
Antihypertensive effects of oleuropein-enriched olive leaf extract in spontaneously hypertensive rats
M.
Romero
ab,
M.
Toral
a,
M.
Gómez-Guzmán
a,
R.
Jiménez
ab,
P.
Galindo
a,
M.
Sánchez
a,
M.
Olivares
c,
J.
Gálvez
abd and
J.
Duarte
*ab
aDepartamento de Farmacología, Facultad de Farmacia, Universidad de Granada, 18071, Granada, Spain. E-mail: jmduarte@ugr.es; Fax: +(34)-958248264; Tel: +(34)-958241791
bInstituto de Investigación Biosanitaria de Granada (ibs.GRANADA), Granada, Spain
cLaboratorio de Descubrimiento y Preclínica, Departamento de Investigación BIOSEARCH S.A, Granada, Spain
dCIBER-ehd, Centro de investigaciones biomédicas (CIBM), 18100-Armilla, Granada, Spain
First published on 10th November 2015
Abstract
The effects of chronic consumption of oleuropein-enriched (15% w/w) olive leaf extract (OLE) on blood pressure, endothelial function, and vascular oxidative and inflammatory status in spontaneously hypertensive rats (SHR) were evaluated. Ten Wistar Kyoto rats (WKY) and twenty SHR were randomly assigned to three groups: a control WKY group, a control SHR group and a SHR group treated with OLE (30 mg kg−1) for 5 weeks. Long-term administration of OLE reduced systolic blood pressure, heart rate, and cardiac and renal hypertrophy. OLE treatment reversed the impaired aortic endothelium-dependent relaxation to acetylcholine observed in SHR. OLE restored aortic eNOS phosphorylation at Ser-1177 and Thr-495 and increased eNOS activity. OLE eliminated the increased aortic superoxide levels, and reduced the elevated NADPH oxidase activity, as a result of reduced NOX-1 and NOX-2 mRNA levels in SHR. OLE reduced the enhanced vascular TLR4 expression by inhibition of mitogen-activated protein kinase (MAPK) signaling with the subsequent reduction of proinflammatory cytokines. In conclusion, OLE exerts antihypertensive effects on genetic hypertension related to the improvement of vascular function as a result of reduced pro-oxidative and pro-inflammatory status.
Introduction
Hypertension is one of the most powerful risk factors for cardiovascular events, including myocardial infarction and stroke. Treatment with any commonly used antihypertensive regimen reduces the risk of total major cardiovascular events; the larger the reductions in blood pressure the larger the reductions in risk.1 Nevertheless, most patients with hypertension will require two or more anti-hypertensive medications to achieve their blood pressure goals (<140/90 mmHg or <150/90 mmHg for adults, 60 years and older, and <140/90 mmHg in patients with hypertension and diabetes, regardless of age),2 which, on the other hand, also means the increment of risks of adverse drug reaction and medication costs. One considerable alternative to bridge the efficacy and therapeutic costs is the use of potential herbal medicines.The leaves of the olive tree (Olea europaea L.) have been used since ancient times to combat high blood pressure, atherosclerosis and diabetes and for other medicinal purposes.3 Our group described for the first time the hypotensive effects of a decoction of olive leaf in rats.4 The anti-hypertensive and cholesterol-lowering actions of olive leaves are well-documented in animal and human studies.5–10 In addition, the anti-hypertensive effect of the olive leaf extract (OLE) had demonstrated its superiority over the recommended life-style changes.11 Interestingly, the evaluation of safety parameters and occurrence of adverse events showed that OLE was safe and tolerable in patients with stage-1 hypertension.10 Olive leaf contains active substances such as oleuropein (a polyphenolic iridoid glycoside),12 oleacein13 and oleanolic acid.14 EFLA® 943, a stable olive leaf extract which is standardized to oleuropein, has been preclinically studied for its safety and anti-hypertensive effects.9 However, the mechanism of action by which OLE exerts its antihypertensive effects is not clear. The antihypertensive effects of active constituents in OLE, such as oleuropein and oleacein, might be associated with the inhibition of the angiotensin converting enzyme.14 Oleuropein has been recognized as one of the components of a decoction of olive leaf responsible for acute endothelium-independent vasodilatory effects in isolated rat aorta.4 We also showed that oleuropein decreases the sinus node function and the guinea-pig atria contractile response, which cannot be attributed to the inhibitory effects on calcium entry through L-type channels.15 In contrast, in a latest study using isolated rabbit's heart and rat's cardiomyocytes, it was reported that OLE suppressed the L-type calcium channel directly and reversibly.16 Very recently, chronic oleuropein treatment in rats with simultaneous type 2 diabetes and renal hypertension showed antihypertensive effects which seems to be partly mediated by improving the release of nitric oxide (NO), and antioxidant and sympathoplegic activities.17 Other minor components, such as triterpenic compounds (oleanolic and maslinic acids), induce acute vasorelaxation of the aorta from spontaneously hypertensive rats (SHR) that involves calcium-independent release of endothelial nitric oxide (NO).18,19 Chronic pomace olive oil supplemented with oleanolic acid improves the endothelial function in conductance20 and resistance arteries21 from SHR by increasing endothelial NO synthase (eNOS) protein expression.
Based on the positive results observed in the preclinical and human studies, the current study was designed to primarily confirm the anti-hypertensive effect of the OLE in SHR, an established model of genetic hypertension. The secondary objectives of the study were to investigate the mechanisms involved in its antihypertensive effects focusing on the improvement of the vascular function and the possible antioxidant and anti-inflammatory properties in the vascular wall.
