
Advances in predicting organic contaminant abatement during ozonation of municipal wastewater effluent: reaction kinetics, transformation products, and changes of biological effects
Yunho
Lee
*a and
Urs
von Gunten
*bcd
aSchool of Environmental Science and Engineering, Gwangju Institute of Science and Technology (GIST), Gwangju 500-712, Republic of Korea. E-mail: yhlee42@gist.ac.kr; Fax: +82 62 7152434; Tel: +82 62 7152468
bSchool of Architecture, Civil, and Environmental Engineering (ENAC), École Polytechnique Fédérale de Lausanne, CH-1015, Lausanne, Switzerland
cEawag, Swiss Federal Institute of Aquatic Science and Technology, Ueberlandstrasse 133, P.O. Box 611, 8600 Duebendorf, Switzerland
dInstitute of Biogeochemistry and Pollutant Dynamics, ETH Zurich, CH-8092 Zurich, Switzerland. E-mail: vongunten@eawag.ch; Fax: +41 58 765 5028; Tel: +41 58 765 5270
First published on 3rd March 2016
Abstract
Ozonation of municipal wastewater effluent has been considered in recent years as an enhanced wastewater treatment technology to abate trace organic contaminants (micropollutants). The efficiency of ozonation for micropollutant abatement depends on (1) the reactivity of ozone and OH radical (˙OH) with the target micropollutant, (2) the dosage of ozone and the stability of ozone and ˙OH in a given water matrix, (3) the removal of undesirable effects (e.g., biological activities) of a micropollutant after structural transformation, and (4) the biodegradability of transformation products in biological post-treatment. In this article, recent advances in predicting organic micropollutant abatement during ozonation of municipal wastewater effluents are reviewed with a focus on (i) principle-based approaches for describing and modeling the reaction kinetics of ozone and ˙OH, (ii) transformation products and pathways, (iii) changes of biological activities, and (iv) biodegradation of transformation products in biological post-treatment. Using the chemical kinetics based on ozone and ˙OH rate constants (i.e., compound-specific information) and exposures (i.e., water matrix-specific information), a generalized prediction of the abatement efficiency of various micropollutants in varying water quality appears to be possible. QSAR-type correlations based on Hammett coefficients or quantum chemical energy calculations or (semi)empirical models have been developed for predicting the ozone and ˙OH rate constants and exposures, respectively. Models based on the ozone and ˙OH reaction rules can be used to predict the transformation products of micropollutants by ozone and ˙OH. Reaction rule-based models in combination with the chemical kinetics information will enable the prediction of transformation product evolution during ozonation. The biological activities of transformation products have been assessed by an effect-driven approach using in vitro bioassays. Biological activities with specific modes of action (e.g., receptor-binding activities) were found to be quite efficiently removed, upon slight structural modifications by ozone or ˙OH. The formation of new biological activities has also been observed, which warrants identification of the responsible toxicophore(s) and quantitative exposure-based risk assessment. Finally, there is only limited experimental information on the biodegradability of transformation products; however, biodegradability probability models can be used to make first estimates. In future research, the discussed principle-based approaches can be more actively applied to determine and predict not only the abatement levels of the parent micropollutants but also the formation of transformation products and the consequent changes of biological activities and biodegradability, which determines the overall treatment efficiency.
 Yunho Lee (left) and Urs von Gunten (right) | Yunho Lee is an assistant professor in the School of Environmental Science & Engineering at Gwangju Institute of Science and Technology (GIST), Korea. He received a Ph.D. in the Chemical Engineering Department at Seoul National University, Korea in 2005 and did postdoctoral research at the Swiss Federal Institute of Aquatic Science and Technology (Eawag) before joining GIST in 2011. His research focuses on characterizing and optimizing oxidative water treatment processes for drinking water and municipal and aquaculture wastewater for abating organic and biological contaminants of concern with minimized toxic by-product formation. |
Urs von Gunten is a full professor in the School of Architecture, Civil and Environmental Engineering at the Swiss Federal Institute of Technology, Lausanne (EPFL) since 2011 and a senior scientist at the Swiss Federal Institute of Aquatic Science and Technology (Eawag) since 1995. He received a Ph.D. in chemistry from the Swiss Federal Institute of Technology, Zürich (ETHZ) in 1989 and joined Eawag thereafter as a postdoctoral researcher and later on as a group leader. His research focuses on water quality and water treatment issues related to drinking water and wastewater with a strong emphasis on kinetics and mechanisms of oxidation and disinfection processes. Furthermore, novel tools to characterize oxidation processes and process control systems are developed in his research group. He is also involved in up-scaling of treatment processes from laboratory- to full-scale systems. |
Water impactOzonation, a widely applied technology for drinking water treatment, has received increasing attention as an enhanced municipal wastewater treatment technology to improve the effluent quality by abating various organic contaminants (micropollutants). In this tutorial review, we describe and discuss recent advances in principle-based tools to assess and predict the micropollutant abatement efficiency during ozonation of municipal wastewater effluents. A wealth of information on the reaction kinetics of ozone and ˙OH with micropollutants and wastewater effluent matrix components has been accumulated, which allows generalized prediction of the abatement levels of micropollutants. Substantial progress has also been made in identifying and predicting the formation of transformation products of micropollutants and the consequences for changes of bioactivities and biodegradability, which can be applied for emerging micropollutants. The described current knowledge and future research needs on these topics will be very useful for researchers and practitioners who consider applications of ozonation for micropollutant abatement in municipal wastewater treatment. |
1. Introduction
Municipal wastewater treatment plants (WWTPs) receive various industrially synthesized or naturally produced compounds such as hormones, pharmaceuticals, personal care products, as well as biocides and their abiotic or biotic transformation products.1–3 These contaminants can be transformed or removed during biological wastewater treatment processes; however, the effluents still contain trace levels (from ng L−1 to μg L−1) of these contaminants,4,5 some of which can negatively impact the health of aquatic ecosystems or the quality of drinking water resources. The presence of steroid estrogens in sewage effluents has received great attention as these compounds can induce fish feminization.6,7 The presence of various classes of antibiotics has also raised concern as they can cause spreading of antibiotic resistance among the bacterial population in receiving waters, which in turn can affect the antibiotic resistance rate of human pathogens.8 These trace chemical contaminants have been termed differently depending on research groups or communities such as TOrC (trace organic contaminants), CEC (contaminants of emerging concern) or PPCP (pharmaceuticals and personal care products). The term ‘micropollutant’ has been widely used in some European communities and will be mainly used throughout this paper.1Advanced water treatment processes based on oxidation, adsorption, and membrane separation have been tested as options to upgrade the quality of municipal wastewater effluents by eliminating organic micropollutants.2,5 This paper will focus on oxidation by ozone and hydroxyl radicals (˙OH). Ozonation for wastewater treatment can be considered an advanced oxidation process (AOP) because ozone is quickly transformed into ˙OH due to the reaction of ozone with dissolved effluent organic matter.9,10 Ozonation has recently been intensively tested as an enhanced wastewater treatment technology in laboratory-, pilot- and full-scale studies.11–20 In Switzerland, ozonation followed by biological filtration has recently been implemented and operated in full-scale treatment for the purpose of micropollutant abatement.21 Previous studies have shown that significant abatement of a large array of micropollutants and a decrease in a range of biological effects can be achieved.22 It has also been demonstrated that in certain wastewaters, undesired oxidation by-products (e.g., aldehydes, bromate, and N-nitrosodimethylamine (NDMA)) or cytotoxicity/genotoxicity were formed upon ozonation, which has to be considered for an upgrade of a WWTPs.23–33 It should be noted that some of these oxidation by-products such as aldehydes can be readily removed in biological post-treatment.17,18,34 NDMA has also been shown to be biodegradable35 and significant abatement of NDMA was found in biological post-treatment steps in some full-scale ozonation studies.13,17 Nevertheless, since the formation of NDMA is still unpredictable and its degradation in biological post-treatment can be quite variable, an assessment of the fate of NDMA is important.32 The formation of bromate can be important especially for indirect potable reuse settings as bromate does not typically degrade in receiving water bodies under aerobic conditions. Bromate formation is typically lower than 40 μg L−1 during ozonation of wastewater effluents at typical specific ozone doses (e.g., 1 gO3/gDOC). In these cases, the dilution of wastewater effluents in receiving waters would be usually sufficient to reduce the bromate concentration below the drinking water guidelines (i.e., 10 μg L−1).32 However, in any case, ozonation should be optimized to keep the bromate level as low as possible. Overall, because the formation of bromate or NDMA strongly depends on the presence of specific precursors (i.e., bromide or NDMA precursors), the suitability of ozonation should be tested in the planning stage of a WWTP upgrade.32
The suitability of ozonation for micropollutant abatement depends on the following four aspects (Scheme 1). Firstly, the reactivity of oxidizing agents (O3 and ˙OH) with the target micropollutant and the stability of the oxidizing agents in water matrices determine the transformation efficiency of the parent micropollutant. To this end, quantification of the reactivity of the oxidizing agents with wastewater matrices such as effluent organic matter is more important as compared to cleaner drinking water sources such as groundwater. Secondly, organic micropollutants are not mineralized but only partially transformed under typical treatment conditions. Therefore, it is important to assess the removal of undesired effects of a micropollutant after structural transformation. If the transformation products contain some residual or even higher bioactivity compared to the parent compound, the structural identification of the transformation products can be of interest. Thirdly, the biodegradability of transformation products should be assessed experimentally or with biodegradability probability models. Finally, the possible formation of toxic oxidation by-products such as bromate, NDMA, or other unidentified cytotoxic/genotoxic compounds from the reaction of the oxidizing agents with water matrix components should be considered.
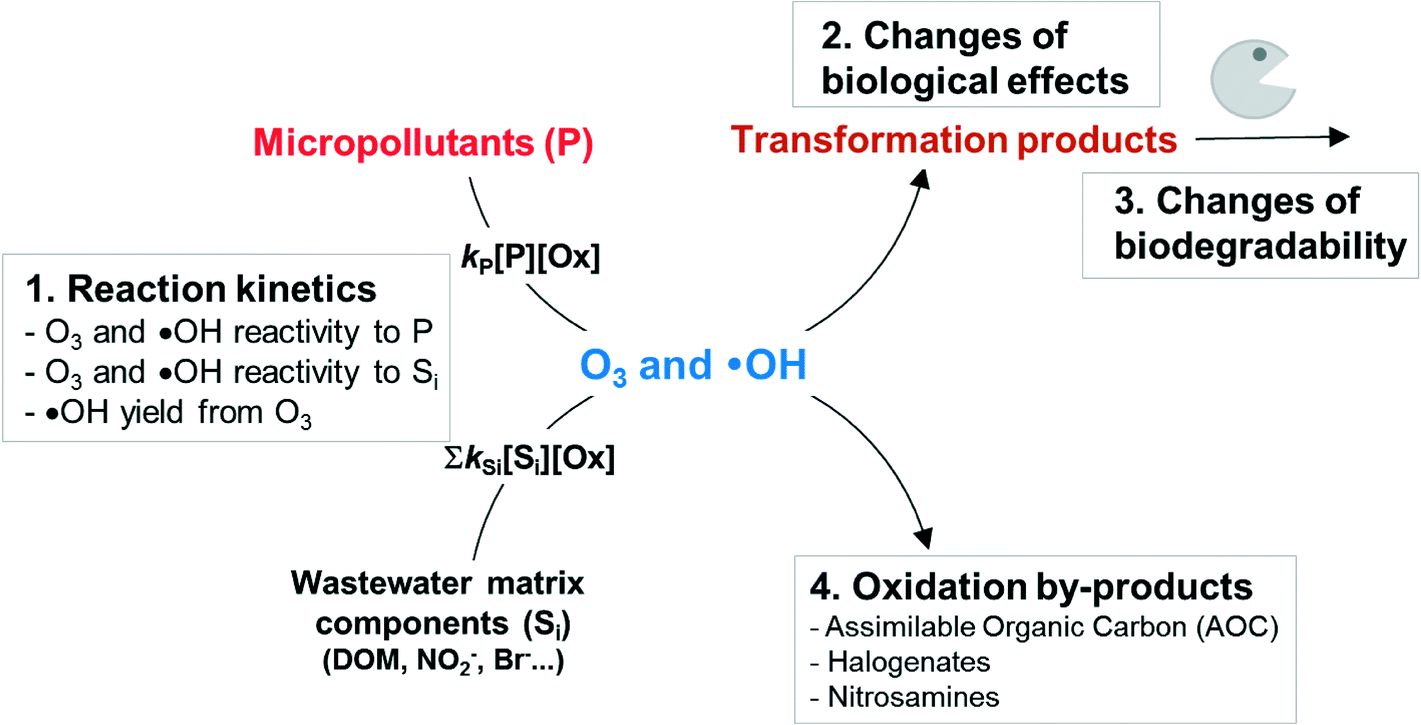 | ||
| Scheme 1 The four critical aspects that should be considered for the efficiency/feasibility of the ozonation process for micropollutant abatement (adapted from von Gunten, 2003):36 (1) reactivity of the target compound with ozone and/or hydroxyl radical, and the stability of these oxidants in a given water matrix, (2) formation of transformation products and their biological effects, (3) biodegradability of transformation products, and (4) formation of oxidation by-products from the reaction of ozone and/or hydroxyl radicals with water matrix components. | ||
This tutorial review aims to provide recent advances in predicting organic micropollutant abatement during ozonation of municipal wastewater effluents. The focus will be on principle-based prediction tools for the reaction kinetics of ozone and ˙OH with micropollutants and wastewater effluent matrix components (section 2) and the transformation products of micropollutants and the consequences for changes of bioactivities and biodegradability (section 3). The formation and mitigation of oxidation by-products from the wastewater matrix components will only be covered for the formation of NDMA. Bromate formation and mitigation is covered elsewhere.32,33,37 Future research needs for optimizing the ozonation process for micropollutant abatement in municipal wastewater treatment will also be discussed.
2. Kinetic models and parameters
2.1. Master kinetic equations
The abatement of a micropollutant (MP) during ozonation occurs by oxidation by both ozone and ˙OH. The kinetics of MP abatement can be formulated by eqn (1). An integration of eqn (1) over the reaction time in an ideal batch or plug-flow reactor yields eqn (2).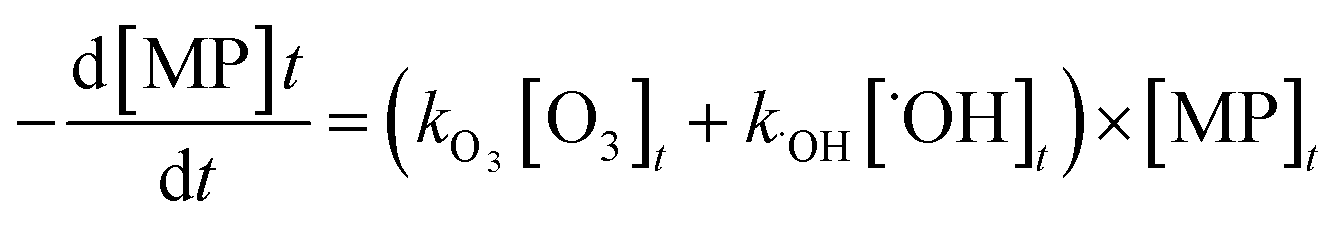 | (1) |
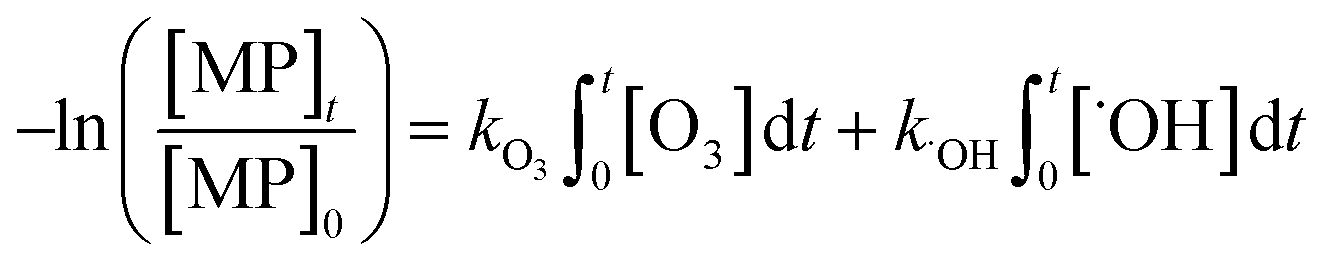 | (2) |
According to eqn (2), the abatement rate of a MP (e.g., % abatement rate = 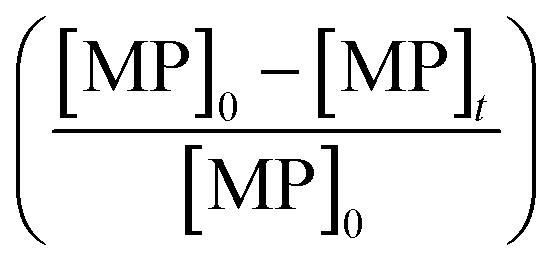 ×100) can be predicted if the two second-order rate constants (i.e., kO3 and k˙OH) and the two exposures (i.e.,
×100) can be predicted if the two second-order rate constants (i.e., kO3 and k˙OH) and the two exposures (i.e., 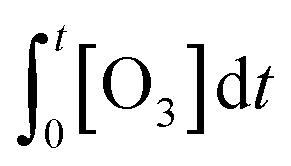 and
and 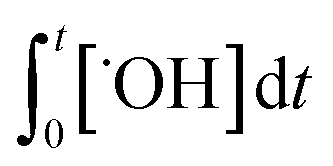 ) are known. kO3 and k˙OH are physical chemical constants and reflect the reactivity of MP towards ozone and ˙OH, respectively, whereas,
) are known. kO3 and k˙OH are physical chemical constants and reflect the reactivity of MP towards ozone and ˙OH, respectively, whereas, 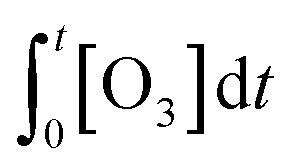 and
and 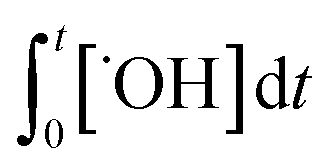 are related to the stability of ozone and ˙OH, respectively, in a given water matrix. In eqn (2), the rate constant parameters are independent of the oxidant exposure parameters. This implies that the second order rate constants can be obtained independently of the water matrices, by laboratory experiments in well-defined model systems or by predictions based on quantitative structure–activity relationships (QSARs) or quantum chemical calculations (see below). It is also important to predict the ozone and ˙OH exposures as a function of ozone dose and reaction time, which are the two major process operation parameters. Further details of each kinetic parameter will be discussed below.
are related to the stability of ozone and ˙OH, respectively, in a given water matrix. In eqn (2), the rate constant parameters are independent of the oxidant exposure parameters. This implies that the second order rate constants can be obtained independently of the water matrices, by laboratory experiments in well-defined model systems or by predictions based on quantitative structure–activity relationships (QSARs) or quantum chemical calculations (see below). It is also important to predict the ozone and ˙OH exposures as a function of ozone dose and reaction time, which are the two major process operation parameters. Further details of each kinetic parameter will be discussed below.
