
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Control of vesicle membrane permeability with catalytic particles†
Ross W.
Jaggers
,
Rong
Chen
and
Stefan A. F.
Bon
*
Department of Chemistry, University of Warwick, Coventry, C47 7AL, UK. E-mail: S.Bon@warwick.ac.uk; Web: http://www.bonlab.info Tel: +44 (0)2476 574009
First published on 20th August 2015
Abstract
Giant polymer vesicles which have membrane-embedded catalytically active manganese oxide particles are made using droplet-based microfluidics. It is demonstrated that these colloidal particles can regulate the membrane permeability of the polymersomes upon their exposure to, and catalytic reaction with, small amounts of dissolved hydrogen peroxide.
Conceptual insightsThe ability to control membrane permeability in vesicles allows for regulated transport of matter across the vesicular wall. Vesicles can be seen as microscopic sacs containing a compartmentalized volume of liquid dispersed in a bulk liquid environment. Nature has devised sophisticated strategies to accomplish control of transmembrane transport, including endo- and exocytosis as well as the incorporation of transmembrane proteins into cell membranes. A variety of synthetic approaches have been explored by scientists in order to accomplish such control in manmade systems. Examples include hybrid systems whereby transmembrane proteins were incorporated as part of synthetic vesicles, and the use of responsive macromolecular building blocks to regulate membrane porosity upon an external trigger in polymer vesicles, also referred to as polymersomes. Here we show for the first time that the permeability of the membrane of polymer vesicles can be controlled by membrane-embedded catalytically active manganese oxide particles. The ability to chemically trigger activity of the catalytic particle hereby inducing a temporary increase of membrane permeability offers precise time-specific control of transmembrane transport. It is our belief that this concept can be applied to a wide variety of membrane-based systems. |
A vesicle is a self-contained body of liquid surrounded by a lamellar, often bilayer-type, membrane dispersed in a liquid environment. Compartmentalization of small and finite volumes of liquids and the consecutive emergence of membrane bioenergetics are identified as being of key importance in the evolution of cells, and hence the origins of life.1 The ability to mediate transport across a cell membrane is crucial for cellular activity, from regulation of ion transport to uptake of nutrients.2 In eukaryotic and prokaryotic cells, functional proteins embedded in the cell membrane regulate transport of ions and molecules and are known as transmembrane proteins (TMPs). This concept of having the ability to regulate and control membrane permeability on demand is being explored in a variety of scientific fields, using a range of both biological and chemical methods.3–29
A pioneering example where biology, in the form of TMPs, meets synthetic macromolecular chemistry, in the form of polymer vesicles, was reported by Meier and coworkers in which polymersome nanoreactors were permeabilized to small solutes by the inclusion of a nonspecific channel protein, bacterial porin OmpF, into the membrane wall.3 In a different study Meier and coworkers demonstrated the incorporation of Aquaporin Z, a water channel protein, into the walls of polymeric vesicles.4 Inclusion of this protein resulted in a significantly increased water permeability, almost 90 times greater than vesicles without. Choi and Montemagno elegantly demonstrated that several proteins working in conjunction with one another could be inserted into a vesicle membrane, creating a multi-protein, light-driven adenosine triphosphate (ATP) engine.5 Moreover, channel proteins have also been used to mediate deoxyribonucleic acid (DNA) translocation by acting as a receptor for binding bacteriophage viruses, creating polymeric vehicles for DNA.6
Synthetic membrane channels have also been used to modify the permeability of polymeric vesicles, for example, Hammer and colleagues showed that the inclusion of dendritic helical pores facilitates proton transport across a membrane.7 Perrier, Jolliffe and coworkers8 demonstrated the use of cyclic peptide–poly(N-isopropylacrylamide) conjugates as thermoresponsive channels, whilst Gin et al.9 showed that aminocyclodextrins can be used as anion-selective channels.