Results
Effects of OLE on blood pressure, and morphological variables
At the end of the 5 weeks of administration, OLE induced a progressive reduction in both systolic blood pressure (SBP) (−21.6 ± 5.5 mmHg) (Fig. 1A), and heart rate (HR) (−47.0 ± 11.6 bpm) (Fig. 1B). The effect on SBP was only seen after 3 weeks and that of HR at 4 weeks.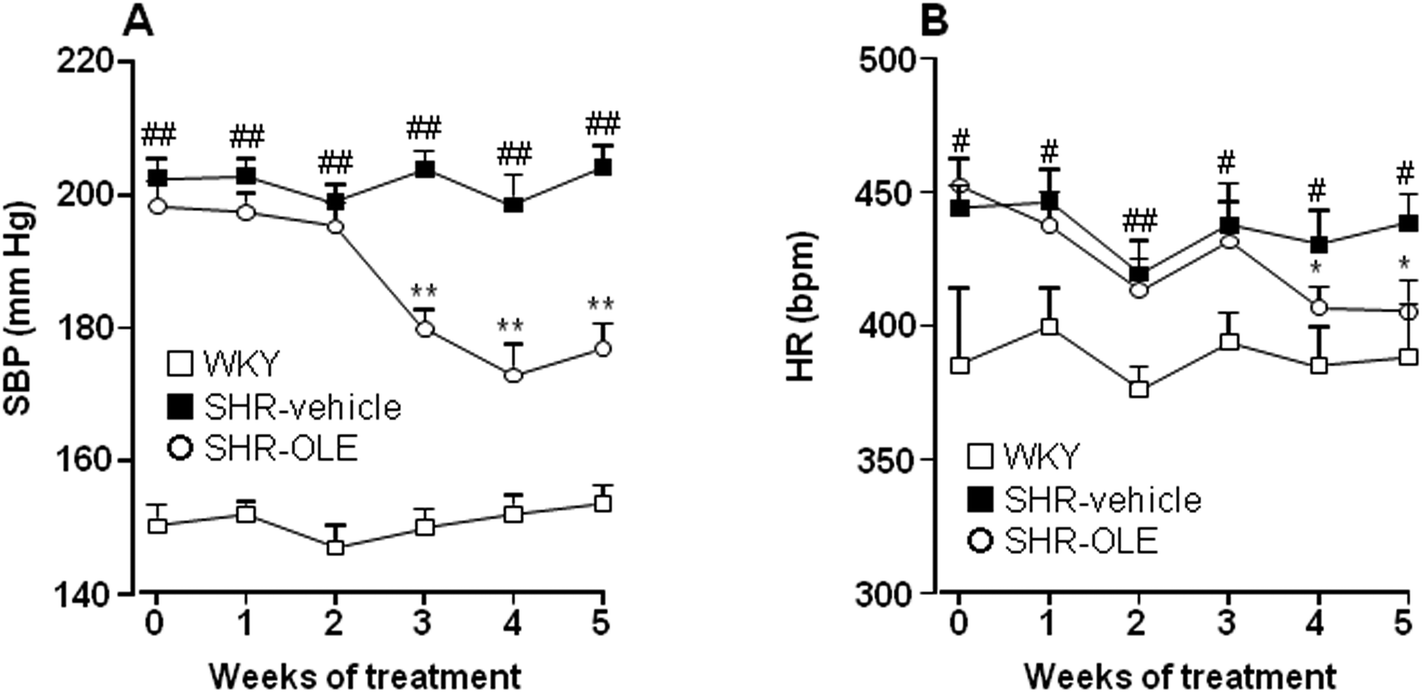 | ||
| Fig. 1 Effects of long-term OLE administration on (A) systolic blood pressure (SBP) and (B) heart rate (HR) as measured by tail-cuff plethysmography in SHR rats. Values are expressed as mean ± SEM (n = 10 rats). # and ## indicate P < 0.05 and P < 0.01, respectively, compared with the WKY control group, * and ** P < 0.05 and P < 0.01, respectively, compared with the SHR control group. | ||
The body weight (BW) increased in both Wistar Kyoto rats (WKY) and SHR control groups after 5 weeks (7.2 ± 0.5% and 6.2 ± 0.6%, respectively). SHR treated with OLE showed a similar final BW to control SHR (Table 1). The absolute heart weight (HW) and left ventricle weight (LVW), as well as HW, LVW, and kidney (KW) relative to the tibia length (TL) were higher in the SHR control group as compared with the WKY control group. Treatment with OLE weakly, but significantly, reduced HW/TL and LVW/TL, and eliminated the increase in KW/TL (Table 1).
| WKY (n = 10) | SHR (n = 9) | SHR-OLE (n = 9) | |
|---|---|---|---|
| BW, body weight; HW, heart weight; LVW, left ventricle weight; KW, kidney weight. Values are expressed as mean ± SEM of n rats. #P < 0.05 and ##P < 0.01 vs. WKY and *P < 0.05 vs. SHR-control. | |||
| BW (g) | 397 ± 6 | 374 ± 7# | 368 ± 5## |
| HW (mg) | 1169 ± 29 | 1300 ± 24## | 1271 ± 30# |
| LVW (mg) | 879 ± 24 | 1075 ± 18## | 1037 ± 24## |
| KW (mg) | 945 ± 23 | 965 ± 16 | 927 ± 16 |
| HW/TL (mg cm−1) | 2.98 ± 0.06 | 3.39 ± 0.04## | 3.26 ± 0.04##, * |
| LVW/TL (mg cm−1) | 2.24 ± 0.05 | 2.80 ± 0.03## | 2.64 ± 0.04##, * |
| KW/TL (mg cm−1) | 2.38 ± 0.05 | 2.52 ± 0.03# | 2.38 ± 0.06* |
OLE treatment improves endothelial function in SHR
Aortae from control SHR showed significant reduced endothelium-dependent vasodilator responses to acetylcholine in arteries stimulated by phenylephrine as compared with the aortae from control WKY (Emax = 31 ± 5% vs. 57 ± 6%, respectively). OLE consumption produced a significant increase in the relaxation induced by acetylcholine in SHR rats (Emax = 50 ± 7%, P < 0.05 vs. SHR control) (Fig. 2A). The relaxant response induced by acetylcholine was fully inhibited by NG-nitro-L-arginine methyl ester (L-NAME) in all experimental groups (Fig. 2B), showing that in this vessel acetylcholine-induced relaxation in both WKY and SHR was entirely dependent on endothelium-derived NO.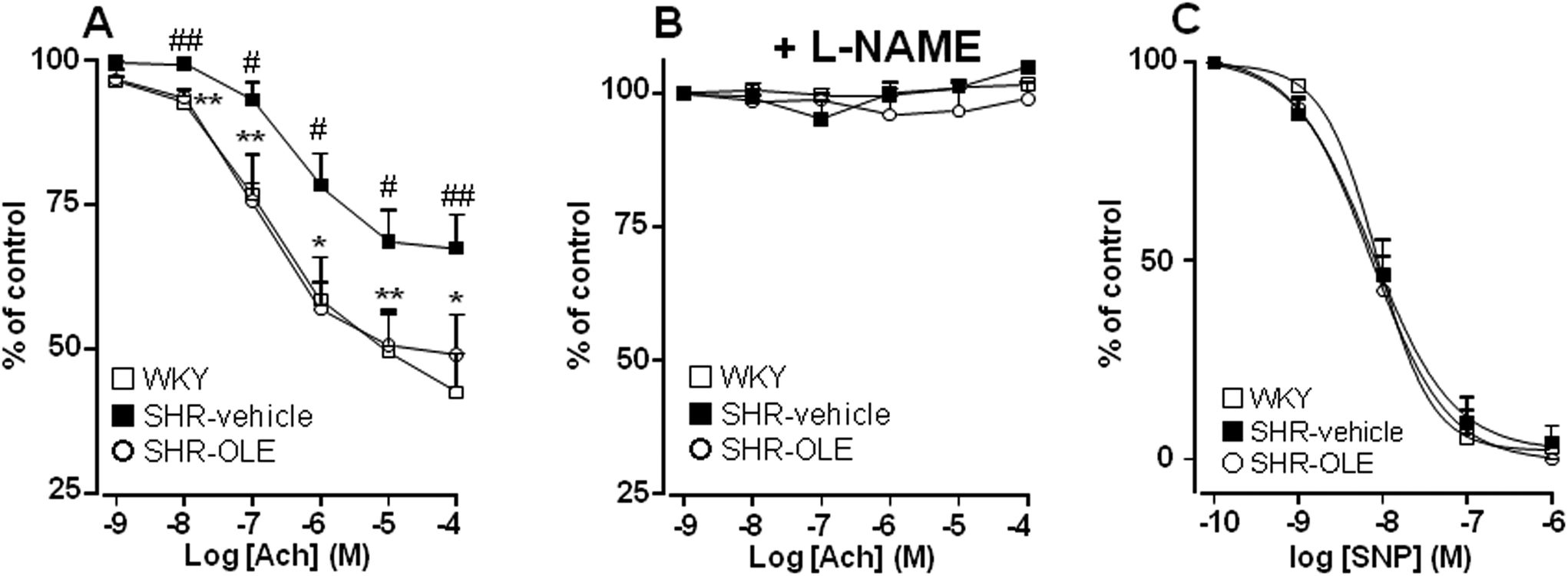 | ||
| Fig. 2 Effects of OLE on endothelial function. Vascular relaxant responses induced by acetylcholine (ACh) (A, B) and by sodium nitroprusside (SNP) (C) in aortae pre-contracted by 10−6 M phenylephrine in the absence (A, C) and presence (B) of L-NAME (10−4 M) in rings from all the experimental groups. Values are expressed as mean ± SEM (n = 8–9 rings from different rats). # and ## indicate P < 0.05 and P < 0.01, respectively, compared with the respective WKY control group. * and ** indicate p < 0.05 and p < 0.01, respectively, compared with SHR control rats. | ||
To analyze whether the impaired response to endothelial-derived NO is due to a defect in the signaling of NO in the vascular smooth muscle, we analyzed the effects of nitroprusside, which directly activates soluble guanylyl cyclase in vascular smooth muscle, mimicking the effects of endogenous NO. The endothelium-independent vasodilator responses to nitroprusside were not different among the groups (Fig. 2C).