2.2. Second-order rate constants for the reactions of micropollutants with ozone (kO3)
Most aqueous ozone reactions follow second-order reaction kinetics, first order in ozone and first order in the target compound.38,39 Currently, a few hundred kO3 values are known for various organic compounds including contaminants found in municipal wastewaters.22 Ozone is a selective oxidant and reacts rapidly only with compounds containing electron-rich moieties (ERMs), such as phenols, anilines, olefins, reduced sulfur, and neutral amine moieties.40,41Fig. 1 shows the kO3 values as a function of pH for phenol, aniline, ethene, dimethylsulfide, trimethylamine, dimethylamine, and methylamine, which represent the ozone-reactive organic functional groups encountered in micropollutants. For ionizable compounds such as phenol and amines, the kO3 values significantly increase with increasing pH due to the enhanced ozone reactivity of the deprotonated/neutral forms of phenolic and amine moieties.38 The pH-dependent second-order rate constant (kO3) for monoprotic phenols can be calculated based on eqn (3):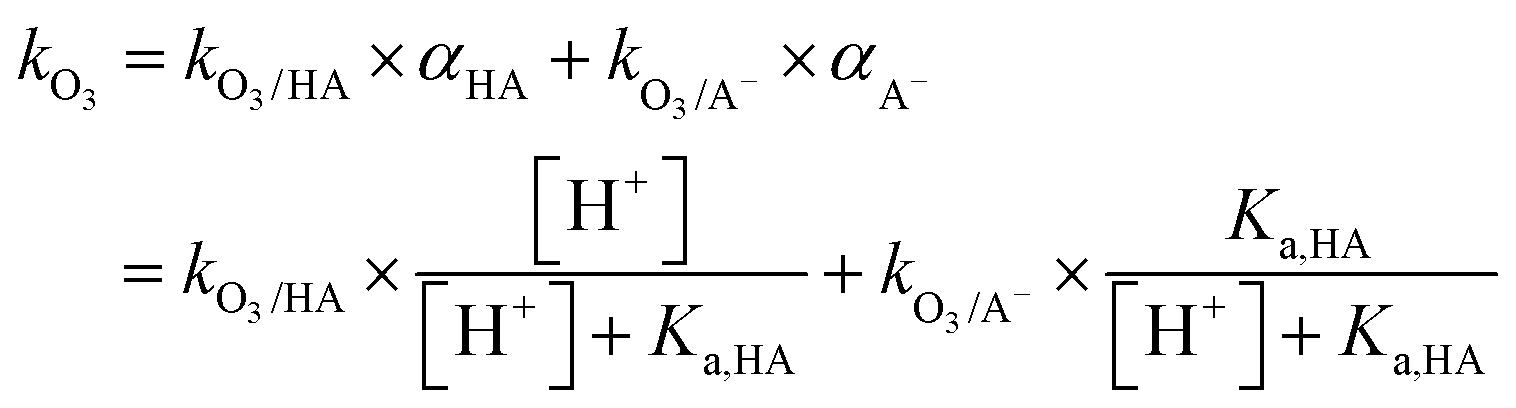 | (3) |
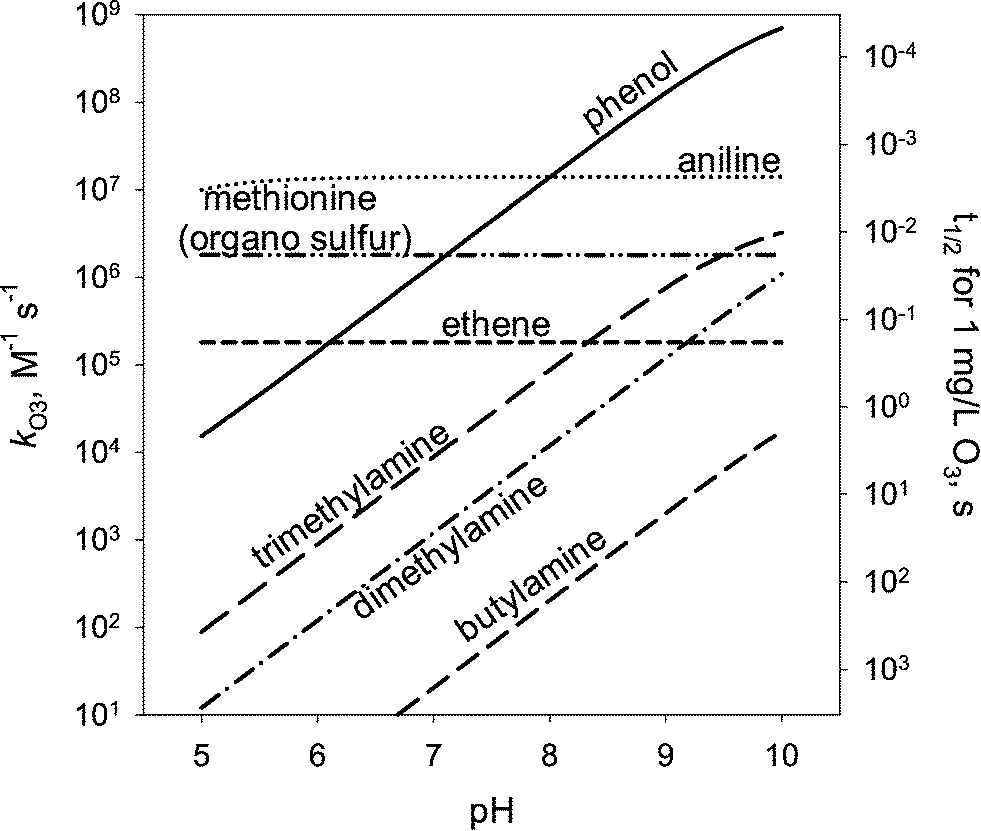 | ||
| Fig. 1 pH-dependent second-order rate constants and half-lives (t1/2) for the reaction of ozone (kO3) with electron-rich organic compounds: phenol, aniline, ethene (olefin), methionine (organic sulfur), trimethylamine (tertiary amine), dimethylamine (secondary amine), and butylamine (primary amine). The half-lives are calculated for an ozone concentration of 1 mg L−1. The kO3 values were taken from von Sonntag and von Gunten, 2012.22 | ||
The kO3 values in Fig. 1 for the simple structure of ERMs can serve as a good basis to predict the kO3 values for micropollutants with more complex structures. Good correlations such as quantitative structure–activity relationships (QSARs) have been found between the kO3 values for compounds having a common ERM (phenols, anilines, olefins, and amines) and substituent descriptor variables such as Hammett (σ) or Taft sigma (σ*) constants.41 These (semi)empirically-obtained sigma constants express quantitatively the electron-withdrawing or electron-donating properties of substituents.42 Hammett σ and Taft σ* constants have been typically applied to aromatic and aliphatic compounds, respectively. Table 1 summarizes the QSARs established for the reaction of ozone with phenols, phenolates, anilines, benzene derivatives, olefins, and amines based on σ or σ*. These sigma constant-based QSARs were found to predict the kO3 values within a factor of 1/3 to 3 compared to the measured values.41 QSARs using Hammett or Taft constants, while providing good predictions, are often restricted to a set of compounds with similar structures (e.g., a series of substituted phenols), requiring several QSARs to be developed. For aromatic compounds, the QSARs for phenols, anilines, and substituted benzenes had to be separately developed (see Table 1). In addition, Hammett or Taft constants are not available for certain substituents, which limits the QSAR applications.
| Compound class | QSAR equation | r 2 | S y.x | n |
|---|---|---|---|---|
| a Coefficient of correlation. b Standard deviation of the linear regression. c Number of data points. | ||||
| Hammett/Taft descriptors | ||||
| Non-dissociated phenols | log(kO3) = 3.53 − 3.24∑σ+o,m,p | 0.81 | 0.38 | 24 |
| Dissociated phenols | log(kO3) = 8.80 − 2.27∑σ+o,m,p | 0.96 | 0.26 | 13 |
| Anilines | log(kO3) = 7.15 − 1.54∑σ−o,m,p | 0.85 | 0.33 | 14 |
| Benzene derivatives | log(kO3) = −0.04 − 3.35∑σ+p | 0.93 | 0.72 | 50 |
| Olefins | log(kO3) = 6.18 − 0.49∑σ* | 0.86 | 0.51 | 48 |
| Aliphatic amines | log(kO3) = 6.13 − 1.00∑σ* | 0.86 | 0.61 | 54 |
| Quantum chemical descriptors | ||||
| Phenols | log(kO3) = 27.37 + 2.69EHOMO | 0.94 | 0.53 | 35 |
| Anilines | log(kO3) = 34.60 + 3.41EHOMO | 0.86 | 0.45 | 16 |
| Benzene derivatives | log(kO3) = 23.13 + 2.45EHOMO | 0.81 | 0.63 | 40 |
| Olefins | log(kO3) = 18.54 + 1.32ENBO,C–C(π) | 0.82 | 0.70 | 45 |
| Aliphatic amines | log(kO3) = 15.80 + 0.83ENBO,LP-N | 0.83 | 0.87 | 59 |
Instead of empirical σ or σ* constants, quantum chemical molecular orbital descriptors have recently been tested for their correlation with kO3 with an aim of developing QSARs.43,44 Good correlations were found between the kO3 values for aromatic compounds and the energy of the highest occupied molecular orbital (HOMO). For olefins and amines as aliphatic compounds, the kO3 values were well correlated with the energy of a localized molecular orbital such as the natural bond orbital (NBO) energy of the C–C π bond and the NBO energy of the lone pair electrons of amines, respectively (Table 1).44 It was found that the quantum molecular descriptors are useful surrogates for the traditional σ or σ* constants for QSARs of the ozone reactions, especially when σ or σ* are not available for substituents. The kO3 values predicted by the quantum molecular descriptor-based QSARs were found to be within a factor of 1/4 to 4 compared to the measured values.44
2.3. Second-order rate constants for the reactions of target compounds with ˙OH (k˙OH)
More than a thousand k˙OH values are known for various organic compounds and organic contaminants in waters.45–48 ˙OH is a less selective oxidant compared to other drinking water oxidants and reacts rapidly with most organic compounds. The reaction of ˙OH occurs by (1) addition to an aromatic system, olefin, or lone electron pairs of nitrogen or sulfur, (2) hydrogen abstraction, and (3) electron transfer (e.g., halogenated phenols). Among these reaction mechanisms, the addition reactions are usually the fastest with a close to diffusion-controlled rate constants (≥5 × 109 M−1 s−1). H-abstraction reactions are typically slower than addition reactions; however, the overall rate constants via H-abstraction can be significant for organic compounds with multiple C–H bonds. Therefore, k˙OH values in the range of 5 × 109–1010 M−1 s−1 are observed for compounds containing aromatic rings, olefins, organic sulfur, free amine, or aliphatic groups with multiple H-atoms.A group contribution method (GCM) can be used to predict the aqueous k˙OH values of organic compounds.47 In this method, the k˙OH per reaction mechanism is estimated for (1) H-abstraction, (2) addition to olefins, (3) addition to aromatic compounds, and (4) addition to N, P, and S moieties. Each of these reaction mechanisms has a ‘group rate constant’ and a ‘group contribution factor’, which represent the reactivity of a reference reaction and the effect of neighboring groups (or substituents) on the reactivity of a reference reaction, respectively. Currently, 66 group rate constants and 80 group contribution factors are available for the aforementioned four ˙OH reaction mechanisms, which were determined based on 310 k˙OH values.47 The total k˙OH is obtained by the sum of individual k˙OH per reaction mechanism, which corresponds to the substructures of a compound. The GCM method has been shown to predict the experimental k˙OH values within a factor of 2 compared to the measured k˙OH values.47
Recent studies have shown that the k˙OH per reaction mechanism can also be predicted by linear free energy relationships (LFERs) based on a quantum chemical calculation method. LFERs were found between the k˙OH per reaction mechanism such as H abstraction from C–H bonds, addition to alkenes, and addition to aromatic compounds vs. the corresponding quantum mechanically calculated aqueous phase free energy of activation (ΔGactrxn) (Minakata and Crittenden, 2011; Minakata et al., 2014; Minakata et al., 2015).49–51 The LFER method has been shown to predict the experimental k˙OH values within a factor of 2–5 compared to the measured k˙OH values. The LFER method is also a useful alternative to the GCM method when the group rate constants or group contribution factors are not available. Furthermore, the electronic, steric, and solvation effects of neighboring groups can be considered in the quantum chemical calculation LFER method, which is lacking in the GCM method based on empirical correlations. The GCM and LFER methods have been used to predict the k˙OH values of emerging organic contaminants.52–54
2.4. Ozone exposure
Ozone exposure can be determined by integrating the ozone decay curves over time (von Gunten and Hoigné, 1994).55 The ozone decay characteristics in real waters and the effect of water quality parameters such as dissolved organic matter (DOM), alkalinity, pH, and temperature on the ozone decay have been investigated over several decades.22,36 Ozone decomposition in DOM-containing waters typically shows multi-phasic kinetics; however, it is often approximated by an empirical bi-phasic approach, i.e., a rapid consumption of added ozone within a few seconds to a minute (typically termed as instantaneous or spontaneous ozone demand) followed by a relatively slower ozone decrease during the remaining reaction time (von Sonntag and von Gunten, 2012).22 The instantaneous (spontaneous) ozone demand is attributed to the reaction of ozone with reactive moieties present in DOM such as phenols, neutral amines, or olefins (Buffle and von Gunten, 2006).56For typical ozone doses applied to wastewater effluent matrices, ozone is usually completely consumed in 5–20 min, which is more rapid compared to many relatively clean natural waters57 due to the higher concentration of DOM. High levels of nitrite are formed during partial nitrification in biological wastewater treatment, which can be a critical factor in ozone applications, since it consumes ozone quickly with a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 molar stoichiometry without generating ˙OH, NO2− + O3 → NO3− + O2, kO3 = 3.7 × 105 M−1 s−1 (Naumov et al., 2010).58 As DOM and nitrite usually control the ozone consumption characteristics in wastewater effluents, it is of interest to compare the ozone exposure as a function of specific ozone dose, i.e., the DOC-normalized and nitrite-corrected ozone doses (i.e., gO3/gDOC* = gO3/gDOC − (48/14)(gNO2-N/gDOC) to compare the variability/consistency of the ozone exposure across wastewater matrices.
:1 molar stoichiometry without generating ˙OH, NO2− + O3 → NO3− + O2, kO3 = 3.7 × 105 M−1 s−1 (Naumov et al., 2010).58 As DOM and nitrite usually control the ozone consumption characteristics in wastewater effluents, it is of interest to compare the ozone exposure as a function of specific ozone dose, i.e., the DOC-normalized and nitrite-corrected ozone doses (i.e., gO3/gDOC* = gO3/gDOC − (48/14)(gNO2-N/gDOC) to compare the variability/consistency of the ozone exposure across wastewater matrices.
Fig. 2 summarizes the ozone exposure data available in the literature as a function of the specific ozone dose during ozonation of municipal wastewater effluents. In all cases, the ozone exposures were determined after complete depletion of ozone (usually less than 20 min). The ozone exposures for the same specific ozone dose were quite variable (more than a factor of 4 variations), indicating that differences in the DOM characteristics and other water quality parameters such as carbonate (1.0–7.7 mM as HCO3−) and pH (6.9–8.2) affect the ozone decay characteristics, in addition to the quantity of DOM (DOC or TOC = 2.4–15 mgC L−1) or nitrite (0–0.77 mgN L−1). Furthermore, analytical uncertainties in DOC or TOC measurements due to differing sample preparation methods or oxidation efficiency in the organic carbon analyzer can also be responsible for part of the observed variability in the ozone exposures (note that both DOC or TOC were used in the studies presented in Fig. 2).
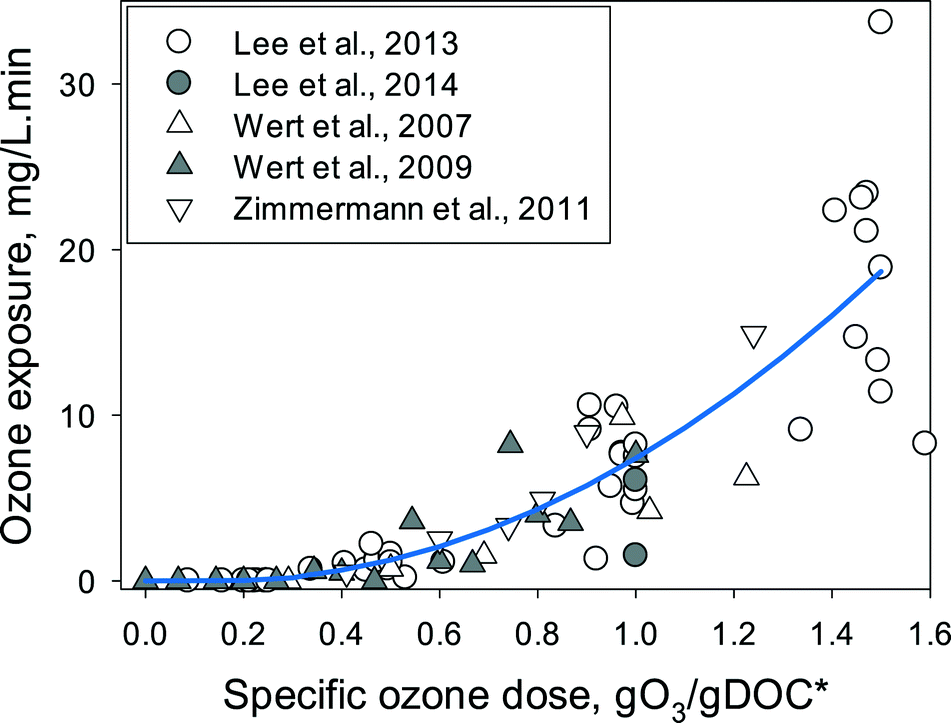 | ||
| Fig. 2 Ozone exposures determined for various specific ozone doses (gO3/gDOC*) in municipal wastewater effluents (DOC or TOC = 2.4–15 mgC L−1, pH = 6.9–8.2, alkalinity = 1.0–7.7 mM as HCO3−, and nitrite = 0–0.77 mgN L−1). The specific ozone dose represents the DOC-normalized and nitrite-corrected ozone doses, i.e., gO3/gDOC* = gO3/gDOC − (48/14)(gNO2-N/gDOC). Ozone exposure data were taken from Lee et al., 2013 (ref. 52) and 2014,53 Wert et al., 2007 (ref. 23) and 2009,59 and Zimmermann et al., 2011.17 The line represents the fitting of the data with the empirical equations described in the main text (eqn (4)). | ||
The ozone exposures remained zero at low specific ozone doses ≲0.3 gO3/gDOC* and started to increase with increasing specific ozone doses. The ‘zero’ ozone exposure at low ozone doses is due to the complete consumption of ozone before the first experimental measurement in a conventional kinetic method (10–30 s, spontaneous ozone demand). Nevertheless, even at such negligible ozone exposures, significant elimination of micropollutants was found during wastewater ozonation.52 This indicates that the ozone exposure at low ozone doses is observed to be zero, only due to the limited time resolution of the experimental method, and the actual ozone exposure is not zero. Thus, accurate determination of the ozone exposures requires ozone measurements with higher time resolutions in the range of < seconds, which can be achieved by e.g., quench-flow systems (Buffle et al. 2006).10 As an alternative method, ozone exposure can be estimated from the decrease in ozone-reactive compounds which has been demonstrated in some studies albeit large differences in the estimated ozone exposures were observed depending on the tested ozone reactive compound.11,53,60 The following empirical equation can be derived from the fitting of the measured data from the literature (Fig. 2, line) and used for a rough estimation of the ozone exposure as a function of the specific ozone dose (gO3/gDOC*). It should be noted that due to the complex ozone chemistry in real waters and the non-linear relationship between the ozone exposure and the specific ozone dose, the prediction of the ozone exposure is currently possible only empirically by curve fitting with large uncertainties (more than a factor of 4).