The use of membrane channels is not the only route towards gaining control of transmembrane transport; the inherent permeability of a vesicle membrane itself can be influenced by physical phenomena. Near their liquid-crystalline transition temperatures, liposomes exhibit an enhanced leakiness of their membranes,10 a property exploited by Yatvin and coworkers in their design of temperature-controlled, drug-releasing vesicles.11 Battaglia and coworkers demonstrated shear-rate induced modulation of polymersome membrane permeability.12
The amphiphilic balance of the macromolecular building blocks of polymer vesicles can be altered to regulate permeability. Oxidative responsive polymersomes in which the hydrophobic propylene sulfide units were oxidised by hydrogen peroxide into their more hydrophilic propylene sulfoxide analogues were reported by Tirelli, Hubbell and co-workers.13 A variation on this concept using glucose oxidase to generate hydrogen peroxide in situ was reported by Hubbell et al.14 Ahmed and Discher showed that triggered partial hydrolysis of the block copolymer building blocks of vesicular structures induced self-porating membranes capable of enhanced release.15 Du and Armes showed a pH dependence in polymersome membrane permeability.16 Eisenberg et al. interestingly showed that an alternating pH in the surrounding solution of a vesicle can trigger a “breathing” behaviour, resulting in the diffusion of species in and out of the vesicle upon a reversible change in vesicle volume.17 Van Hest and coworkers incorporated phenyl boronic acid based stimuli responsive block copolymers into vesicular structures which allowed for pore formation at both high pH and sugar exposure.18 De Geest and coworkers created core–shell capsules consisting of a degradable microgel core surrounded by a semi-permeable membrane. Upon degradation of the microgel core, the accompanied increase in osmotic pressure ruptures the surrounding membrane, and triggers the release of the encapsulated species.19
One interesting approach is the use of functional nanoparticles on or in the membrane, as well as inside, of manmade vesicles. In these hybrid supracolloidal structures it has been shown that control of membrane permeability is possible using the characteristic features of the nanoparticles.20–28
Oh and coworkers incorporated nanoparticles of silver20 and gold21 into the membranes of phospholipid vesicles and showed enhanced membrane fluidity and thus permeability. Paasonen and colleagues used the concept of hyperthermia to show that exposure to light of vesicles with gold nanoparticles on or in their membranes enhanced their permeability.22 Weitz et al. created giant polymersomes from poly(N-isopropyl acrylamide) containing diblock copolymers and gold nanoparticles using microfluidics.23 These polymersomes exhibited thermo- and photo-responsive behaviour and tunable permeabilities. Lecommandoux and others prepared magneto-responsive polymer vesicles by embedding iron oxide nanoparticles into the lamellar membranes of vesicles.24–29 Upon exposure to an externally applied magnetic field the vesicles could undergo magnetotaxis (gradient field), deform, be used as contrast agents in magnetic resonance imaging and as drug delivery vehicles that delivered their payload by means of radiofrequency magnetic hyperthermia.
In this manuscript, we show that manganese oxide particles that are embedded into the membranes of polymer vesicles are able to enhance permeability upon activation and reaction of the catalytic particles with small amounts of hydrogen peroxide. To the best of our knowledge this concept of using chemical heterogeneous catalysis as a strategy to influence membrane permeability has not been previously reported. We believe that our method offers an interesting tactic to control and regulate release from vesicular structures dispersed in a liquid.
Polymer vesicles of low dispersity in size distribution were prepared using a microfluidics technique from water-in-oil-in-water double-emulsion droplets, with a middle phase of chloroform containing poly(n-butyl methacrylate)94-block-poly(N,N-dimethyl-amino ethyl methacrylate)37 and manganese oxide particles, as shown in Fig. 1. For membrane permeability and release studies, which are described later, the inner aqueous phase contained either sodium sulfate, sodium fluoride, or congo red dye.
 | ||
| Fig. 1 Schematic drawings of (a) the formation of double emulsion droplets of low dispersity in size distribution using a flow-focussing microfluidic device. An inner aqueous phase (pink, b) is contained within an intermediate organic phase containing amphiphilic block-copolymer and catalyst particles (yellow, a) in an aqueous outer phase (blue, c); (b) the formation of vesicles from the double emulsion droplets. After evaporation of the volatile organics from the middle phase, vesicular polymer capsules are formed, containing catalyst, that is manganese oxide, particles in their walls. | ||
Dark field microscopy indicates that manganese oxide particles are part of the membrane of the vesicles as scattering is observed (see Fig. 2a). Cryogenic scanning electron microscopy confirms that manganese oxide particles are embedded into the membrane of the polymer vesicle (see Fig. 3). The addition of small amounts of hydrogen peroxide (0.1 wt%) to sodium sulfate loaded vesicles dispersed in a barium chloride solution showed a steady release of their content in the form of observed precipitation of barium sulfate particles (see Fig. 2b).
