef CAS PubMed.
- A. G. Vlyssides, P. K. Karlis and G. Mahnken, J. Appl. Electrochem., 2003, 33, 155 CrossRef CAS.
- D. Rajkumar and K. Palanivelu, J. Hazard. Mater., 2004, 113, 123 CrossRef CAS PubMed.
- M. Panizza and G. Cerisola, Water Res., 2006, 40, 1179 CrossRef CAS PubMed.
- C. Belaid, M. Kallel, M. Khadhraou, G. Lalleve, B. Elleuch and J. F. Fauvarque, J. Appl. Electrochem., 2006, 36, 1175 CrossRef CAS.
- A. Giannis, M. Kalaitzakis and E. Diamadopoulos, J. Chem. Technol. Biotechnol., 2007, 82, 663 CrossRef CAS.
- N. N. Rao, K. M. Somasekhar, S. N. Kaul and L. Szpyrkowicz, J. Chem. Technol. Biotechnol., 2001, 76, 1124 CrossRef CAS.
- L. Szpyrkowicz, G. H. Kelsall, S. N. Kaul and M. De Faveri, Chem. Eng. Sci., 2001, 56, 1579 CrossRef CAS.
- L. Szpyrkowicz, S. N. Kaul and R. N. Neti, J. Appl. Electrochem., 2005, 35, 381 CrossRef CAS.
- L. Szpyrkowicz, S. N. Kaul, R. N. Neti and S. Satyanarayan, Water Res., 2005, 39, 1601 CrossRef CAS PubMed.
- C. R. Costa, C. M. R. Botta, E. L. G. Espindola and P. Olivi, J. Hazard. Mater., 2008, 153, 616 CrossRef CAS PubMed.
- D. Rajkumar, B. J. Song and J. G. Kim, Dyes Pigm., 2007, 72, 1 CrossRef.
- D. Rajkumar and J. G. Kim, J. Hazard. Mater., 2006, 136, 203 CrossRef CAS PubMed.
- G. R. P. Malpass, D. W. Miwa, S. A. S. Machado and A. J. Motheo, J. Hazard. Mater., 2008, 156, 170 CrossRef CAS PubMed.
- A. G. Vlyssides, D. Papaioannou, M. Loizidoy, P. K. Karlis and A. A. Zorpas, Waste Manage., 2000, 20, 569 CrossRef CAS.
- L. Szpyrkowicz and M. Radaelli, Sep. Sci. Technol., 2007, 42, 1493 CrossRef CAS.
- L. Szpyrkowicz and M. Radaelli, J. Appl. Electrochem., 2006, 36, 1151 CrossRef CAS.
- L. Szpyrkowicz, C. Juzzolino, S. Daniele and M. De Faveri, Catal. Today, 2001, 66, 519 CrossRef CAS.
- L. Szpyrkowicz, C. Juzzolino, S. N. Kaul, S. Daniele and M. De Faveri, Ind. Eng. Chem. Res., 2000, 39, 3241 CrossRef CAS.
- L. Szpyrkowicz, M. Radaelli and S. Daniele, Catal. Today, 2005, 100, 425 CrossRef CAS.
- E. Chatzisymeon, N. P. Xekoukoulotakis, A. Coz, N. Kalogerakis and D. Mantzavinos, J. Hazard. Mater., 2006, 137, 998 CrossRef CAS PubMed.
- N. Mohan and N. Balasubramanian, J. Hazard. Mater., 2006, 136, 239 CrossRef CAS PubMed.
- N. Mohan, N. Balasubramanian and C. A. Basha, J. Hazard. Mater., 2007, 147, 644 CrossRef CAS PubMed.
- E. Turro, A. Giannis, R. Cossu, E. Gidarakos, D. Mantzavinos and A. Katsaounis, J. Hazard. Mater., 2011, 190, 460 CrossRef CAS PubMed.
- T. Matsue, M. Fujihira and T. Osa, J. Electrochem. Soc., 1981, 128, 2565 CrossRef CAS.
- E. Brillas, R. M. Bastida and E. Llosa, J. Electrochem. Soc., 1995, 142, 1733 CrossRef CAS.
- E. Brillias, E. Mur and J. Casado, J. Electrochem. Soc., 1996, 143, 49 CrossRef.
- E. Brillas, R. Sauleda and J. Casado, J. Electrochem. Soc., 1997, 144, 2374 CrossRef CAS.
- E. Brillas, R. Sauleda and J. Casado, J. Electrochem. Soc., 1998, 145, 759 CrossRef CAS.
- E. Brillas, E. Mur, R. Sauleda, L. Sanchez, F. Peral, X. Domenech and J. Casado, Appl. Catal., B, 1998, 16, 31 CrossRef CAS.
- M. Sudoh, T. Kodera, K. Sakai, J. Q. Zhang and K. Koide, J. Chem. Eng., 1986, 19, 513 CrossRef CAS.