| O3 exposure (M s) = fO3(gO3/gDOC*) = (1.3 × 10−7) exp(25gO3/gDOC*) for 0 < gO3/gDOC* ≤ 0.3 = (1.3 × 10−2)(gO3/gDOC* − 0.3)2 + (3.8 × 10−3)(gO3/gDOC* − 0.3) + (3.0 × 10−4) for gO3/gDOC* > 0.3 | (4) |
2.5. ˙OH exposure
˙OH exposure can be calculated by integrating the ˙OH concentration over the reaction time.61 With ˙OH as a short-lived compound (lifetime of μs), its transient concentration can be calculated as the ratio of ˙OH formation rate (ν˙OH, M s−1) to the first-order ˙OH consumption rate by the water matrix components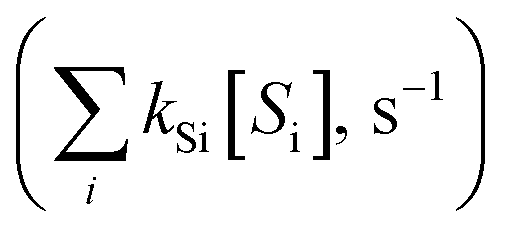 , i.e., [˙OH] =
, i.e., [˙OH] = 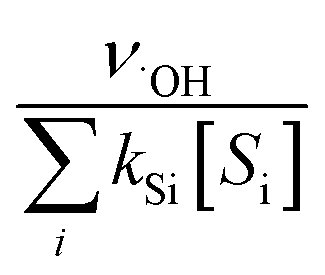 . The term,
. The term,  , can be calculated by getting the sum of the product of the concentration of each water matrix component (Si) and the corresponding second-order rate constant for its reaction with ˙OH (kSi). The ˙OH formation rate is related to the ozone decomposition rate by ν˙OH = ηνO3 in which η is the molar ˙OH yield from ozone decomposition. Integration of the ˙OH concentration over time yields eqn (5), which relates ˙OH exposure to the molar ˙OH yield (η), the concentration of ozone consumption (ΔO3), and the ˙OH consumption rate constant by the matrix components
, can be calculated by getting the sum of the product of the concentration of each water matrix component (Si) and the corresponding second-order rate constant for its reaction with ˙OH (kSi). The ˙OH formation rate is related to the ozone decomposition rate by ν˙OH = ηνO3 in which η is the molar ˙OH yield from ozone decomposition. Integration of the ˙OH concentration over time yields eqn (5), which relates ˙OH exposure to the molar ˙OH yield (η), the concentration of ozone consumption (ΔO3), and the ˙OH consumption rate constant by the matrix components 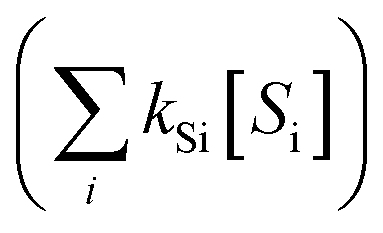 . ˙OH exposure can also be experimentally determined from the decrease in an ozone-recalcitrant compound such as para-chlorobenzoic acid (pCBA, k˙OH,pCBA = 5 × 109 M−1 s−1) as shown in eqn (5).22,61
. ˙OH exposure can also be experimentally determined from the decrease in an ozone-recalcitrant compound such as para-chlorobenzoic acid (pCBA, k˙OH,pCBA = 5 × 109 M−1 s−1) as shown in eqn (5).22,61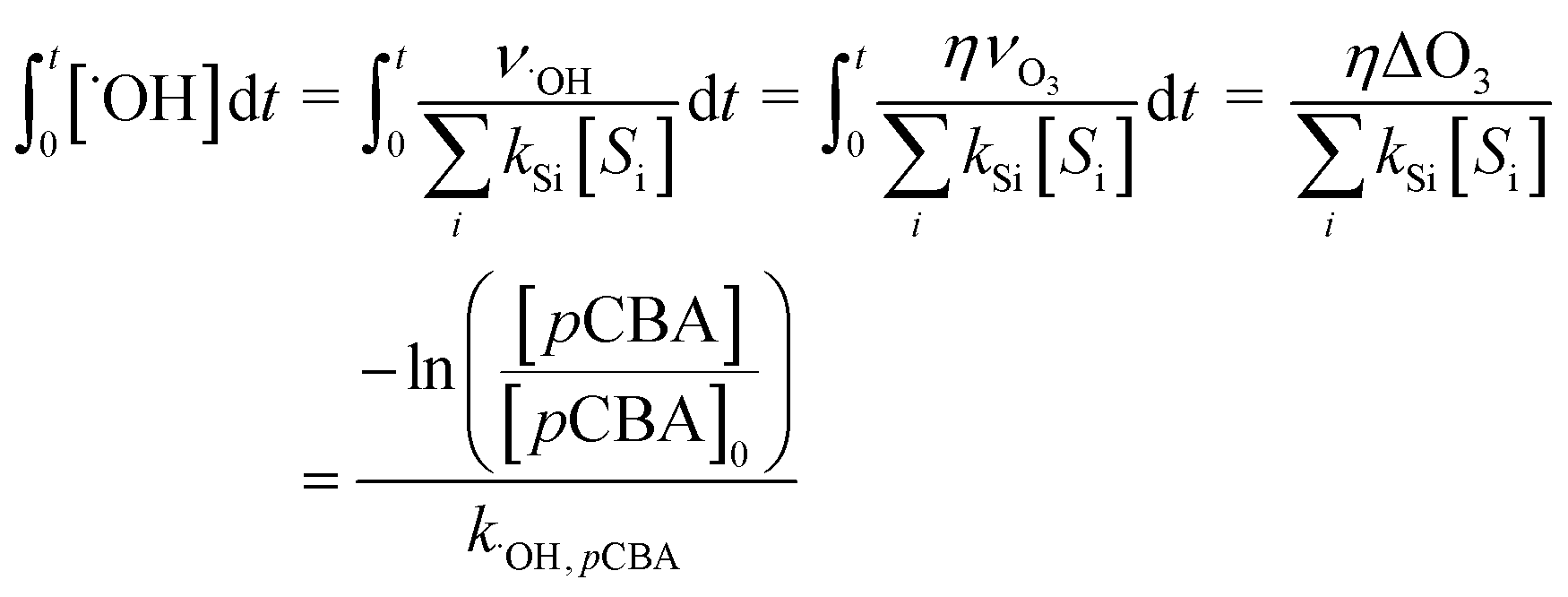 | (5) |
Fig. 3 summarizes the ˙OH exposures determined in various wastewater effluents as a function of the specific ozone dose. The ˙OH exposure increases minimally within the specific ozone dose range of 0–0.2 gO3/gDOC* and then starts to increase significantly with further increasing specific ozone doses. The lower ˙OH yield for low specific ozone doses could be due to the ozone consumption by DOM moieties with less ˙OH formation such as olefins or tertiary amines.22 Alternatively, it could be a reflection of the higher ˙OH yield at the larger specific ozone doses. It is expected that for low ozone doses the ozone decomposition occurs mainly by the ozone reaction with DOM moieties such as phenols, activated aromatics, olefins, or neutral amines in which the primary molar ˙OH yields from the reaction of ozone with these moieties are reported to be in the range of 0–40%.22 For larger ozone doses, the ozone reactive DOM moieties are exhausted, and thus the ozone decomposition is more driven by the superoxide radical-mediated propagation reaction, i.e., ˙OH + DOM + O2 → → O2˙− and O3 + O2˙− → → ˙OH.36,62 As the theoretical ˙OH yield from the ozone reaction with the superoxide radical (O2˙−) is 100%, the ˙OH yields in the propagation reactions for ozone consumption are expected to be higher compared to those of ozone reaction with the DOM moieties.63 It should be noted that the ˙OH scavenging rate by DOM or carbonate species does not decrease during ozonation at typical ozone doses because the ozone or ˙OH degradation products of DOM exhibit the same or similar reactivity to ˙OH compared to the respective unreacted DOM. Furthermore, the carbonate concentration is not affected by its reaction with ˙OH.40,63
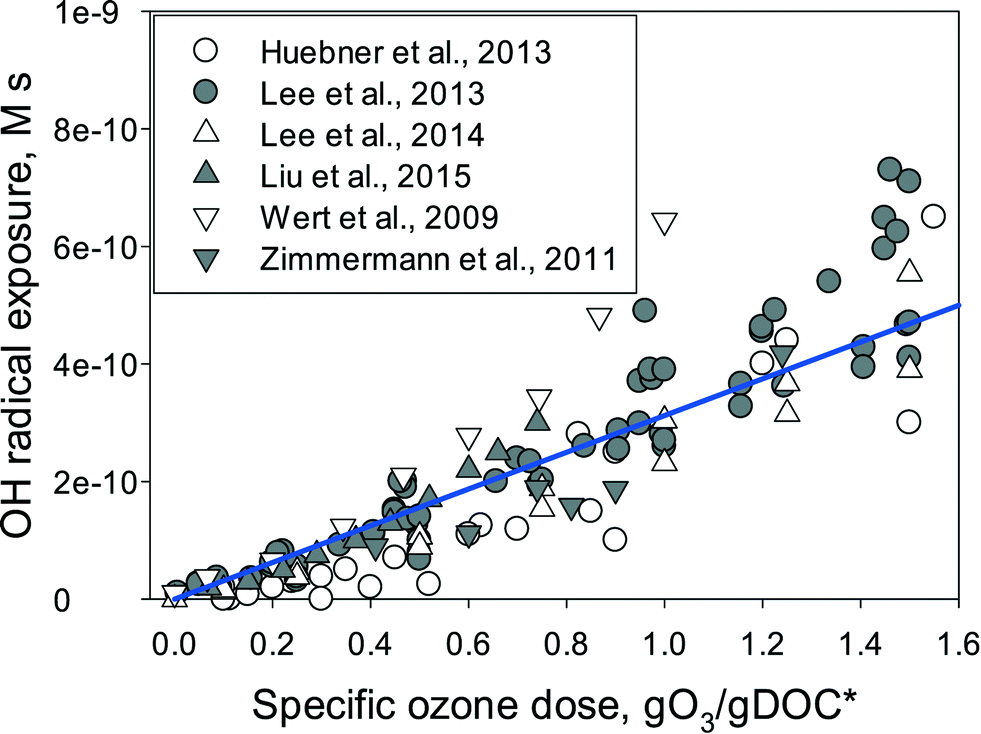 | ||
| Fig. 3 ˙OH exposure as a function of specific ozone dose (gO3/gDOC*) for ozonation of wastewater effluents. Symbols represent the measured data, which were calculated from the decrease in para-chlorobenzoic acid (pCBA) as a ˙OH probe compound. The line indicates the model prediction with eqn (6) using a molar ˙OH yield of 0.5 (η = 0.5) and under the assumption that DOM is the dominant ˙OH consumer with a k˙OH,DOC of 4 × 108 Mc−1 s−1, which is in the range of measured rate constants for the reaction of ˙OH with effluent DOM.40,52,63–66 | ||
The ˙OH yield (η) can be estimated based on eqn (5) (i.e., 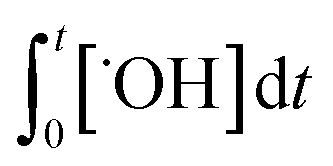 =
= 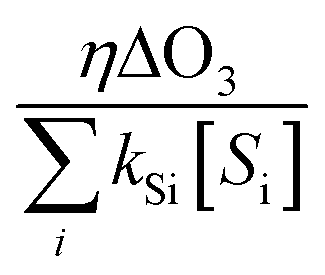 ) if the ˙OH consumption rate by the water matrix components, i.e.,
) if the ˙OH consumption rate by the water matrix components, i.e., 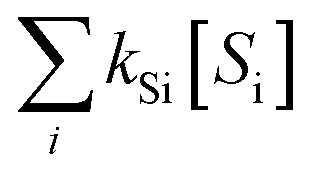 , s−1, is known. In typical municipal wastewater effluents, DOM and carbonate species are the major ˙OH scavengers. Nitrite (NO2−), despite its high reaction rate with ˙OH (k˙OH,NO2− = 1.0 × 1010 M−1 s−1),46 is quickly consumed by its rapid reaction with ozone, thus it is not a significant ˙OH scavenger during ozonation. The rate constants for the reaction of ˙OH with wastewater DOM have been measured on the basis of the dissolved organic carbon concentration (k˙OH,DOC) and range from 1.2 × 108 Mc−1 s−1 to 14.1 × 108 Mc−1 s−1.40,52,63–66 The average value from these studies is (4.0 ± 3.2) × 108 Mc−1 s−1. The ˙OH reaction rate constants with HCO3− and CO32− are: k˙OH,HCO3− = 8.5 × 106 M−1 s−1 and k˙OH,CO32− = 3.9 × 108 M−1 s−1, respectively.46 Taking a wastewater effluent containing 6 mg L−1 of DOC, 150 mg L−1 of alkalinity (as CaCO3), and pH 7.5 (as a representative effluent quality), the overall ˙OH scavenging rate is calculated to be 2.2 × 105 s−1 in which the relative contributions of DOM and carbonate are 92% and 8%, respectively. This exercise indicates that DOM is the major ˙OH scavenger during ozonation of typical municipal wastewater effluents. By assuming that DOM is the dominant ˙OH scavenger in the wastewater effluents in Fig. 3 and using a k˙OH,DOC of 4 × 108 Mc−1 s−1, ˙OH yields (η) were calculated to be 49(±18)% for specific ozone doses ≥0.3 gO3/gDOC*. This indicates that ∼0.5 mole of ˙OH is produced on average per mole of ozone during wastewater effluent ozonation (η = 0.5). This is similar to a molar ˙OH yield of 0.55 which was determined during ozonation of benzoic acid at pH 10.5 in an earlier ozonation study.67 ˙OH yields much lower than 0.5 (e.g., 9–24%) were also reported in some studies in which the ˙OH yield was determined by measuring formaldehyde formation in the presence of excess tert-butanol.52,68 It should be noted, however, that the presence of tert-butanol suppresses subsequent ˙OH radical reactions that may have otherwise increased the ˙OH radical yield substantially through the ˙O2−induced chain reaction via promoters.22 Therefore, the ˙OH radical yields determined by the tert-butanol method should be considered as the primary ˙OH radical yields mainly from the direct reaction of ozone with DOM in wastewater effluents. Finally, eqn (6) can be used for a rough estimate of ˙OH exposure as a function of the specific ozone dose (shown as the line in Fig. 3).
, s−1, is known. In typical municipal wastewater effluents, DOM and carbonate species are the major ˙OH scavengers. Nitrite (NO2−), despite its high reaction rate with ˙OH (k˙OH,NO2− = 1.0 × 1010 M−1 s−1),46 is quickly consumed by its rapid reaction with ozone, thus it is not a significant ˙OH scavenger during ozonation. The rate constants for the reaction of ˙OH with wastewater DOM have been measured on the basis of the dissolved organic carbon concentration (k˙OH,DOC) and range from 1.2 × 108 Mc−1 s−1 to 14.1 × 108 Mc−1 s−1.40,52,63–66 The average value from these studies is (4.0 ± 3.2) × 108 Mc−1 s−1. The ˙OH reaction rate constants with HCO3− and CO32− are: k˙OH,HCO3− = 8.5 × 106 M−1 s−1 and k˙OH,CO32− = 3.9 × 108 M−1 s−1, respectively.46 Taking a wastewater effluent containing 6 mg L−1 of DOC, 150 mg L−1 of alkalinity (as CaCO3), and pH 7.5 (as a representative effluent quality), the overall ˙OH scavenging rate is calculated to be 2.2 × 105 s−1 in which the relative contributions of DOM and carbonate are 92% and 8%, respectively. This exercise indicates that DOM is the major ˙OH scavenger during ozonation of typical municipal wastewater effluents. By assuming that DOM is the dominant ˙OH scavenger in the wastewater effluents in Fig. 3 and using a k˙OH,DOC of 4 × 108 Mc−1 s−1, ˙OH yields (η) were calculated to be 49(±18)% for specific ozone doses ≥0.3 gO3/gDOC*. This indicates that ∼0.5 mole of ˙OH is produced on average per mole of ozone during wastewater effluent ozonation (η = 0.5). This is similar to a molar ˙OH yield of 0.55 which was determined during ozonation of benzoic acid at pH 10.5 in an earlier ozonation study.67 ˙OH yields much lower than 0.5 (e.g., 9–24%) were also reported in some studies in which the ˙OH yield was determined by measuring formaldehyde formation in the presence of excess tert-butanol.52,68 It should be noted, however, that the presence of tert-butanol suppresses subsequent ˙OH radical reactions that may have otherwise increased the ˙OH radical yield substantially through the ˙O2−induced chain reaction via promoters.22 Therefore, the ˙OH radical yields determined by the tert-butanol method should be considered as the primary ˙OH radical yields mainly from the direct reaction of ozone with DOM in wastewater effluents. Finally, eqn (6) can be used for a rough estimate of ˙OH exposure as a function of the specific ozone dose (shown as the line in Fig. 3).
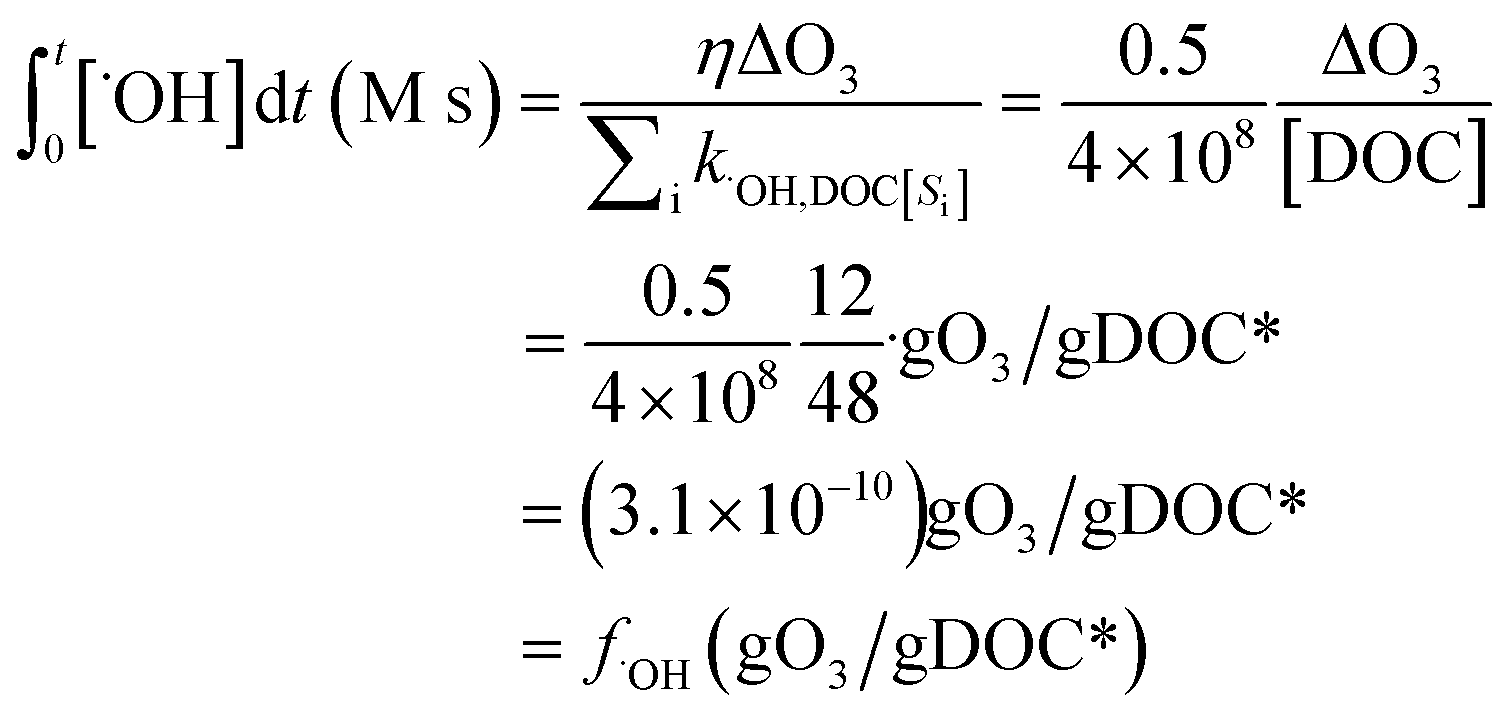 | (6) |
2.6. Generalized abatement efficacy of micropollutants
The % abatement of a micropollutant (MP) as a function of the specific ozone dose (gO3/gDOC*) can be calculated by eqn (7) in which the information for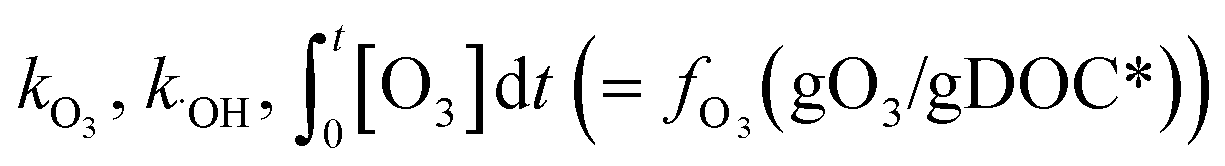 and
and 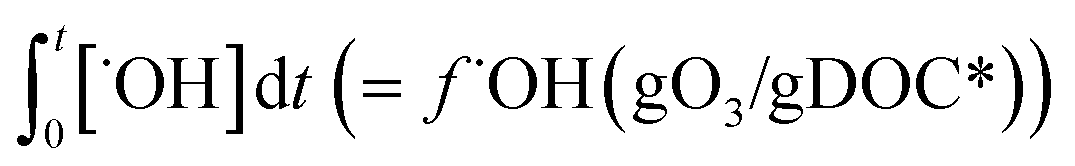 can be obtained from the discussion described in previous sections.
