
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Nanogels as imaging agents for modalities spanning the electromagnetic spectrum
Minnie
Chan
a and
Adah
Almutairi
*b
aDepartment of Chemistry and Biochemistry, University of California, San Diego, La Jolla, CA 92093-0600, USA
bSkaggs School of Pharmacy and Pharmaceutical Sciences, KACST-UCSD Center of Excellence in Nanomedicine, Laboratory of Bioresponsive Materials, University of California, 9500 Gilman Dr., 0600, PSB 2270, La Jolla, San Diego, CA 92093-0600, USA. E-mail: aalmutairi@ucsd.edu; Tel: +1 (858) 246 0871
First published on 25th September 2015
Abstract
In the past few decades, advances in imaging equipment and protocols have expanded the role of imaging in in vivo diagnosis and disease management, especially in cancer. Traditional imaging agents have rapid clearance and low specificity for disease detection. To improve accuracy in disease identification, localization and assessment, novel nanomaterials are frequently explored as imaging agents to achieve high detection specificity and sensitivity. A promising material for this purpose are hydrogel nanoparticles, whose high hydrophilicity, biocompatibility, and tunable size in the nanometer range make them ideal for imaging. These nanogels (10 to 200 nm) can circumvent uptake by the reticuloendothelial system, allowing longer circulation times than small molecules. In addition, their size/surface properties can be further tailored to optimize their pharmacokinetics for imaging of a particular disease. Herein, we provide a comprehensive review of nanogels as imaging agents in various modalities with sources of signal spanning the electromagnetic spectrum, including MRI, NIR, UV-vis, and PET. Many materials and formulation methods will be reviewed to highlight the versatility of nanogels as imaging agents.
 Minnie Chan | Minnie Chan graduated with a Bachelor of Science degree in Biomolecular Science at New York University's Polytechnic School of Engineering in 2010. She then began her graduate studies in the Department of Chemistry and Biochemistry at the University of California, San Diego under the guidance of Professor Adah Almutairi. Her PhD thesis focused on nanogel formulation and their use in MRI. |
 Adah Almutairi | Adah Almutairi is an associate professor at UC San Diego and is co-director of the Center of Excellence in Nanomedicine and Engineering. Prof. Almutairi has eight issued or pending patents, several of which have been licensed and one of which is being developed by the company she founded, eLux Medical. Her lab's research has been recognized by Chemical & Engineering News and Materials Today, among other scientific media outlets. She completed her PhD in Materials Chemistry at UC Riverside and came to UC San Diego in 2008 from UC Berkeley, where she worked with Professor Jean Fréchet to develop several nanoprobes for in vivo imaging. |
1. Introduction
Imaging is an essential part of clinical protocols that can provide morphological, structural, metabolic, functional and molecular information, as a minimally invasive procedure, for disease identification and assessment. Computed tomography (CT), positron emission tomography (PET), magnetic resonance imaging (MRI) and ultrasound are the most commonly used imaging modalities.1,2 To generate signal or enhance contrast, contrast agents are often used in the imaging regime. However, most clinically approved contrast agents are small molecules, which are rapidly cleared from the body, limiting the imaging window. In addition, these agents only provide overall whole-body contrast,3,4 and often lack disease specificity. An imaging agent that has optimal clearance, can be targeted to or respond to disease biomarkers would enable diagnosis and assessment with higher sensitivity, specificity, and efficiency.3–5Nanomaterials, materials of submicron size, have opened up a new opportunity to overcome these challenges. Various studies have been performed on the development of micelles,6–10 polymeric nanoparticles,11–13 and dendrimers14,15 as imaging agents. The larger size of these macromolecular structures prevents their rapid clearance from the body through the renal system. The size and surface properties of nanoparticles can be modified to optimize their pharmacokinetics for imaging a specific disease. Increased imaging specificity would significantly improve accuracy in diagnosis, allowing better disease management planning and an improved prognosis. Additionally, nanoparticles have been demonstrated to improve the stability of encapsulated or attached probes.16–19
Hydrogel nanoparticles (nanogels) are hydrophilic nanosized polymeric networks that are held together in three dimensions through physical or chemical crosslinking.20–23 Nanogels have unique advantages over other nanoparticle imaging systems. They are highly biocompatible due to their high water content and consequent low interfacial tension with biological fluids, resulting in physical properties that resemble those of living tissues.21–26 Their size, ranging from 20 to 200 nm, can be controlled systematically through optimization of formulation parameters.27 In addition, nanogels can be designed to respond to environmental changes, such as temperature, pH, magnetic field, and ionic strength, with changes in their physiochemical properties, such as volume, water content, refractive index, interior network permeability, and hydrophilicity.28 This stimuli responsiveness can be exploited to design disease-responsive imaging nanogels. Nanogels also exhibit high loading efficiencies of both hydrophobic and hydrophilic molecules, including proteins, nucleic acids and quantum dots, and even act as chaperones to protect fragile molecules against degradation.29–31 Last but not least, release kinetics of nanogels can also be regulated by varying crosslinking density or incorporating stimulus-responsive crosslinkers. These properties make nanogels a promising platform for theranostic agents. Finally, nanogels possess high colloidal stability, essential for in vivo imaging/delivery agents.