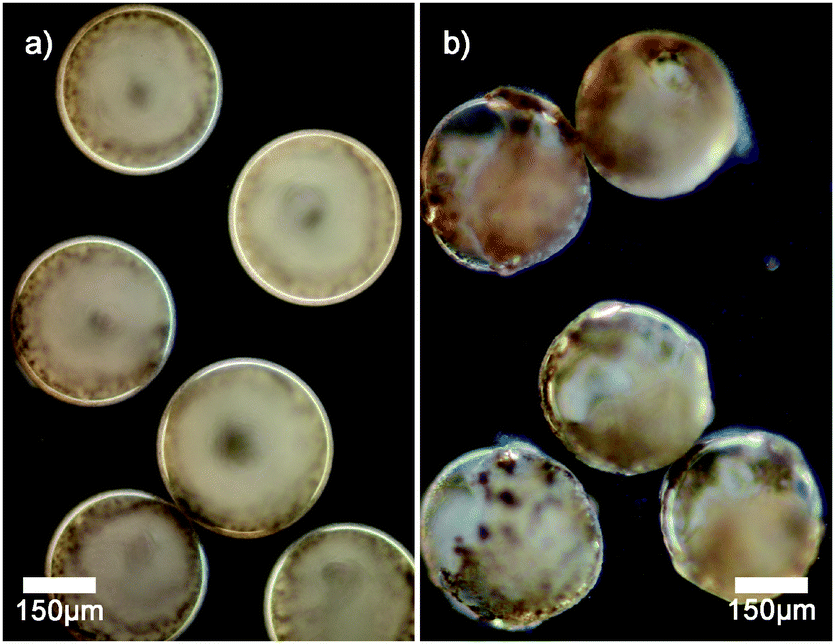 | ||
| Fig. 2 Dark field microscopy images of (a) vesicles with membrane embedded manganese oxide particles; (b) vesicles undergoing slow release of encapsulated sulfate ions upon addition of a small amount of hydrogen peroxide. The outer water phase contains Ba2+ cations, and hence the precipitation of barium sulfate occurs, which is observed as a haze. | ||
 | ||
| Fig. 3 Cryo-SEM images of vesicles with membrane embedded manganese oxide particles: (a) overview image of a single frozen vesicle in ice; (b) magnified observation of the region in blue in panel (a); (c) magnified view of the region in yellow in panel (a). The manganese oxide particles can be clearly observed to be part of the vesicular membrane. | ||
This observation implied a dramatic change in permeability of the vesicle membranes when exposed to a chemical trigger and prompted us to study the release characteristics of the catalytic manganese oxide containing membranes quantitatively.
Fluoride ion selective electrode measurements were carried out in the continuous phase of sodium fluoride loaded vesicles. To provide reference, the release profiles of vesicles with and without embedded manganese oxide particles were compared (see Fig. 4, ESI†). Complete release of fluoride ions results in a concentration of 62 ppm.
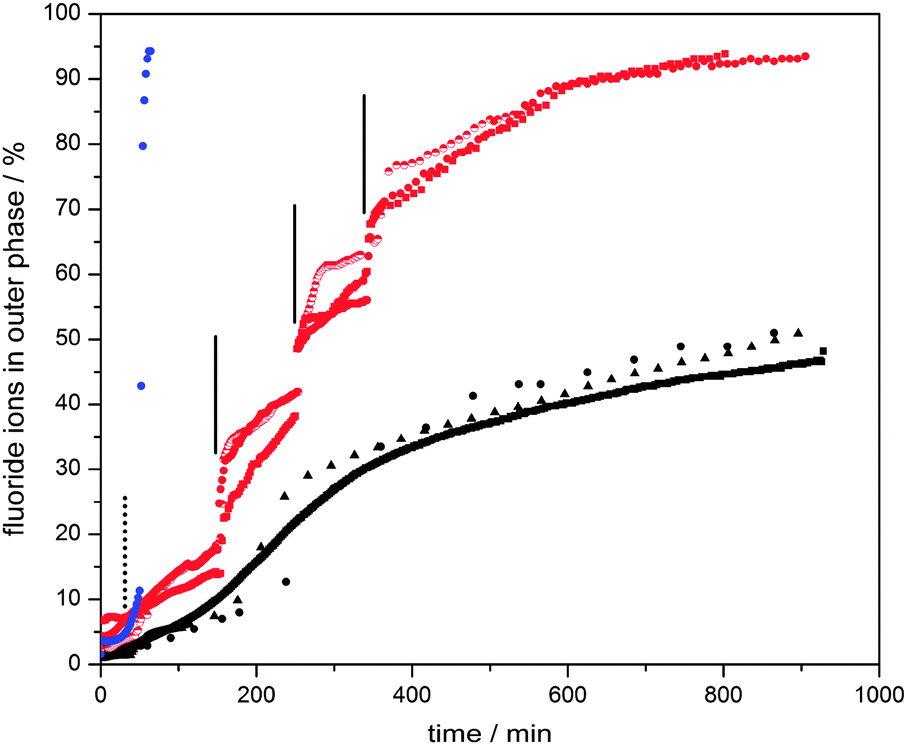 | ||
| Fig. 4 Fluoride ion release profiles of water-based dispersions of ca. 6000 MnO2 particle embedded polymersomes containing 0.2 M sodium fluoride (6 mL of water outer phase, 0.1 mL of inner aqueous phase) with (a) 3.0 mL of 30 wt% H2O2 added at 50 min (blue, dashed vertical line indicates time of hydrogen peroxide addition), (b) aliquots of 0.2 mL of 3 wt% H2O2 added at 150, 250 and 340 min (red, solid vertical lines indicate time of hydrogen peroxide addition), and (c) no H2O2 stimulus (black). This process of “catalytic” enhanced release was visualized clearly under dark field microscopy observations in which release of sulfate ions triggered the formation of insoluble barium sulfate particles (Fig. 5, also see Movie in ESI†). | ||
 | ||
| Fig. 5 Dark microscopy sequence of video stills (see MovieCatalyticVesicle.mp4, ESI†) of a water-based (containing barium cations) dispersion of vesicles with membrane embedded manganese oxide particles and loaded with sodium sulfate, after the addition of a large aliquot of hydrogen peroxide. The stills show the formation of oxygen bubbles, leading to deformation and rupture of the vesicle membranes hereby releasing the sulfate ion content, as visualized by the formation of a stream of in situ formed barium sulfate particles (a white haze). Scale bar: 150 μm. | ||
The vesicles without manganese particles showed a significantly higher release rate of fluoride ions than those with particles embedded in their membranes. It is worth noting that the experiments start when chloroform has not fully evaporated from the membrane phase. Its presence led to retardation, as initially a thicker hydrophobic barrier has to be overcome by the ions. For vesicles without particles in their walls, this retardation period was 20–30 min. Once chloroform had evaporated, the release rate reached its maximum and started to decelerate as the concentration difference between