
Control of nitrosamines during non-potable and de facto wastewater reuse with medium pressure ultraviolet light and preformed monochloramine†
Daniel L.
McCurry
a,
Stuart W.
Krasner
b and
William A.
Mitch
*a
aDepartment of Civil and Environmental Engineering, Stanford University, Stanford, CA 94305, USA. E-mail: wamitch@stanford.edu; Fax: +650 723 7058; Tel: +650 725 9298
bMetropolitan Water District of Southern California, La Verne, CA 91750, USA
First published on 18th March 2016
Abstract
Water utilities practicing chlorination of poorly nitrified wastewater effluents for non-potable reuse, or of ammonia-containing, wastewater-impacted source waters during de facto reuse may form N-nitrosodimethylamine (NDMA) by reactions between wastewater-derived NDMA precursors and inorganic chloramines formed in situ when chlorine reacts with ammonia. Treatment with moderate doses (186 mJ cm−2 germicidal fluence) of UV from medium pressure mercury lamps (MPUV) upstream of chloramination minimized NDMA formation by destroying ambient NDMA (i.e., the NDMA occurring in wastewater) by >70% and deactivating chloramine-reactive NDMA precursors by 46%. UV from low pressure mercury lamps (LPUV) was less effective for NDMA precursor deactivation. Ozonation does not destroy ambient NDMA and formed significant levels of NDMA from ozone-reactive precursors. For waters with low ammonia concentrations, breakpoint chlorination deactivated NDMA precursors, but does not destroy ambient NDMA, and formed an array of regulated and unregulated halogenated byproducts. Even over a 3 d contact time, application of preformed monochloramine reduced NDMA formation by 39% by minimizing the exposure to inorganic dichloramine, the inorganic chloramine precursor to NDMA. However, the benefit of preformed monochloramine was restricted to waters with low to moderate ammonia concentrations (<20 mg L−1 as N), because dichloramine formation during in situ chloramination is less important for high ammonia waters. Overall, the combination of MPUV with preformed monochloramine reduced final NDMA concentrations by 73–93% relative to in situ chloramination, a common current practice for non-potable reuse.
Water ImpactFor wastewater reuse scenarios employing chlorine disinfection of ammonia-containing waters, the chloramines that form can produce the carcinogen, NDMA. For de facto or non-potable reuse scenarios involving prolonged chloramine contact, MPUV combined with preformed monochloramine may serve as a cost-effective treatment alternative to minimize NDMA concentrations. |
1. Introduction
Increasing populations in arid regions coupled with the low availability of pristine water sources is driving many water utilities to consider the reuse of municipal wastewater.1 Three categories of wastewater reuse can be distinguished: use of source waters impacted by upstream municipal wastewater discharges (de facto reuse), recycling of wastewater for non-potable purposes (e.g., irrigation), and potable reuse. In addition to pathogens, chemical contaminants represent a key concern for reuse operations. For all three reuse categories, the National Research Council indicated that byproducts of disinfection, particularly N-nitrosodimethylamine (NDMA), and perfluorinated chemicals occurred at concentrations closer to levels of potential human health concern than other chemical contaminants (e.g., pharmaceuticals).1Among disinfection byproducts, the importance of N-nitrosodimethylamine (NDMA) was highlighted.1 According to the U.S. EPA, a 0.6 ng L−1 NDMA concentration in drinking water is associated with an age-adjusted 10−6 lifetime excess cancer risk.2 California has instituted a 10 ng L−1 notification level for NDMA,3 which has also been applied to potable reuse waters. The U.S. EPA is considering regulating N-nitrosamines in drinking water as part of the 6 year review of the disinfection byproduct rule.4
NDMA is particularly important for reuse facilities because both it and its precursors occur in sewage. NDMA was measured in sewage and primary effluent at a median concentration of 88 ng L−1, with concentrations up to 790 ng L−1 measured in sewage impacted by industrial discharges.5 The median concentration decreased to 46 ng L−1 in secondary effluents prior to disinfection,5 reflecting partial removal during biological treatment. U.S. wastewater treatment plants with poorly nitrified effluents had a median concentration of 11 ng L−1 of NDMA (75th and 90th percentiles were 24 and 122 ng L−1, respectively), whereas wastewater treatment plants with well-nitrified effluents had a median concentration of 3.0 ng L−1 (maximum of 8.2 ng L−1).6 That is because when utilities employ free chlorine to disinfect ammonia-containing source waters for de facto reuse scenarios or non-nitrified municipal wastewater effluents for wastewater reuse scenarios, chloramines are rapidly formed in situ. NDMA forms when inorganic dichloramine reacts with secondary and tertiary amines,7–9 which are enriched in municipal wastewater effluents.10,11 Although in situ chloramination is frequently applied upstream of the membrane systems in certain potable reuse trains to control biofouling, NDMA concentrations are mitigated by the short contact time (minutes) before NDMA12 and its precursors are rejected by reverse osmosis membranes. Even though NDMA rejection is only partial (∼50%), it is efficiently removed by the UV-based advanced oxidation processes generally employed following reverse osmosis.13,14 NDMA formation may be greater for de facto reuse and non-potable or reuse scenarios where reverse osmosis membranes and advanced oxidation are not employed, and chloramine contact times can be on the order of days.
For de facto or non-potable reuse scenarios with ammonia-containing source waters, application of alternative disinfectants upstream of chloramination could mitigate nitrosamine formation. Previous research with drinking water sources that did not contain ammonia indicated that application of disinfectant exposures relevant to Giardia control reduced NDMA formation during subsequent chloramination in the order ozone > free chlorine ∼ ultraviolet light from a medium pressure mercury (MPUV) lamp > UV from a low pressure mercury lamp (LPUV).9,15 For de facto or non-potable reuse waters containing low ammonia, free chlorine contact may be feasible via application of sufficient free chlorine to break out the ammonia, but the implications of the free chlorine contact for halogenated byproduct formation relative to nitrosamine control need clarification. While no significant NDMA formation was observed for ozone application to drinking water supplies9,15 research has indicated significant NDMA formation upon ozone application to wastewaters,16–19 particularly when applied to control biofouling upstream of membrane systems in potable reuse trains.20,21 Whether the NDMA formation associated with ozonation negates the gains achieved via precursor deactivation when ozonation is followed by chloramination in non-potable reuse systems is unclear. While UV disinfection is an increasingly popular option for wastewater discharged to surface waters, UV disinfection often is bypassed for non-potable reuse applications where maintenance of a chlorine-based disinfectant residual is required.22 Unlike ozone or free chlorine, UV treatment could significantly degrade the N-nitrosamines occurring in sewage13,23 while deactivating NDMA precursors and pathogens, but this option needs further evaluation.