Despite these advantages and the versatility of nanogels, there are limited reports of nanogel imaging agents. Two reviews on this topic have been published; however, they mainly focus on nanogels as drug carriers.27,32 Here, we provide an updated overview of nanogel imaging agents for various modalities spanning the electromagnetic (EM) spectrum (Fig. 1). Nanogel imaging agents will be categorized according to the wavelength of the imaging source: radiowave (MRI), near infrared (NIR), visible light, ultraviolet (UV), and gamma ray (PET) radiation (Fig. 1). Nanogel agents with imaging modalities other than the EM waves, such as ultrasound, have been reported but they are not discussed in this review.33 We will also provide an overview of the commonly used formulation methods, and discuss recent works on multimodal nanogel imaging agents, exploring how various materials and formulation methods can be employed to formulate nanogels for either in vitro or in vivo applications (Tables 1 to 4).
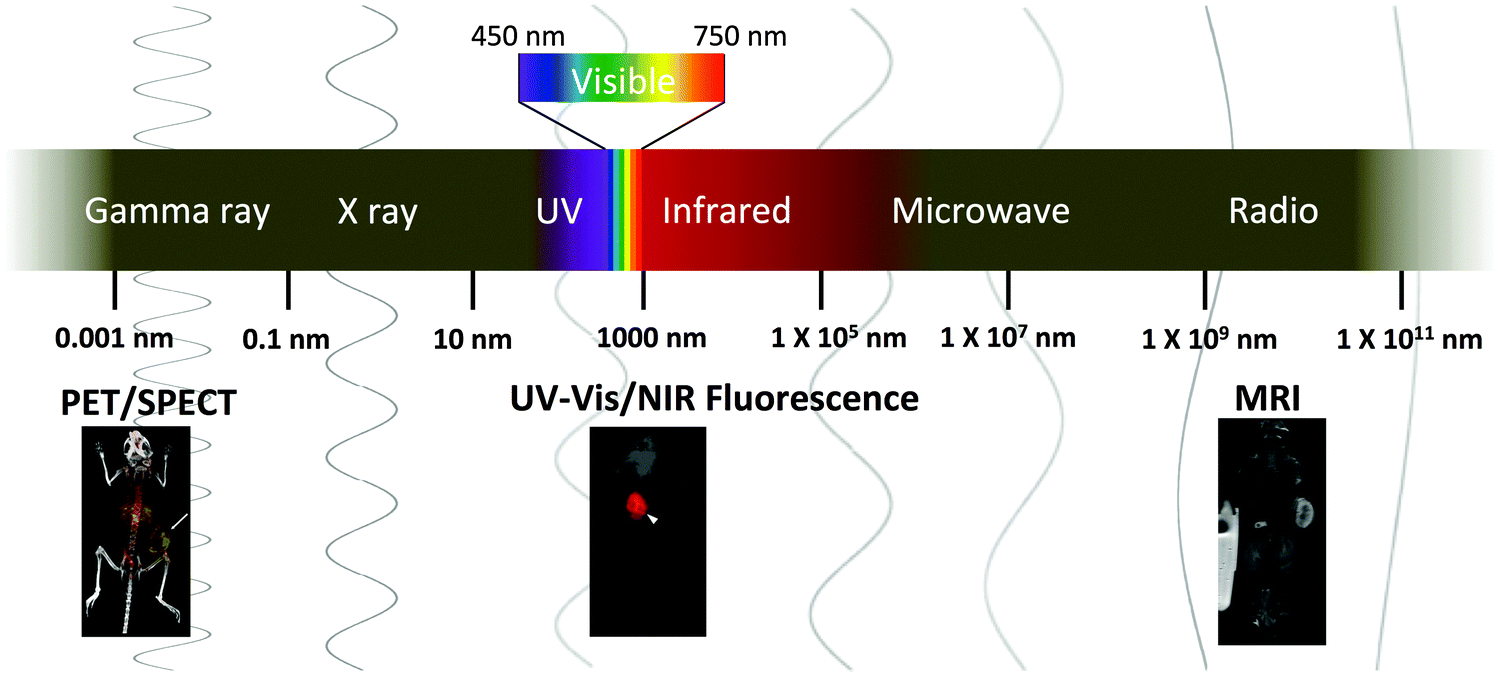 | ||
| Fig. 1 Imaging modalities with sources of radiation/signal spanning the electromagnetic spectrum. | ||
| Imaging modality | Dye/contrast agent | Means of incorporation | Polymer components | Responsive size change | Responsive imaging property | Formulation methoda | Size (nm) | Targeting group | Cells/in vivo work | Therapeutics | Ref. |
|---|---|---|---|---|---|---|---|---|---|---|---|
| a Formulation methods: refer to Fig. 1. | |||||||||||
| (a) | |||||||||||
| MRI (Gd-based) | Gd-DOTA | As chemical crosslinkers | PAA chemically crosslinked by DTPA/DOTA crosslinkers | N/A | N/A | A | 54–85 | N/A | N/A | N/A | 20 |
| MRI (Gd-based) | Gd-DTPA | Conjugation | Poly(PEGMA)-crosslink by ethylene glycol bisacrylamide | N/A | N/A | B | 10 | N/A | Imaging of blood vessel in mice | N/A | 54 |
| MRI (Gd-based) | Gd-DTPA | Electrostatic | PAA + chitosan | pH (zeta potential) | N/A | A + D | 220 | N/A | Imaging of rabbit brain and liver | N/A | 56 |
| MRI (Gd-based) + NIR fluorescence |
Gd3+
Cy 5.5 |
As physical crosslinkers (electrostatic)
Conjugation |
PEI | N/A | N/A | B | 159 ± 62 | N/A | Imaging of SCC7 tumor of mice | N/A | 55 |
| MRI (Fe3O4) | Fe3O4 | Encapsulation | Dextrin-VA-SC16 | N/A | N/A | C | 100 | N/A | Uptake in Murine bone marrow-derived macrophages | N/A | 61 |
| MRI (Fe3O4) | Fe3O4 | As core from which polymer matrix was built on | Poly(PEGMA) crosslinked with MBA | N/A | N/A | A + E | 68 | N/A | N/A | Doxorubicin | 63 and 64 |
| MRI (Fe3O4) | Fe3O4 | As core from which polymer matrix was built on | Poly(AEM·HCl) crosslinked with MBA | N/A | N/A | A + E | 19 | N/A | Healthy mice | N/A | 65 |
| MRI (Fe3O4) | Fe3O4 | Encapsulation | Poly(NIPAAm-co-AA) crosslink by MBA | Temperature and pH | N/A | A + E | 237 (pH 2.7) to 387 (7.6) 25 °Ca | N/A | Cytotoxicity on HeLa cells | Doxorubicin | 62 |