the inside and outside of the vesicles decreased. Strikingly, the chloroform evaporation process took much longer for the vesicles with manganese dioxide particles (about 100–110 min, see Fig. 4, ESI†). It is plausible that both the reduced effective surface area due to the presence of impermeable manganese oxide particles (ca. 20 vol% of the membranes) and particle-induced light scattering (protecting the vesicles from heating up due to light exposure under the microscope) explain this observation. We speculate that the slow down of release after ca. 300 minutes is due to the fading out of the chloroform plasticization effect, making the vesicle membranes less fluid-like.
The release behaviour of the manganese oxide vesicles exposed to different hydrogen peroxide solutions was studied. For this we prepared water-based dispersions of ca. 6000 vesicles containing 0.2 M sodium fluoride (6 mL of water outer phase, 0.1 mL of inner aqueous phase). Again, release profiles were measured with a fluoride selective electrode. When a diluted hydrogen peroxide solution was added after 150 min, 0.2 mL of 3 wt% H2O2 (establishing a bulk H2O2 concentration of ca. 0.1 wt%), a clear increase in membrane permeability and thus enhanced release rate of fluoride ions was observed (Fig. 4). Approximately 20–30 minutes after this stimulus, the release rate dropped to a similar level to that of the sample without H2O2 (in the region between 200 minutes and 250 minutes), indicating that the majority of the hydrogen peroxide trigger had been consumed. The same treatment was repeated at both 250 minutes and 340 minutes in order to confirm the initial observation. Again, a temporary increase in membrane permeability was observed. This shows that the catalytic reaction between hydrogen peroxide and the manganese oxide particles increases membrane permeability until the hydrogen peroxide is depleted (see Fig. 4). The exact mechanism of this temporary increased permeability is not fully understood. We hypothesise that the catalytic breakdown of hydrogen peroxide to dioxygen and water at the site of the particles induces the formation of transient openings at the interface between the particles and the membrane, thus increasing membrane permeability. It is important to note that the driving force for release depends on the difference between the concentrations of fluoride ions inside and outside of the polymer vesicles. Therefore, we also carried out blank experiments in which aliquots of water were added. Indeed, small enhancements of release rates were observed in line with the induced concentration change. However, this effect was minor in comparison with the hydrogen peroxide catalytic trigger. Another point worth noting, which could plausibly explain enhanced release, is the potential self-destruction of a sub set of vesicles upon addition of the hydrogen peroxide trigger. We therefore carried out smaller scale experiments with 200 vesicles and counted the vesicles before and after our release experiments. Less than 4 vesicles (2%) were found to be destroyed in each of these runs. The majority of vesicles survive the peroxide trigger and therefore we can conclude that that enhanced membrane permeability was indeed achieved by the catalytic activity of the manganese oxide particles.