- Y. Sun and J. J. Pignatello, Environ. Sci. Technol., 1993, 27, 304 CrossRef CAS.
- J. De Laat and H. H. Gallard, Environ. Sci. Technol., 1999, 33, 2726 CrossRef CAS.
- F. J. Rivas, F. J. Beltrán, O. Gimeno and P. Alvarez, J. Hazard. Mater., 2003, 96, 259 CrossRef CAS PubMed.
- W. Gernjak, T. Krutzler, A. Glaser, S. Malato, J. Caceres, R. Bauer and A. R. FernandezAlba, Chemosphere, 2003, 50, 71 CrossRef CAS PubMed.
- G. R. Agladze, G. S. Tsurtsumia, B. I. Jung, J. S. Kim and G. Gorelishvili, J. Appl. Electrochem., 2007, 37, 385 CrossRef CAS.
- M. Pimentel, N. Oturan, M. Dezotti and M. A. Oturan, Appl. Catal., B, 2008, 83, 140 CrossRef CAS.
- S. H. Yang and X. H. Li, J. Hazard. Mater., 2005, 118, 85 CrossRef PubMed.
- L. Zhou, M. H. Zhou, Z. X. Hu, Z. H. Bi and K. G. Serrano, Electrochim. Acta, 2014, 140, 376 CrossRef CAS.
- A. Babuponnusami and K. Muthukumar, Chem. Eng. J., 2012, 183, 1 CrossRef CAS.
- M. A. Fernández de Dios, O. Iglesias, E. Bocos, M. Pazos and M. A. Sanromán, Ind. Eng. Chem., 2014, 20, 3754 CrossRef.
- F. E. F. Rêgo, A. M. S. Solano, I. C. da C. Soares, D. R. da Silva, C. A. Martinez-Huitle and M. Panizza, J. Environ. Chem. Eng., 2014, 2, 875 CrossRef.
- E. Rosales, O. Iglesias, M. Pazos and M. A. Sanromán, J. Hazard. Mater., 2012, 213, 369 CrossRef PubMed.
- R. Salazar and M. S. Ureta-Zañartu, Water, Air, Soil Pollut., 2012, 223, 4199 CrossRef CAS.
- J. J. Aaron and M. A. Oturan, Turk. J. Chem., 2001, 25, 509 CAS.
- A. Dhaouadi, L. Monser and N. Adhoum, Electrochim. Acta, 2009, 54, 4473 CrossRef CAS.
- A. Özcan, Y. Sahin, A. S. Koparal and M. A. Oturan, Appl. Catal., B, 2009, 89, 620 CrossRef.
- A. D. Pozzo, C. Merli, I. Sirs, J. A. Garrido, R. M. Rodriguez and E. Brillas, Environ. Chem. Lett., 2005, 3, 7 CrossRef.
- Y. J. Wang, X. Y. Li, L. M. Zhen, H. Q. Zhang, Y. Zhang and C. W. Wang, J. Hazard. Mater., 2012, 229, 115 CrossRef PubMed.
- E. Atmaca, J. Hazard. Mater., 2009, 163, 109 CrossRef CAS PubMed.
- S. Mohajeri, H. A. Aziz, M. H. Isa, M. A. Zahed and M. N. Adlan, J. Hazard. Mater., 2010, 176, 749 CrossRef CAS PubMed.
- I. Sirés, J. A. Garrido, R. M. Rodríguez, E. Brillas, N. Oturan and M. A. Oturan, Appl. Catal., B, 2007, 72, 382 CrossRef.
- E. Guinea, J. A. Garrido, R. M. Rodríguez, P. C. C. Arias and F. C. E. Brillas, Electrochim. Acta, 2010, 55, 2101 CrossRef CAS.
- E. Isarain-Chávez, C. Arias, P. L. Cabot, F. Centellas and R. M. Rodríguez, Appl. Catal., B, 2010, 96, 361 CrossRef.
- M. Zhou, Q. Tan, Q. Wang, Y. Jiao, N. Oturan and M. A. Oturan, J. Hazard. Mater., 2012, 215, 287 CrossRef PubMed.
- N. Oturan, E. Brillas and M. A. Oturan, Environ. Chem. Lett., 2012, 10, 165 CrossRef CAS.
- S. Ricardo, E. Brillas and I. Sirés, Appl. Catal., B, 2012, 115, 107 Search PubMed.
- S. Garcia-Segura, J. A. Garrido, R. M. Rodríguez, P. L. Cabot, F. Centellas, C. Arias and E. Brillas, Water Res., 2012, 46, 2067 CrossRef CAS PubMed.
- F. C. Moreira, S. Garcia-Segura, V. J. P. Vilar, R. A. R. Boaventura and E. Brillas, Appl. Catal., B, 2013, 142, 877 CrossRef.