| MRI (Fe3O4) | Fe3O4 | Encapsulation | Self-assembly of poly(AAc-co-DSA); further coated by (poly(γ-GA-co-γ-GAOSu)-g-PEG-FA) | pH and magnetic hyperthermia | N/A | C + D | 243 (pH 4.7) to 221 (pH 7.4) 20 °Ca | Folate acid | Therapeutic efficacy on tumor-bearing mice | Doxorubicin | 77 |
| (b) | |||||||||||
| MRI (Fe3O4) + UV fluorescence |
Fe3O4
fluorescein |
Encapsulation
Conjugation |
PBMA-g-(C12/fluorescein) | N/A | N/A | C | 131–250 | N/A | N/A | N/A | 60 |
| MRI (Fe3O4) + UV fluorescence |
Fe3O4
fluorescein |
Encapsulation
Conjugation |
pCBMA with disulfide crosslinks | Disulfide: reducing environment | N/A | A + E | 110 | RGD | Uptake in Macrophage cell | Fluorescently labeled dextran (model) | 75 |
| MRI (Fe3O4) + NIR fluorescence |
Fe3O4
Cy5.5 |
Encapsulation
Conjugation |
Poly(NIPAAm-co-AA) | pH and/temperature | N/A | A + E | 100 (pH 6.8) to 85 (pH 7.4) at 37 °Ca | Lactoferrin | Imaging of C6 glioma in rats | N/A | 66 |
| MRI (Fe3O4) + UV fluorescence |
Fe3O4
Dil |
Encapsulation
Encapsulation |
Self-assembled poly(NIPAAm-co-AA) coated with PEI through electrostatic interaction | Temperature | N/A | C + D | 200 | N/A | Imaging of mice injected with nanogels-loaded hMSC | EPFG expression plasmid | 68 |
| MRI (Fe3O4) + UV fluorescence | MnFe2O4 fluorescent Schiff base bond (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N) and double bond (CC) N) and double bond (CC) |
As core from which polymer graft on
Part of polymer matrix |
Self-assembled PLL crosslinked by poly-GA | N/A | N/A | B + E | <200 | N/A | Imaging of mice injected with nanogels-loaded DC | N/A | 69 |
| MRI (19F) | 19F-TFEMA | Part of polymer matrix | Poly-(DEAMA-co-TFEMA-EGDMA-PEG) crosslinker with ethylene glycol | pH | Signal is turned “On” as pH decrease | A | 63 (pH 7.4) and 90 (pH 6.5) | N/A | N/A | N/A | 72 and 73 |
| Imaging modality | Dye/contrast agent | Means of incorporation | Polymer components | Responsive size change | Responsive imaging property | Formulation methoda | Size (nm) | Targeting group | Cells/in vivo work | Therapeutics | Ref. |
|---|---|---|---|---|---|---|---|---|---|---|---|
| a Formulation methods: refer to Fig. 1. | |||||||||||
| PET | 99mTc labeled 1-hexylcarbamoyl-5-fluorouracil (HCFU) | Encapsulation | Poly-(NIPAAM-co-VP) crosslinked by MBA; coated with h polysorbate 80 | N/A | N/A | A | 50 | N/A | Biodistribution imaging of rabbits | HCFU | 91 |
| PET + UV fluorescence |
68Ga-NODAGA
Alexa Fluor 488 |
Conjugation
Conjugation |
Star-shaped PEG crosslinked by disulfide bond | Reducing environment | N/A | B | 290 | N/A | Uptake in monocytes | N/A | 92 |
| PET | 64Cu-NOTA | As crosslinker | PAA chemically crosslinked with NOTA crosslinkers | N/A | N/A | A | 63 | N/A | Imaging of 4T1 tumor and metastasis of mice | N/A | 93 |