can be obtained from the discussion described in previous sections.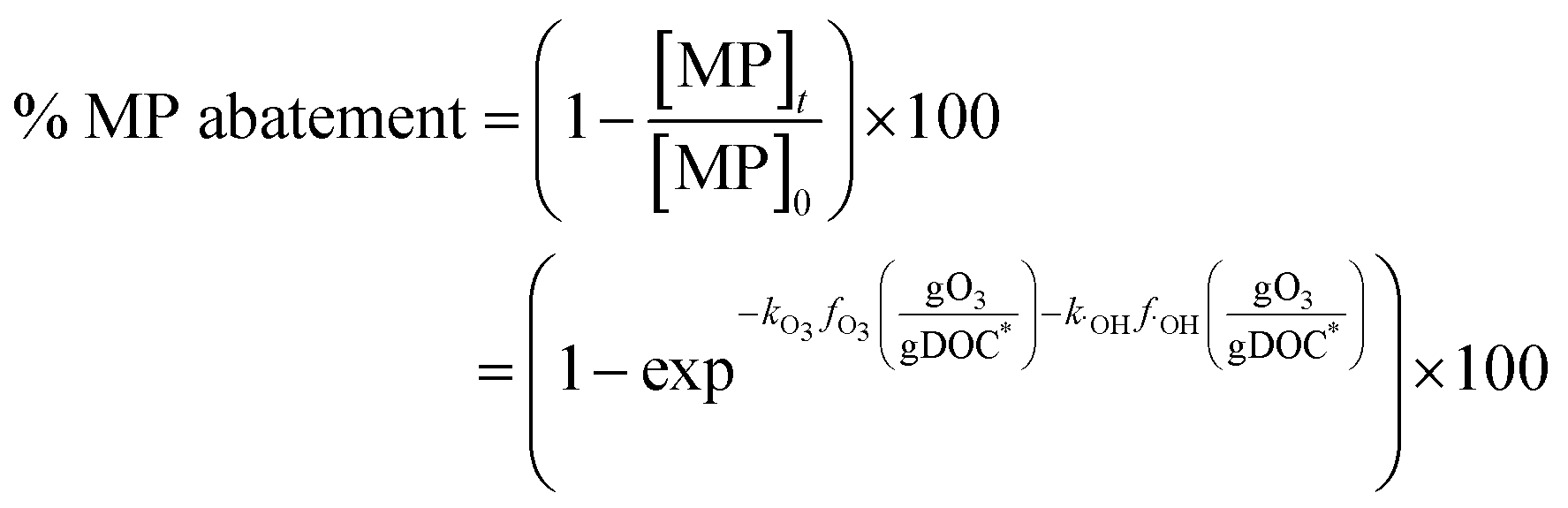 | (7) |
Eqn (7) was used to calculate the %abatement of model micropollutants with a range of ozone and ˙OH reactivities. The model micropollutants were classified into five categories according to their kO3 and k˙OH values at pH 7: MP-I (kO3 = 105 M−1 s−1 and k˙OH = 1010 M−1 s−1), MP-II (kO3 = 103 M−1 s−1 and k˙OH = 1010 M−1 s−1), MP-III (kO3 = 5 M−1 s−1 and k˙OH = 6 × 109 M−1 s−1), MP-IV (kO3 = 1 M−1 s−1 and k˙OH = 3 × 109 M−1 s−1), and MP-V (kO3 = 0.1 M−1 s−1 and k˙OH = 109 M−1 s−1). The five categories were selected to represent ozone and OH reactivities that show characteristic abatement efficiencies for a wide range of micropollutants. The ozone and ˙OH exposures as a function of specific ozone doses were calculated by eqn (4) and (6), respectively, which represent the average values determined in municipal wastewater effluents.
Fig. 4 shows the calculated % abatement levels of micropollutants as a function of the specific ozone dose during ozonation of a municipal wastewater effluent. The abatement of MP-I is quite efficient. For a specific ozone dose of 0.2 gO3/gDOC*, more than 90% and for 0.25 gO3/gDOC* > 99% could be abated. Micropollutants containing phenols, anilines, olefins, tertiary amines, organic sulfur, or combinations thereof (activated by electro-positive substituents such as alkyl or alkoxy groups) usually exhibit kO3 ≥ 105 M−1 s−1 at pH 7 and k˙OH = ∼1010 M−1 s−1 (ref. 41) and their abatement efficiency is expected to be similar to MP-I. It was found that a further increase of kO3 to >105 M−1 s−1 did not result in a more efficient abatement due to the limit of the mixing rate between the ozone stock solution and the treated sample or ozone transfer efficiency.53,60 The % abatement contribution of O3 (CO3,%) and ˙OH (C˙OH,%) to the overall abatement of a MP at a given ozone dose can be estimated by eqn (8) and (9), respectively:
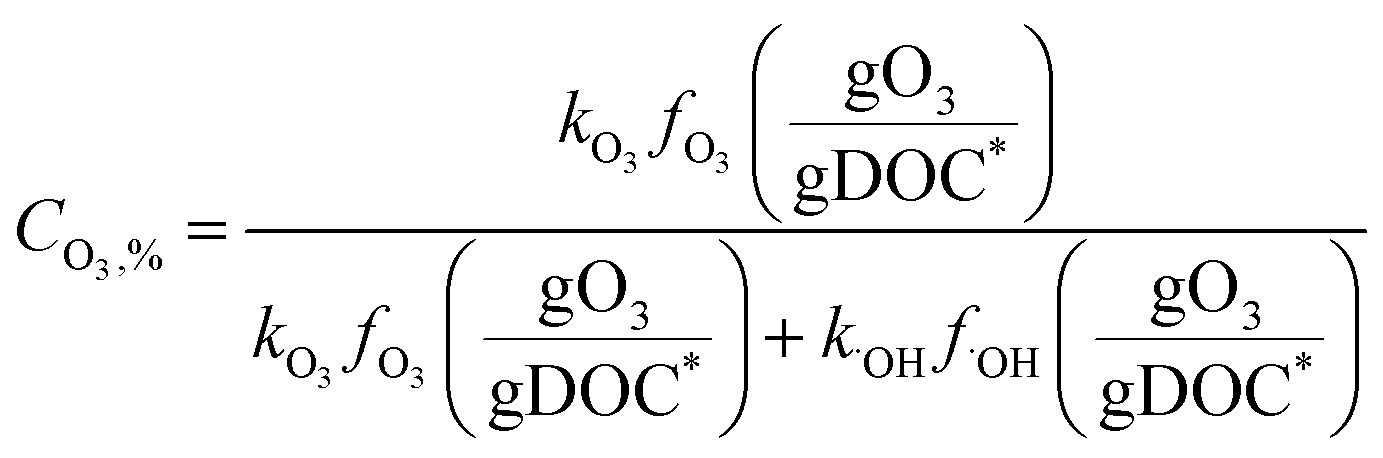 | (8) |
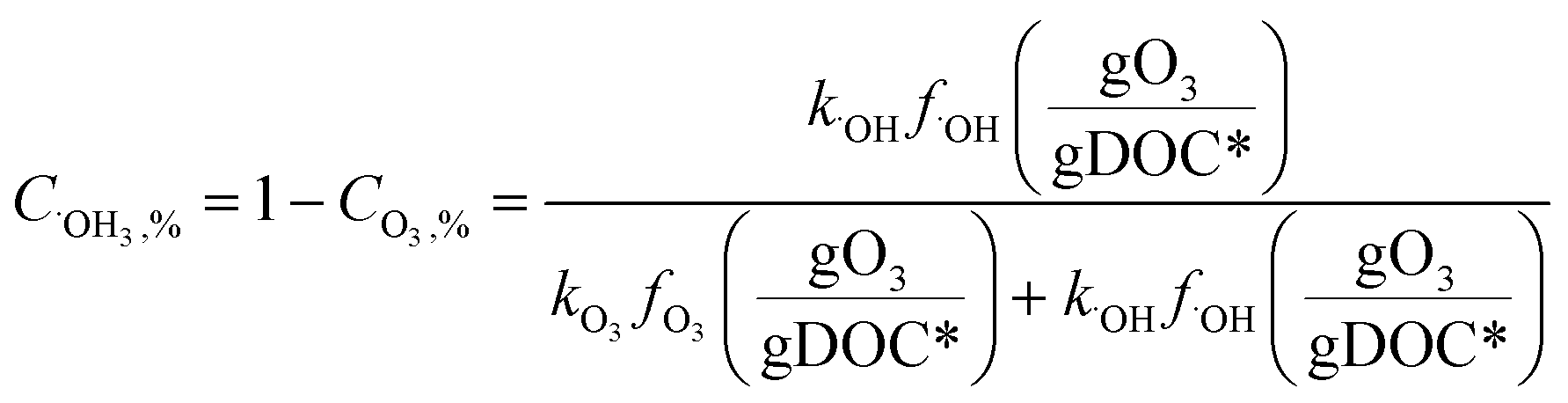 | (9) |
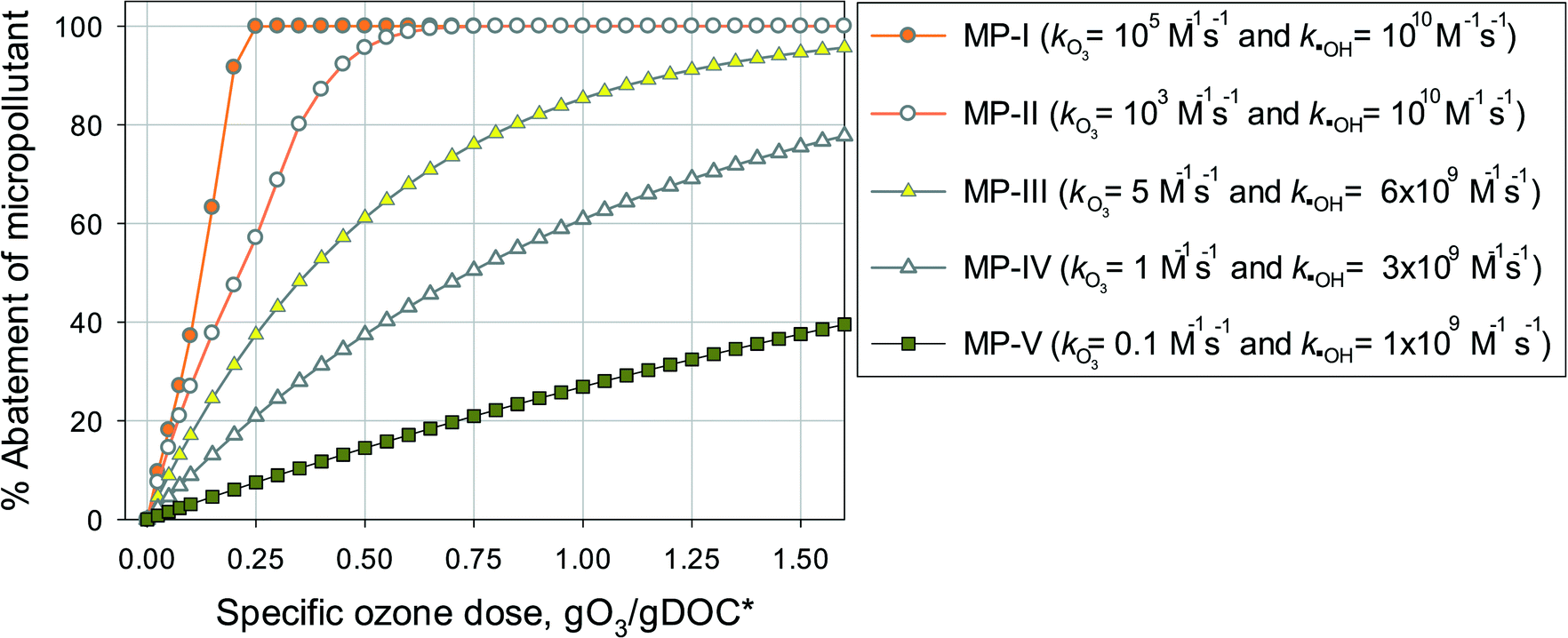 | ||
| Fig. 4 Calculated % abatement of micropollutants (MPs) with a range of ozone and ˙OH reactivities as a function of the specific ozone dose (gO3/gDOC*) during ozonation of a municipal wastewater effluent. The details of the model calculations are described in the main text. | ||
Due to the relatively high ozone reactivity, the direct reaction with ozone is found to be mainly responsible (more than 90%) for the abatement of MP-I.
The abatement efficiency of MP-II was lower than that of MP-I due to its lower ozone reactivity. The abatement level was 90% for a specific ozone dose of 0.42 gO3/gDOC* and >99% at 0.6 gO3/gDOC*. Micropollutants composed of deactivated (by electronegative substituents such as halogens, carbonyl, or nitro groups) phenols and anilines, activated benzenes, and tertiary, secondary and primary amines exhibit kO3 in the range of 102–105 M−1 s−1 at pH 7 and k˙OH in the range of 5 × 109–1010 M−1 s−1.41 Due to the wide range of kO3, the abatement efficiency of these micropollutants would vary depending on the magnitude of kO3. With decreasing kO3 from 105 M−1 s−1 to 10 M−1 s−1, the pattern for the abatement efficiency will change from that of MP-I to that of MP-III. For MP-II, 55% of the abatement was achieved by its direct reaction with ozone and the rest was obtained by its reaction with ˙OH yielding an overall 90% abatement of MP-II.