- S. Garcia-Segura, E. B. Cavalcanti and E. Brillas, Appl. Catal., B, 2014, 144, 588 CrossRef CAS.
- M. A. Oturan, I. Sirés, N. Oturan, S. Perocheau, J. L. Laborde and S. Trevin, J. Electroanal. Chem., 2008, 624, 329 CrossRef CAS.
- W. S. Chen and C. P. Huang, Ultrason. Sonochem., 2014, 21, 840 CrossRef CAS PubMed.
- B. Boye, M. M. Dieng and E. Brillas, Electrochim. Acta, 2003, 48, 781 CrossRef CAS.
- E. Brillas, B. Boye, M. A. Banos, J. C. Calpe and J. A. Garrido, Chemosphere, 2003, 51, 227 CrossRef CAS PubMed.
- S. Vasudevan, J. Water Process Eng., 2014, 2, 53 CrossRef.
- G. Dubpernel, Selected Topics in the History of Electrochemistry, The Electrochemical Society, Princeton, 1978 Search PubMed.
- B. Fleet, in Electrochemistry, Past and Present, ed. J. T. Stock and M. V. Orna, America Chemical Society, Washington, DC, 1989 Search PubMed.
- G. Issabayeva, M. K. Aroua and N. M. Sulaiman, Desalination, 2006, 194, 192 CrossRef CAS.
- M. Spitzer and R. Bertazzoli, Hydrometallurgy, 2004, 74, 233 CrossRef CAS.
- Y. Oztekin and Z. Yazicigil, Desalination, 2006, 190, 79 CrossRef CAS.
- J. H. Chang, A. V. Ellis, C. T. Yan and C. H. Tung, Sep. Purif. Technol., 2009, 68, 216 CrossRef CAS.
- J. E. D. V. Segundo, G. R. Salazar-Banda, A. C. O. Feitoza, E. O. Vilar and E. B. Cavalcanti, Sep. Purif. Technol., 2012, 88, 107 CrossRef CAS.
- J. R. Hernández-Tapia, J. Vazquez-Arenas and I. González, J. Hazard. Mater., 2013, 262, 709 CrossRef PubMed.
- N. J. Bunce, S. G. Merica and J. Lipkowski, Chemosphere, 1997, 35, 2719 CrossRef CAS.
- P. Dabo, A. Cyr, F. Laplante, F. Jean, H. Menard and J. Lessard, Environ. Sci. Technol., 2000, 34, 1265 CrossRef CAS.
- N. L. Stock and N. J. Bunce, Can. J. Chem., 2002, 80, 200 CrossRef CAS.
- N. Hoshi, K. Sasaki, S. Hashimoto and Y. Hori, J. Electroanal. Chem., 2004, 568, 267 CrossRef CAS.
- A. A. Jalil, N. F. A. Panjang, S. Akhbar, M. Sundang, N. Tajuddin and S. Triwahyono, J. Hazard. Mater., 2007, 148, 1 CrossRef CAS PubMed.
- J. J. Li, H. L. Liu, X. W. Cheng, Y. J. Xin, W. X. Xu, Z. P. Ma, J. Ma, N. Q. Ren and Q. Li, Ind. Eng. Chem. Res., 2012, 51, 15557 CrossRef CAS.
- S. Rondinini, P. R. Mussini, F. Crippa and G. Sello, Electrochem. Commun., 2000, 2, 491 CrossRef CAS.
- B. Huang, A. A. Isse, C. Durante, C. Wei and A. Gennaro, Electrochim. Acta, 2012, 70, 50 CrossRef CAS.
- A. A. Isse, G. Sandonà, C. Durante and A. Gennaro, Electrochim. Acta, 2009, 54, 3235 CrossRef CAS.
- C. Bellomunno, D. Bonanomi, L. Falciola, M. Longhi, P. R. Mussini, L. M. Doubova and G. Silvestro Di, Electrochim. Acta, 2005, 50, 2331 CrossRef CAS.
- G. Fiori, S. Rondinini, G. Sello, A. Vertova, M. Cirja and L. Conti, J. Appl. Electrochem., 2005, 35, 363 CrossRef CAS.
- Y. H. Xu, Y. H. Zhu, F. M. Zhao and C. A. Ma, Appl. Catal., A, 2007, 324, 83 CrossRef CAS.
- Y. H. Xu, C. P. Zhang, C. P. Chu and C. A. Ma, J. Electroanal. Chem., 2012, 664, 39 CrossRef CAS.
- A. A. Isse, S. Gottardello, C. Durante and A. Gennaro, Phys. Chem. Chem. Phys., 2008, 10, 2409 RSC.
- A. A. Isse, S. Gottardello, C. Durante and A. Gennaro, Electrochim. Acta, 2009, 54, 3235 CrossRef CAS.