One could argue that the manganese oxide particles act as stoppers, which upon peroxide activation, could pop out, hereby creating a temporary pore allowing for the observed enhancement in release of fluoride ions. ICP-OES measurements of the outer water phase post peroxide treatment showed no sign of presence of manganese. We therefore can confirm that the manganese oxide particles remained associated with the vesicle membranes (see ESI† for details).
Triggered release of the full vesicle content from hybrid vesicles is possible upon exposure to high concentrations of hydrogen peroxide (3.0 mL, 30 wt%). The formation of oxygen bubbles upon catalytic reaction causes imminent membrane deformation and rupture, followed by immediate release and self-destruction of the vesicles (Fig. 4, blue, and Fig. 5).
A question one could ask is if there is a variation between release profiles of different compounds from the polymersomes. The vesicle membrane can be seen as a barrier for transport phenomena. We therefore decided to look at the release profile of the diazo dye congo red, which is the disodium salt of 3,3′-([1,1′-biphenyl]-4,4′-diyl)bis(4-aminonaphthalene-1-sulfonic acid). In equivalent experiments to those of the aforementioned fluoride release ones, polymer vesicles were loaded with an aqueous solution of congo red (3.0 mM) and the dye concentration in the outer phase was monitored using UV-Vis spectroscopy (wavelength maximum = 505 nm). The results are displayed in Fig. 6.
 | ||
| Fig. 6 Congo red release profiles of water-based dispersions of ca. 6000 MnO2 particle embedded polymersomes containing 3.0 mM congo red dye (6 mL of water outer phase, 0.1 mL of inner aqueous phase) with (a) 3.0 mL of 30 wt% H2O2 (blue), or (b) 0.2 mL of 3 wt% H2O2 (red) added at 150 min (solid vertical line indicate time of hydrogen peroxide addition), and (c) no H2O2 stimulus (black). | ||
As seen in the fluoride release experiments, treatment with 3.0 mL of 30 wt% hydrogen peroxide causes rupture of all vesicles triggering instant and “complete” release, whilst a smaller 0.2 mL of 3 wt% of H2O2 treatment caused temporary acceleration of dye release. When we compare the release profiles of fluoride ions with congo red dye from manganese oxide loaded polymersomes in the absence of any trigger, the fraction of congo red released after 900 minutes is substantially lower, that is 11% versus ca. 48% of the fluoride ions. It should be noted, however, that over the time period of 400 to 900 minutes, release rates appear not to differ substantially.
The addition of 0.2 mL of 3 wt% of H2O2 after 150 min clearly enhances the release rate of congo red. What is striking is that in both catalytic scenarios the total amount released was limited to approximately 50% of the encapsulated dye expected to be released. We found that the reason for this was the strong physisorption of the dye to the amphiphilic block copolymer, that is poly(n-butyl methacrylate)94-block-poly(N,N-dimethyl-amino ethyl methacrylate)37. This strong adsorption, and therefore accumulation of congo red into the vesicle membrane wall, was confirmed by confocal microscopy (see Fig. 7). For this we purposely buckled a polymer vesicle using an NaCl solution. As can be seen from the image, a higher intensity and thus, concentration, of the dye was observed in the wrinkled parts of the membrane.
 | ||
| Fig. 7 Confocal microscope image of a partially buckled polymer vesicle loaded with 0.003 M congo red dye solution, the polymersome being dispersed in an aqueous NaCl environment. Scale bar: 50 μm. | ||
Conclusions
We have demonstrated that the permeability of vesicular structures containing “active” colloidal particles as part of their membrane can be regulated by use of a chemical trigger. This model system offers an exciting method of regulating and controlling transmembrane transport, which may be of interest to a range of scientific fields.One comment we would like to make and relates to the area of the evolution of life is that a hybrid vesicular structure, which has “active” colloidal particles either adhered to the surface of the membrane, or embedded into it, can achieve chemically activated control of membrane permeability. This is what we have shown here.