During the chloramination process, NDMA formation could be mitigated by reducing the occurrence of inorganic dichloramine, the chloramine species responsible for NDMA formation.7,24 Dichloramine formation is partially controlled via equilibrium with monochloramine (eqn (1)), increasing with decreasing pH and ammonia concentrations.
| NHCl2 + NH3 + H+ ⇌ 2NH2Cl + H+ | (1) |
However, due to the rapidity of inorganic chloramine formation reactions from free chlorine and ammonia, injection of concentrated free chlorine into an ammonia-containing solution may promote higher dichloramine formation than expected based upon equilibrium considerations due to the relatively high free chlorine![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :ammonia molar ratios at the point of chlorine injection.25 In a previous pilot-scale investigation, we demonstrated that injection of monochloramine, preformed under conditions inhibiting dichloramine formation, could reduce NDMA formation relative to injection of free chlorine into an ammonia-containing wastewater over ∼4 minute contact times upstream of membrane units in a potable reuse plant.26 However, the relative prevalence of monochloramine to dichloramine should tend towards equilibrium over longer contact times (eqn (1)), raising questions as to the significance of the preformed monochloramine strategy for de facto or non-potable reuse scenarios. Others found that application of preformed monochloramine to one well-nitrified wastewater effluent (∼0.1 mg L−1 as N ammonia) reduced NDMA formation over a 24 h contact period to 16 ng L−1 compared to the 310 ng L−1 NDMA formed when chloramines were formed in-line by application of 2.4 mg L−1 as N ammonia followed by 10 mg L−1 as Cl2 free chlorine (a 1.2:1 ammonia:chlorine molar ratio).27 However, the benefits of preformed monochloramine application needs validation over a broader range of waters, particularly non-nitrified wastewater effluents, where higher ammonia concentrations may reduce dichloramine formation associated with the high chlorine concentration at the site of chlorine injection.
:ammonia molar ratios at the point of chlorine injection.25 In a previous pilot-scale investigation, we demonstrated that injection of monochloramine, preformed under conditions inhibiting dichloramine formation, could reduce NDMA formation relative to injection of free chlorine into an ammonia-containing wastewater over ∼4 minute contact times upstream of membrane units in a potable reuse plant.26 However, the relative prevalence of monochloramine to dichloramine should tend towards equilibrium over longer contact times (eqn (1)), raising questions as to the significance of the preformed monochloramine strategy for de facto or non-potable reuse scenarios. Others found that application of preformed monochloramine to one well-nitrified wastewater effluent (∼0.1 mg L−1 as N ammonia) reduced NDMA formation over a 24 h contact period to 16 ng L−1 compared to the 310 ng L−1 NDMA formed when chloramines were formed in-line by application of 2.4 mg L−1 as N ammonia followed by 10 mg L−1 as Cl2 free chlorine (a 1.2:1 ammonia:chlorine molar ratio).27 However, the benefits of preformed monochloramine application needs validation over a broader range of waters, particularly non-nitrified wastewater effluents, where higher ammonia concentrations may reduce dichloramine formation associated with the high chlorine concentration at the site of chlorine injection.
The purpose of this work was to evaluate combinations of UV, free chlorine or ozone disinfectants and preformed monochloramine for the mitigation of NDMA formation over prolonged chloramine contact times within ammonia-containing de facto or non-potable reuse waters. First, we evaluated the benefits of MPUV treatment for both the destruction of sewage-associated NDMA, and deactivation of NDMA precursors. Second, we assessed the extent to which application of preformed monochloramine could complement MPUV treatment over prolonged chloramine contact times, and how the efficacy of preformed monochloramine depends upon the ammonia concentration. Third, we compared free chlorine application, achieved via breakpoint chlorination of a de facto reuse water exhibiting low ammonia concentrations, to MPUV for control of NDMA formation, while considering halogenated DBP formation associated with the free chlorine contact time. Fourth, we assessed ozonation to control NDMA formation for non-potable reuse scenarios relative to MPUV.