- C. Durante, A. A. Isse, S. Gottardello and A. Gennaro, Appl. Catal., B, 2009, 88, 479 CrossRef CAS.
- Y. H. Xu, H. Zhang, C. P. Chu and C. A. Ma, J. Electroanal. Chem., 2012, 664, 39 CrossRef CAS.
- S. Ardizzone, G. Cappelletti, L. M. Doubova, P. R. Mussini, S. Passeri and S. Rondinini, Electrochim. Acta, 2003, 48, 3789 CrossRef CAS.
- S. Rondinini, P. R. Mussini, P. Muttini and G. Sello, Electrochim. Acta, 2001, 46, 3245 CrossRef CAS.
- S. Agarwal, Environ. Sci. Technol., 2009, 43, 915 CrossRef CAS PubMed.
- K. R. Zhu, S. A. Baig, J. Xu, T. T. Sheng and X. H. Xu, Electrochim. Acta, 2012, 69, 389 CrossRef CAS.
- B. B. Huang, A. A. Isse, C. Durante, C. H. Wei and A. Gennaro, Electrochim. Acta, 2012, 70, 50 CrossRef CAS.
- Y. H. Xu, Q. Q. Cai, H. X. Ma, Y. He, H. Zhang and C. A. Ma, Electrochim. Acta, 2013, 96, 90 CrossRef CAS.
- Z. Q. He, K. J. Lin, J. J. Sun, L. N. Wen, C. Gao, J. M. Chen, S. Song, Y. Y. Qian and W. P. Liu, Electrochim. Acta, 2013, 109, 502 CrossRef CAS.
- Z. R. Sun, H. Ge, X. Hu and Y. Z. Peng, Sep. Purif. Technol., 2010, 72, 133 CrossRef CAS.
- I. F. Cheng, Q. Fernando and N. Korte, Environ. Sci. Technol., 1997, 31, 1074 CrossRef CAS.
- M. M. Baizer and J. L. Chruma, J. Org. Chem., 1972, 37, 1951 CrossRef CAS.
- J. He, A. E. Saez, W. P. Ela, E. A. Betterton and R. G. Arnold, Ind. Eng. Chem. Res., 2004, 43, 913 CrossRef CAS.
- Z. Liu, R. G. Arnold, E. A. Betterton and E. Smotkin, Environ. Sci. Technol., 2001, 35, 4320 CrossRef CAS PubMed.
- J. R. Stromberg, J. D. Wnuk, R. A. F. Pinlac and G. J. Meyer, Nano Lett., 2006, 6, 1284 CrossRef CAS PubMed.
- C. Durante, B. B. Huang, A. A. Isse and A. Gennaroa, Appl. Catal., B, 2012, 126, 355 CrossRef CAS.
- A. A. Isse, B. B. Huang, C. Durante and A. Gennaroa, Appl. Catal., B, 2012, 126, 347 CrossRef CAS.
- Y. L. Jiao, D. L. Wu, H. Y. Ma, C. C. Qiu, J. T. Zhang and L. M. Ma, Electrochem. Commun., 2008, 44, 1474 CrossRef.
- H. Ma, Y. P. Huang, M. W. Shen, R. Guo, X. Y. Cao and X. Y. Shi, J. Hazard. Mater., 2012, 211, 349 CrossRef PubMed.
- X. H. Mao, A. Ciblak, K. Baek, M. Amiri, R. Loch-Caruso and A. N. Alshawabkeh, Water Res., 2012, 46, 1847 CrossRef CAS PubMed.
- H. Y. Jeonga, K. Anantharaman, S. P. Hyun, M. Son and K. F. Hayes, Water Res., 2013, 47, 6639 CrossRef PubMed.
- A. Tsyganok, K. Otsuka, I. Yamanaka, V. Plekhanov and S. Kulikov, Chem. Lett., 1996, 25, 261 CrossRef.
- C. H. Lin and S. K. Tseng, Water Sci. Technol., 2000, 42, 167 CAS.
- A. Tsyganok, K. Otsuka, I. Yamanaka, V. Plekhanov and S. Kulikov, Chem. Lett., 1996, 25, 261 CrossRef.
- S. Rondinini, P. R. Mussini, M. Specchia and A. Vertova, J. Electrochem. Soc., 2001, 148, 102 CrossRef.
- Z. R. Sun, X. F. Wei, H. T. Shen and X. Hu, Electrochim. Acta, 2014, 129, 433 CrossRef CAS.
- Y. H. Xu, Q. Q. Cai, H. X. Ma, Y. He, H. Zhang and C. A. Ma, Electrochim. Acta, 2013, 96, 90 CrossRef CAS.
- H. Wang, Z. Y. Bian, G. Lu, L. Pang, Z. P. Zeng and D. Z. Sun, Appl. Catal., B, 2012, 125, 449 CrossRef CAS.