Endothelial NO synthase gene (Fig. 3A) and protein (Fig. 3B) expression in the aorta were similar in control SHR as compared with WKY rats. However, lower eNOS at Ser1177 phosphorylation (Fig. 3B) and higher eNOS phosphorylation at Thr495 (Fig. 3C) were found in the aorta from SHR animals as compared to control WKY. In addition, the phosphorylation of Akt at Ser473 was also reduced in SHR (Fig. 3D). OLE treatment restored the levels of eNOS phosphorylation.
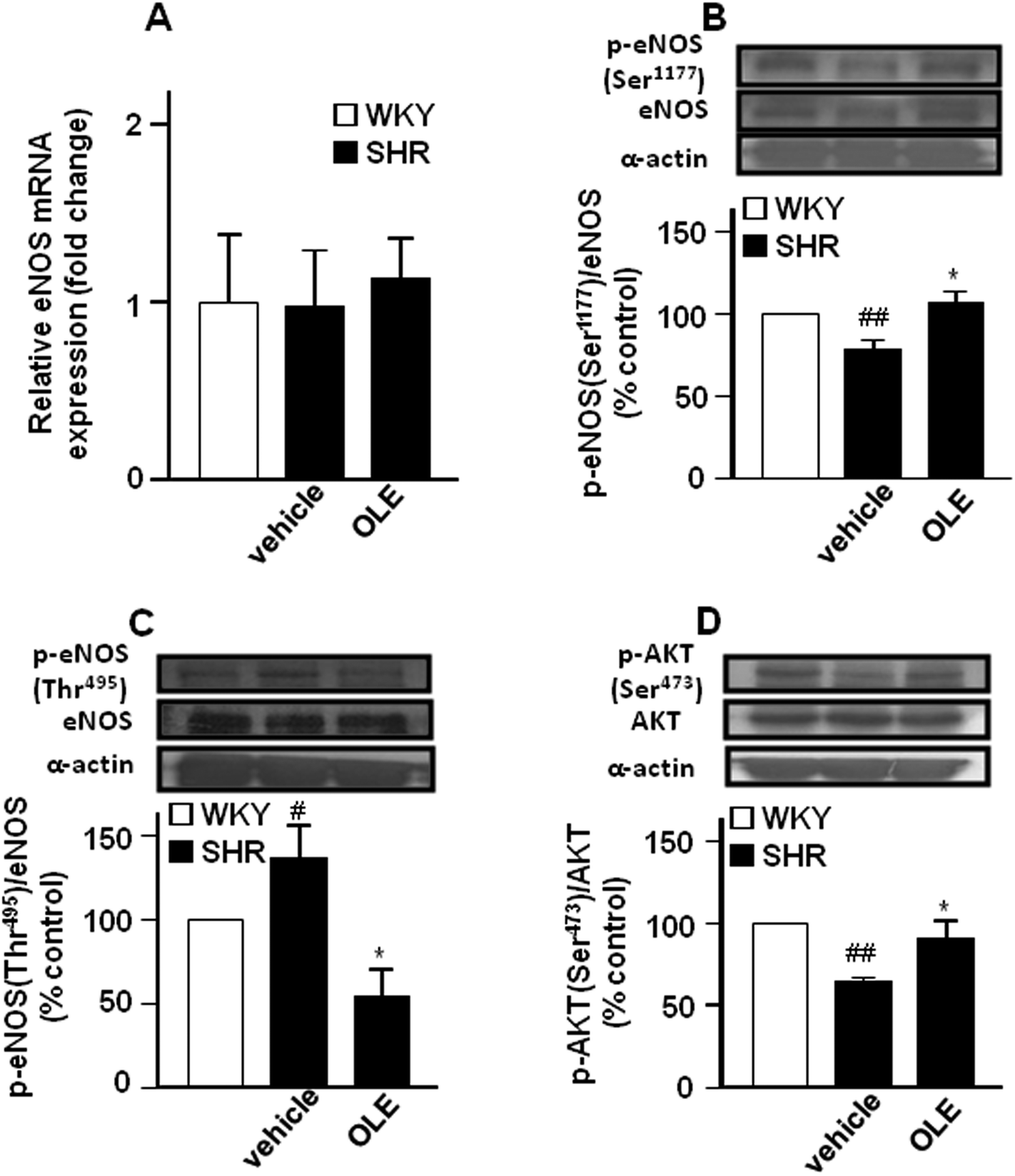 | ||
| Fig. 3 Effects of OLE on the expression of eNOS at the level of mRNA by RT-PCR (A) and protein phosphorylation of eNOS at Ser1177 (B) and at Thr495 (C), and AKT at Ser473 (D) by western blot in SHR. Values are expressed as mean ± SEM (n = 8–9 rings from different rats for RT-PCR and 4–6 for western blot). # and ## indicate P < 0.05 and P < 0.01, respectively, compared with the respective WKY control group. * indicates P < 0.05 compared with SHR control rats. | ||
OLE reduces vascular reactive oxygen species (ROS) levels in SHR by reducing NADPH oxidase activity
Red fluorescence could be observed in adventitial, medial and endothelial cells from sections of the aorta incubated with dihydroethidium (DHE) (Fig. 4A). Nuclear red fluorescence was quantified and normalized to the blue fluorescence of the nuclear stain 4,6-diamidino-2-phenylindole dichlorohydrate (DAPI), allowing comparisons between different sections. Rings from SHR showed marked increased staining in the adventitial, medial and endothelial cells as compared with WKY rats which was significantly reduced by OLE treatment (Fig. 4A and B). | ||
| Fig. 4 Effects of OLE on the O2˙− levels and NADPH oxidase pathway. (A) Pictures show arteries incubated in the presence of DHE which produces a red fluorescence when oxidized to ethidium by O2˙−, blue fluorescence of the nuclear stain DAPI. (B) Averaged values, mean ± SEM (n = 5–7 rings from different rats) of the red ethidium fluorescence normalized to the blue DAPI fluorescence. NADPH oxidase activity measured by lucigenin-enhanced chemiluminescence (C), and mRNA levels of NADPH oxidase subunits NOX-1 (D), NOX-2 (E), and NOX-4 (F) in aorta from all the experimental groups. Values are expressed as mean ± SEM (n = 8–9 rings from different rats). # and ## indicate P < 0.05 and P < 0.01, respectively, compared with the WKY control group. * and ** indicate P < 0.05 and P < 0.01, respectively, compared with the SHR control group. | ||
NADPH oxidase activity was increased in the aortic rings from SHR as compared with WKY rats (Fig. 4C). OLE prevented this increase in NADPH oxidase activity in SHR. A significant increase in the mRNA levels of NADPH oxidase subunits, NOX-1 (Fig. 4D) and NOX-2 (Fig. 4E) was observed in aortic tissue from SHR as compared with WKY rats, without change in NOX-4 (Fig. 4F). Again, OLE treatment reduced the gene expression of both NOX-1 and NOX-2 in SHR.