2. Materials and methods
2.1. Sampling and source characterization
Grab samples from four municipal wastewater treatment plant effluents in California and one drinking water source in the upper midwest (impacted by wastewater and other watershed sources during the spring runoff) were collected upstream of disinfection in 10 L HDPE containers and transported on ice to the laboratory. Upon arrival, samples were filtered through 0.7 μm pre-combusted (450 °C, 4 h) glass-fiber filters, and stored at 4 °C until use. Samples were adjusted to pH 8 (with 1 M NaOH/HCl) before experimentation. One water (WW1B) contained a small (0.2 mg L−1) chloramine residual due to partial recycling of chloraminated effluent back into the influent stream; this water was quenched with stoichiometric ascorbic acid within 3 h of sample collection to minimize chloramine exposure. Total organic carbon was measured with a Shimadzu (Kyoto, Japan) TOC-L instrument. Inorganic nitrogen species (NH4+, NO2−, NO3−) were measured with a WestCo SmartChem 200 Discrete Analyzer. Bromide (as total bromine) was measured with a Thermo Scientific (Waltham, MA) XSERIES 2 ICP-MS. Source water quality data are given in Table 1. Not all samples were used for each experiment. For example, for certain experiments evaluating the effect of ammonia concentration, ammonia was spiked into samples exhibiting low ammonia concentrations so that the ammonia concentration could be systematically varied; for these experiments, samples exhibiting high ammonia concentrations were not employed.| Sample code | Ambient NDMA (ng L−1) | NDMA UFC (ng L−1) | DOC (mg L−1) | UV254 (cm−1) | SUVA254 (L mg−1 m) | Bromide (μg L−1) | Ammonia (mg L−1 as N) | Nitrite (mg L−1 as N) | Nitrate (mg L−1 as N) |
|---|---|---|---|---|---|---|---|---|---|
| NM = not measured; NDMA UFC = NDMA formed after uniform formation condition chloramination. | |||||||||
| DW | <2 | 57 | 5.0 | 0.067 | 1.34 | 40 | 0.53 | 0.03 | 1.4 |
| WW1 | 26 | 39 | 9.5 | 0.189 | 1.98 | 1400 | 22 | 0.04 | <0.1 |
| WW2A | 40 | 46 | 7.5 | 0.353 | 4.70 | 510 | 0.05 | 0.22 | 13 |
| WW2B | 15 | 43 | 9.0 | 0.185 | 2.06 | NM | 0.04 | 0.18 | 14 |
| WW3 | 30 | 38 | 18.8 | 0.185 | 0.98 | NM | 57 | 0.08 | <0.1 |
| WW4 | 14 | 128 | 8.4 | 0.199 | 2.36 | 696 | 3.1 | 0.61 | 15 |
2.2. Pretreatment
Pretreatments with MPUV, LPUV, or ozone were applied at exposures relevant to Giardia inactivation and advanced oxidation treatment, as described previously.15 Briefly, for UV treatments, incident fluence rates was determined using iodide/iodate chemical actinometry.28 Low (186 mJ cm−2) and high (1000 mJ cm−2) germicidal fluence doses were calculated by correcting the incident fluence by the solution absorbance (i.e., average fluence), and normalizing the average fluence by the absorption spectrum of DNA, as described elsewhere.28 MPUV treatments were applied with a 450 W Hanovia medium-pressure mercury bulb and LPUV treatments were applied with four 12 W General Electric low-pressure mercury bulbs. Light was applied using semi-collimated beam apparatuses; light from the UV lamps shone down through a shutter onto 0.5 L samples in 0.7 L borosilicate crystallization dishes, continuously mixed by a magnetic stir bar.Ozone was generated by passing oxygen gas (>99%, Praxair, Danbury, CT) at ∼1 L min−1 through a Triogen LAB2B ozone generator (East Kilbride, Scotland). The resulting ozone-enriched oxygen stream was passed through a buffer trap (50 mM phosphate, pH 6) and continuously bubbled into 1 L of deionized water, chilled to ∼0 °C using an ice-water bath. Ozone stock concentrations were standardized by combining 1 mL of ozone stock with 2 mL of 0.5 M H3PO4, and directly measuring UV absorbance (ε260 = 3200 M−1 cm−1).29 Ozone residual concentrations were measured by the indigo method.30 To determine ozone dosing requirements, a range of ozone concentrations were added to each water. Ozone concentrations after dosing were monitored until no longer detectable (<0.1 mg L−1) and cumulative exposures were calculated using a trapezoidal Riemann sum of concentrations over time, as described previously.15 Ozone doses were selected such that low (0.2 mg min L−1) or high (1.0 mg min L−1) exposure was achieved just as the ozone residual was exhausted, to avoid the need to quench residual ozone.
2.3. Chloramination
Water samples were chloraminated in one of two ways: either free chlorine was added directly to form chloramines in situ by reaction with ambient ammonia, or preformed monochloramine was added. Working stocks of free chlorine (∼50 mM) were prepared from a ∼0.8 M stock (Fisher Scientific) and standardized daily by UV spectroscopy (ε292 = 365 M−1 cm−1).31 Preformed monochloramine stocks were prepared as previously described.7 Briefly, 10 mL of ammonium chloride (24 mM) was prepared and adjusted to pH 8.5–9 with 1 M NaOH. 10 mL of a 40 mM free chlorine stock was added dropwise to the continuously stirred ammonium chloride solution. The concentration was standardized spectrophotometrically at 245 nm and 295 nm, the absorbance maxima for monochloramine and dichloramine, respectively, as described previously;25 dichloramine was not measureable in the monochloramine stocks. Water samples (0.5 L) were buffered prior to chloramination (4 mM borate; pH 8) in borosilicate volumetric flasks. Free chlorine or chloramines were added to water samples at 5 mg L−1 as Cl2 (wastewater effluent) or 2.5 mg L−1 as Cl2 (drinking water), both chosen to target a 1–2 mg L−1 as Cl2 chlorine residual after three days, to mimic typical distribution system conditions (uniform formation conditions (UFC)).32 For WW2, a well-nitrified wastewater effluent, the addition of 5 mg L−1 as Cl2 free chlorine would exceed the breakpoint. To evaluate in situ chloramination for this water, the sample was spiked with ammonium chloride to achieve 1.06 mg L−1 as N and mixed prior to the addition of free chlorine. Samples were stored for 3 d at 21 ± 1 °C in the dark, followed by pH and chlorine residual (DPD colorimetric method)33 measurements. Samples were quenched with 33 mg L−1 ascorbic acid and stored at 4 °C until analysis.2.4. Analytical methods
Nitrosamines were measured with a modified version of EPA Method 521. Samples (0.5 L) were spiked with deuterated d6-NDMA and d8-N-nitrosomorpholine (d8-NMOR) at 20 ng L−1 each, for isotope dilution analysis. Samples were extracted on solid phase coconut shell activated carbon cartridges (Enviro-Clean, United Chemical, Bristol, PA) and eluted with 12 mL of dichloromethane (Fisher, Optima grade), which was then dried with magnesium sulfate heptahydrate (∼1 g) and blown down to 1 mL under forced N2 gas. Samples were injected on an Agilent (Santa Clara, CA) 7890 A gas chromatograph, separated by an Agilent DB-1301 column (60 m × 0.25 mm × 1 μm), and detected on an Agilent 240 ion trap mass spectrometer. Retention times, parent ions, and quantification ions are given in Table S1.† Sucralose was measured as a conservative tracer of wastewater input in the wastewater-impacted drinking water, according to a previously developed method.34 Sucralose in U.S. wastewater treatment plant effluents is typically ∼20–30 μg L−1,35 and the measured value in our drinking water sample was 0.3 μg L−1, indicating ∼1–1.5% wastewater by volume. Halogenated DBPs were measured as described previously.363. Results and discussion