- K. Miyoshi, Y. Kamegaya and M. Matsumura, Chemosphere, 2004, 56, 187 CrossRef CAS PubMed.
- A. A. Jalil, S. Triwahyono, N. Aini, M. Razali, N. H. H. Hairom, A. Idris, M. Nazlan, M. Muhid, A. Ismail, N. Aisyah, M. Yahaya, N. Akma, L. Ahmad and H. Dzinun, J. Hazard. Mater., 2010, 174, 581 CrossRef CAS PubMed.
- M. Paidar, I. Rousar and K. Bouzek, J. Appl. Electrochem., 1999, 29, 611 CrossRef CAS.
- C. Lu, S. G. Lu and W. H. Qiu, Electrochim. Acta, 1999, 44, 2193 CrossRef CAS.
- O. W. J. S. Rutten, A. Van Sandwijk and G. Van Weert, J. Appl. Electrochem., 1999, 29, 87 CrossRef.
- S. Ureta-Zanartu and C. Yanez, Electrochim. Acta, 1997, 42, 1725 CrossRef CAS.
- E. E. Kalu, R. E. White and D. T. Hobbs, J. Electrochem. Soc., 1996, 143, 3094 CrossRef CAS.
- I. Katsounaros, D. Ipsakis, C. Polatides and G. Kyriacou, Electrochim. Acta, 2006, 52, 1329 CrossRef CAS.
- M. Li, C. P. Feng, Z. Y. Zhang and N. Sugiura, Electrochim. Acta, 2009, 54, 4600 CrossRef CAS.
- I. Katsounaros, M. Dortsioua, C. Polatides, S. Preston, T. Kypraios and G. Kyriacou, Electrochim. Acta, 2012, 71, 270 CrossRef CAS.
- G. E. Dima, A. C. A. de Vooys and M. T. M. Koper, J. Electroanal. Chem., 2003, 554, 15 CrossRef.
- M. A. Hasnat, R. Agui, S. Hinokuma, T. Yamaguchi and M. Machida, Catal. Commun., 2009, 10, 1132 CrossRef CAS.
- N. Comisso, S. Cattarin, S. Fiameni, R. Gerbasi, L. Mattarozzi, M. Musiania, L. Vázquez-Gómez and E. Verlato, Electrochem. Commun., 2012, 25, 91 CrossRef CAS.
- L. Mattarozzi, S. Cattarin, N. Comisso, P. Guerriero, M. Musiania, L. Vázquez-Gómez and E. Verlato, Electrochim. Acta, 2013, 89, 488 CrossRef CAS.
- Q. Zhang, L. Ding, H. Cui, J. P. Zhai, Z. B. Wei and Q. Li, Appl. Surf. Sci., 2014, 308, 113 CrossRef CAS.
- I. G. Casella and M. Contursi, Electrochim. Acta, 2014, 138, 447 CrossRef.
- A. Anastasopoulos, L. Hannah and B. E. Hayden, J. Catal., 2013, 305, 27 CrossRef CAS.
- Y. Y. Birdja, J. Yang and M. T. M. Koper, Electrochim. Acta, 2014, 6, 11 Search PubMed.
- Y. Lou, Y. T. Shao, P. Li, Z. L. Li and Z. J. Niu, J. Electroanal. Chem., 2008, 624, 33 CrossRef CAS.
- Y. Wang, J. H. Qua, R. C. Wu and P. J. Lei, Water Res., 2006, 40, 1224 CrossRef CAS PubMed.
- G. Innocenzo and C. M. Contursi, Electrochim. Acta, 2014, 05, 125 Search PubMed.
- L. Ma, B. Y. Zhang, H. L. Li and J. Q. Chambers, J. Electroanal. Chem., 1993, 362, 201 CrossRef CAS.
- F. Bouamrane, A. Tadjeddine, J. E. Butler, R. Tenne and C. L. Clement, J. Electroanal. Chem., 1996, 405, 95 CrossRef.
- I. G. Casella and M. Gatta, J. Electroanal. Chem., 2004, 568, 183 CrossRef CAS.
- M. T. de Groot and M. T. M. Koper, J. Electroanal. Chem., 2004, 562, 81 CrossRef CAS.
- N. Owaga, H. Kodaiku and S. Ikeda, J. Electroanal. Chem., 1986, 208, 117 CrossRef.
- G. Horanyi and E. M. Rizmayer, J. Electroanal. Chem., 1985, 188, 265 CrossRef CAS.
- I. Katsounaros and G. Kyriacou, Electrochim. Acta, 2008, 53, 5477 CrossRef CAS.
- S. Loghambal and L. Rajendran, J. Electroanal. Chem., 2011, 661, 137 CrossRef CAS.
- E. A. Vik, D. A. Carlson, A. S. Eikum and E. T. Gjessin, Water Res., 1984, 18, 1355 CrossRef CAS.