| PET | [18F]-labelled BoHc/A | Conjugation to encapsulated drugs | Amino group- and cholesteryl-group-bearing pullulan (cCHP) | N/A | N/A | C | 40 | N/A | Imaging delivery to nasal mucosa of mice | BoHc/A | 84 |
| PET + visible light |
68Ga-DOTA
NaYF4:Yb/Er/Tm UCNP |
Conjugation
As core from wit polymer was build on |
PEI (coated on UCNP), conjugated with PEG | N/A | N/A | E | 136 | RGD | Imaging of M21 tumor bearing mice | N/A | 94 |
| PET + NIR |
64
Cu-DOTA
Cy5.5 |
Conjugation
Conjugation |
5β-Cholanic acid and azide groups modified glycol chitosan | N/A | NIR turned ON upon cleavage of MMP-sensitive peptide | C | ∼300 | MMP-sensitive peptide | Imaging of A549 tumor in mice | N/A | 95 |
| PET + NIR |
124I-labeled chlorophyll-a
cyanine dye |
Encapsulation
Conjugation |
PAA | N/A | — | A | 18–25 | N/A | Imaging of Colon26 tumors in mice | Chlorophyll-a (PDT) | 96 |
| Imaging modality | Dye/contrast agent | Means of incorporation | Polymer components | Responsive size change | Responsive imaging property | Formulation methoda | Size (nm) | Targeting group | Cells/in vivo work | Therapeutics | Ref. |
|---|---|---|---|---|---|---|---|---|---|---|---|
| a Formulation methods: refer to Fig. 1. | |||||||||||
| UV-vis |
Fe3O4
QD |
As core from which polymer was built on | Chitosan | pH | N/A | E | 160 | N/A | Uptake in L02 cells | Insulin | 67 |
| UV-vis | QD | Electrostatic interaction and chemical crosslinks with polymer matrix | CM-dextran and PLL | N/A | N/A | D + E | 190 | N/A | N/A | N/A | 100 |
| UV-vis | QD | Electrostatic interaction between HA and QD | HA | N/A | N/A | E | 50–120 | HA | Imaging of lymphatic vessels on ears of mice | N/A | 99 |
| UV-vis | QD | In situ synthesis inside the nanogels | Chitosan and PMAA crosslinked with MBA | pH | Photoluminescence (PL) increase as pH decreases | A + D | 85–175 | N/A | Uptake and cytoxicity on mouse melanoma B16F10 cells | Temozolomide | 105 |
| UV-vis | QD | Electrostatic interaction | Cholesterol-bearing amino-group-modified Pullulan | N/A | N/A | C + E | 38 | N/A | Uptake in HeLa cells | N/A | 102 |
| UV-vis | QD | Electrostatic interaction between lysosome, QD and polymer | Carboxymethyl cellulose | N/A | PL decreases as pH decreases | D + E | 285 | — | Uptake and cytotoxicity on HepG2 & MCF-7 cells | Methotrexate | 101 |
| NIR | QD | In situ synthesis inside the nanogels | HPC and PAA crosslinked wit MBA | pH and temperature | PL increases as pH decreases | A + D | 32–50 (pH 4.5) to 75–83 (pH 7.4) | N/A | Cytotoxicity on B16F10 cells | Temozolomide | 104 |
| UV-vis | QD | As core from which polymer was built on | His-tagged polypeptides | pH, temperature and competitors | N/A | E | 40.2 | RGD | Uptake and cytotoxicity on HeLa cells | Dye ABDPSP & fluorescein sodium (model drugs) | 103 |