3.1. MPUV destruction of ambient NDMA and NDMA precursors
There was no measureable ambient NDMA in the wastewater-impacted drinking water. However, all four wastewaters contained ambient NDMA (Table 1) and treatment with a modest (186 mJ cm−2 germicidal fluence) dose of MPUV light reduced ambient NDMA by >70% (Fig. 1), as previously observed in recycled waters or drinking waters.13,15 Subsequent addition of free chlorine to all five ammonia-containing waters (or after addition of 1.06 mg L−1 as N ammonia to the well-nitrified WW2A), forming chloramines in situ, increased NDMA concentrations, but to below the initial ambient levels in three out of four wastewaters. This reduction in NDMA reflects the balance between NDMA formation during chloramination and the destruction of ambient NDMA and NDMA precursors by MPUV. To isolate the impact of MPUV on NDMA precursors, Fig. 2 plots NDMA precursors assessed as NDMA measured after UFC chloramination minus the NDMA measured pre-chloramination (i.e., net NDMA formation). MPUV pre-treatment deactivated NDMA precursors by an average of 46% across the wastewater-impacted drinking water and the four wastewaters (Fig. 2). While MPUV was largely ineffective for deactivating NDMA precursors in one wastewater (WW2A), pre-treatment with MPUV still achieved a net reduction in NDMA concentrations relative to the ambient NDMA at this facility (Fig. 1) due to the effective degradation of ambient NDMA. | ||
| Fig. 1 NDMA concentrations in untreated samples (ambient), samples treated with 186 mJ cm−2 MPUV germicidal fluence only, or samples treated with 186 mJ cm−2 MPUV and chloraminated by application of free chlorine (2.5 mg L−1 as Cl2 for DW; 5 mg L−1 as Cl2 for others). Experimental conditions: pH = 8, [borate buffer] = 4 mM, T = 21 ± 1 °C. Error bars represent the range (n = 2) or standard deviations (n = 3) of experimental replicates. | ||
 | ||
| Fig. 2 Net NDMA formation (i.e., post-chloramination NDMA minus pre-chloramination NDMA) after in situ chloramination (5 [or 2.5 for DW] mg L−1 as Cl2) for 3 d with or without pre-treatment with MPUV (186 mJ cm−2 germicidal fluence). Experimental conditions: pH = 8, [borate buffer] = 4 mM, T = 21 ± 1 °C. Error bars represent the range (n = 2) or standard deviations (n = 3) of experimental replicates. | ||
3.2. Comparison of MPUV to LPUV and ozone for pre-treatment
Without subsequent chloramination, MPUV reduced NDMA concentrations to a somewhat greater degree than LPUV alone at 186 mJ cm−2 germicidal fluence, in contrast with previous results suggesting equal efficiency13 in deionized water (Fig. 3A). In contrast, ozone formed additional NDMA in one wastewater, as has been previously observed in certain full-scale wastewater treatment plants,20 and did not affect the NDMA concentration in the other wastewater. | ||
| Fig. 3 NDMA concentrations after pretreatment without and with post-treatment with chloramines formed in situ by addition of 5 mg L−1 as Cl2 free chlorine. Pre-treatment exposures were 0.2 mg min L−1 O3 or 186 mJ cm−2 LPUV or MPUV germicidal fluence. Experimental conditions: pH = 8, [borate buffer] = 4 mM, T = 21 ± 1 °C, chloramine exposure time = 3 d. Error bars represent the range (n = 2) or standard deviations (n = 3) of experimental replicates. | ||
When no pre-treatment was applied, NDMA measured in the same samples after 3 d of treatment with chloramines formed in situ by addition of free chlorine was substantially higher in the wastewater containing 3.1 mg L−1 as N ammonia (WW4), but not the wastewater containing 56 mg L−1 as N ammonia (WW3) (Fig. 3B). While WW4, a nitrified effluent, featured higher nitrite concentrations than WW3, previous research has demonstrated that chloramines do not react appreciably with nitrite to form NDMA via a N2O4 intermediate.37 Given that inorganic dichloramine has been demonstrated to serve as the predominant inorganic chloramine precursor for NDMA formation,7 the higher ammonia concentration in the second wastewater would be expected to reduce NDMA formation by favoring monochloramine over dichloramine. The dependence on ammonia concentration is evaluated further below. Pre-treatment with MPUV reduced net NDMA concentrations measured after in situ chloramination to a greater degree than LPUV or ozone.
The poor performance of ozone contrasts with wastewater-impacted drinking waters, where ozone was frequently the most effective pre-treatment for reducing NDMA formation during subsequent chloramination.9,15 Two factors explain these differences. First, wastewaters typically contain ambient NDMA, unlike wastewater-impacted source waters9,15 (Table 1). While UV treatment degrades ambient NDMA, ozone does not.38 Second, fresh wastewaters frequently feature precursors capable of producing NDMA during ozonation20 (Fig. 3A). Wastewater-impacted drinking water sources rarely feature ozone-reactive precursors,9,15 although such precursors have been found in certain watersheds impacted by specific chemicals (e.g., a metabolite of the fungicide, tolylfluanid17 and industrial anti-yellowing agents18).
Deactivation of NDMA precursors (calculated as NDMA measured after chloramination minus the NDMA measured pre-chloramination) by MPUV and LPUV was compared over 80–1000 mJ cm−2 germicidal fluence in WW4 (Fig. 4A). MPUV was much more effective for deactivation of NDMA precursors at low germicidal fluence. However, for MPUV, NDMA precursor concentrations rebounded somewhat near 400 mJ cm−2 germicidal fluence, and this rebound was repeatable. The rebound suggests a tradeoff between photo-inactivation of certain precursors and photo-generation of others. Regardless, NDMA precursor concentrations were still lower after MPUV than LPUV treatment at every germicidal fluence level evaluated. Germicidal fluence reflects the incident fluence corrected for light screening, and normalized to the absorption spectrum of DNA. Incident fluence better reflects the energy input (relevant to operational costs) associated with UV treatment. When analyzed against incident fluence, MPUV treatment was even more efficient for NDMA precursor deactivation than LPUV treatment (Fig. 4B).