OLE reduces vascular inflammatory response in SHR
The expression of phospho-ERK1/2 (Fig. 5A) and phospho-p38 (Fig. 5B) proteins was increased in the aorta from the SHR control group as compared with WKY control rats. Chronic OLE treatment reduced mitogen-activated protein kinase (MAPK) phosphorylation in SHR.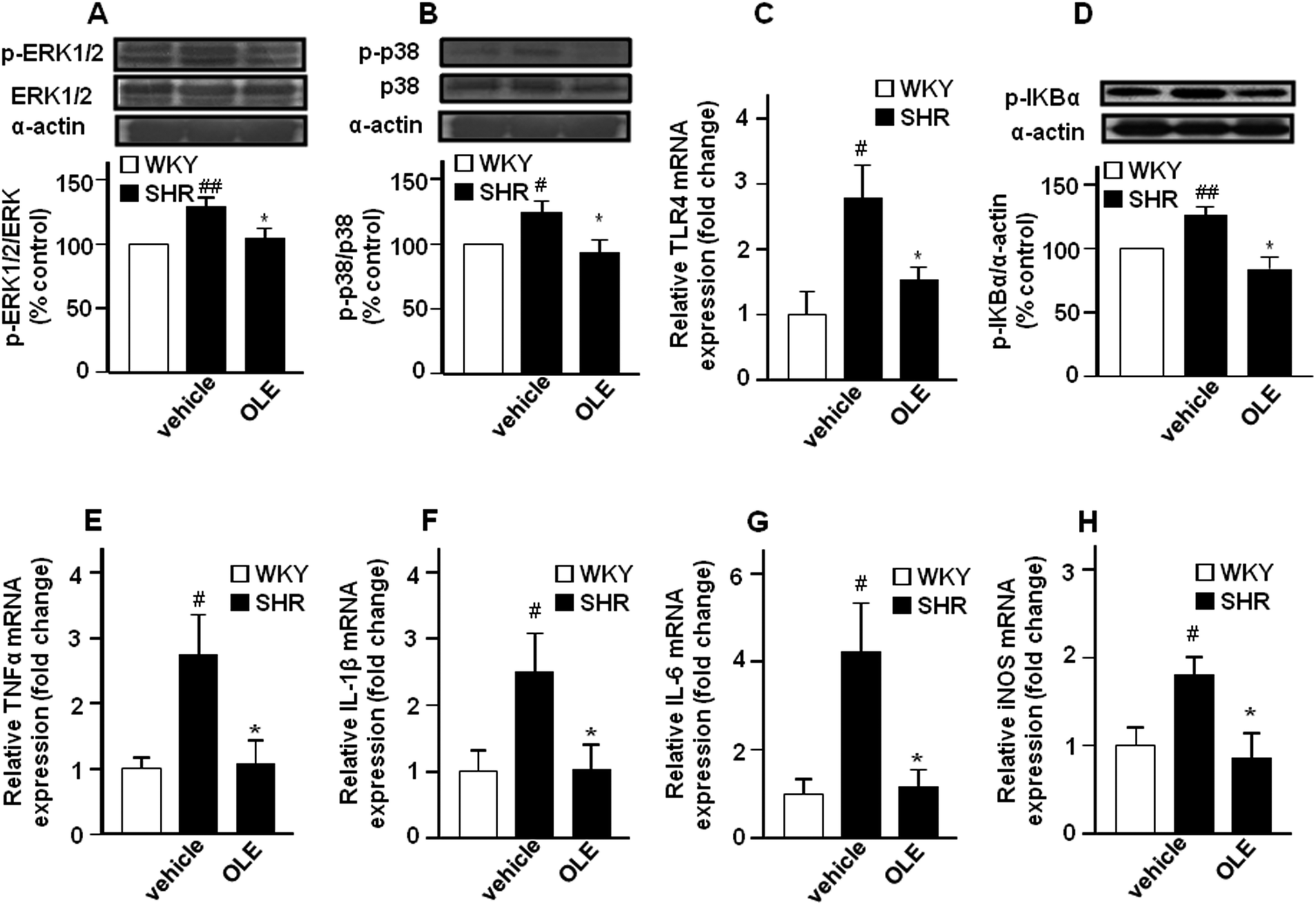 | ||
| Fig. 5 Effects of OLE on vascular inflammation in SHR. Effects on phosphorylation of ERK1/2 (A) and p38 (B) MAPKs and IkBα (D) measured by western blots, and aortic mRNA levels of TLR4 (C) and pro-inflammatory cytokines TNFα (E), IL-1β (F), IL-6 (G), and iNOS (H) by RT-PCR. Data presented as a ratio of the arbitrary units of mRNA (2−ΔΔCt). Values are expressed as mean ± SEM (n = 8–9 rings from different rats for RT-PCR and 4–6 for western blot). # and ## indicate P < 0.05 and P < 0.01, respectively, compared with the respective WKY control group. * indicates P < 0.05 compared with SHR control rats. | ||
The mRNA levels of the toll-like receptor 4 (TLR4) mRNA level (Fig. 5C), and its downstream phospho-IκBα (Fig. 5D) in aortic homogenates were higher in SHR as compared with WKY. In addition, the expression of the proinflammatory cytokine tumor necrosis factor α (TNFα) (Fig. 5E), interleukin 1β (IL-1β) (Fig. 5F) and interleukin 6 (IL-6) (Fig. 5G), as well as of inducible NOS (iNOS) (Fig. 5H) were also increased in SHR. OLE consumption also reduced the higher TLR4 mRNA level and IκBα phosphorylation and restored the mRNA levels of proinflammatory cytokines and iNOS to values similar to WKY.