| UV-vis | Carbon shell and magnetic core nanoparticles | As core from which polymer was built on | Poly(NIPAM-AAm) crosslinked with MBA | NIR/magnetic induced thermal responsive | PL increases as temperature increase | A + E | 320 (at 24 °C) | — | Cytotoxicity of B16F10 cells | Curcumin | 106 |
| UV-vis | Ag NP | As core from which polymer was built on | Poly(NIPAM-AA) crosslinked with MBA | pH | Blue shift and increase in absorption intensity as pH decreases | A + E | ∼77 (pH 5.0) to ∼137 (pH 7.4) at 37 °C | — | Uptake and cytotoxicity on B16F10 cells | Dipyridamole | 114 |
| UV-vis | Au NP | As core from which polymer was built on | P(NIPAAM-co-AAm) crosslinked with MBA | Visible light induced thermal responsive | N/A | A + E | >80 (37 °C) and ∼53 (25 °C) | N/A | Uptake and cytotoxicity of HeLa cells | 5-Fluorouracil | 110 |
| UV-vis | Au–Ag NP | As core from which polymer was built on | PEG crosslinked with PEGMA | NIR induced thermal responsive | PL increases as temperature increases | A + E | 18–43 (37 °C) | HA | Uptake and cytotoxicity on B16F10 cells | Temozolomide | 111 |
| UV-vis | Au–Ag NP | As core from which polymer was built on | PS crosslinked with DVB and PEG crosslinked with PEGMA | NIR induced thermal responsive | N/A | A + E | 22–37 (37 °C) | N/A | Uptake and cytotoxicity on B16F10 cells | Curcumin | 112 |
| UV-vis | Au NP | In situ synthesis inside the nanogel | Lysosome–dextran | N/A | N/A | C | 190 | N/A | Uptake and cytotoxicity on KB cells | Doxorubicin | 114 |
| UV-vis | Au NP | As core from which polymer was built on | Chitosan and PAA crosslinked with glutaldehyde | N/A | N/A | D + A | ∼120 | N/A | Uptake and cytotoxicity on HepG2 cells | N/A | 113 |
| UV-vis | Au NP | In situ synthesis inside the nanogel | Chitosan | N/A | N/A | B | 80–230 | N/A | — | N/A | 115 |
| Imaging modality | Dye/contrast agent | Means of incorporation | Major polymer backbone | Responsive size change/degradability | Responsive imaging property | Formulation methoda | Size (nm) | Targeting group | Cells/in vivo work | Therapeutics | Ref. |
|---|---|---|---|---|---|---|---|---|---|---|---|
| a Formulation methods: refer to Fig. 1. | |||||||||||
| UV-vis | 5-Aminofluorescein (5-AF), 7-amino-4-methyl coumarin (AMC), or QDs | Conjugation | Chitosan and PAA crosslinked with MBA | N/A | N/A | A + D | 180 | N/A | In vivo lymphatic node mapping of Balb/c mice | Doxorubicin, or Vascular endothelial growth factor C | 117 |
| UV-vis | Fluorescein isothiocyanate | Conjugation | Poly(amidoamine) dendrimers and alginate crosslinked with Ca2+ | pH | N/A | E + B | 433 | N/A | Uptake and cytotoxicity on CAL-72 cells | Doxorubicin | 118 |
| UV-vis | 8-Hydroxypyrene-1-carbaldehyde (HPC) | Encapsulation | Polyurethane | — | Change in emission wavelength as a function of pH | B | 92 | N/A | Uptake in NIH/3T3 fibroblast cells | N/A | 19 |