- M. Y. A. Mollah, R. Schennach, J. R. Parga and D. L. Cocke, J. Hazard. Mater., 2001, 84, 29 CrossRef CAS PubMed.
- G. Chen, Sep. Purif. Technol., 2004, 38, 11 CrossRef CAS.
- P. Cañizares, M. Carmona, J. Lobato, F. Martínez and M. A. Rodrigo, Ind. Eng. Chem. Res., 2005, 44, 4178 CrossRef.
- P. K. Holt, G. W. Barton and C. A. Mitchell, Chemosphere, 2005, 59, 355 CrossRef CAS PubMed.
- M. Kobya, E. Demirbas, A. Dedeli and M. T. Sensoy, J. Hazard. Mater., 2010, 173, 326 CrossRef CAS PubMed.
- S. Zhao, G. H. Huang, G. H. Cheng, Y. F. Wang and H. Y. Fu, Desalination, 2014, 344, 454 CrossRef CAS.
- M. H. Isaa, E. H. Ezechi, Z. Ahmed, S. F. Magram and S. R. M. Kutty, Water Res., 2014, 51, 113 CrossRef PubMed.
- M. M. Emamjomeh and M. Sivakumar, J. Environ. Manage., 2009, 90, 1663 CrossRef CAS PubMed.
- M. G. Cora, Int. J. Environ. Eng., 2009, 1, 3 CrossRef.
- S. Vasudevan and J. Lakshmi, Environ. Eng. Sci., 2012, 29, 563 CrossRef CAS.
- Y. Meas, J. A. Ramirez, M. A. Villalon and T. W. Chapman, Electrochim. Acta, 2010, 55, 8165 CrossRef CAS.
- C. H. Gong, Z. G. Zhang, H. T. Li, D. Li, B. C. Wu, Y. W. Sun and Y. J. Cheng, J. Hazard. Mater., 2014, 274, 465 CrossRef CAS PubMed.
- Y. O. Fouad, Alexandria Eng. J., 2014, 53, 199 CrossRef.
- P. Cañizares, F. Martínez, J. Lobato and M. A. Rodrigo, J. Hazard. Mater., 2007, 145, 233 CrossRef PubMed.
- A. S. Koparal, Y. S. Yildiz, B. Keskinler and N. Demirciŏglu, Sep. Purif. Technol., 2008, 59, 175 CrossRef CAS.
- S. K. Verma, V. Khandegar and A. K. Saroha, J. Hazard., Toxic Radioact. Waste, 2013, 17, 146 CrossRef CAS.
- S. Vasudevan and J. Lakshmi, Environ. Eng. Sci., 2012, 29, 563 CrossRef CAS.
- S. Zodi, B. Merzouk, O. Potier, F. Lapicque and J. P. Leclerc, Sep. Purif. Technol., 2013, 108, 215 CrossRef CAS.
- P. K. Holt, G. W. Barton, M. Wark and C. A. Mitchell, Colloids Surf., A, 2002, 211, 233 CrossRef CAS.
- E. Mohora, S. Rončević, B. Dalmacija, J. Agbaba, M. Watson, E. Karlović and M. Dalmacija, J. Hazard. Mater., 2012, 235, 257 CrossRef PubMed.
- M. Bayramoglu, M. Eyvaz and M. Kobya, Chem. Eng. J., 2008, 128, 155 CrossRef.
- K. Charoenlarp and W. Choyphan, Asian J. Energy Environ., 2009, 10, 250 Search PubMed.
- M. C. Wei, K. S. Wang, C. L. Huang, C. W. Chiang, T. J. Chang, S. S. Lee and S. H. Chang, Chem. Eng. J., 2012, 192, 37 CrossRef CAS.
- V. Khandegar and A. K. Saroha, Indian Chem. Eng., 2013, 55, 1 CrossRef.
- V. Khandegar and A. K. Saroha, J. Hazard., Toxic Radioact. Waste, 2016, 20(1) DOI:10.1061/(ASCE)HZ.2153-5515.0000278.
- M. Carmona, M. Khemis, J. P. Leclerc and F. Lapicque, Chem. Eng. Sci., 2006, 61, 1237 CrossRef CAS.
- M. Asselin, P. Drogui, S. K. Brar, H. Benmoussa and J. F. Blais, J. Hazard. Mater., 2008, 151, 446 CrossRef CAS PubMed.
- T. Coskun, F. Ilhan, N. M. Demir, E. Debik and U. Kurt, Environ. Technol., 2012, 33, 801 CrossRef CAS PubMed.
- R. Mahajan, V. Khandegar and A. K. Saroha, Int. J. Chem. Environ. Eng., 2013, 4, 104 CAS.