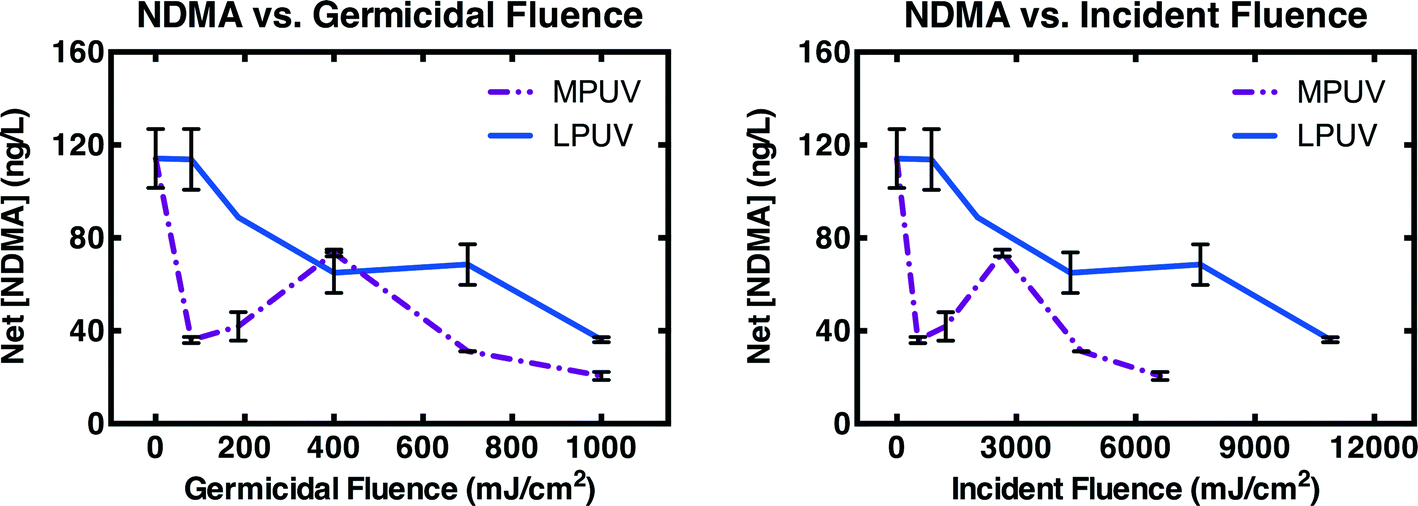 | ||
| Fig. 4 Net NDMA formation (i.e., post-chloramination NDMA minus pre-chloramination NDMA) after pretreatment with MPUV or LPUV fluence reported as germicidal fluence, and incident fluence, followed by post-treatment with chloramines formed in situ by addition of 5 mg L−1 as Cl2 free chlorine in WW4. Experimental conditions: pH = 8, [borate buffer] = 4 mM, T = 21 ± 1 °C, chloramine exposure time = 3 d. Error bars represent the range (n = 2) or standard deviations (n = 3) of experimental replicates. | ||
3.3. Evaluation of breakpoint chlorination
Breakpoint chlorination was evaluated as an alternative to chloramination in the wastewater-impacted drinking water source (DW) with relatively low ammonia (0.53 mg L−1 as N) compared to wastewaters but relatively high ammonia for drinking waters. Note, most drinking water treatment plants will need to achieve a free chlorine residual for obtain pre-disinfection credit before post-chloramination. The high chlorine doses needed to break out ammonia in the non-nitrified wastewaters render breakpoint chlorination infeasible. Like MPUV, exposure to free chlorine can deactivate NDMA precursors,9,15 but free chlorine produces an array of halogenated disinfection byproducts (DBPs).9 Several options were evaluated for the drinking water. First, free chlorine (2.5 mg L−1 as Cl2) was added to form chloramines in situ (0 mg min L−1 free chlorine exposure in Fig. 5). Second, 7.4 as Cl2 free chlorine was added, a dose which exceeds the 4.0 mg L−1 as Cl2 breakpoint for this water. After dosing free chlorine, total chlorine residual concentrations were monitored to compute free chlorine exposure, and after fixed exposures (4–225 mg min L−1), sufficient chlorine and ammonia were added to reach 2.5 mg L−1 as Cl2 as chloramines. Third, preformed monochloramine (2.5 mg L−1 as Cl2) was applied to control samples. NDMA concentrations measured after 3 d of chloramination were highest for in situ chloramination (0 mg min L−1 free chlorine exposure) and decreased with increasing free chlorine exposure, particularly at or above 93 mg min L−1, consistent with previous work.15 However, formation of halogenated DBPs, including the four regulated trihalomethanes (THM4), and unregulated haloacetaldehydes (HAlds), and dichloroacetonitrile (DCAN), all increased with increasing free chlorine exposure. Addition of preformed monochloramine simultaneously minimized formation of NDMA and halogenated DBPs.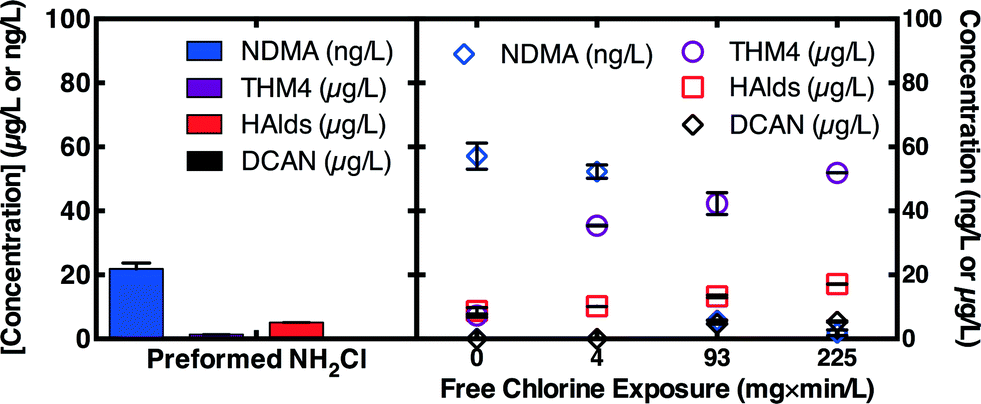 | ||
| Fig. 5 NDMA, sum of 4 trihalomethanes (THM4) (chloroform, bromodichloromethane, dibromochloromethane, and bromoform), sum of 7 haloaldehydes (HAlds) (dichloroacetaldehyde, bromochloroacetaldehyde, dibromoacetaldehyde, trichloroacetaldehyde, bromodichloroacetaldehyde, dibromochloroacetaldehyde, and trichloroacetaldehyde), and dichloroacetonitrile (DCAN) concentrations in the wastewater-impacted drinking water (DW) after application of preformed monochloramine, in situ chloramination (2.5 mg L−1 as Cl2) or breakpoint chlorination by free chlorine addition (7.4 mg L−1). Breakpoint samples were supplemented with ammonia (0.53 mg L−1 as N) after the indicated free chlorine exposures and held for 3 d. Experimental conditions: pH = 8, [borate buffer] = 4 mM, T = 21 ± 1 °C. Error bars represent the range (n = 2) or standard deviations (n = 3) of experimental replicates. Krasner, S. W., R. Shirkhani, P. Westerhoff, D. Hanigan, W. A. Mitch, D. L. McCurry, C. Chen, J., Skadsen, and U. von Gunten. 2015. Controlling the Formation of Nitrosamines During Water Treatment. Denver, Colo: Water Research Foundation. Reprinted with permission. | ||