| UV-vis/NIR | DMDP-M | Encapsulation | Crosslinked Pluronic F127 | — | Fluorescence turned “on” in presence of thiols | B | 45 | N/A | Uptake in NIH/3T3 fibroblasts | N/A | 123 |
| UV-vis | Self-fluorescent abietane | As part of polymer matrix | Poly(abietane methacrylate) crosslinked with PEGDA | N/A | N/A | A | 90–135 | Folic acid | Uptake in MCF-7 cells | Doxorubicin | 120 |
| UV-vis | Self-fluorescent ALC linker | Electrostatic encapsulation | Branched PEI crosslinked by aldehyde-L-cystine (ALC) linker | Thiols | Decrease in pH and presence of reducing agents lowers the fluorescence | B | 200 | N/A | Transfection in multiple cell lines | pDNA | 121 |
| UV-vis | Rhodamine B | As part of polymer matrix | P(NIPAM-co-RhBUA) crosslinked with MBA | Temperature | Increased in fluorescence in presence of Cr3+ and increased temperature | A | 117 (20 °C) and 50 (40 °C) | N/A | N/A | N/A | 122 |
| NIR fluorescence | Cyanine dye | Conjugation | Azide-functionalized PEG methyl ether crosslinked with L-cystine-N-carboxy anhydride | Reducing environment | N/A | B | 210 | N/A | N/A | Doxorubicin | 127 |
| NIR-797 isothiocyanate | Conjugation | Methacrylated carboxymethyl cellulose crosslinked with disulfide linkage-containing crosslinkers | Reducing environment | N/A | B | 192 | N/A | NIR fluorescence imaging of H22 tumor-bearing mice | Doxorubicin | 126 | |
| IRDye800 | Conjugation | Amine-cholesteryl-group-bearing pullulan | N/A | N/A | C | 30 | N/A | SLN Mapping in mice and pigs | N/A | 130 and 131 | |
| ICG | Conjugation | ICG–HA conjugate | HAdase | Fluorescence turn “On” in presence of HAdase | C | 189 | HA | Tumor and SLN imaging of MDA-MB-231 tumor bearing mice | N/A | 134 | |
| ICG | Conjugation | Self-assembled ICG–HA conjugate with surface further crosslinked with disulfide linkage-containing crosslinkers | HAdase | Fluorescence turn “On” in presence of HAdase | C + B | 293 | N/A | Imaging of front paw of mice that are injected with nanogels pre-incubated with HAdase | N/A | 135 | |
| ICG | Conjugation | HA + poly(beta-amino)ester (PBAE) + ICG | pH | Fluorescence turn “On” in presence of HAdase | D | 73 (pH 7.4) and 15 (pH 5.5) | HA | Uptake in MDA-MB-231 breast cancer cells | N/A | 136 | |
| Ga-porphyrin | As crosslinkers | α,ω-Diazide/hydroxyl PEG and α,ω-difolate/azidePEG crosslinked by Ga-TPPPP | N/A | — | B | 110 | Folate | — | N/A | 137 | |
2. Nanogels formulation methods
Nanogels can be formulated using various materials and methods (Fig. 2). One of the most common is radical polymerization in an inverse emulsion25,26,34,35 (Fig. 2A), in which monomers, crosslinkers, and catalysts dispersed in aqueous droplets are stabilized in a continuous organic phase by surfactants; droplets become nanogels upon polymerization. Examples of commonly used monomers include acrylamide (AAm), pH-sensitive acrylic acids (AA) and thermoresponsive N-isopropylacrylamide (NIPAM). Inverse emulsion polymerization employs commercially available monomers and crosslinkers, can generate smart constructs if stimulus-responsive monomers and crosslinkers are incorporated, and allows control over mesh size by varying the crosslinking density. However, it requires surfactants that are difficult to completely remove and provides limited control over uniformity of nanogel size, often resulting in polydisperse particles.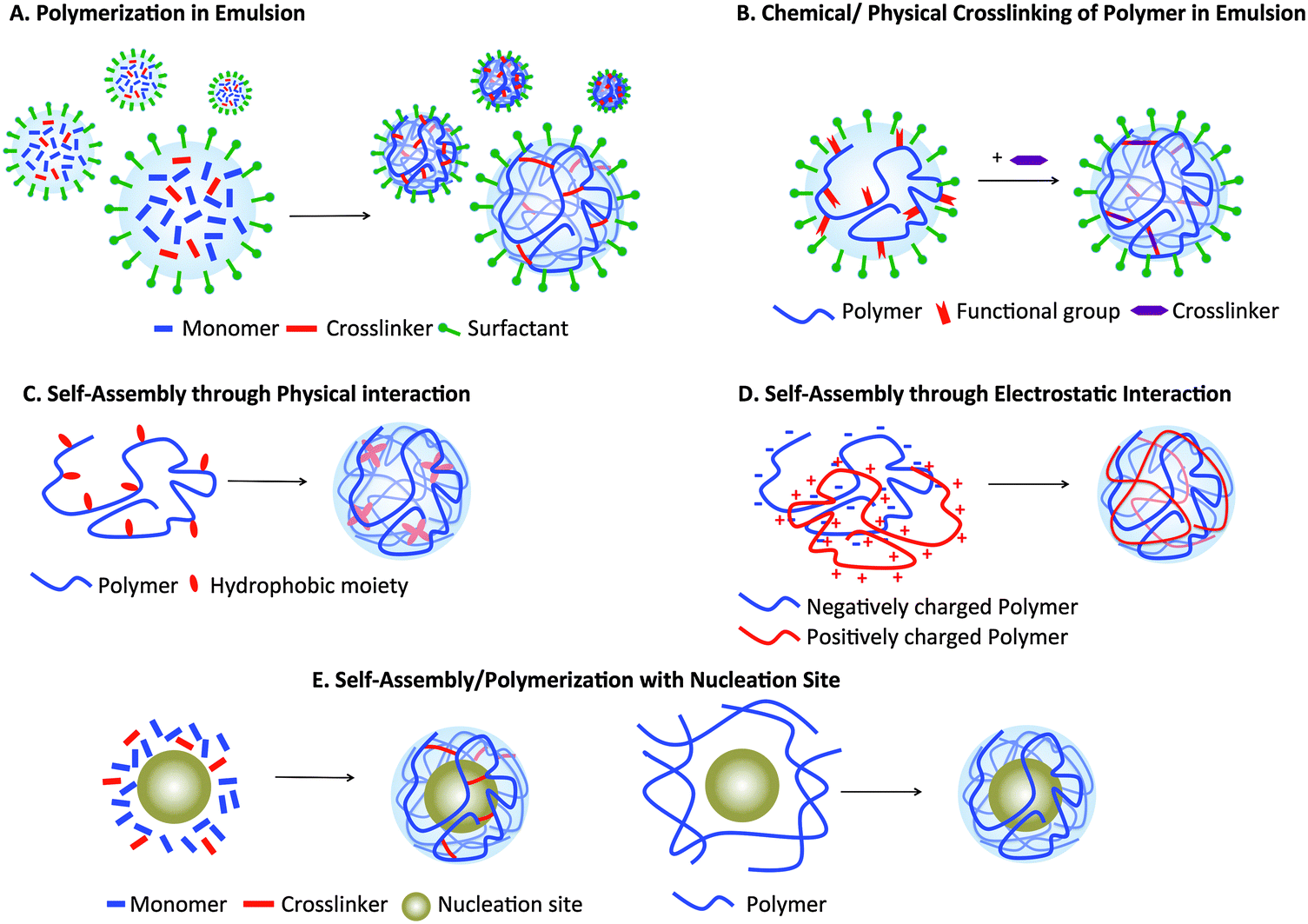 | ||
| Fig. 2 Nanogel formulation strategies. (A) Hydrophilic monomers and crosslinkers in a water-in-oil emulsion, stabilized by surfactants. Upon the addition of a catalyst, polymerization occurs within the emulsion droplets, forming nanogels. (B) Hydrophilic polymer modified with functional groups that allow physical/chemical crosslinking to form nanogels. (C) Polymer modified with hydrophobic moieties for self-assembly into nanogels. (D) Positively and negatively charged polymer self-assembly through electrostatic interaction. (E) Polymerization of monomers and crosslinkers shell or self-assembly of polymer modified with hydrophobic moieties in presence of nucleation sites. | ||
Nanogels can also be formulated through crosslinking (either physical or chemical) of hydrophilic polymers that are modified with functional groups, such as thiols and acrylates35–37 (Fig. 2B), while amphiphilic polymer networks, such as cholesterol-modified pullulan, can self-assemble into nanogels (Fig. 2C). Although both methods require chemical modification of polymers, they allow formulation of nanogels using natural polymers, such as non-immunogenic polysaccharides (chitosan, hyaluronan, and dextran). The latter method does not require a surfactant, permitting a robust and facile formulation. However, since physical crosslinks are not stable, further chemical crosslinks are necessary for their in vivo application. Fig. 2D illustrates another formulation technique using a combination of oppositely charged polymer chains held together by electrostatic interactions. This approach does not require polymer modification, unlike the previous two methods, adding convenience and versatility. However, nanogels formed using this method are held together solely through physical interaction and so are not stable. Finally, nanogels can also be formed in the presence of nucleation sites, often inorganic nanoparticles such as iron oxide nanoparticles or quantum dots, on which the polymers or monomers are adsorbed and polymerization occurs (Fig. 2E). Nucleation sites work as a “template” on which nanogels are built, therefore generating nanogels of higher monodispersity. More details on each method of nanogel preparation have been published in other reviews.23,38,39
3. Magnetic resonance imaging (MRI)