- F. Janpoor, A. Torabian and V. Khatibikamal, J. Chem. Technol. Biotechnol., 2011, 86, 1113 CrossRef CAS.
- C. Ricordel and H. Djelal, J. Environ. Chem. Eng., 2014, 2, 1551 CrossRef CAS.
- X. M. Chen, G. H. Chen and P. L. Yue, Sep. Purif. Technol., 2000, 19, 65 CrossRef CAS.
- D. D. Nguyen, H. H. Ngo and Y. S. Yoona, Bioresour. Technol., 2014, 153, 116 CrossRef CAS PubMed.
- A. Akyol, Desalination, 2012, 285, 91 CrossRef CAS.
- C. H. Gong, Z. G. Zhang, H. T. Li, D. Li, B. C. Wu, Y. W. Sun and Y. J. Cheng, J. Hazard. Mater., 2014, 274, 465 CrossRef CAS PubMed.
- C. Barrera-Díaz, B. Frontana-Uribe and B. Bilyeu, Chemosphere, 2014, 105, 160 CrossRef PubMed.
- S. Vasudevan, Journal of Water Process Engineering, 2014, 2, 53 CrossRef.
- B. A. Aji, Y. Yavuz and A. S. Koparal, Sep. Purif. Technol., 2012, 86, 248 CrossRef.
- F. Akbal and S. Camci, Chem. Eng. Technol., 2010, 33, 1655 CrossRef CAS.
- F. Akbal and S. Camci, Environ. Prog. Sustainable Energy, 2011, 31, 340 CrossRef.
- F. Akbal and S. Camci, Desalination, 2011, 269, 214 CrossRef CAS.
- M. H. Isa, E. H. Ezechi, Z. Ahmed, S. F. J. Magram and S. R. M. Kutty, Water Res., 2014, 51, 113 CrossRef CAS PubMed.
- G. Moussavi, F. Majidi and M. Farzakia, Desalination, 2011, 280, 127 CrossRef CAS.
- M. Behbahani, M. R. AlaviMoghaddam and M. Arami, Desalination, 2011, 271, 209 CrossRef CAS.
- E. Lacasa, P. Cañizares, C. Sáez, F. J. Fernández and M. A. Rodrigo, J. Chem. Eng., 2011, 171, 1012 CrossRef CAS.
- S. Vasudevan, J. Jayaraj, J. Lakshmi and G. Sozhan, Korean J. Chem. Eng., 2009, 26, 1058 CrossRef CAS.
- M. M. Emamjomeha and M. Sivakumar, J. Environ. Manage., 2009, 90, 1663 CrossRef PubMed.
- V. Khandegar and A. K. Saroha, J. Environ. Manage., 2013, 128, 949 CrossRef CAS PubMed.
- S. Farhadi, B. Aminzadeh, A. Torabian, V. Khatibikamal and M. A. Fard, J. Hazard. Mater., 2012, 219, 35 CrossRef PubMed.
- S. Cotillas, J. Llanos, O. G. Mirand, G. C. Díaz-Trujillo, P. Cañizarea and M. A. Rodrigo, Electrochim. Acta, 2014, 140, 396 CrossRef CAS.
- K. Bani-Melhem and E. Smith, Chem. Eng. J., 2012, 198, 201 CrossRef.
- S. Song, Z. Q. He, J. P. Qiu, L. J. Xu and J. M. Chen, Sep. Purif. Technol., 2007, 55, 238 CrossRef CAS.
- C. Barrera-Díaz, B. Frontana-Uribe and B. Bilyeu, Chemosphere, 2014, 105, 160 CrossRef PubMed.
- H. Strathmann, in Synthetic membranes: science, engineering and applications, ed. P. M. Bungay, H. K. Lonsdale and M. N. de Pinho, Reidel, Dordrecht/Holland, 1986 Search PubMed.
- C. H. Huang, T. W. Xu, Y. P. Zhang, Y. H. Xue and G. W. Chen, J. Membr. Sci., 2007, 288, 1 CrossRef CAS.
- C. H. Huang and T. W. Xu, Environ. Sci. Technol., 2006, 40, 5233 CrossRef CAS PubMed.
- T. W. Xu, Resour., Conserv. Recycl., 2002, 37, 1 CrossRef.
- W. Y. Ye, J. Huang, J. Y. Lin, X. Zhang, J. G. Shen, P. Luis and B. V. der Bruggen, Sep. Purif. Technol., 2015, 144, 206 CrossRef CAS.
- R. Ibáñez, A. Pérez-González, P. Gómez, A. M. Urtiaga and I. Ortiz, Desalination, 2013, 309, 165 CrossRef.
- B. van der Bruggen, R. Milis, C. Vandecasteele, P. Bielen, E. van San and K. Huysman, Water Res., 2003, 37, 3867 CrossRef CAS PubMed.