For MP-III, MP-IV, and MP-V, ˙OH is mainly responsible for their abatement and the contribution of ozone is minimal (below 10%). The abatement efficiency of these compounds is controlled by the magnitude of their k˙OH values. Compared to MP-I and MP-II for which the % abatement levels increase linearly with an increasing specific ozone dose, the % abatement of MP-III, MP-IV, and MP-V follows a negative exponential curve as a function of the specific ozone dose. In other words, the relative residual concentration for these micropollutants follows a pseudo-first-order decrease with respect to the specific ozone dose. As explained before, this is a consequence of the constant competition for ˙OH between the micropollutant and DOM as the major ˙OH scavenger. This leads to a less efficient abatement of the target micropollutant as its concentration decreases in terms of % abatement levels due to constant half-lives for the concentration decrease. At a specific ozone dose of 1.25 gO3/gDOC* (higher end of practical specific ozone doses), the % abatement levels of MP-III, MP-IV, and MP-V were 91%, 69%, and 32%, respectively. Compounds with non-activated aromatics or olefins or multiple activated C–H bonds have a k˙OH of >5 × 109 M−1 s−1. Compounds with strongly deactivated aromatics or deactivated aliphatic structures have a k˙OH of <5 × 109 M−1 s−1.47
3. Transformation products
In the previous section, the kinetics and abatement efficacy of target micropollutants were discussed. For typical ozone treatment conditions, organic micropollutants are not mineralized but transformation product(s) are generated. The type of transformation products depends on the reaction kinetics, which determines whether the pathway is mainly driven by ozone or ˙OH, the reaction mechanisms of each pathway or a combination thereof, and the structure and properties of the micropollutants. The relative contribution of ozone and ˙OH to the reaction pathways can be assessed based on the chemical kinetics discussed in section 2 (i.e., eqn (8) and (9)). Below, the reaction mechanisms of ozone and ˙OH are discussed depending on the structure and properties of micropollutants. This section is not intended to provide a comprehensive review on the transformation products from the reaction of organic compounds with ozone and ˙OH. For a complete review on these topics, readers can refer to Hoigné, 1998;69 von Gunten, 2003;36 von Sonntag and von Gunten, 2012;22 and Hübner et al. 2015.70 Rather we focus on some recent studies on the transformation products of wastewater-derived organic contaminants in water treatment with ozone and ˙OH, which are summarized in Table 2.| Compound | Application | Transforming agent | Reactive Moieties | Transformation byproducts | Reference |
|---|---|---|---|---|---|
| Acesulfame | Sweetener | O3 | Olefin | Cleavage of double bond | Scheurer et al., 2012 (ref. 89) |
| Amoxicillin | Antibiotic | O3 | Phenol | Hydroxylated phenol | Andreozzi et al., 2005 (ref. 121) |
| ˙OH (γ-radiolysis) | Phenol | Mono- and di-hydroxylated phenol products | Song et al., 2008 (ref. 122); Jung et al., 2012 (ref. 123) | ||
| Atenolol | O3 (unclear separation of ozone vs. ˙OH) | Activated benzene | Hydroxylated aromatic, ring opening products | Tay et al., 2011 (ref. 118) | |
| ˙OH (γ-radiolysis) | Hydroxylated aromatic; hydroxylated aliphatic chain | Song et al., 2008 (ref. 114) | |||
| Bezafibrate | Lipid regulator | O3 (unclear separation of ozone vs. ˙OH) | Aromatic | Hydroxylated benzene, benzene-ring opening products | Dantas et al., 2007 (ref. 73) |
| ˙OH (γ-radiolysis) | Hydroxylated benzene (including chloro- and ipso-alkoxy substitution), | Razavi et al., 2009 (ref. 111) | |||
| Benzotriazole | Chelating agent | O3 | Aromatic | Benzene-ring opening products | Mawhinney et al., 2012 (ref. 78) |
| O3 (unclear separation of ozone vs. ˙OH) | Aromatic | Hydroxylated products | Benitez et al., 2015 (ref. 124) | ||
| Bisphenol-A | Plasticizer | O3 | Phenol | Quinone-, catechol-, ring opening products | Deborde et al., 2008 (ref. 74) |
| Carbamazepine | Anti-epileptic | O3 | Olefin | Cleavage of double bond, secondary ring formation | McDowell et al., 2005 (ref. 86); Hübner et al., 2014 (ref. 91) |
| ˙OH (UV/H2O2) | Hydroxylated aromatic, carbonyl formation at olefin moiety | Keen et al., 2012 (ref. 125) | |||
| Carboxy-acyclovir | Antiviral | O3 | Olefin | Epoxide intermediate, 1,2-acyl shift and hydrolytic opening of imidazole ring | Prasse et al., 2012 (ref. 93) |
| Cephalexin | Antibiotic | O3 | Thioether; olefin | Cephalexin sulfoxide; scission of double bond | Dodd et al., 2010 (ref. 87) |
| Cetirizine | Antihistamine | O3 | Tertiary amine | Cetirizine N-oxide | Borowska et al., 2016 (ref. 98) |
| Clofibric acid | ˙OH (γ-radiolysis) | Hydroxylated aromatic dechlorination (or substitution of chlorine by hydroxyl) | Razavi et al., 2009 (ref. 111) | ||
| Ciprofloxacin | Antibiotic | O3 | Secondary amine | Dealkylated ciprofloxacin | Dewitte et al., 2008 (ref. 99) |
| ˙OH (γ-radiolysis) | Hydroxylated aromatics | An et al., 2010 (ref. 117) | |||
| Destruction of piperazine moiety | |||||
| Clarithromycin | Antibiotic | O3 | Tertiary amine | N-Oxide (>90%) or demethylated clarithromycin | Lange et al., 2006 (ref. 95) |
| DEET | Insect repellent | O3 (unclear separation of ozone vs. ˙OH) | Hydroxylated benzene, ring opening products, dealkylated amine | Tay et al., 2009 (ref. 126) | |
| ˙OH (γ-radiolysis) | Hydroxylated benzene, dealkylated amine | Song et al., 2009 (ref. 115) | |||
| N-Desmethyl-levofloxacin | Antibiotic | O3 | Olefin | Cleavage of double bond | El Najjar et al., 2013 (ref. 90) |
| Diclofenac | Anti-inflammatory | O3 | Aniline | 5-Hydroxydiclofenac, diclofenac-2,5-iminoquinone, 2,6-dichloroaniline | Coelho et al., 2009 (ref. 84); Sein et al., 2008 (ref. 83) |
| Enrofloxacin | ˙OH (γ-radiolysis) | Hydroxylated aromatic ring, destruction of piperazine side chain | Santoke et al., 2009 (ref. 116) | ||
| Estrone | Steroid hormone | O3 | Phenol | Ring opening products | Huber et al., 2004 (ref. 72); de Oliveira Pereira et al., 2011 (ref. 77); Segura et al., 2013 (ref. 79) |
| 17α-Ethinylestradiol | Steroid hormone | O3 | Phenol | Quinone-, catechol-, ring opening products | Huber et al., 2004 (ref. 72) |
| Fexofenadine | Antihistamine | O3 | Tertiary amine | Fexofenadine N-oxide | Borowska et al., 2016 (ref. 98) |
| Hydrochlorothiazide | Diuretic drug | O3 | N of cyclic sulfonamide | Imine product | Borowska et al., 2016 (ref. 98) |
| Imazalil | Fungicide | O3 | Olefin | Cleavage of double bond | Genena et al., 2011 (ref. 88) |
| Iopromide (iomeprol, iopamidol) | Contrast media | ˙OH (γ-radiolysis or UV/H2O2) | De-iodination and hydroxylation of benzene, alcohol oxidation to ketone | Jeong et al., 2010 (ref. 112); Singh et al., 2015 (ref. 113) | |
| Levofloxacin | Antibiotic | O3 | Tertiary amine; olefin | N-Oxide or demethylated levofloxacin; cleavage of double bond | El Najjar et al., 2013 (ref. 90) |
| Methyl-benzotriazole | Chelating agent | O3(unclear separation of ozone vs. ˙OH) | Aromatic | Benzene-ring opening products, hydroxylated or de-methylated benzene | Müller et al., 2012 (ref. 127) |
| Metoprolol | β-Blocker | O3 | Secondary amine | Hydroxylamine or dealkylated metoprolol | Benner and Ternes, 2009 (ref. 76); Sein et al., 2009 (ref. 128); Tay et al., 2013 (ref. 129) |
| ˙OH (γ-radiolysis) | Hydroxylated aromatic dealkylated products | Song et al., 2008 (ref. 114) | |||
| Norfloxacin | Antibiotic | O3 | Secondary amine | Dealkylated norfloxacin | Liu et al., 2012 (ref. 130) |
| Octylphenol | Alkylphenol surfactant | O3 | Phenol | Hydroxylated phenol | Ning et al., 2007 (ref. 131) |
| Parabens | Antimicrobial | O3 (unclear separation of ozone vs. ˙OH) | Phenol | Hydroxylated phenol | Tay et al., 2010 (ref. 132) |
| Paracetamol | Analgesic | O3 (unclear separation of ozone vs. ˙OH) | Phenol | Hydroquinone, catechol, ring opening products | Andreozzi et al., 2003 (ref. 133); El Najjar et al., 2014 (ref. 134) |
| Penicillin G | Antibiotic | O3 | Thioether | Sulfoxide | Dodd et al., 2010 (ref. 87) |
| ˙OH (γ-radiolysis) | Hydroxylation of aromatic ring | Song et al., 2008 (ref. 122) | |||
| Phenazone | Analgesic | O3 | Olefin | Cleavage of double bond | Favier et al., 2015 (ref. 92) |
| Progesterone | Hormone | O3 | Olefin | Cleavage of double bond | Barron et al., 2006 (ref. 135) |
| Propranolol | Beta-blocker | O3 | Secondary amine; naphthalene | Hydroxylamines; ring opening products | Benner and Ternes, 2009 (ref. 75); Dantas et al., 2011 (ref. 136) |
| ˙OH (γ-radiolysis) | Hydroxylation of aromatic ring, cleavage of alkoxy-naphthalene to hydroxyl-naphthalene and alcohol | Song et al., 2008 (ref. 114) | |||
| Roxithromycin | Antibiotic | O3 | Tertiary amine | Demethylated roxithromycin | Radjenovic et al., 2009 (ref. 96) |
| Sucralose | Artificial sweetener | ˙OH (UV/H2O2) | Dechlorination by hydroxyl substitution | Keen and Linden, 2013 (ref. 66) | |
| Sulfamethoxazole | Antibiotic | O3(unclear separation of ozone vs. ˙OH) | Benzene ring | Hydroxylated aniline, nitro-benzene | Abellan et al., 2008 (ref. 82); Gómez-Ramos et al., 2011 (ref. 85) |
| Tetracycline | Antibiotic | O3 | Phenol; tertiary amine; olefin | Hydroxylation of phenol; demethylated; scission of double bond | Khan et al., 2010 (ref. 81) |
| ˙OH (γ-radiolysis) | Addition to rings or H-abstraction product | Jeong et al., 2010 (ref. 137) | |||
| Tramadol | Pain killer | O3 | Tertiary amine | N-Oxide (90%) or demethylated (10%) tramadol | Zimmermann et al., 2012 (ref. 97) |
| Triclosan | Antimicrobial | O3 (unclear separation of ˙OH) | Phenol | Hydroxylated triclosan, dichloro-phenol, chloro-catechol | Chen et al., 2012 (ref. 138) |
| Trimethoprim | Antibiotic | O3 | Aromatic ring | Ring hydroxylation | Kuang et al., 2013 (ref. 139) |
| Venlafaxine | Anti-depressant | O3 | Tertiary amine | N-Oxide venlafaxine | Lester et al., 2013 (ref. 140) |
3.1. Transformation product formation from the reactions of target compounds with ozone
Ozone reacts rapidly with electron-rich moieties such as phenols, anilines, olefins, neutral amines, and reduced sulfur species. Therefore, these moieties present in micropollutants will mainly be attacked and transformed by ozone. The primary reaction of ozone with a compound (S) proceeds via the formation of a transient ozone adduct (S + O3 → S+–O–O–O−) and subsequent decomposition by different pathways such as (1) oxygen atom transfer (S+–O–O–O− → S+–O− + 1O2 (3O2)), (2) electron transfer (S+–O–O–O− → S˙+ + O3˙−), (3) formation of an oxyl radical (S+–O–O–O− → S+–O˙ + O2˙−).36,71 In addition, the reaction of ozone with a C–C double bond proceeds via the formation of ozonide (1,3-cycloadduct of ozone) that leads to the products from the cleavage of the C–C bond (Criegee mechanism). The ozone transformation products summarized in Table 2 were determined in the presence of excess of a ˙OH scavenger such as tert-butanol to exclude ˙OH reactions. However, many product studies with ozone were conducted under conditions for which the contributions of ozone and ˙OH are not clearly separated, which makes it difficult to differentiate between ozone- and ˙OH-induced transformation products. These studies were not included in Table 2.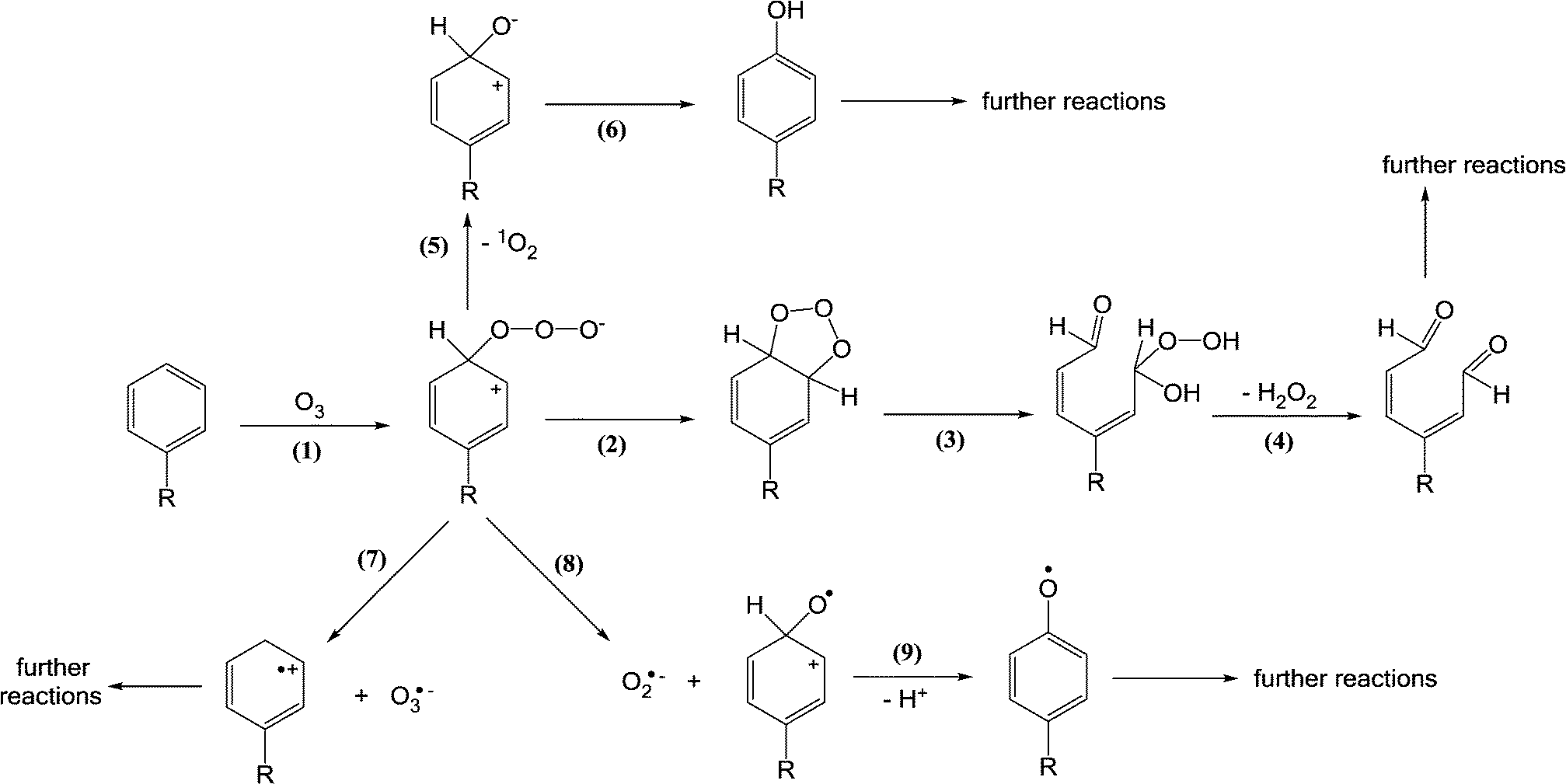 | ||
| Scheme 2 Reactions of ozone with aromatic compounds (adapted from von Sonntag and von Gunten, 2012).22 | ||
The reaction of ozone with anilines as aromatic amines can occur via ozone attack on the nitrogen or the benzene ring. The ozone attack on the nitrogen can form nitroso- and nitro-benzene as primary products [reactions (1)–(4), Scheme 3] albeit the yields of these products are low (less than 1%). The ozone attack on the benzene ring leads to the formation of the ozone-adduct followed by the release of an ozonide radical anion (O3˙−) and an aminyl radical (C6H5–N˙) [reactions (5) and (6), Scheme 3]. The ozone-adduct can also undergo 1O2 elimination and this leads to the formation of hydroxylated benzene products [reaction (7), Scheme 3]. The ozone attack on the benzene ring can also lead to the formation of ozonide and cleavage of the ring [reactions (8)–(11), Scheme 3]. From the ozone reaction with diclofenac and sulfamethoxazole (aromatic amine micropollutants), benzene ring hydroxylated or quinone products were observed.82–85 Overall, the identified products from the ozone reaction with anilinic compounds explain only a small fraction of the total pool of products.
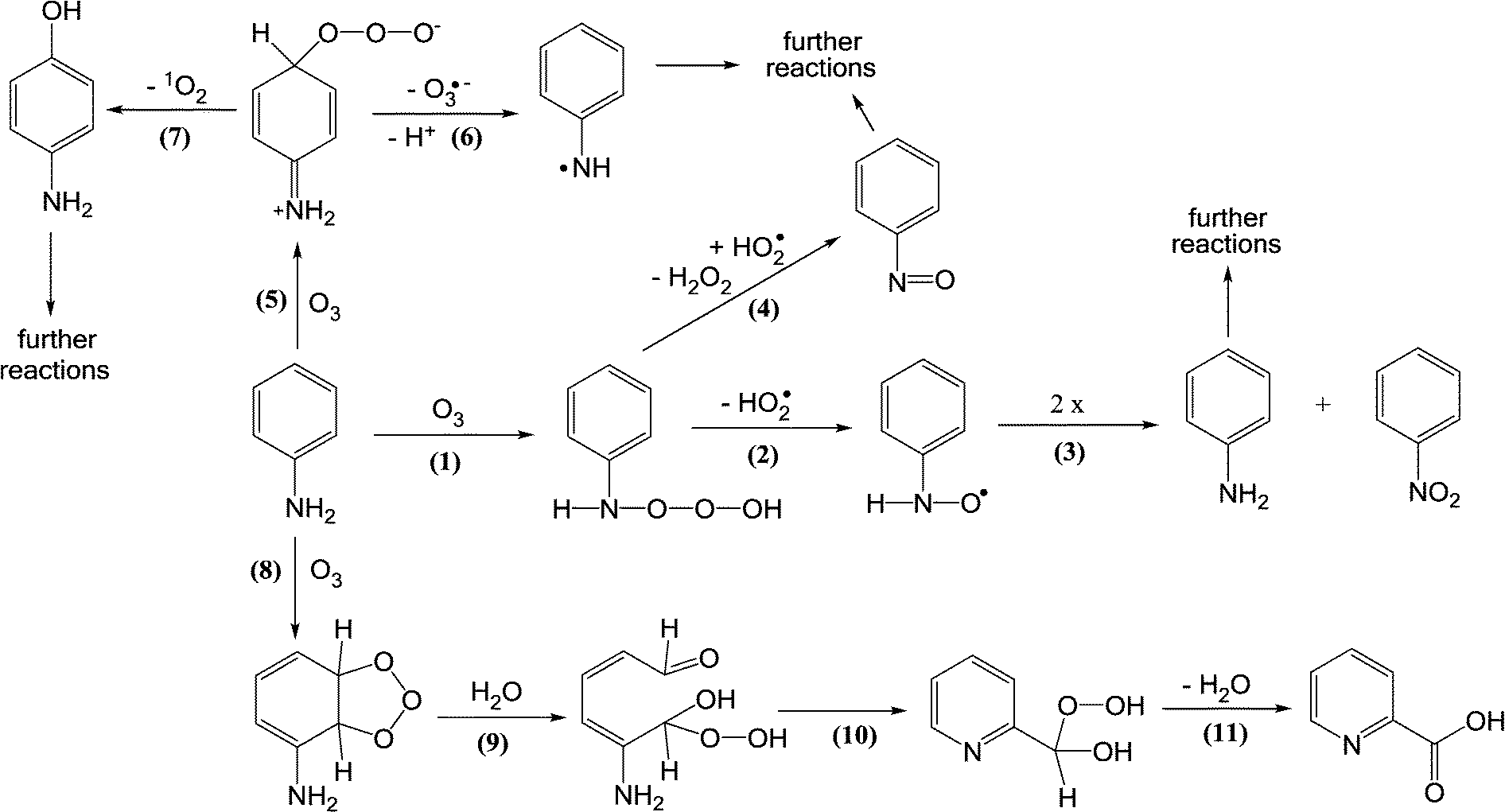 | ||
| Scheme 3 Reactions of ozone with aniline as an aromatic amine compound (adapted from von Sonntag and von Gunten, 2012).22 | ||
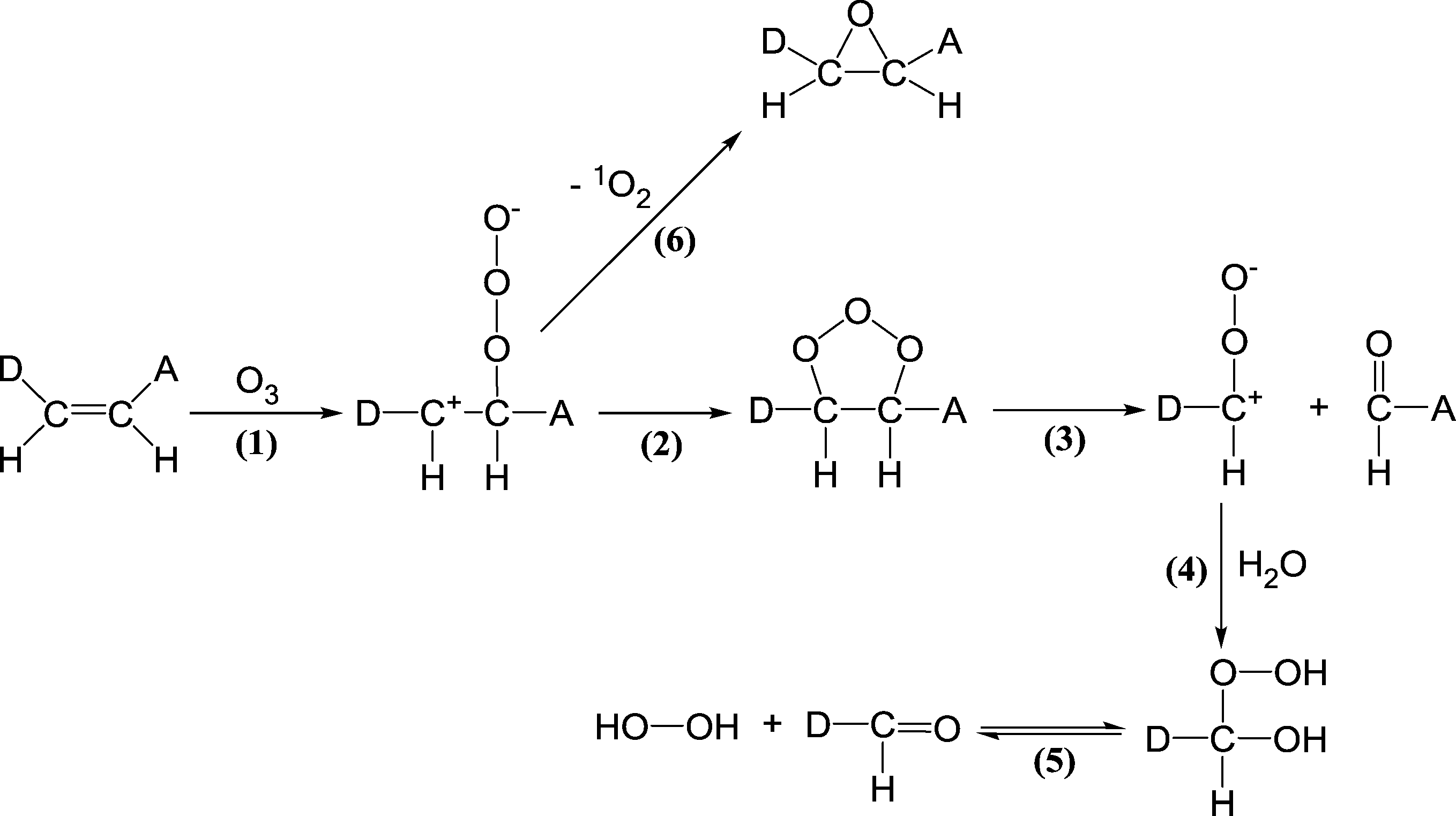 | ||
| Scheme 4 Reactions of ozone with olefinic compounds (adapted from von Sonntag and von Gunten, 2012).22 | ||
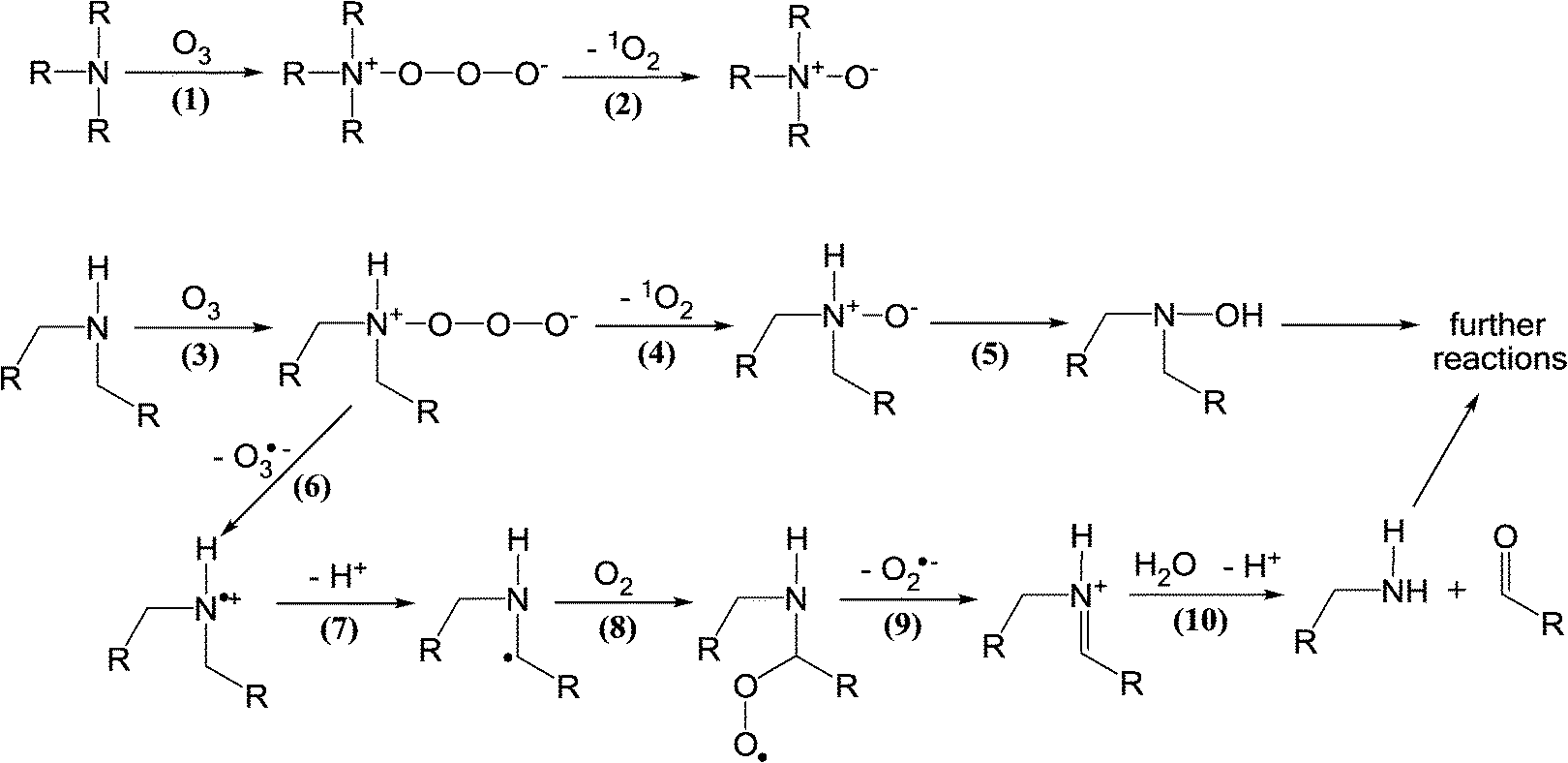 | ||
| Scheme 5 Reactions of ozone with aliphatic amine compounds (adapted from von Sonntag and von Gunten, 2012).22 | ||
In addition to the N,N-dimethylsulfamide functional group, hydrazine, hydrazides, and hydrazones with N,N-dimethyl groups have been identified as potent NDMA precursors during ozonation with molar NDMA yields of up to 94%.24,26,100,103,104 Some of these compounds are used in specific industries and have the potential to be discharged to municipal wastewater treatment plants and sewage-impacted receiving waters, which was observed, e.g., in Japan.24,26 Due to the high NDMA yields, these derivatives of hydrazines might explain some of the observed and sometimes significant NDMA formation during ozonation. However, there is no sufficient information to assign NDMA formation during ozonation to specific precursors. Furthermore, the kinetics and mechanisms for the ozone reactions with the known NDMA precursors are currently poorly understood.