3.4. Evaluation of preformed monochloramine
Preformed monochloramine was further evaluated as an alternative to in situ chloramination in all four wastewaters and the wastewater-impacted drinking water. Preformed monochloramine would minimize exposure to dichloramine (and therefore NDMA formation) by avoiding the dichloramine formed when concentrated free chlorine is injected into an ammonia-containing solution. Without pre-treatment, preformed monochloramine reduced final NDMA concentrations relative to in situ chloramination in all waters by an average 39 ± 7% (Fig. 6). The average reduction in NDMA formation (i.e., after subtracting off pre-chloramination NDMA) due to preformed monochloramine was 74 ± 19%. Coupling MPUV pre-treatment (186 mJ cm−2 germicidal fluence) with preformed monochloramine reduced NDMA by between 73% and 93% (Fig. 6), demonstrating that this combination substantially reduces NDMA formation compared to the current standard practice of in situ chloramination alone with no pre-treatment. | ||
| Fig. 6 NDMA concentrations measured after application of 5 [or 2.5 for DW] mg L−1 as Cl2 either as free chlorine for in situ chloramination, or as preformed monochloramine, or after 186 mJ cm−2 MPUV germicidal fluence followed by preformed monochloramine. Experimental conditions: pH = 8, [borate buffer] = 4 mM, T = 21 ± 1 °C, chloramine exposure time = 3 d. Error bars represent the range (n = 2) or standard deviations (n = 3) of experimental replicates. | ||
However, the effectiveness of preformed monochloramine as an alternative to in situ chloramination appeared to be somewhat dependent on ambient ammonia concentrations. Preformed monochloramine was most effective in two of the three low ammonia waters, and was somewhat effective in the third low ammonia water (Fig. 6). Excess ammonia would be expected to promote monochloramine at the expense of dichloramine according to eqn (1). To directly evaluate the effect of ammonia, ammonia was spiked into a well-nitrified wastewater effluent (WW2B) to achieve a range of concentrations prior to addition of 5 mg L−1 as Cl2. Addition of preformed monochloramine to WW2B without ammonia amendment was also evaluated. Preformed monochloramine formed the least NDMA (Fig. 7). For in situ chloramination, NDMA formation increased with ammonia concentrations up through 15 mg L−1 as N. Organic amine precursors to NDMA tend to be more reactive than ammonia with free chlorine.39 At low ammonia concentrations, NDMA precursors can compete with ammonia to reaction with free chlorine, enabling partial deactivation of NDMA precursors. As ammonia concentrations increase, reaction of organic amine precursors with free chlorine becomes less important. At a much higher ammonia concentration (50 mg L−1 as N), NDMA formation declined as dichloramine formation became less important. At the highest ammonia concentration evaluated (50 mg L−1 as N), NDMA formation was similar to the formation observed with pre-formed monochloramine. While these results broadly concur with the greater efficacy of preformed monochloramine for wastewaters featuring low ammonia concentrations (Fig. 6), they indicate that there is a concentration dependency within these low ammonia wastewaters, such that preformed monochloramine may be of greatest benefit for wastewaters with moderately low ammonia concentrations.