Discussion
The present study has demonstrated that chronic oral administration of OLE improved endothelial dysfunction, vascular inflammation, vascular oxidative stress, cardiac and renal hypertrophy and reduced high BP in genetic hypertension.Endothelial dysfunction is a hallmark underlying vascular disease. In the aorta, NO is the major factor accounting for endothelium-dependent relaxation22 as denoted by the almost full inhibitory action of L-NAME in this study. Thus, the diminished acetylcholine-induced relaxation indicates an impaired agonist-induced NO bioactivity. OLE treatment eliminated the altered responses to acetylcholine observed in aortae from SHR, indicating a protective role in agonist-induced NO bioactivity. These results are in agreement with previous data showing that chronic oleuropein treatment, the main component of OLE, reversed the hypertension/diabetes-induced impairment of NO release in rat aorta.17 Additionally, hydroxytyrosol, the main metabolite of oleuropein,23 also reversed the reduced intracellular NO levels stimulated by acetylcholine in endothelial cells incubated in vitro with high glucose and free fatty acids.24 eNOS is a constitutive enzyme, controlled at the transcriptional and post-transcriptional levels. The post-transcriptional eNOS regulation is dependent on the phosphorylation state, mainly on a serine residue (Ser-1177) on the reductase domain and on a threonine residue (Thr-495) within the calcium/calmodulin residue.25 In fact, in our experiment, changes in eNOS activity correlate with simultaneous changes in eNOS phosphorylation at the activator site Ser-1177 and the inhibitor site Thr-495, since in control SHR, with lower eNOS activity, the level of Ser-1177 and Thr-495 phosphorylation was reduced and increased, respectively. In addition, lower eNOS phosphorylation at Ser-1177 also correlates with the lower activity of its activator Akt in SHR. In vitro, oleanolic acid, a minor component of OLE, evoked endothelium-derived NO release by increasing the phosphorylation of Akt kinase at Ser-473 and eNOS at Ser-1177.21 The eNOS Thr-495 residue is constitutively phosphorylated in all endothelial cells and is a negative regulatory site. The link between phosphorylation and NO production can be explained by interference with the binding of calmodulin to calmodulin-binding domain. The constitutively active kinase which phosphorylates eNOS Thr-495 is most probably protein kinase C,25 and under inflammatory conditions Rho-kinase.26 When endothelial cells are stimulated with agonists like acetylcholine, substantially more calmodulin binds to eNOS if Thr-495 is dephosphorylated.27 The impaired relaxant response to acetylcholine found in SHR might be related, at least in part, to higher eNOS Thr-495 phosphorylation, which was reversed by OLE treatment via eNOS Thr-495 dephosphorylation. Also, protein kinase C inhibition by hydroxytyrosol28 might be involved in the reduced eNOS Thr-495 phosphorylation induced by OLE. The present study demonstrates that chronic OLE consumption stimulates eNOS activity, primarily through non-genomic changes in eNOS expression, but through post-transcriptional eNOS phosphorylation at Ser-1177, via Akt, and eNOS dephosphorylation at Thr-495.
In the present study, the relaxant response to the activator of soluble guanylyl cyclase nitroprusside was similar in the aorta from all experimental groups, indicating that alterations in cyclic GMP do not seem to contribute to the endothelial dysfunction in SHR. Thus, the functional changes observed in endothelium-dependent relaxation should be attributed to an alteration in NO synthesis and/or its bioavailability. A key mechanism of endothelial dysfunction in hypertension involves the vascular production of ROS, particularly superoxide (O2˙−), which reacts rapidly with NO and inactivates it.29–31 In the aorta from the SHR group incubated with DHE there was a marked increase in red fluorescence indicating increased vascular O2˙− levels. We described that OLE treatment eliminated this increase in SHR, showing antioxidant properties in vivo. These data are in agreement with the reduced serum levels of malondialdehyde, a marker of systemic oxidative stress, found in rat fed high carbohydrate-high fat diet treated with an olive leaf extract enriched in oleuropein,32 and in hypertensive/diabetic rats treated with oleuropein.17 In addition, reduced intracellular ROS levels were found in endothelial cells exposed to hyperglycemia and free fatty acids and treated with hydroxytyrosine.24 These data might also be consistent with the potent O2˙− scavenging effect of hydroxytyrosol and oleuropein described previously under in vitro conditions.33
Increased O2˙− production via NADPH oxidase is thought to contribute to hypertension and endothelial dysfunction in SHR and to essential hypertension.34–36 We found increased NADPH oxidase activity in the aorta from SHR as compared to WKY, which was suppressed by OLE treatment. The NADPH oxidase is a multi-enzymatic complex formed by gp91phox or its vascular homologues NOX-1, NOX-2 and NOX-4, rac, p22phox, p47phox, and p67phox. We found a marked increase in the expression of the catalytic NADPH oxidase subunits NOX-1 and NOX-2 in the aortas from SHR, which was also eliminated by OLE consumption, and this could be involved in the NADPH oxidase inhibition and in the reduction of the vascular ROS level induced by OLE. Protein kinase C inhibition by hydroxytyrosol28 might also be involved in the reduced NADPH oxidase activity.
The enhancement of ROS production, in particular O2˙−, affects the endothelial function not only by reducing NO bioavailability, but also by promoting inflammation.37 MAPK signaling pathways are associated with the vascular inflammation that is modulated by ROS.38 However, in vascular tissue, angiotensin II, thrombin or fibroblast growth factor-2 (bFGF)-induced MAPK phosphorylation of cJun-N-terminal kinase (JNK) and p38, but not ERK1/2, is modulated by NOX enzymes. Responses in smooth muscle cells are mediated by NOX-1 and in endothelial cell by NOX-2.39 Our data are in agreement with the fact that vascular ROS increase links with amplified pro-inflammatory vascular status through MAPK activation, since the higher aortic ROS content, from NOX-1 and NOX-2, found in SHR correlates with the higher p38 phosphorylation and higher mRNA level of the proinflammatory cytokines TNFα, IL-1β, and IL-6 as compared to the WKY control group. In addition, TNFα and IL-1β are able to induce iNOS in the blood vessel, which was also increased in the aorta from SHR. Moreover, OLE administration, which reduced the ROS level, also reduced p38 activation as well as the higher mRNA levels of these cytokines and the iNOS enzyme found in the aorta from SHR. Of note, ERK1/2 signaling is also upregulated in SHR. ERK1/2 activation is associated with vascular contraction and growth, important determinants of vascular dysfunction and arterial remodeling in hypertension.40 In fact, normalization of ERK1/2 activity improved endothelial-dependent relaxation in SHR.40 In the present study we show that the phosphorylation of ERK1/2 in the aorta is higher in SHR compared with WKY, which was normalized after OLE treatment. This reduced ERK1/2 phosphorylation might be involved in the improvement of acetylcholine-induced relaxation shown by OLE in SHR.
It has been reported that olive oil polyphenols, including oleuropein and hydroxytyrosol, inhibited NF-κB activation induced by bacterial lipopolysaccharide (LPS) in endothelial cells.41 LPS stimulates the expression of TLR4 in the vasculature, which resulted in increased NADPH oxidase-dependent O2˙− production and inflammation.42,43 TLR4 activation contributes to increased BP and low-grade vascular inflammation displayed by SHR. In fact, in the present study, the aortic mRNA levels of TLR4 where higher in SHR than in WKY, and were reduced by OLE treatment. Enhanced TLR4 expression and mRNA stabilization in human aortic smooth muscle cells is mediated by NADPH oxidase-related ROS production and MAPK signalling pathway in vitro.44 In agreement with these in vitro results we found that the protective effects of OLE on vascular inflammation might be related to the interference in TLR4 expression, as a result of reduced vascular NADPH oxidase-mediated ROS production and p38 MAPK activation, and TLR4 signaling, shown by lower IκBα phosphorylation and pro-inflammatory cytokine production. Our data strongly suggests that antioxidants act downstream on any membrane receptor, at a step common to all proinflammatory agents, reducing ROS-mediated MAPK activation and subsequent inflammation.
Sustained high blood pressure is one of the most powerful determinants of the development of cardiac and renal hypertrophy.45 In our study, SHR showed increased left ventricular and renal weight indices as compared to normotensive WKY rats. Chronic OLE treatment also significantly reduced this organ target damage. Thus, the beneficial effects of OLE on the cardiovascular structure seem to be related to its blood pressure lowering effect. However, other effects beyond the antihypertensive properties, such as the decrease in the heart rate, the antioxidant effects and the protection from NO break-down, might also play a role in the prevention of morphological changes observed in SHR rats treated with OLE.