Dimethylamine and compounds with a dimethylamino group have also been shown to produce NDMA during ozonation with very low NDMA yields, i.e., molar yields of less than 0.05%.105–107 Therefore, these compounds can be significant NDMA precursors only when their concentrations are sufficiently high (e.g., mM concentration range) such as in dye-containing industrial wastewaters. Amine-based polymers, used as water treatment coagulants or sludge treatment, have also been found to be responsible for significant NDMA formation during ozonation via dimethylamine release from polymer degradation by ozone or ˙OH.28,108
3.2. ˙OH transformation products
˙OH reacts with organic compounds mainly by addition (olefinic, aromatic, free amine, and sulfur moieties) and H-abstraction (mainly C–H bonds) mechanisms. The GCM method discussed in the previous section can be used to estimate the rate constant for each ˙OH reaction pathway. The reaction of ˙OH with organic compounds can generate carbon-centered radicals (R˙). Carbon-centered radicals react with O2 very rapidly (k = ∼2 × 109 M−1 s−1) generating organic peroxyl radicals (ROO˙). The chemistry of organic peroxyl radicals in aqueous solution has been extensively studied as peroxyl radicals play important roles in oxidation processes occurring in biological and environmental systems. Good reviews on this topic can be found elsewhere109,110 and only a brief overview is given here. Peroxyl radicals can decay via unimolecular or bimolecular processes. Unimolecular decays are typically found for α-hydroxyperoxyl radicals, which generate carbonyl compounds and superoxide radicals for alkylperoxy radicals (˙OOCR2O− → R2C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O + O2˙−) or phenol and superoxide radicals for hydroxylcyclohexadienylperoxyl radicals (˙OOC6H6O− → C6H5OH + O2˙−). In the bimolecular decay process, peroxyl radicals form tetroxides as transient intermediates, which decay via different pathways depending on their structure. Tertiary peroxyl radicals decay to oxyl radicals and O2 (2R3COO˙ → R3COOOOCR3 → 2R3CO˙ + O2). For primary and secondary peroxyl radicals, the decay pathway to carbonyls, alcohols, and oxygen (2R2HCOO˙ → R2HCOOOOCR2H → R2CO + R2CH(OH) + O2, Russel mechanism) or to carbonyls and H2O2 (2R2HCOO˙ → R2HCOOOOCR2H → 2R2CO + H2O2, Bennett mechanism) is also possible in addition to the oxyl radical pathway. The oxyl radicals formed from the decay of the peroxyl radicals undergo β-fragmentation (tertiary oxyl radicals) or 1,2-H shift (primary and secondary oxyl radicals) into carbon centered radicals and/or carbonyl compounds as products. The carbon-centered radicals then again react with oxygen forming peroxyl radicals, which constitutes radical chain reactions leading to the fragmentation of the carbon structure.
O + O2˙−) or phenol and superoxide radicals for hydroxylcyclohexadienylperoxyl radicals (˙OOC6H6O− → C6H5OH + O2˙−). In the bimolecular decay process, peroxyl radicals form tetroxides as transient intermediates, which decay via different pathways depending on their structure. Tertiary peroxyl radicals decay to oxyl radicals and O2 (2R3COO˙ → R3COOOOCR3 → 2R3CO˙ + O2). For primary and secondary peroxyl radicals, the decay pathway to carbonyls, alcohols, and oxygen (2R2HCOO˙ → R2HCOOOOCR2H → R2CO + R2CH(OH) + O2, Russel mechanism) or to carbonyls and H2O2 (2R2HCOO˙ → R2HCOOOOCR2H → 2R2CO + H2O2, Bennett mechanism) is also possible in addition to the oxyl radical pathway. The oxyl radicals formed from the decay of the peroxyl radicals undergo β-fragmentation (tertiary oxyl radicals) or 1,2-H shift (primary and secondary oxyl radicals) into carbon centered radicals and/or carbonyl compounds as products. The carbon-centered radicals then again react with oxygen forming peroxyl radicals, which constitutes radical chain reactions leading to the fragmentation of the carbon structure.
Table 2 summarizes the ˙OH-induced transformation products of selected wastewater-derived organic micropollutants. These micropollutants and their transformation products were selected based on the following criteria: (1) Studies with ˙OH as the main oxidizing agent from γ-radiolysis or UV/H2O2. The γ-radiolysis in N2O/O2 (80%/20%) saturated solution can generate ˙OH as the major reactive species (90%), thus the ˙OH-induced transformation pathways can be studied exclusively. For UV/H2O2, only micropollutants with low direct UV photolysis efficiency were selected. Ozonation at basic pH or O3/H2O2 (peroxone) can also be a source of ˙OH and studies for ozone-resistant micropollutants were selected. (2) The ˙OH-induced transformation products of ozone-reactive micropollutants (e.g., kO3 > 104 M−1 s−1) are included in Table 2 despite the fact that they are not likely to be relevant during ozonation because the ozone-reactive moieties of these compounds are mainly transformed by ozone. They are included to compare the ozone- and ˙OH-induced transformation products. (3) Initial transformation products (i.e., primary or secondary transformation products) are mainly considered as they are usually relevant for practical ozonation systems.
The product studies in Table 2 show that the reaction of ˙OH with aromatic moieties leads to the formation of hydroxylated aromatic compounds as identified initial products. For the micropollutants with halogenated aromatic moieties (clofibric acid and iopromide), products with dehalogenated aromatic moieties with substitution by the hydroxyl group were observed.111–113 The relevant hydroxylation mechanisms of aromatic moieties by ˙OH reactions are well established.109,110 For micropollutants with ozone-reactive aromatic moieties such as amoxicillin, propranolol, and tetracycline, however, it should be noted that the aromatic moieties (phenolic or naphthalene moiety) could have been already deactivated by ozone before an attack of ˙OH could occur. For the micropollutants with aromatic moieties of low ozone reactivity (atenolol, bezafibrate, clofibric acid, ciprofloxacin, DEET, enrofloxacin, iopromide, metoprolol, and penicillin G), the initial hydroxylated aromatic products (i.e., phenols) will be quickly further transformed by their reactions with ozone during ozonation, which is different from the product evolution patterns obtained in γ-radiolysis or UV/H2O2 systems (relatively slower further transformation by ˙OH). Table 2 also includes the cases of ˙OH reaction with aliphatic amines (ciprofloxacin, DEET, and enrofloxacin) and aliphatic hydrocarbons with neighboring (hydr)oxyl or halogen groups (atenolol, iopromide, metoprolol, and sucralose). Deaminated, dealkylated, or dehalogenated products were observed,112–119 which can be explained by the ˙OH reaction mechanisms with these moieties.
It should be noted that a very large number of initial transformation products can be formed during ozonation of organic contaminants especially from ˙OH reactions due to the non-selective nature of this oxidant and only a fraction of the initial transformation products has been usually identified in the studies in Table 2. In addition, the major transformation products, which can be expected from the proposed reaction mechanisms, are sometimes missing. In future studies, it is recommended that the identified transformation products are compared with the predicted ones based on the discussed ozone and ˙OH reaction mechanisms and vice versa. It is also worthwhile to mention the importance of using appropriate analytical tools for detecting and identifying transformation products. Chromatographic separation methods combined with mass spectrometry such as LC-MS or GC-MS have become the most common tools for transformation product analyses. Often, the mass spectrometry data alone are not enough for exact structural elucidation and complementary methods such as nuclear magnetic resonance (NMR) or infrared spectroscopy (IR) are necessary. High-resolution tandem mass spectrometry is increasingly available for environmental chemical analysis and has opened new opportunities for transformation product analyses. Required confidence levels in structural identification by high resolution mass spectrometric analyses have recently been proposed and discussed.120 This should be more actively applied for transformation product identification. Finally, complete mass balances or yield determination for transformation products would be desirable; however, this has rarely been achieved due to a lack of chemical standards for transformation products. Considering the high cost required for a complete mass balance analysis, such a level of investigations can be done after it has been demonstrated that the identified transformation products are toxicologically relevant (see the section below).
3.3. Automated prediction system for transformation product formation during ozonation
The ozone and ˙OH reaction mechanisms in combination with the corresponding methods for rate constant prediction discussed in the previous sections can be used to predict the formation and evolution of transformation products during ozonation of organic contaminants. The prediction processes can be done manually but it often requires time-consuming calculation steps, especially for ˙OH reactions. For ˙OH-based AOPs, computer-aided kinetic models have been developed to simulate the degradation pathways and the transformation product formation of organic contaminants.141–144 These models are composed of the following components: (1) chemical structure input and recognition: the computer model recognizes chemical molecules as graphical objects, which allows a mathematical representation of structural information. (2) Pathway generator: the reaction rules based on ˙OH reaction mechanisms that are built in the model can enumerate all possible ˙OH reaction pathways of target organic chemicals. (3) Rate constant estimator: the reaction rate constant estimation methods for ˙OH (e.g., GCM method) and other peroxyl radicals are then applied for the generated reaction pathways. (4) Mechanistic reduction of model size: as ˙OH reaction can generate very high numbers of products and pathways, unimportant reactions and species are screened out to reduce the model size and thus improve the computational efficiency. (5) Model solver: the obtained reaction pathways with the corresponding rate constants are then used to build a set of ordinary differential equations (ODE) which can be solved numerically (deterministic approach). Alternatively, the model can be solved using the Monte Carlo methods (stochastic approach) in cases that the model is too large to be solved using the ODE solvers.It has been demonstrated that computer-aided AOP kinetic models can successfully predict the ˙OH-induced degradation pathways of low molecular organic contaminants such as acetone and trichloroethene (TCE), for which the experimental data for the full degradation pathways are available.142 Additionally, the models have been tested to simulate the variation of the molecular weight distribution during AOP treatment of structurally simple polymers such as polyacrylamide and polyethylene glycol.143,144 It would be interesting to apply these computer-aided kinetic models to predict the formation and evolution of initial transformation products of structurally complex organic contaminants during AOP treatment. Currently, a computer-aided, automated prediction system to simulate ozone-induced transformation of organic compounds is under development.145 The structure of the model is very similar to the model for ˙OH. Finally, ozone and ˙OH kinetic models should be properly combined to be able to predict the transformation pathways and product evolutions of organic contaminants during ozonation of real waters, in which both ozone and ˙OH will contribute to product formation.
3.4. Biological activity of transformation products and their biodegradability
Full mineralization of micropollutants is rarely achieved during ozonation; therefore, it is important to consider the biological activities (e.g., antimicrobial or estrogenic activity) of transformation products compared to the parent compounds. Transformation products have received increasing attention in environmental risk assessment of micropollutants and reviews are available on this topic.3,146,147 Here, a brief introduction to this topic is given with a discussion on the recent research progress in the assessment of biological activity changes from ozone and ˙OH transformation products of wastewater-derived organic micropollutants.Exposure- or effect-driven approaches can be applied for risk assessment of transformation products.147 In the exposure-driven approach, individual ozone or ˙OH transformation products are identified, synthesized, and then subjected to biological activity measurements. As experimental identification and synthesis of individual transformation products are highly demanding tasks, this approach has been rarely used for ozone or ˙OH transformation product studies. Few studies are available for which all transformation products have been identified and quantified with a complete mass balance for ozone- or ˙OH-induced transformations. In one study, two stereoisomeric (R)- and (S)-sulfoxides as major products from penicillin G transformation by ozone were identified. These two products were synthesized and analyzed for their antibacterial activity, allowing a prediction of the evolution of the antimicrobial activity during ozonation of penicillin G.87
In the effect-driven approach, the target micropollutant is treated with a range of ozone or ˙OH doses. The resulting mixtures of a parent micropollutant and its transformation products are analyzed by quantitative biological activity tests as well as the residual concentration of the parent micropollutant. If the bioactivity and the concentration of the parent micropollutant are abated to the same relative extent, the mixtures of the transformation products are considered to have negligible biological activity compared to the parent micropollutant. Only when the relative decrease in biological activity is lower than the relative decrease of the parent micropollutant concentration transformation products with significant residual biological activity are formed and need to be identified.147
The effect-driven approach using in vitro bioassays has been applied to assess the ozone- and ˙OH-induced changes of biological activities of antibacterial compounds,95,148–150 an antiviral compound,151N-nitrosoamines,152 and steroid estrogens.72,153,154 It should be noted that the tested bioassays are based on the specific mode of action such as receptor binding or damage to protein/DNA. In most cases, the relative biological activity and the relative concentration of the parent micropollutant decrease in a 1:1 relationship, indicating that the structural transformation induced by ozone and/or ˙OH is sufficient to remove the biological activities of the parent micropollutants. Often the ozone reactive moieties such as phenols or amines are structurally responsible for the biological activities (i.e., toxicophore) and the transformation of these toxicophores by ozone and/or ˙OH reactions leads to a full elimination of their bioactivities. Even the less selective ˙OH-induced modifications in other parts of the micropollutant's toxicophore are sufficient to remove the bioactivities of the parent compound. This indicates that not only toxicophores but also other structural parts are important for the bioactivities based on specific mode of action (e.g., receptor binding).
Transformation products with some residual biological activities were found for ozone reactions with two β-lactam antibiotics, penicillin G and cephalexin.87,149 The transformation of doxycycline and erythromycin by UV/H2O2 treatment in a wastewater effluent matrix showed transient intermediate products with residual antibacterial activity.150 The residual antibacterial activity was not observed during experiments in ultra-purified water, suggesting that complex secondary reactions in the wastewater matrix were responsible for the formation of active transformation products. The transformation of N-nitroso-di-n-propylamine (NDPA) and N-nitropyrrolidine (NPYR) by ˙OH generates products with some residual mutagenic activity.152 It was explained that the nitroso moiety is the toxicophore for the mutagenic activity and structural modifications in the alkyl chains lead to only partial removal of the mutagenic activity.
In a recent study, 23 compounds of the USEPA contaminant candidate list 3 (CCL3) were subjected to treatment with ozone, ˙OH and direct photolysis and the mixtures of parent compounds and transformation products as a function of the degree of transformation were tested for mutagenicity and estrogenicity by in vitro assays.155 From the 23 compounds tested, mostly a reduction of the biological effects was observed; however, for 6 compounds an increase in mutagenicity and in one case (quinoline) estrogenicity could be observed for one or several of the oxidative treatments. Typically, these effects increased with increasing transformation of the parent compound, but then they decreased again for a full transformation of the target compound.155 Weakly estrogenic transformation product(s) (potency of 3 × 10−4 relative to 17β-estradiol) from the non-estrogenic quinoline were found during UV/H2O2 treatment (by ˙OH oxidation), but not by ozone and UV treatment. Hydroxylated quinoline(s) at the 6, 7, or 8 position are proposed to be responsible for the estrogenicity formation.155 As quinoline shows low reactivity to ozone while considerable reactivity to ˙OH (kO3 = 51 M−1 s−1 and k˙OH = 7 × 109 M−1 s−1),156 its initial transformation during ozonation will be mainly driven by ˙OH oxidation, forming hydroxylated quinolines which in turn will be quickly further transformed by ozone due to their enhanced ozone reactivity.