- J. F. Koprivnjak, E. M. Perdue and P. H. Pfromm, Water Res., 2006, 40, 3385 CrossRef CAS PubMed.
- C. Wisniewski, F. Persin, T. Cherif, R. Sandeaux, A. Grasmick and C. Gavach, Desalination, 2001, 139, 199 CrossRef CAS.
- C. Wisniewski, F. Persin, T. Cherif, R. Sandeaux, A. Grasmick, C. Gavach and F. Lutin, Desalination, 2002, 149, 331 CrossRef CAS.
- L. Pinhedo, R. Pelegrini, R. Bertazzoli and A. Motheo, Appl. Catal., B, 2004, 57, 75 CrossRef.
- M. Catanho, G. R. P. Malpass and A. Motheo, Appl. Catal., B, 2005, 62, 193 CrossRef.
- R. T. Pelegrini, R. S. Freire, N. Duran and R. Bertazzoli, Environ. Sci. Technol., 2001, 35, 2849 CrossRef CAS PubMed.
- G. R. P. Malpass, D. W. Miwa, A. C. P. Miw, S. A. S. Machado and A. J. Motheo, Environ. Sci. Technol., 2007, 41, 7120 CrossRef CAS PubMed.
- G. R. P. Malpass, D. W. Miwa, A. C. P. Miw, S. A. S. Machado and A. J. Motheo, J. Hazard. Mater., 2009, 167, 224 CrossRef CAS PubMed.
- M. Catanho, G. R. P. Malpass and A. J. Motheo, Appl. Catal., B, 2006, 62, 193 CrossRef CAS.
- S. Xiao, J. Qu, X. Zhao, H. Liu and D. Wan, Water Res., 2009, 43, 1432 CrossRef CAS PubMed.
- M. V. B. Zanoni, J. J. Sene, H. Selcuk and M. Anderson, Environ. Sci. Technol., 2004, 38, 3203 CrossRef CAS PubMed.
- F. L. Souza, J. M. Aquina, D. W. Miwa, M. A. Rodrigob and A. J. Motheo, J. Environ. Chem. Eng., 2014, 2, 811 CrossRef CAS.
- M. Rivera, M. Pazos and M. A. Sanromán, J. Chem. Technol. Biotechnol., 2009, 84, 1118 CrossRef CAS.
- Z. Ai, J. Li, L. Zhang and S. Lee, Ultrason. Sonochem., 2010, 17, 370 CrossRef CAS PubMed.
- R. H. Lima Leite, P. Cognet, A. M. Wilhelm and H. Delmas, Chem. Eng. Sci., 2002, 7, 76 Search PubMed.
- G. Zhao, S. Shen, M. Li, M. Wu, T. Cao and D. Li, Chemosphere, 2008, 73, 1407 CrossRef CAS PubMed.
- V. O. Abramov, A. E. Gekhman, V. M. Kuznetsov and G. J. Price, Ultrason. Sonochem., 2006, 13, 303 CrossRef CAS PubMed.
- M. D. Esclapez, V. Sáez, D. Milán-Yáñez, I. Tudela, O. Louisnard and J. González-García, Ultrason. Sonochem., 2010, 17, 1010 CrossRef CAS PubMed.
- M. D. Esclapez, M. I. Díez-García, V. Sáez, I. Tudela, J. M. Pérez, J. González-García and P. M. Bonete, Electrochim. Acta, 2011, 56, 8138 CrossRef CAS.
- M. J. M. de Vidales, S. Barba, C. Sáez, P. Cañizares and M. A. Rodrigo, Electrochim. Acta, 2014, 140, 20 CrossRef.
- H. Park, K. H. Choo, H. S. Park, J. Choi and M. R. Hoffmann, Chem. Eng. J., 2013, 215, 802 CrossRef.
- J. Y. Jiang, J. L. Hu, M. X. Cui and H. Tian, Renewable Energy, 2012, 43, 179 CrossRef CAS.
- F. Kargi and E. C. Catalkaya, Int. J. Hydrogen Energy, 2011, 36, 8252 CrossRef CAS.
- C. S. Peng, Y. Y. Liu, J. J. Bi, H. Z. Xu and A. Ahmed, J. Hazard. Mater., 2011, 189, 814 CrossRef CAS PubMed.
- P. Li, C. Peng, F. Li, S. Song and A. Ahmed, Int. J. Environ. Res., 2011, 5, 797 CAS.
- C. S. Peng, R. J. Jin, G. Y. Li, F. S. Li and Q. B. Gu, Sep. Purif. Technol., 2014, 136, 42 CrossRef CAS.
- H. Strathmann, Ion-Exchange Membrane Separation Processes, Elsevier, Amsterdam, 2004 Search PubMed.
- J. Llanos, S. Cotillas, P. Cañizares and M. A. Rodrigo, Sep. Purif. Technol., 2014, 132, 362 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2016 |