Experimental
This study was carried out in accordance with the regulations and requirements of the European Union concerning the protection of animals used for scientific purposes, and the experimental protocol was approved by the Ethics Committee of Laboratory Animals of the University of Granada (Spain; permit number 459-bis-CEEA-2012).Animals and experimental groups
Twelve-week old, male SHR and WKY were obtained from Harlan Laboratories (Barcelona, Spain). All rats were fed ad libitum throughout the whole experiment period and on standard rat chow. An adaptation period of two weeks for blood pressure measurements was allowed before the initiation of the experimental protocols. Ten WKY and twenty SHR were randomly assigned to three groups (n = 10): a control WKY group and a control SHR group, both received water as vehicle, and a SHR group treated with OLE (30 mg per kg per day by oral gavage) for 5 weeks. OLE consisting of an optimized mixture of compounds derived from olive leaf, standarized for its oleuropein content (15% w/w), medial triterpenic acid content (10% w/w), and hydroxytyrosol content (1% w/w), supplied by Biosearch S.A. (Granada, Spain). OLE is manufactured in accordance with food grade and food safety standards embraced by the Global Food Safety Initiative (FSSC 22000). The OLE treatment was stopped 24 hours before the end of the experiment in order to study the long-term effects of OLE without the involvement of the effects of acute administration. Food and water intake was recorded daily for all groups. During the experimental periods rats had free access to tap water and chow. The body weight was measured every week.Blood pressure measurements
The SBP and heart rate, HR, were measured weekly at room temperature using tail-cuff plethysmography as described previously.46Cardiac and renal weight indices
When the experimental period was complete, 18 h fasting animals were anaesthetized with 2.5 mL per kg equitensin (i.p.) and blood was collected from the abdominal aorta. Finally, the rats were killed by exsanguination. The kidneys and ventricles were then removed and weighed. The heart was divided into right ventricle and left ventricle plus septum.Vascular reactivity studies
Segments of thoracic aortic rings were mounted in an organ chamber filled with Krebs solution (composition in mM: NaCl 118, KCl 4.75, NaHCO3 25, MgSO4 1.2, CaCl2 2, KH2PO4 1.2 and glucose 11) at 37 °C and gassed with 95% O2 and 5% CO2 and maintained at a resting tension of 2 g. Isometric tension was recorded using an isometric force-displacement transducer (Letigraph 2000) connected to an acquisition system, as previously described.46The concentration–relaxation response curves to acetylcholine (10−9 M–10−4 M) were obtained in rings pre-contracted to the same tension with phenylephrine (0.3 × 10−6 and 10−6 M, in WKY and SHR, respectively). The concentration–relaxation response curves to nitroprusside (10−10–10−6 M) were obtained in the dark in rings without endothelium pre-contracted to the same tension with phenylephrine.
Measurement of ex vivo vascular ROS levels
We used DHE, an oxidative fluorescent dye, to localize ROS in aortic segments in situ, as previously described.47 Briefly, the aorta segments were added into the optimum cutting temperature compound medium (Tissue-Tek; Sakura Finetechnical, Tokyo, Japan), quickly frozen, and cut into 10 μm thick sections in a cryostat (Microm International Model HM500 OM). The sections were incubated at room temperature for 30 min with 10−5 M DHE in the dark, counterstained with the nuclear stain DAPI (3 × 10−7 M) and in the following 24 h examined using a fluorescence microscope (Leica DM IRB, Wetzlar, Germany). Sections were photographed and ethidium and DAPI fluorescence were quantified using ImageJ (version 1.32j, NIH, http://rsb.info.nih/ij/). ROS production was estimated from the ratio of ethidium/DAPI fluorescence.NADPH oxidase activity
The lucigenin-enhanced chemiluminescence assay was used to determine NADPH oxidase activity in intact aortic rings, as previously described.48 Aortic rings from all the experimental groups were incubated for 30 minutes at 37 °C in HEPES-containing physiological salt solution (pH 7.4) of the following composition (in mM): NaCl 119, HEPES 20, KCl 4.6, MgSO4 1, Na2HPO4 0.15, KH2PO4 0.4, NaHCO3 1, CaCl2 1.2 and glucose 5.5. Aortic production of O2˙− was stimulated by addition of NADPH (100 μM). Rings were then placed in tubes containing physiological salt solution, with or without NADPH, and lucigenin was injected automatically at a final concentration of 5 μM. NADPH oxidase activity was determined by measuring luminescence over 200 s in a scintillation counter (Lumat LB 9507, Berthold, Germany) in 5 s intervals and was calculated by subtracting the basal values from those in the presence of NADPH. The vessels were then dried, and the dry weight was determined. NADPH oxidase activity is expressed as relative luminescence units (RLU) per min per mg dry aortic tissue.Western blotting analysis
We examined the state of eNOS, protein kinase B (Akt), ERK1/2, and IκBα phosphorylation in aortae homogenates. Aortic homogenates were run on a sodium dodecyl sulphate (SDS)-polyacrylamide electrophoresis (25 μg of protein per lane), then proteins were transferred to polyvinylidene difluoride membranes (PVDF). Phosphorylated eNOS (Ser1177, and Thr495), Akt (Ser473), ERK1/2 (Thr183 and Tyr185), IκBα (Ser32/36), eNOS, Akt, and ERK1/2, were detected after the membranes were incubated with the respective primary antibodies: rabbit monoclonal anti-p-eNOS-ser-1177 (Cell Signalling Technology, MA, USA), rabbit monoclonal anti-p-eNOS-thr-495 (Millipore, Darmstadt, Germany), mouse monoclonal anti-eNOS (Transduction Laboratories, San Diego, California, USA), rabbit anti-p-Akt-ser-473, rabbit anti-Akt, rabbit anti-ERK1/2 (Cell Signalling Technology, MA, USA), mouse anti-p-ERK1/2-Thr183 and Tyr185 (Sigma-Aldrich, Barcelona, Spain), and rabbit polyclonal anti-p-IκBα-Ser-32/36 (Santa Cruz Biotechnology, Santa Cruz, USA). All were used at 1/1000 dilution and incubated overnight at 4 °C. The membranes were then incubated with secondary peroxidase conjugated goat anti-rabbit or goat anti-mouse antibodies (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3000, Santa Cruz Biotechnology, Santa Cruz, USA), respectively. Antibody binding was detected by an ECL system (Amersham Pharmacia Biotech, Amersham, UK) and densitometric analysis was performed using Scion Image-Release Beta 4.02 software (http://www.scioncorp.com).49 The phospho-eNOS/eNOS, p-Akt/Akt, pERK1/2/ERK1/2, and p-IκBα/α-actin abundance ratios were calculated and data is expressed as a percentage of the values in the control aorta from the same gel. Samples were re-probed for expression of smooth muscle α-actin.
:3000, Santa Cruz Biotechnology, Santa Cruz, USA), respectively. Antibody binding was detected by an ECL system (Amersham Pharmacia Biotech, Amersham, UK) and densitometric analysis was performed using Scion Image-Release Beta 4.02 software (http://www.scioncorp.com).49 The phospho-eNOS/eNOS, p-Akt/Akt, pERK1/2/ERK1/2, and p-IκBα/α-actin abundance ratios were calculated and data is expressed as a percentage of the values in the control aorta from the same gel. Samples were re-probed for expression of smooth muscle α-actin.