The origin of biological membranes remains ambiguous. Wächtershäuser30,31 and Russell and coworkers,32 for example, argue that porous pyrite (FeS) structures found in deep-sea hydrothermal vents contain small pockets of water and have the ability to accumulate organic matter due to the relative hydrophobic nature of pyrite. The hypothesis is that these compartmentalized structures are suitable microenvironments for life to evolve and, therefore, can be seen as semi-permeable predecessors to protocells.‡ They are formed through precipitation and assembly of metal sulfides on the surface of hydrogen sulfide rich gas bubbles, another example being based on zinc sulfide.33 A modern synthetic analogue of such semipermeable compartmentalized structures would be a colloidosome,34 which can be built from colloidal particles that adhere to liquid–liquid, or liquid–gas interfaces, so-called Pickering stabilizers.35 The next step in evolution of these early compartmentalised structures describes the formation of a cell membrane in the form of liposomes, and here is where the problem lies. How would a primitive cell get its nutrients when modern model lipid-based cell membranes are effectively non-permeable for desired molecules on an acceptable time scale, if membrane-proteins are left out?36 Mansy and coworkers suggested that protocells made with fatty acid components were able to take up nucleotides through concerted flipping,37 fast at high temperatures, and that in addition DNA strand separation processes benefited from thermal fluctuations.38 We would like to suggest here that a hybrid vesicular structure, which has “active” colloidal particles as part of its membrane, may have controlled permeability in primitive cells.
Notes and References
- Origins of Life: The Primal Self-Organization, ed. R. Egel, D. H. Lankenau, A. Y. Mulkidjanian and Y. Armen, Springer, Berlin, 2011, ch. 1 Search PubMed.
- S. J. Singer and G. L. Nicolson, Science, 1972, 175, 720–731 CAS.
- C. Nardin, S. Thoeni, J. Widmer, M. Winterhalter and W. Meier, Chem. Commun., 2000, 1433–1434 RSC.
- M. Kumar, M. Grzelakowski, J. Zilles, M. Clark and W. Meier, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 20719–20724 CrossRef CAS PubMed.
- H. Choi and C. D. Montemagno, Nano Lett., 2005, 5, 2538–2542 CrossRef CAS PubMed.
- A. Graff, M. Sauer, P. Van Gelder and W. Meier, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 5064–5068 CrossRef CAS PubMed.
- A. J. Kim, M. S. Kaucher, K. P. Davis, M. Peterca, M. R. Imam, N. A. Christian, D. H. Levine, F. S. Bates, V. Percec and D. A. Hammer, Adv. Funct. Mater., 2009, 19, 2930–2936 CrossRef CAS.
- M. Danial, C. M. N. Tran, K. A. Jolliffe and S. Perrier, J. Am. Chem. Soc., 2014, 136, 8018–8026 CrossRef CAS PubMed.
- N. Madhavan, E. C. Robert and M. S. Gin, Angew. Chem., Int. Ed., 2005, 44, 7584–7587 CrossRef CAS PubMed.
- D. Papahadjopoulos, K. Jacobson, S. Nir and T. Isac, Biochim. Biophys. Acta, 1973, 311, 330–348 CrossRef CAS.
- M. B. Yatvin, J. N. Weinstein, W. H. Dennis and R. Blumenthal, Science, 1978, 202, 1290–1293 CAS.
- J. Gaitzsch, D. Appelhans, L. Wang, G. Battalgia and B. Voit, Angew. Chem., 2012, 51, 4448–4451 CrossRef CAS PubMed.
- A. Napoli, M. Valentini, N. Tirelli, M. Müller and J. A. Hubbell, Nat. Mater., 2004, 3, 183–189 CrossRef CAS PubMed.
- A. Napoli, M. J. Boerakker, N. Tirelli, R. J. M. Nolte, N. A. J. M. Sommerdijk and J. A. Hubbell, Langmuir, 2004, 20, 3487–3491 CrossRef CAS PubMed.
- F. Ahmed and D. E. Discher, J. Controlled Release, 2004, 96, 37–53 CrossRef CAS PubMed.