It has been reported that acute toxicity increases in bioassays (e.g., bioluminescence assay with V. fischeri) during transformation of some micropollutants by ozone or ˙OH.85,93,134,136,157,158 Often the toxicity observed at low ozone and/or ˙OH exposure decreases again with a further increase of the ozone and/or ˙OH exposure. For an interpretation of these results, the following aspects should be considered. (1) The mode of toxic action was usually not identified in these studies; it was not determined whether the parent micropollutant is a baseline toxicant or a specific toxicant. To elucidate these aspects, QSAR-based estimation methods of the baseline toxicity can be applied.146 (2) Upon oxidation by ozone and/or ˙OH, organic compounds usually become oxygen-rich and hydrophilic, thus the baseline toxicity is expected to decrease. Therefore, an increase of the toxicity during the transformation can be explained by activation of the existing toxicophore or by the formation of new toxicophore(s). Hydroxylated aromatic-, aldehyde-, or nitro-moieties, which are commonly generated from ozone- and/or ˙OH-induced transformation of organic compounds, can be considered as new toxicophores causing membrane, protein, or DNA damage.159,160 (3) Not only the relative increase in the toxicity but also the absolute toxicity of the transformation products should be considered for the risk assessment because very high concentrations (e.g., mM range) of the parent micropollutants were mostly used in the bioassays especially for baseline toxicants. Even though an increase of the toxic potential was observed (e.g., quinoline, see above), the overall toxic effects from the transformation products can be insignificant considering their low concentrations under realistic conditions (e.g., nM range).
A range of in vitro bioassays based on specific mode of toxic action have been applied to assess the changes of biological activities during ozonation of real wastewater effluents. Significant reductions of estrogenic activity, arylhydrocarbon receptor response, neurotoxicity, phytotoxicity, or genotoxicity have been observed after ozone treatment of real wastewater effluents.12,161,162 In contrast, increases in the in vitro mutagenicity or in vivo genotoxicity or developmental toxicity were also found after ozonation while interestingly the formed toxicities decreased significantly for most of the tested cases after subsequent biological filtration.25,27,34,163 This indicates the formation of biodegradable toxic oxidation by-products such as aldehydes.23 In one case, the ozone induced mutagenicity decreased only partly even in a biological post sand filtration.27 It should also be noted that these biologically active products are expected to be mainly produced from the transformation of bulk dissolved organic matter (mg L−1 range), not necessarily from the trace organic micropollutants (ng L−1–μg L−1 range). These oxygen-rich compounds, which are formed from the reaction of ozone and/or ˙OH with the effluent/natural organic matter, can significantly contribute to the assimilable organic carbon (AOC) or biodegradable organic carbon (BDOC), which is easily removed in biological post-treatment steps.17,164 Therefore, for the planned upgrade of municipal wastewater treatment with an ozonation step, a biological post-treatment is required in Switzerland.
In addition to the degradation of oxidation by-products from the bulk dissolved organic matter during biological post-treatment steps, the biodegradability of the transformation products from micropollutants is also of interest. Research progress in the biodegradability of micropollutant-derived transformation products has been recently reviewed.70 Only limited experimental information is available to assess the degradation of transformation products during biological post-treatment. One case in point is carbamazepine, for which four main transformation products after cleavage of the double bond (named as BQM, BaQM, BQD, and BaQD) have been measured during ozonation.91 Three of these products were biodegradable, whereas one product (i.e., BaQD) was persistent in biological sand filtration.91 Therefore, in terms of risk assessment, this transformation product should be the main target for toxicity evaluation. In another study, three transformation products with a hydroxyl or carbonyl group were identified during UV/H2O2 treatment of carbamazepine (˙OH oxidation) and these products could be fully mineralized by a post treatment with a mixed bacterial inoculum.125
Because of the lack of experimental information for the biodegradability of transformation products, biodegradability probability or pathway prediction models can be used for a first assessment.70 To this end, it could be shown that the probability of biodegradability of ozone transformation products increases relative to the parent compounds for olefins and aromatic compounds with ring opening. For transformation products other than ring opening for aromatic compounds no enhancement of biodegradability is predicted. The transformation of amines leading to N-oxides or hydroxylamines is not expected to improve the biodegradability of ozone transformation products.70
4. Summary and outlook
Principle-based approaches (i.e., chemical kinetics and mechanisms of ozone and ˙OH reactions) have been shown to be very useful for the prediction of the elimination efficiency of various micropollutants and their transformation products during ozonation of municipal wastewater effluents. It has been demonstrated that a generalized prediction of the elimination of various micropollutants is possible using a chemical kinetics approach based on ozone and ˙OH second order rate constants (i.e., compound-specific information) and exposures (i.e., water matrix-specific information). Second order rate constants can be experimentally determined or predicted based on Hammett/Taft-type or quantum chemical-type QSARs. Oxidant exposures can be assessed by (semi)empirical models. Regarding the second order rate constants for the reactions of micropollutants with ozone and ˙OH, more reliable measurements with compounds having a wide range of structural variations or less studied structural moieties (e.g., heterocyclic compounds) are recommended for developing new or upgrading existing QSAR models for the rate constant prediction. Quantum chemical calculation tools are expected to play an increasingly important role in developing more generally applicable rate constant prediction QSAR models. More measurements of ozone and ˙OH exposures as a function of the (specific) ozone doses for individual wastewaters covering a wider range of water matrix characteristics are also required. This data will lead to a better understanding of the reactivity of ozone and/or ˙OH with effluent organic matter (as the main oxidant consumer) and help to bring ozone and ˙OH exposure prediction models to the next level. Due to the relatively low selectivity of ˙OH, the ˙OH exposures can be readily predicted by a model consisting of the ozone dose, the ˙OH yield from ozone consumption, and the ˙OH reaction rate with water matrix components, which can be useful in predicting the ˙OH exposure when an experimental determination of ˙OH exposure is not possible.Compared to the research progress in reaction kinetics for predicting the abatement efficiency of parent micropollutants, less information is available for the formation of transformation products. The established ozone reaction mechanisms with ozone-reactive moieties such as phenols, olefins, amines, organo sulfur compounds, etc. can serve as a basis to develop ozone reaction rules for predicting ozone-induced transformation pathways. Similarly, ˙OH reaction rules based on the ˙OH addition or H-abstraction mechanisms can be used for predicting the ˙OH-induced transformation pathways. To apply these reaction rules, the main oxidation pathways by ozone and/or ˙OH should be determined for the parent micropollutant and its transformation products. This can be assessed by using the kinetic information, that is, the ozone and ˙OH second order rate constants and the ozone and ˙OH exposures for the given treatment conditions. As a rule of thumb, the initial transformation products of ozone-reactive micropollutants (i.e., kO3 > 105 M−1 s−1) will be mainly controlled by the ozone reaction rules. Similarly, the initial transformation products of ozone-refractory micropollutants (i.e., kO3 < 10 M−1 s−1) will be mainly controlled by the ˙OH reaction rules. However, the reaction of ozone-refractory moieties with ˙OH can produce ozone-reactive moieties (e.g., phenols from benzenes) and further transformation of these activated moieties is then driven by ozone reactions. The ozone and ˙OH reaction rules and the QSARs for the rate constant estimation can be implemented in computer-based tools, which allow automated prediction of transformation pathways of organic contaminants during ozonation. This has been demonstrated for predicting the transformation products and pathways of small organic contaminants during treatment with the advanced oxidation process UV/H2O2. A similar system for ozone reactions is currently under development. Finally, more experimental data for ozone- and ˙OH-induced transformation products are required, which can be used to confirm and refine the reaction rules. For acquiring these data, it is highly desirable to clearly separate the ozone- from ˙OH-induced transformation products and to consider quantitative aspects of the transformation products (i.e., yield and mass balance).
The reactions of ozone with N,N-dimethyl-sulfamide or N,N-dimethyl-hydrazine derivative functional groups are found to produce NDMA, which might be relevant for NDMA formation during wastewater ozonation. Further information on NDMA precursors and the corresponding kinetics and mechanisms of NDMA formation during ozonation are necessary to better assess and mitigate NDMA formation.
Biological activities of transformation products can be systematically assessed using exposure-and/or effect-driven approaches. These approaches are complementary to each other. The effect-driven approach using in vitro bioassays has been shown to be more practical for screening purposes. Biological activities based on specific modes of action such as antibacterial, antiviral, or estrogenic activity, etc. have been found to decrease significantly upon slight structural modifications of the parent compound by reaction with ozone and/or ˙OH. It appears that both toxicophores and other structural parts are responsible for the specific mode of biological activities (e.g., receptor binding). To this end, ozone and/or ˙OH are quite efficient in eliminating the effects of some important (eco)toxic micropollutants such as antibiotics and hormones. The formation of new biological activities has also been observed, which can be mostly related to the activation of the existing toxicophore or by the formation of new toxicophore(s). In this case, identification of the toxicophore(s) and quantitative risk assessment are required. An exposure-driven approach can be used when the transformation products retain the biological activities of the parent compound or obtain new biological activities. Transformation product and pathway prediction models can be useful tools for identifying target transformation products of toxicological concern. Research on identifying and quantifying transformation products from micropollutants covering a wide range of structural variations will warrant a further advancement in the field of transformation product and pathway prediction modeling.
In principle, individual transformation products have to be synthesized and tested for biological activities, which is usually expensive and time-consuming. In this respect, in silico biological activity prediction models might have potential to be used as screening tools for assessing the bioactivities of transformation products. These computational techniques, some of which are originally developed for drug discovery, have been shown to be quite useful for high throughput screening of the adverse effects of chemicals including environmental contaminants,165–168 while their application to biological effect assessment for transformation products is still limited.3
The trend in ozone research to not only investigate parent compound abatement but also include the formation of transformation products and assess their biological effects should be the way forward. Furthermore, there is a lack of experimental information on the biodegradability of transformation products and its consequences for biological effects. Thus, this issue should also get more attention in future research.
Acknowledgements
This study was supported by the National Research Foundation funded by the Ministry of Science ICT & Future Planning (NRF-2013R1A2A2A03068929) and the Korea Institute of Marine Science & Technology Promotion funded by the Ministry of Oceans and Fisheries, Korea (KIMST-20150307).References
- R. Schwarzenbach, B. I. Escher, K. Fenner, T. B. Hofstetter, C. A. Johnson and U. von Gunten, Science, 2006, 313, 1072–1077 CrossRef CAS PubMed.
- T. A. Ternes and A. Joss, Human Pharmaceuticals, Hormones, and Fragrances: The Challenge of Micropollutants in Urban Water Management, IWA Publishing, London, 2006 Search PubMed.
- D. M. Cwiertny, S. A. Snyder, D. Schlenk and E. P. Kolodziej, Environ. Sci. Technol., 2014, 48, 11737–11745 CrossRef CAS PubMed.
- A. Joss, S. Zabczynski, A. Göbel, B. Hoffmann, D. Löffler, C. S. McArdell, T. A. Ternes, A. Thomsen and H. Siegrist, Water Res., 2006, 40, 1686–1696 CrossRef CAS PubMed.
- R. L. Oulton, T. Kohn and D. M. Cwiertny, J. Environ. Monit., 2010, 12, 1956–1978 RSC.
- S. Jobling, M. Nolan, C. R. Tyler, G. Brighty and J. P. Sumpter, Environ. Sci. Technol., 1998, 32, 2498–2506 CrossRef CAS.
- K. A. Kidd, P. J. Blanchfield, K. H. Mills, V. P. Palace, R. E. Evans, J. M. Lazorchak and R. W. Flick, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 8897–8901 CrossRef CAS PubMed.
- A. Pruden, Environ. Sci. Technol., 2014, 48, 5–14 CrossRef CAS PubMed.
- M.-O. Buffle, J. Schumacher, S. Meylan, M. Jekel and U. von Gunten, Ozone: Sci. Eng., 2006, 28, 247–259 CrossRef CAS.
- M.-O. Buffle, J. Schumacher, E. Salhi, M. Jekel and U. von Gunten, Water Res., 2006, 40, 1884–1894 CrossRef CAS PubMed.
- M. M. Huber, A. Gőbel, A. Joss, N. Hermann, D. Lőffler, C. S. McArdell, A. Ried, H. Siegrist, T. A. Ternes and U. von Gunten, Environ. Sci. Technol., 2005, 39, 4290–4299 CrossRef CAS PubMed.
- B. I. Escher, N. Bramaz and C. Ort, J. Environ. Monit., 2009, 11, 1836–1846 RSC.
- J. Hollender, S. G. Zimmermann, S. Koepke, M. Krauss, C. S. McArdell, C. Ort, H. Singer, U. von Gunten and H. Siegrist, Environ. Sci. Technol., 2009, 43, 7862–7869 CrossRef CAS PubMed.
- B. I. Escher, M. Lawrence, M. Macova, J. F. Müller, Y. Poussade, C. Robillot, A. Roux and W. Gernjak, Environ. Sci. Technol., 2011, 45, 5387–5394 CrossRef CAS PubMed.
- D. Gerrity, S. Gamage, J. C. Holady, D. B. Mawhinney, O. Quinones, R. A. Trenholm and S. A. Snyder, Water Res., 2011, 45, 2155–2165 CrossRef CAS PubMed.
- X. Yang, R. C. Flowers, H. S. Weinberg and P. C. Singer, Water Res., 2011, 45, 5218–5228 CrossRef CAS PubMed.
- S. G. Zimmermann, M. Wittenwiler, J. Hollender, M. Krauss, C. Ort, H. Siegrist and U. von Gunten, Water Res., 2011, 45, 605–617 CrossRef CAS PubMed.
- J. Reungoat, B. I. Escher, M. Macova, F. X. Argaud, W. Gernjak and J. Keller, Water Res., 2012, 46, 863–872 CrossRef CAS PubMed.
- J. Margot, C. Kienle, A. Magnet, W. Mirco, L. Rossi, L. F. de Alencastro, C. Abegglen, D. Thonney, N. Chèvre, M. Schärer and D. A. Barry, Sci. Total Environ., 2013, 461–462, 480–498 CrossRef CAS PubMed.
- J. Blackbeard, J. Lloyd, M. Magyar, J. Mieog, K. G. Linden and Y. Lester, Environ. Sci.: Water Res. Technol., 2016, 2, 213–222 CAS.
- R. I. L. Eggen, J. Hollender, A. Joss, M. Schärer and C. Stamm, Environ. Sci. Technol., 2014, 48, 7683–7689 CrossRef CAS PubMed.
- C. von Sonntag and U. von Gunten, Chemistry of Ozone in Water and Wastewater Treatment: From Basic Principles to Applications, IWA Publishing, London, 2012 Search PubMed.
- E. C. Wert, F. L. Rosario-Ortiz, D. D. Drury and S. A. Snyder, Water Res., 2007, 41, 1481–1490 CrossRef CAS PubMed.
- K. Kosaka, M. Asami, Y. Konno, M. Oya and S. Kunikane, Environ. Sci. Technol., 2009, 43, 5236–5241 CrossRef CAS PubMed.
- D. Stalter, D. A. Magdeburg, M. Weil, T. Knacker and J. Oehlmann, Water Res., 2010, 44, 439–448 CrossRef CAS PubMed.
- K. Kosaka, M. Asami, K. Ohkubo, T. Iwamoto, Y. Tanaka, H. Koshino, S. Echigo and M. Akiba, Environ. Sci. Technol., 2014, 48, 11243–11250 CrossRef CAS PubMed.
- A. Magdeburg, D. Stalter, M. Schliusener, T. Ternes and J. Oehlmann, Water Res., 2014, 50, 35–47 CrossRef CAS PubMed.
- M. Sgroi, P. Roccaro, G. L. Oelker and S. A. Snyder, Environ. Sci. Technol., 2014, 48, 10308–10315 CrossRef CAS PubMed.
- D. Gerrity, A. N. Pisarenko, E. Marti, R. A. Trenholm, F. Gerringer, J. Reungoat and E. Dickenson, Water Res., 2015, 72, 251–261 CrossRef CAS PubMed.
- M. Sgroi, P. Roccaro, G. L. Oelker and S. A. Snyder, Water Res., 2015, 70, 174–183 CrossRef CAS PubMed.
- A. N. Pisarenko, E. J. Marti, D. Gerrity, J. R. Peller and E. R. V. Dickenson, Environ. Sci.: Water Res. Technol., 2015, 1, 668–678 CAS.
- Y. S. Wildhaber, H. Mestankova, M. Schärer, K. Schirmer, E. Salhi and U. von Gunten, Water Res., 2015, 75, 324–335 CrossRef PubMed.
- Y. Lee, D. Gerrity, M. Lee, S. Gamage, A. Pisarenko, R. A. Trenholm, S. Canonica, S. A. Snyder and U. von Gunten, Environ. Sci. Technol., 2016 DOI:10.1021/acs.est.5b04904.
- D. Stalter, A. Magdeburg and J. Oehlmann, Water Res., 2010, 44, 2610–2620 CrossRef CAS PubMed.
- J. O. Sharp, T. K. Wood and L. Alvarez-Cohen, Biotechnol. Bioeng., 2005, 89, 608–618 CrossRef CAS PubMed.
- U. von Gunten, Water Res., 2003, 37, 1443–1467 CrossRef CAS PubMed.
- F. Soltermann, C. Abegglen, C. Götz and U. von Gunten, Environ. Sci. Technol. Search PubMed submitted.
- J. Hoigné and H. Bader, Water Res., 1983, 17, 185–194 CrossRef.
- J. Hoigné, H. Bader, W. R. Haag and J. Staehelin, Water Res., 1985, 19, 993–1004 CrossRef.
- Y. Lee and U. von Gunten, Water Res., 2010, 44, 555–566 CrossRef CAS PubMed.
- Y. Lee and U. von Gunten, Water Res., 2012, 46, 6177–6195 CrossRef CAS PubMed.
- C. Hansch, A. Leo and D. Hoekman, Exploring QSAR: Hydrophobic, Electronic, and Steric Constants, American Chemical Society, Washington, DC, 1995 Search PubMed.
- S. Naumov and C. von Sonntag, Ozone: Sci. Eng., 2010, 32, 61–65 CrossRef CAS.
- M. Lee, S. G. Zimmermann, J. S. Arey, K. Fenner and U. von Gunten, Environ. Sci. Technol., 2015, 49, 9925–9935 CrossRef CAS PubMed.
- NDRL/NIST Solution Kinetics Database on the Web, A compilation of kinetics data on solution-phase reactions, available at, http://kinetics.nist.gov/solution/ Search PubMed.
- G. V. Buxton, C. L. Greenstock, W. P. Helman and W. P. Ross, J. Phys. Chem. Ref. Data, 1988, 17, 513–886 CrossRef CAS.
- D. Minakata, K. Li, P. Westerhoff and J. Crittenden, Environ. Sci. Technol., 2009, 43, 6220–6227 CrossRef CAS PubMed.
- B. A. Wols and C. H. M. Hofman-Caris, Water Res., 2012, 46, 2815–2827 CrossRef CAS PubMed.
- D. Minakata and J. Crittenden, Environ. Sci. Technol., 2011, 45, 3479–3486 CrossRef CAS PubMed.
- D. Minakata, S. P. Mezyk, J. W. Jones, B. R. Daws and J. Crittenden, Environ. Sci. Technol., 2014, 48, 13925–13932 CrossRef CAS PubMed.
- D. Minakata, W. Song, S. P. Mezyk and W. J. Cooper, Phys. Chem. Chem. Phys., 2015, 17, 11796–11812 RSC.
- Y. Lee, D. Gerrity, M. Lee, A. E. Bogeat, E. Salhi, S. Gamage, R. A. Trenholm, E. C. Wert, S. A. Snyder and U. von Gunten, Environ. Sci. Technol., 2013, 47, 5872–5881 CrossRef CAS PubMed.
- Y. Lee, L. Kovalova, C. S. McArdell and U. von Gunten, Water Res., 2014, 64, 134–148 CrossRef CAS PubMed.
- R. Xiao, M. Noerpel, H. L. Luk, Z. Wei and R. Spinney, Int. J. Quantum Chem., 2014, 114, 74–83 CrossRef CAS.
- U. von Gunten and J. Hoigné, Environ. Sci. Technol., 1994, 28, 1234–1242 CrossRef CAS PubMed.
- M.-O. Buffle and U. von Gunten, Environ. Sci. Technol., 2006, 40, 3057–3063 CrossRef CAS PubMed.