Gene expression analysis in aorta
The analysis of gene expression in the aorta was performed by RT-PCR, as previously described.49 For this purpose total RNA was extracted from the aorta by homogenization and converted to cDNA by standard methods. Polymerase chain reaction was performed with a Techne Techgene thermocycler (Techne, Cambridge, UK). A quantitative real-time RT-PCR technique was used to analyze the mRNA expression of eNOS, iNOX, NOX-1, NOX-4, TNFα, IL-1β, IL6, and TLR4. The sequences of the sense and antisense primers used for amplification are described in Table 2. The efficiency of the PCR reaction was determined using a dilution series of standard vascular samples. To normalize mRNA expression, the expression of the housekeeping gene β-actin was used. The mRNA relative quantification was calculated using the ΔΔCt method.| mRNA targets | Descriptions | Sense | Antisense |
|---|---|---|---|
| eNOS | Endothelial nitric oxide synthase | ATGGATGAGCCAACTCAAGG | TGTCGTGTAATCGGTCTTGC |
| iNOS | Inducible nitric oxide synthase | TCTGTGCCTTTGCTCATGAC | CATGGTGAACGTTCTTGG |
| NOX-1 | NOX-1 subunit of NADPH oxidase | TCTTGCTGGTTGACACTTGC | TATGGGAGTGGGAATCTTGG |
| NOX-4 | NOX-4 subunit of NADPH oxidase | ACAGTCCTGGCTTACCTTCG | TTCTGGGATCCTCATTCTGG |
| TNF-α | Tumor necrosis factor-alpha | ACGATGCTCAGAAACACACG | CAGTCTGGGAAGCTCTGAGG |
| IL-6 | Interleukin-6 | GATGGATGCTTCCAAACTGG | AGGAGAGCATTGGAAGTTGG |
| IL-1β | Interleukin-1β | GTCACTCATTGTGGCTGTGG | GCAGTGCAGCTGTCTAATGG |
| TLR4 | Toll-like receptor-4 | GCCTTTCAGGGAATTAAGCTCC | AGATCAACCGATGGACGTGTAA |
| Actb | Beta actin | AATCGTGCGTGACATCAAAG | ATGCCACAGGATTCCATACC |
Drugs
All drugs used were obtained from Sigma (Alcobendas, Madrid, Spain). All drugs and chemicals were dissolved in distilled deionized water.Statistical analysis
Results are expressed as means ± SEM of measurements. The evolution of tail SBP with time was compared using the nested design, with treatment and days as fixed factors and the rat as a random factor. When the overall difference was significant, comparisons were made using Bonferroni's method with an appropriate error. Analysis of the nested design was also carried out with groups and concentrations to compare the concentration–response curves to acetylcholine. The remaining variables were compared using a two way factor design, where group and treatment were fixed effect factors with unequal sample sizes in the different groups. When interaction was significant, Bonferroni's method was used for pairwise comparisons. P < 0.05 was considered statistically significant.Conclusion
Chronic OLE treatment reduces high BP in genetic hypertensive rats. This effect was related to endothelial dysfunction improvement, as a result of reduced vascular pro-inflammatory and pro-oxidative status (Fig. 6). | ||
| Fig. 6 Scheme showing the pathways involved in the protective effect of OLE. | ||
Acknowledgements
This work was supported by Grants from Comisión Interministerial de Ciencia y Tecnología (SAF2011-28150, SAF2014-55523-R), Junta de Andalucía (CTS-164, Proyecto de excelencia, P12-CTS-2722 and P10-AGR-6826), by the Ministerio de Ciencia e Innovación, Instituto de Salud Carlos III (RIC, RD12/0042/0011), Spain and funded by Biosearch SA in the framework of the PRONAOS project granted by CENIT program of the Center for Industrial Technological Development (CDTI). M. S. is a postdoctoral fellow of Red HERACLES, and M. R. is a postdoctoral fellow of RIC. The CIBEREHD is funded by the Instituto de Salud Carlos III.Notes and references
- F. Turnbull, Lancet, 2003, 362, 1527–1535 CrossRef CAS.
- C. Armstrong, Am. Fam. Physician, 2014, 90, 503–504 Search PubMed.
- C. Jänicke, J. Grünwald and T. Brendler, Handbuch Phytotherapie, Wissenschaftliche Verlagsgesellschaft, Stuttgart, 2003 Search PubMed.
- A. Zarzuelo, J. Duarte, J. Jiménez, M. Gonzalez and M. P. Utrilla, Planta Med., 1991, 57, 417–419 CrossRef CAS PubMed.
- S. Cherif, N. Rahal, M. Haouala, B. Hizaoui, F. Dargouth, M. Gueddiche, Z. Kallel, G. Balansard and K. Boukef, J. Pharm. Belg., 1996, 51, 69–71 CAS.
- N. Bennani-Kabchi, H. Fdhil, Y. Cherrah, L. Kehel, F. el Bouayadi, A. Amarti, M. Saïdi and G. Marquié, Thérapie, 1999, 54, 717–723 CAS.
- N. Bennani-Kabchi, H. Fdhil, Y. Cherrah, F. El Bouayadi, L. Kehel and G. Marquie, Ann. Pharm. Fr., 2000, 58, 271–277 CAS.
- R. De Pasquale, M. T. Monforte, A. Trozzi, A. Raccuia, S. Tommasini and S. Ragusa, Plantes Med. Phytother., 1991, 25, 134–140 Search PubMed.
- M. T. Khayyal, M. A. El-Ghazaly, D. M. Abdallah, N. N. Nassar, S. N. Okpanyi and M. H. Kreuter, Arzneim. Forsch./Drug Res., 2002, 52, 797–802 CAS.
- E. Susalit, N. Agus, I. Effendi, R. R. Tjandrawinata, D. Nofiarny, T. Perrinjaquet-Moccetti and M. Verbruggen, Phytomedicine, 2011, 18, 251–258 CrossRef CAS PubMed.
- T. Perrinjaquet-Moccetti, A. Busjahn, C. Schmidlin, A. Schmidt, B. Bradl and C. Aydogan, Phytother. Res., 2008, 22, 1239–1242 CrossRef PubMed.
- L. Panizzi, M. L. Scarpati and G. Oriente, Gazz. Chim. Ital., 1960, 90, 1449–1485 CAS.
- L. I. Somova, F. O. Shode, P. Ramnanan and A. Nadar, J. Ethnopharmacol., 2003, 84, 299–305 CrossRef CAS PubMed.
- K. Hansen, A. Adsersen, S. B. Christensen, S. R. Jensen, U. Nyman and U. W. Smitt, Phytomedicine, 1996, 2, 319–325 CrossRef CAS PubMed.
- J. Duarte, O. Pérez, A. Zarzuelo, J. Jiménez, F. Pérez-Vizcaíno and J. Tamargo, Planta Med., 1993, 59, 318–322 CrossRef CAS PubMed.
- A. Scheffler, H. W. Rauwald, B. Kampa, U. Mann, F. W. Mohr and S. Dhein, J. Ethnopharmacol., 2008, 120, 233–240 CrossRef CAS PubMed.
- A. A. Nekooeian, A. Khalili and M. B. Khosravi, J. Asian Nat. Prod. Res., 2014, 16, 953–962 CrossRef CAS PubMed.
- R. Rodriguez-Rodriguez, J. S. Perona, M. D. Herrera and V. Ruiz-Gutierrez, J. Agric. Food Chem., 2006, 54, 2096–2102 CrossRef CAS PubMed.