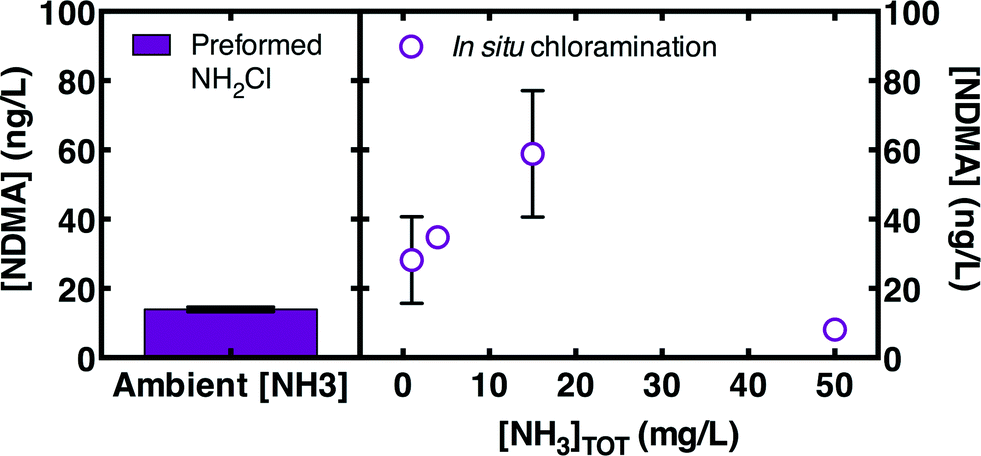 | ||
| Fig. 7 Net NDMA formation (i.e., total NDMA minus influent NDMA) measured in a well-nitrified wastewater (WW2B) spiked with varying ammonia concentrations and treated with 5 mg L−1 as Cl2 chloramines for 3 d. Experimental conditions: pH = 8, [borate buffer] = 4 mM, T = 21 ± 1 °C. Error bars represent the range (n = 2) or standard deviations (n = 3–4) of experimental replicates. | ||
4. Conclusions
For chloraminating utilities practicing de facto reuse or non-potable reuse of municipal wastewater, the combination of MPUV and preformed monochloramine can be effective for minimizing NDMA, despite the long chloramine contact times that can occur in these facilities. Even at moderate germicidal fluence (186 mJ cm−2), MPUV was effective for the destruction of both ambient NDMA, and NDMA precursors. In contrast, LPUV was effective for destruction of ambient NDMA, but less so for deactivation of NDMA precursors. While effective for NDMA precursor deactivation in drinking waters, ozone does not significantly degrade ambient NDMA, and generated significant levels of NDMA in some wastewaters. Free chlorine was effective for the deactivation of NDMA precursors, but does not destroy ambient NDMA. Moreover, significant doses of free chlorine may be needed to exceed the breakpoint in non-nitrified wastewaters, and the resulting free chlorine exposure generates regulated and unregulated halogenated DBPs. By reducing dichloramine concentrations, preformed monochloramine minimized NDMA formation compared with in situ chloramination, but there was a dependence on the ammonia concentration. Preformed monochloramine was most effective for wastewaters featuring moderate ammonia concentrations (∼15 mg L−1 as N). For wastewaters featuring high ammonia concentrations (∼50 mg L−1 as N), there appeared to be no significant benefits. For fully nitrified wastewaters, free chlorine could be added before ammonia, which would minimize dichloramine formation during in situ chloramination, but the concentrations of halogenated DBPs may increase during the free chlorine contact time.The combination of MPUV and preformed monochloramine may represent a low-cost alternative for NDMA control. For example, the conventional wastewater treatment plants feeding some reuse facilities have switched to UV disinfection for the portion of their flow that is discharged to receiving waters, but the portion of the flow sent to the reuse facility may be taken prior to UV disinfection. Using the UV-disinfected wastewater effluent as the influent to the reuse facility would avoid an extra capital cost for a new UV system, and facilitate pathogen removal goals for these non-potable waters. For facilities using wastewater with low to moderate ammonia concentrations, application of preformed monochloramine could be beneficial. Monochloramine formation reactions are fast. Preformed monochloramine could be generated at the facility by injecting ammonia into the existing free chlorine injection line (currently used for in situ chloramination) between the chlorine stock tank and the wastewater flow.
Lastly, there is significant interest in advanced treatment trains for potable reuse that avoid reverse osmosis, due to the problems associated with brine discharge in inland areas. Additionally, there is interest in direct potable reuse (DPR), wherein wastewater purified to potable quality by advanced treatment trains could be routed either to the influents to drinking water treatment plants, or even directly into distribution systems. In the latter case, for a chloraminated distribution system, the potable reuse treatment train effluent would need to carry a chloramine residual, raising concerns about NDMA formation from prolonged chloramine contact times. The sequences of process units that may constitute these advanced treatment trains are still being defined, but may involve a combination of treatment processes to provide multiple barriers. MPUV and preformed monochloramine may also aid NDMA control within these process trains, but additional research is needed to determine the optimal configuration of these processes within the larger process trains.
Acknowledgements
We thank Tiffany Lee of the Metropolitan Water District of Southern California (MWDSC) for analysis of the halogenated DBPs for experiments involving breakpoint chlorination. The authors thank the anonymous participating utilities, and acknowledge funding from the National Science Foundation Engineering Research Center for Re-Inventing the Nation's Urban Water Infrastructure (ReNUWIt). Partial support was provided by the Water Research Foundation (Project 4370; principal investigator was Stuart Krasner [MWDSC], project manager was Djanette Khiari). D. L. M. acknowledges support from the NSF Graduate Research Fellowship Program (Grant No. DGE-11474).References
- National Research Council, Water Reuse: Potential for Expanding the Nation's Water Supply Through Reuse of Municipal Wastewater, National Academies Press, Washington, DC, 2012 Search PubMed.
- U.S. Environmental Protection Agency, U.S. Environmental Protection Agency Announcement of Preliminary Regulatory Determinations for Contaminants on the Third Drinking Water Contaminant Candidate List, 2014, https://federalregister.gov/a/2014-24582, (Accessed August 15, 2015) Search PubMed.
- California Department of Public Health, California Department of Public Health NDMA and Other Nitrosamines - Drinking Water Issues, 2013, http://www.waterboards.ca.gov/drinking_water/certlic/drinkingwater/NDMA.shtml, (Accessed August 15, 2015) Search PubMed.
- U.S. Environmental Protection Agency, Announcement of Preliminary Regulatory Determinations for Contaminants on the Third Drinking Water Contaminant Candidate List, U.S. Environmental Protection Agency, Washington, DC, 2014, https://www.federalregister.gov/articles/2014/10/20/2014-24582/announcement-of-preliminary-regulatory-determinations-for-contaminants-on-the-third-drinking-water (Accessed February 9, 2016) Search PubMed.
- D. L. Sedlak, R. A. Deeb, E. L. Hawley, W. A. Mitch, T. Durbin, S. Mowbray and S. Carr, Water Environ. Res., 2005, 77, 32–39 CrossRef CAS PubMed.