- M. S. Elovitz, U. von Gunten and H.-P. Kaiser, Ozone: Sci. Eng., 2000, 22, 123–150 CrossRef CAS.
- S. Naumov, G. Mark, A. Jarocki and C. von Sonntag, Ozone: Sci. Eng., 2010, 32, 430–434 CrossRef CAS.
- E. C. Wert, F. L. Rosario-Ortiz and S. A. Snyder, Water Res., 2009, 43, 1005–1014 CrossRef CAS PubMed.
- U. Hübner, S. Keller and M. Jekel, Water Res., 2013, 47, 6467–6474 CrossRef PubMed.
- M. S. Elovitz and U. von Gunten, Ozone: Sci. Eng., 1999, 21, 239–260 CrossRef CAS.
- J. Staehelin and J. Hoigné, Environ. Sci. Technol., 1985, 19, 1206–1213 CrossRef CAS PubMed.
- Y. Liu, J. Jiang, J. Ma, Y. Yang, C. Luo, X. Huangfu and Z. Guo, Water Res., 2015, 68, 750–758 CrossRef CAS PubMed.
- F. L. Rosario-Ortiz, S. P. Mezyk, D. F. R. Doud and S. A. Snyder, Environ. Sci. Technol., 2008, 42, 5924–5930 CrossRef CAS PubMed.
- M. M. Dong, S. P. Mezyk and F. L. Rosario-Ortiz, Environ. Sci. Technol., 2010, 44, 5174–5720 CrossRef PubMed.
- O. S. Keen, G. McKay, S. P. Mezyk, K. G. Linden and F. L. Rosario-Ortiz, Water Res., 2014, 50, 408–419 CrossRef CAS PubMed.
- J. Hoigné and H. Bader, Water Res., 1976, 10, 377–386 CrossRef.
- T. Nőthe, H. Fahlenkamp and C. von Sonntag, Environ. Sci. Technol., 2009, 43, 5990–5995 CrossRef.
- J. Hoigné, Chemistry of Aqueous Ozone and Transformation of Pollutants by Ozonation and Advanced Oxidation Processes, in The Handbook of Environmental Chemistry-Quality and Treatment of Drinking Water II, Springer-Verlag, Berlin Heidellberg, 1998, pp. 83–141 Search PubMed.
- U. Hübner, U. von Gunten and M. Jekel, Water Res., 2015, 68, 150–170 CrossRef PubMed.
- U. von Gunten, Water Sci. Technol., 2007, 55, 25–29 CrossRef CAS PubMed.
- M. M. Huber, T. A. Ternes and U. von Gunten, Environ. Sci. Technol., 2004, 38, 5177–5186 CrossRef CAS PubMed.
- R. F. Dantas, M. Canterino, R. Marotta, C. Sans, S. Esplugas and R. Andreozzi, Water Res., 2007, 41, 2525–2532 CrossRef CAS PubMed.
- M. Deborde, S. Rabouan, P. Mazellier, J.-P. Duguet and B. Legube, Water Res., 2008, 42, 4299–4308 CrossRef CAS PubMed.
- J. Benner and T. A. Ternes, Environ. Sci. Technol., 2009, 43, 5086–5093 CrossRef CAS PubMed.
- J. Benner and T. A. Ternes, Environ. Sci. Technol., 2009, 43, 5472–5480 CrossRef CAS PubMed.
- R. de Oliveira Pereira, M. L. de Alda, J. Joglar, L. A. Daniel and D. Barcelo, Chemosphere, 2011, 84, 1535–1541 CrossRef PubMed.
- D. B. Mawhinney, B. J. Vanderford and S. A. Snyder, Environ. Sci. Technol., 2012, 46, 7102–7111 CrossRef CAS PubMed.
- R. A. Segura, P. Kaplan and V. Yargeau, Chem. Cent. J., 2013, 7, 74 CrossRef PubMed.
- M. K. Ramseier and U. von Gunten, Ozone: Sci. Eng., 2009, 31, 201–215 CrossRef CAS.
- M. H. Khan, H. Bae and J.-Y. Jung, J. Hazard. Mater., 2010, 181, 659–665 CrossRef CAS PubMed.
- M. N. Abellan, W. Gebhardt and H. Schroeder, Water Sci. Technol., 2008, 58, 1803–1812 CrossRef CAS PubMed.
- M. M. Sein, M. Zedda, J. Tuerk, T. C. Schmidt, A. Golloch and C. von Sonntag, Environ. Sci. Technol., 2008, 42, 6656–6662 CrossRef CAS PubMed.
- A. D. Coelho, C. Sans, A. Agüera, M. J. Gómez, S. Esplugas and M. Dezotti, Sci. Total Environ., 2009, 407, 3572–3578 CrossRef CAS PubMed.
- M. M. Gómez-Ramos, M. Mezcua, A. Agüera, A. R. Fernández-Alba, S. Gonzalo, A. Rodríguez and R. Rosal, J. Hazard. Mater., 2011, 192, 18–25 Search PubMed.
- D. C. McDowell, M. M. Huber, M. Wagner, U. von Gunten and T. A. Ternes, Environ. Sci. Technol., 2005, 39, 8014–8022 CrossRef CAS PubMed.
- M. C. Dodd, D. Rentsch, H. P. Singer, H.-P. Kohler and U. von Gunten, Environ. Sci. Technol., 2010, 44, 5940–5948 CrossRef CAS PubMed.
- A. K. Genena, D. B. Luiz, W. Gebhardt, R. F. P. M. Moreira, H. J. Jose and H. F. Schroder, Ozone: Sci. Eng., 2011, 33, 308–328 CrossRef CAS.
- M. Scheurer, M. Godejohann, A. Wick, O. Happel and T. Ternes, Environ. Sci. Pollut. Res., 2012, 19, 1107–1118 CrossRef CAS PubMed.
- N. H. El Najjar, A. Touffet, M. Deborde, R. Journel and N. K. Vel Leitner, Chemosphere, 2013, 93, 604–611 CrossRef PubMed.
- U. Hübner, B. Seiwert, T. Reemtsma and M. Jekel, Water Res., 2014, 49, 34–43 CrossRef PubMed.
- M. Favier, R. Devil, K. van Eyck, A. van Schepdael and D. Cabooter, Chemosphere, 2015, 136, 32–41 CrossRef CAS PubMed.
- C. Prasse, M. Wagner, R. Schulz and T. A. Ternes, Environ. Sci. Technol., 2012, 46, 2169–2178 CrossRef CAS PubMed.
- F. Munoz, E. Mvula, S. E. Braslavsky and C. von Sonntag, J. Chem. Soc., Perkin Trans. 1, 2001, 2, 1109–1116 RSC.
- F. Lange, S. Cornelissen, D. Kubac, M. M. Sein, J. von Sonntag, C. B. Hannich, A. Golloch, H. J. Heipieper, M. Möder and C. von Sonntag, Chemosphere, 2006, 65, 17–23 CrossRef CAS PubMed.
- J. Radjenovic, M. Godehardt, M. Petrovic, A. Hein, M. Farre, M. Jekel and D. Barcelo, Environ. Sci. Technol., 2009, 43, 6808–6815 CrossRef CAS PubMed.
- S. G. Zimmermann, A. Schmukat, M. Schulz, J. Benner, U. von Gunten and T. A. Ternes, Environ. Sci. Technol., 2012, 46, 876–884 CrossRef CAS PubMed.
- E. Borowska, M. Bourgin, J. Hollender, C. Kienle, C. S. McArdell and U. von Gunten, Water Res., 2016 DOI:10.1016/j.watres.2016.02.020.
- B. DeWitte, J. Dewulf, K. Demeestere, V. De Vyvere, P. De Wispelaere and H. van Langehove, Environ. Sci. Technol., 2008, 42, 4889–4895 CrossRef CAS PubMed.
- C. K. Schmidt and H.-J. Brauch, Environ. Sci. Technol., 2008, 42, 6340–6346 CrossRef CAS PubMed.
- U. von Gunten, E. Salhi, C. K. Schmidt and W. A. Arnold, Environ. Sci. Technol., 2010, 44, 5762–5768 CrossRef CAS PubMed.
- D. Trogolo, B. K. Mishra, M. B. Heeb, U. von Gunten and J. S. Arey, Environ. Sci. Technol., 2015, 49, 4163–4175 CrossRef CAS PubMed.
- E. J. Marti, A. N. Pisarenko, J. R. Peller and E. R. V. Dickenson, Water Res., 2015, 72, 262–270 CrossRef CAS PubMed.
- K. Kosaka, K. Fukui, Y. Kayanuma, M. Asami and M. Akiba, Ozone: Sci. Eng., 2014, 36, 215–220 CrossRef CAS.
- P. Andrzejewski, B. Kasprzyk-Hordern and J. Nawrocki, Water Res., 2008, 42, 863–870 CrossRef CAS PubMed.
- M. Oya, K. Kosaka, M. Asami and S. Kunikane, Chemosphere, 2008, 73, 1724–1730 CrossRef CAS PubMed.
- L. Yang, Z. Chen, J. Shen, Z. Xu, H. Liang, J. Tian, Y. Ben, X. Zhai, W. Shi and G. Li, Environ. Sci. Technol., 2009, 43, 5481–5487 CrossRef CAS PubMed.
- L. Padhye, Y. Luzinova, M. Cho, B. Mizaikoff, J.-H. Kim and C.-H. Huang, Environ. Sci. Technol., 2011, 45, 4353–4359 CrossRef CAS PubMed.
- C. von Sonntag and H.-P. Schuchmann, Peroxyl radicals in aqueous solutions, in Peroxyl Radicals, ed. Z. Alfassi, John Wiley & Sons, New York, 1997, pp. 172–234 Search PubMed.
- C. von Sonntag, A Chemical Perspective, Free-Radical-Induced DNA Damage and Its Repair, Springer-Verlag, Berlin, Heidelberg, 2006 Search PubMed.
- B. Razavi, W. Song, W. J. Cooper, J. Greaves and J. Jeong, J. Phys. Chem. A, 2009, 113, 1287–1294 CrossRef CAS PubMed.
- J. Jeong, J. Jung, W. J. Cooper and W. Song, Water Res., 2010, 44, 4391–4398 CrossRef CAS PubMed.
- R. R. Singh, Y. Lester, K. G. Linden, N. G. Love, G. E. Atilla-Gokcumen and D. S. Aga, Environ. Sci. Technol., 2015, 49, 2938–2990 Search PubMed.
- W. Song, W. J. Cooper, S. P. Mezyk, J. Greaves and B. M. Peake, Environ. Sci. Technol., 2008, 42, 1256–1261 CrossRef CAS PubMed.
- W. Song, W. J. Cooper, B. M. Peake, S. P. Mezyk, M. G. Nickelsen and K. E. O'Shea, Water Res., 2009, 43, 635–642 CrossRef CAS PubMed.
- H. Santoke, W. Song, W. J. Cooper, J. Greaves and G. E. Miller, J. Phys. Chem. A, 2009, 113, 7846–7851 CrossRef CAS PubMed.
- T. An, H. Yang, G. Li, W. Song, W. J. Cooper and X. Nie, Appl. Catal., B, 2010, 94, 288–294 CrossRef CAS.
- K. S. Tay, N. A. Rahman and M. R. B. Abas, Microchem. J., 2011, 99, 312–326 CrossRef CAS.
- O. Keen and K. G. Linden, Environ. Sci. Technol., 2013, 47, 6799–6805 CrossRef CAS PubMed.
- E. L. Schymanski, J. Jeon, R. Gulde, K. Fenner, M. Ruff, H. Singer and J. Hollender, Environ. Sci. Technol., 2014, 48, 2907–2908 Search PubMed.
- R. Andreozzi, M. Canterino, R. Marotta and N. Paxeus, J. Hazard. Mater., 2005, 122, 243–250 CrossRef CAS PubMed.
- W. Song, W. Chen, W. J. Cooper, J. Greaves and G. E. Miller, J. Phys. Chem. A, 2008, 112, 7411–7417 CrossRef CAS PubMed.
- Y. J. Jung, W. G. Kim, Y. Yoon, J.-W. Kang, Y. M. Hong and H. W. Kim, Sci. Total Environ., 2012, 420, 160–167 CrossRef CAS PubMed.
- F. J. Benitez, J. L. Acero, F. J. Real, G. Roldán and E. Rodríguez, J. Hazard. Mater., 2015, 282, 224–232 CrossRef CAS PubMed.
- O. Keen, S. Baik, K. G. Linden, D. S. Aga and N. G. Love, Environ. Sci. Technol., 2012, 46, 6222–6227 CrossRef CAS PubMed.
- K. S. Tay, N. A. Rahman and M. R. B. Abas, Chemosphere, 2009, 76, 1296–1302 CrossRef CAS PubMed.
- A. Müller, S. C. Weiss, J. Beiβwenger, H. G. Leukhardt, W. Schulz, W. Seitz, W. K. L. Ruck and W. H. Weber, Water Res., 2012, 46, 679–690 CrossRef PubMed.
- M. M. Sein, T. C. Schmidt, A. Golloch and C. von Sonntag, Water Sci. Technol., 2009, 59, 1479–1485 CrossRef CAS PubMed.
- K. S. Tay, N. A. Rahman and M. R. B. Abas, Environ. Sci. Pollut. Res., 2013, 20, 3115–3121 CrossRef CAS PubMed.
- C. Liu, V. Nanaboina, G. V. Korshin and W. Jiang, Water Res., 2012, 46, 5235–5246 CrossRef CAS PubMed.
- B. Ning, N. J. D. Graham and Y. Zhang, Chemosphere, 2007, 68, 1163–1172 CrossRef CAS PubMed.
- K. S. Tay, N. A. Rahman and M. R. B. Abas, Chemosphere, 2010, 81, 1446–1453 CrossRef CAS PubMed.
- R. Andreozzi, V. Caprio, R. Marotta and D. Vogna, Water Res., 2003, 37, 993–1004 CrossRef CAS PubMed.
- N. H. El Najjar, A. Touffet, M. Deborde, R. Journel and N. K. Vel Leitner, Sep. Purif. Technol., 2014, 136, 137–143 CrossRef.
- E. Barron, M. Deborde, S. Rabouan, P. Mazellier and B. Legube, Water Res., 2006, 40, 2181–2189 CrossRef CAS PubMed.
- R. F. Dantas, S. Sans and S. Esplugas, J. Environ. Eng., 2011, 137, 754–759 CrossRef CAS.
- J. Jeong, W. Song, W. J. Cooper, J. Jung and J. Greves, Chemosphere, 2010, 78, 533–540 CrossRef CAS PubMed.
- X. Chen, J. Richard, Y. Liu, E. Dopp, J. Tuerk and K. Bester, Water Res., 2012, 46, 2247–2256 CrossRef CAS PubMed.
- J. Kuang, J. Huang, B. Wang, Q. Cao, S. Deng and G. Yu, Water Res., 2013, 47, 2863–2872 CrossRef CAS PubMed.
- Y. Lester, H. Mamane, I. Zucker and D. Avisar, Water Res., 2013, 47, 4349–4356 CrossRef CAS PubMed.
- K. Li and J. Crittenden, Environ. Sci. Technol., 2009, 43, 2831–2837 CrossRef CAS PubMed.
- X. Guo, D. Minakata, J. Niu and J. Crittenden, Environ. Sci. Technol., 2014, 48, 5718–5725 CrossRef CAS PubMed.
- X. Guo, D. Minakata and J. Crittenden, Environ. Sci. Technol., 2014, 48, 10813–10820 CrossRef CAS PubMed.
- X. Guo, D. Minakata and J. Crittenden, Environ. Sci. Technol., 2015, 49, 9230–9236 CrossRef CAS PubMed.
- M. Lee, L. Blum, K. Fenner and U. von Gunten, unpublished work..
- B. I. Escher, R. Baumgartner, J. Lienert and K. Fenner, Predicting the Ecotoxicological Effects of Transformation Products, in The Handbook of Environmental Chemistry, Reaction and Processes, Part P-Transformation Products of Synthetic Chemicals in the Environment, ed. A. B. A. Boxall, 2009, vol. 2, pp. 205–244 Search PubMed.
- B. I. Escher and K. Fenner, Environ. Sci. Technol., 2011, 45, 3835–3847 CrossRef CAS PubMed.
- S. Suarez, M. C. Dodd, F. Omil and U. von Gunten, Water Res., 2007, 41, 2481–2490 CrossRef CAS PubMed.
- M. C. Dodd, H.-P. Kohler and U. von Gunten, Environ. Sci. Technol., 2009, 43, 2498–2504 CrossRef CAS PubMed.
- O. Keen and K. G. Linden, Environ. Sci. Technol., 2013, 47, 13020–13030 CrossRef CAS PubMed.
- H. Mestankova, K. Schirmer, B. I. Escher, U. von Gunten and S. Canonica, Environ. Pollut., 2012, 161, 30–35 CrossRef CAS PubMed.
- H. Mestankova, K. Schirmer, S. Canonica and U. von Gunten, Water Res., 2014, 66, 399–410 CrossRef CAS PubMed.
- E. J. Rosenfeldt, P. J. Chen, S. Kullman and K. G. Linden, Sci. Total Environ., 2007, 377, 105–113 CrossRef CAS PubMed.
- Y. Lee, B. I. Escher and U. von Gunten, Environ. Sci. Technol., 2008, 42, 6333–6339 CrossRef CAS PubMed.
- H. Mestankova, A. M. Parker, N. Bramaz, S. Canonica, K. Schirmer, U. von Gunten and K. G. Linden, Water Res., 2016, 93, 110–120 CrossRef CAS PubMed.
- X. Wang, X. Huang, C. Zuo and H. Hu, Chemosphere, 2004, 55, 733–741 CrossRef CAS PubMed.
- H. Yu, E. Nie, J. Xu, S. Yan, W. J. Cooper and W. Song, Water Res., 2013, 47, 1909–1918 CrossRef CAS PubMed.
- E. Illés, E. Szabó, E. Takács, L. Wojnárovits, A. Dombi and K. Gajda-Schrantz, Sci. Total Environ., 2014, 472, 178–184 CrossRef PubMed.
- B. I. Escher and J. L. M. Hermens, Environ. Sci. Technol., 2002, 36, 4201–4217 CrossRef CAS PubMed.
- J. A. H. Schwöbel, Y. K. Koleva, S. J. Enoch, F. Bajot, M. Hewitt, J. C. Madden, D. W. Roberts, T. W. Schultz and M. T. D. Cronin, Chem. Rev., 2011, 111, 2562–2596 CrossRef PubMed.
- M. Macova, B. I. Escher, J. Reungoat, S. Carswell, K. L. Chue, J. Keller and J. F. Mueller, Water Res., 2010, 44, 477–492 CrossRef CAS PubMed.
- J. Reungoat, M. Macova, B. I. Escher, S. Carswell, J. F. Mueller and J. Keller, Water Res., 2010, 44, 625–637 CrossRef CAS PubMed.
- A. Magdeburg, D. Stalter and J. Oehlmann, Chemosphere, 2012, 88, 1008–1014 CrossRef CAS PubMed.
- F. Hammes, E. Salhi, O. Koster, H. P. Kaiser, T. Egli and U. von Gunten, Water Res., 2006, 40, 2275–2286 CrossRef CAS PubMed.
- R. Judson, K. Houck, M. Martin, T. Knudsen, R. S. Thomas, N. Sipes, I. Shah, J. Wambaugh and K. Crofton, Basic Clin. Pharmacol. Toxicol., 2014, 115, 69–76 CrossRef CAS PubMed.
- T. B. Knudsen, D. A. Keller, M. Sander, E. W. Carney, N. G. Doerrer, D. L. Eaton, S. C. Fitzpatrick, K. L. Hastings, D. L. Mendrick, R. R. Tice, P. B. Watkins and M. Whelan, Toxicol. Sci., 2015, 143, 256–267 CrossRef CAS PubMed.
- OpenTox, available at http://www.opentox.org/.
- VirtualToxLab, available at http://www.biograf.ch/.
| This journal is © The Royal Society of Chemistry 2016 |