- R. Rodriguez-Rodriguez, E. Stankevicius, M. D. Herrera, L. Ostergaard, M. R. Andersen, V. Ruiz-Gutierrez and U. Simonsen, Br. J. Pharmacol., 2008, 155, 535–546 CrossRef CAS PubMed.
- R. Rodriguez-Rodriguez, M. D. Herrera, M. A. de Sotomayor and V. Ruiz-Gutierrez, Am. J. Hypertens., 2007, 20, 728–734 CrossRef CAS PubMed.
- R. Rodriguez-Rodriguez, M. D. Herrera, M. A. de Sotomayor and V. Ruiz-Gutierrez, Br. J. Nutr., 2009, 102, 1435–1444 CrossRef CAS PubMed.
- P. M. Vanhoutte and V. M. Miller, J. Cardiovasc. Pharmacol., 1985, 7, S12–S23 CrossRef CAS PubMed.
- P. Del Boccio, A. Di Deo, A. De Curtis, N. Celli, L. Iacoviello and D. Rotilio, J. Chromatogr. B Analyt. Technol. Biomed. Life Sci., 2003, 785, 47–56 CrossRef CAS.
- C. E. Storniolo, J. Roselló-Catafau, X. Pintó, M. T. Mitjavila and J. J. Moreno, Redox Biol., 2014, 2C, 971–977 CrossRef PubMed.
- I. Fleming, Pflugers Archiv., 2010, 459, 793–806 CrossRef CAS PubMed.
- H. Nguyen, V. L. Chiasson, P. Chatterjee, S. E. Kopriva, K. J. Young and B. M. Mitchell, Cardiovasc. Res., 2013, 97, 696–704 CrossRef CAS PubMed.
- I. Fleming, B. Fisslthaler, S. Dimmeler, B. E. Kemp and R. Busse, Circ. Res., 2001, 88, E68–E75 CrossRef CAS PubMed.
- E. Scoditti, A. Nestola, M. Massaro, N. Calabriso, C. Storelli, R. De Caterina and M. A. Carluccio, Atherosclerosis, 2014, 232, 17–24 CrossRef CAS PubMed.
- M. R. Tschudi, S. Mesaros, T. F. Lüscher and T. Malinski, Hypertension, 1996, 27, 32–35 CrossRef CAS PubMed.
- R. Kobayasi, E. H. Akamine, A. P. Davel, M. A. Rodrigues, C. R. Carvalho and L. V. Rossoni, J. Hypertens., 2010, 28, 2111–2119 CrossRef CAS PubMed.
- C. K. Roberts, R. J. Barnard, R. K. Sindhu, M. Jurczak, A. Ehdaie and N. D. Vaziri, J. Appl. Physiol., 2005, 98, 203–210 CrossRef CAS PubMed.
- H. Poudyal, F. Campbell and L. Brown, J. Nutr., 2010, 140, 946–953 CrossRef CAS PubMed.
- F. Visioli, G. Bellomo and C. Galli, Biochem. Biophys. Res. Commun., 1998, 247, 60–64 CrossRef CAS PubMed.
- S. Grunfeld, C. A. Hamilton, S. Mesaros, S. W. McClain, A. F. Dominiczak, D. F. Bohr and T. Malinski, Hypertension, 1995, 26, 854–857 CrossRef CAS PubMed.
- H. Suzuki, A. Swei, B. W. Zweifach and G. W. Schmid-Schönbein, Hypertension, 1995, 25, 1083–1089 CrossRef CAS PubMed.
- S. Wind, K. Beuerlein, M. E. Armitage, A. Taye, A. H. Kumar, D. Janowitz, C. Neff, A. M. Shah, K. Wingler and H. H. Schmidt, Hypertension, 2010, 56, 490–497 CrossRef CAS PubMed.
- E. Vila and M. Salaices, Am. J. Physiol. Heart Circ. Physiol., 2005, 288, H1016–H1021 CrossRef CAS PubMed.
- K. K. Griendling, D. Sorescu, B. Lassègue and M. Ushio-Fukai, Arterioscler. Thromb. Vasc. Biol., 2000, 20, 2175–2183 CrossRef CAS.
- R. P. Brandes, N. Weissmann and K. Schröder, J. Mol. Cell Cardiol., 2014, 73, 70–79 CrossRef CAS PubMed.
- R. M. Touyz, C. Deschepper, J. B. Park, G. He, X. Chen, M. F. Neves, A. Virdis and E. L. Schiffrin, J. Hypertens., 2002, 20, 1127–1134 CrossRef CAS PubMed.
- M. A. Carluccio, L. Siculella, M. A. Ancora, M. Massaro, E. Scoditti, C. Storelli, F. Visioli, A. Distante and R. De Caterina, Arterioscler. Thromb. Vasc. Biol., 2003, 23, 622–629 CrossRef CAS PubMed.
- M. Toral, M. Gómez-Guzmán, R. Jiménez, M. Romero, M. Sánchez, M. P. Utrilla, N. Garrido-Mesa, M. E. Rodríguez-Cabezas, M. Olivares, J. Gálvez and J. Duarte, Clin. Sci., 2014, 127, 33–45 CrossRef PubMed.
- C. F. Liang, J. T. Liu, Y. Wang, A. Xu and P. M. Vanhoutte, Arterioscler. Thromb. Vasc. Biol., 2013, 33, 777–784 CrossRef CAS PubMed.
- F. Y. Lin, Y. H. Chen, J. S. Tasi, J. W. Chen, T. L. Yang, H. J. Wang, C. Y. Li, Y. L. Chen and S. J. Lin, Arterioscler. Thromb. Vasc. Biol., 2006, 26, 2630–2637 CrossRef CAS PubMed.
- E. D. Frohlich, C. Apstein, A. V. Chobanian, R. B. Devereux, H. P. Dustan, V. Dzau, F. Fauad-Tarazi, M. J. Horan, M. Marcus, B. Massie, M. A. Pfeffer, R. N. Re, E. J. Roccella, D. Savage and C. Shub, N. Engl. J. Med., 1992, 327, 998–1008 CrossRef CAS PubMed.
- R. Vera, R. Jiménez, F. Lodi, M. Sánchez, M. Galisteo, A. Zarzuelo, F. Pérez-Vizcaíno and J. Duarte, Menopause, 2007, 14, 933–940 CrossRef PubMed.
- M. J. Zarzuelo, R. Jiménez, P. Galindo, M. Sánchez, A. Nieto, M. Romero, A. M. Quintela, R. López-Sepúlveda, M. Gómez-Guzmán, E. Bailón, I. Rodríguez-Gómez, A. Zarzuelo, J. Gálvez, J. Tamargo, F. Pérez-Vizcaíno and J. Duarte, Hypertension, 2011, 58, 733–743 CrossRef CAS PubMed.
- P. Galindo, S. González-Manzano, M. J. Zarzuelo, M. Gómez-Guzmán, A. M. Quintela, A. González-Paramás, C. Santos-Buelga, F. Pérez-Vizcaíno, J. Duarte and R. Jiménez, Food Funct., 2012, 3, 643–650 CAS.
- A. M. Quintela, R. Jiménez, M. Gómez-Guzmán, M. J. Zarzuelo, P. Galindo, M. Sánchez, F. Vargas, A. Cogolludo, J. Tamargo, F. Pérez-Vizcaíno and J. Duarte, Free Radic. Biol. Med., 2012, 53, 730–741 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2016 |