- S. W. Krasner, P. Westerhoff, B. Chen, B. E. Rittmann and G. Amy, Environ. Sci. Technol., 2009, 43, 8320–8325 CrossRef CAS PubMed.
- I. M. Schreiber and W. A. Mitch, Environ. Sci. Technol., 2006, 40, 6007–6014 CrossRef CAS PubMed.
- W. A. Mitch and I. M. Schreiber, Environ. Sci. Technol., 2008, 42, 4811–4817 CrossRef CAS PubMed.
- A. D. Shah, S. W. Krasner, C. F. T. Lee, U. von Gunten and W. A. Mitch, Environ. Sci. Technol., 2012, 46, 4809–4818 CrossRef CAS PubMed.
- W. A. Mitch and D. L. Sedlak, Environ. Sci. Technol., 2004, 38, 1445–1454 CrossRef CAS PubMed.
- I. M. Schreiber and W. A. Mitch, Environ. Sci. Technol., 2006, 40, 3203–3210 CrossRef CAS PubMed.
- M. H. Plumlee, M. López-Mesas, A. Heidlberger, K. P. Ishida and M. Reinhard, Water Res., 2008, 42, 347–355 CrossRef CAS PubMed.
- C. M. Sharpless and K. G. Linden, Environ. Sci. Technol., 2003, 37, 1933–1940 CrossRef CAS PubMed.
- N. Dai and W. A. Mitch, Environ. Sci. Technol., 2015, 49, 8878–8886 CrossRef CAS PubMed.
- D. L. McCurry, S. W. Krasner, U. von Gunten and W. A. Mitch, Water Res., 2015, 84, 161–170 CrossRef CAS PubMed.
- U. von Gunten, E. Salhi, C. K. Schmidt and W. A. Arnold, Environ. Sci. Technol., 2010, 44, 5762–5768 CrossRef CAS PubMed.
- C. K. Schmidt and H. J. Brauch, Environ. Sci. Technol., 2008, 42, 6340–6346 CrossRef CAS PubMed.
- K. Kosaka, M. Asami, Y. Konno, M. Oya and S. Kunikane, Environ. Sci. Technol., 2009, 43, 5236–5241 CrossRef CAS PubMed.
- T. Zeng and W. A. Mitch, Environ. Sci. Technol., 2015, 49, 13158–13167 CrossRef CAS PubMed.
- D. Gerrity, A. N. Pisarenko, E. Marti, R. A. Trenholm, R. Gerringer, J. Reungoat and E. Dickenson, Water Res., 2014, 72, 251–261 CrossRef PubMed.
- M. Sgroi, P. Roccaro, G. Oelker and S. A. Snyder, Chemosphere, 2016, 144, 1618–1623 CrossRef CAS PubMed.
- California Department of Public Health, Regulations Related to Recycled Water, 2014, http://www.waterboards.ca.gov/drinking_water/certlic/drinkingwater/documents/lawbook/RWregulations_20140618.pdf, (Accessed January 15th, 2016) Search PubMed.
- A. D. Shah, N. Dai and W. A. Mitch, Environ. Sci. Technol., 2013, 47, 2799–2808 CrossRef CAS PubMed.
- A. D. Shah and W. A. Mitch, Environ. Sci. Technol., 2012, 46, 119–131 CrossRef CAS PubMed.
- I. M. Schreiber and W. A. Mitch, Environ. Sci. Technol., 2005, 39, 3811–3818 CrossRef CAS PubMed.
- W. A. Mitch, G. L. Oelker, E. L. Hawley, R. A. Deeb and D. L. Sedlak, Environ. Eng. Sci., 2005, 22, 882–890 CrossRef CAS.
- M. J. Farré, K. Döderer, L. Hearn, Y. Poussade, J. Keller and W. J. Gernjak, J. Hazard. Mater., 2011, 185, 1575–1581 CrossRef PubMed.
- S. Jin, A. A. Mofidi and K. G. Linden, J. Environ. Eng., 2006, 132, 831–841 CrossRef CAS.
- C. von Sonntag and U. von Gunten, Chemistry of Ozone in Water and Wastewater Treatment, IWA Publishing, London, 2012 Search PubMed.
- H. Bader and J. Hoigne, Water Res., 1981, 15, 449–456 CrossRef CAS.
- Y. Feng, D. W. Smith and J. R. Bolton, J. Environ. Eng. Sci., 2007, 6, 277–284 CrossRef CAS.
- S. W. Krasner, C. F. T. Lee, W. A. Mitch and U. von Gunten, Development of a Bench-Scale Test to Predict the Formation of Nitrosamines, Water Research Foundation, Denver, 2012 Search PubMed.
- Standard Methods for the Examination of Water and Wastewater, ed. A. D. Eaton, L. S. Clesceri and A. E. Greenberg, American Public Health Association, American Water Works Association, Environment Federation Publishers, Washington, DC, 20th edn, 1998 Search PubMed.
- S. W. Krasner, Y. C. Guo, M. Prescott and S. Leavey, Presented in part at AWWA Water Quality Technology Conference, Long Beach, CA, November 2013 Search PubMed.
- J. Oppenheimer, A. Eaton, M. Badruzzaman, A. W. Haghani and J. G. Jacangelo, Water Res., 2011, 45, 4019–4027 CrossRef CAS PubMed.
- R. Chinn, C. F. T. Lee, S. Krasner, M. Dale, S. Richardson, J. Pressman, T. Speth, R. Miltner and J. E. Simmons, In Proceedings AWWA Water Quality Technology Conference, Denver, CO, 2007 Search PubMed.
- T. Zeng and W. A. Mitch, Environ. Sci. Technol., 2016, 50, 2964–2973 CrossRef CAS PubMed.
- W. A. Mitch, J. O. Sharp, R. R. Trussell, R. L. Valentine, L. Alvarez- Cohen and D. L. Sedlak, Environ. Eng. Sci., 2003, 20, 389–404 CrossRef CAS.
- L. Abia, X. L. Armesto, M. Canle, M. V. Garcia and J. A. Santaballa, Tetrahedron, 1998, 54, 521–530 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ew00044d |
| This journal is © The Royal Society of Chemistry 2016 |