- J. Du and S. P. Armes, J. Am. Chem. Soc., 2005, 127, 12800–12801 CrossRef CAS PubMed.
- S. Yu, T. Azzam, I. Rouiller and A. Eisenberg, J. Am. Chem. Soc., 2009, 131, 10557–10566 CrossRef CAS PubMed.
- K. T. Kim, J. J. L. M. Cornelissen, R. J. M. Nolte and J. C. M. van Hest, Adv. Mater., 2009, 21, 2787–2791 CrossRef CAS.
- B. G. De Geest, S. De Koker, J. Demeester, S. C. De Smedt and W. E. Hennink, Polym. Chem., 2010, 1, 137–148 RSC.
- S. H. Park, S. G. Oh, J. Y. Mun and S. S. Han, Colloids Surf., B, 2005, 44, 117–122 CrossRef CAS PubMed.
- S. H. Park, S. G. Oh, J. Y. Mun and S. S. Han, Colloids Surf., B, 2006, 48, 112–118 CrossRef CAS PubMed.
- L. Paasonen, T. Laaksonen, C. Johans, M. Yliperttula, K. Kontturi and A. Urtti, J. Controlled Release, 2007, 122, 86–93 CrossRef CAS PubMed.
- E. Amstad, S. H. Kim and D. A. Weitz, Angew. Chem., Int. Ed., 2012, 51, 12499–12503 CrossRef CAS PubMed.
- S. Lecommandoux, O. Sandre, F. Chécot, J. Rodriguez-Hernandez and R. Perzynski, Adv. Mater., 2005, 17, 712–718 CrossRef CAS.
- S. Lecommandoux, O. Sandre, F. Chécot and R. Perzynski, Prog. Solid State Chem., 2006, 34, 171–179 CrossRef CAS.
- M. Krack, H. Hohenberg, A. Kornowski, P. Lindner, H. Weller and S. Förster, J. Am. Chem. Soc., 2008, 130, 7315–7320 CrossRef CAS PubMed.
- C. Sanson, O. Diou, J. Thévenot, E. Ibarboure, A. Soum, A. Brûlet, S. Miraux, E. Thiaudière, S. Tan, A. Brisson, V. Dupuis, O. Sandre and S. Lecommandoux, ACS Nano, 2011, 5, 1122–1140 CrossRef CAS PubMed.
- E. Amstad, J. Kohlbrecher, E. Müller, T. Schweizer, M. Textor and E. Reimhult, Nano Lett., 2011, 11, 1664–1670 CrossRef CAS PubMed.
- Y. Chen, A. Bose and G. D. Bothun, ACS Nano, 2010, 4, 3215–3221 CrossRef CAS PubMed.
- G. Wächtershäuser, Syst. Appl. Microbiol., 1988, 10, 207–210 CrossRef.
- G. Wächtershauser, Proc. Natl. Acad. Sci. U. S. A., 1994, 91, 4283–4287 CrossRef.
- M. J. Russell, A. J. Hall, A. G. Cairns-Smith and P. S. Braterman, Nature, 1988, 336, 117 CrossRef.
- A. Y. Mulkidjanian, Biol. Direct, 2009, 4, 26 CrossRef PubMed.
- A. D. Dinsmore, M. F. Hsu, M. G. Nikolaides, M. Marquez, A. R. Bausch and D. A. Weitz, Science, 2002, 298, 1006–1009 CrossRef CAS PubMed.
- S. U. Pickering, J. Chem. Soc., Trans., 1907, 91, 2001–2021 RSC.
- D. W. Deamer, Nature, 2008, 454, 37–38 CrossRef CAS PubMed.
- S. S. Mansy, J. P. Schrum, M. Krishnamurthy, S. Tobé, D. A. Treco and J. W. Szostak, Nature, 2008, 454, 122–125 CrossRef CAS PubMed.
- S. S. Mansy and J. W. Szostak, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 13351–13355 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5mh00093a |
| ‡ Protocells, the predecessors to modern cells, possess a self-replicating genome encapsulated by a membrane that can grow and divide, thus allowing it replicate. Unlike a cell, however, a protocell mediates its cell uptake not with sophisticated membrane transport control, but by diffusion through the membrane alone. The evolution of these primitive cell analogues is a hot topic of debate amongst evolutionary scientists, with particular focus on the requirements of self-replication and cell function.32,33 |
| This journal is © The Royal Society of Chemistry 2016 |