MRI is one of the most frequently used clinical imaging techniques, employing a relatively low energy EM wave with wavelength ∼2.5 m (3 T) to 5 m (1.5 T). In addition to low ionizing radiation exposure, it also offers high anatomical resolution and great soft tissue contrast.40,41 Despite these advantages, MRI is limited by its low inherent contrast, a problem that can be overcome by contrast agents. Current clinically used MRI contrast agents can be divided into two categories: T1 and T2 contrast agents.42,43T1 contrast agents are usually gadolinium (Gd) chelates, while T2 contrast agents are mainly iron oxide particles, including superparamagnetic iron oxide nanoparticle (SPIONs), ultra-small superparamagnetic iron oxide nanoparticles (uSPIONs) and very small superparamagnetic iron oxide nanoparticles (vSPIONs).
T
1 agents enhance contrast by reducing the longitudinal relaxation time of the surrounding endogenous water, thus increasing the signal intensity and providing positive contrast. Conversely, T2 contrast agents provide negative contrast as they enhance water's transverse relaxation.44 The ability of an MRI contrast agent to provide contrast enhancement is characterized by its relaxivity, the measure of the change in the relaxation property of water per concentration of the contrast agent:  , where r1 corresponds to the longitudinal relaxivity, r2 corresponds to the transverse relaxivity, and [ion] corresponds to the concentration of the ions of the contrast agent.44
, where r1 corresponds to the longitudinal relaxivity, r2 corresponds to the transverse relaxivity, and [ion] corresponds to the concentration of the ions of the contrast agent.44
3.1 T 1 agents
Most current clinical T1 contrast agents are small molecules, limiting relaxivity at the clinically relevant magnetic field of an MRI scanner (approximately 3 to 6 mM−1 s−1 at 1.5 T) due to their fast tumbling frequency.42,45–47 Thus, high-molecular-weight contrast agents,45,47–49 produced by encapsulation or conjugation of Gd-chelates with polymeric nanoparticles, dendrimers, micelles or polymers, that have higher relaxivities would be useful.50–53 Another advantage of nanogels for MRI is their high water content, allowing Gd ions to “relax” more water molecules in a given period than would be possible in more hydrophobic assemblies.Currently, most strategies for the formulation of nanoparticles with Gd involve conjugation of Gd chelates to particles (Table 1a). Soleimani et al. conjugated Gd chelates on the surface of poly(ethylene glycol) methyl ether methacrylate nanogels crosslinked by ethylene glycol dimethacrylate.54 The nanogels, with a relaxivity of 17.5 mM−1 s−1 at 1.5 T, enhanced the overall signal intensity (blood vessels) in tumor-bearing mice substantially more than Magnevist at 20 minutes after injection. Yet, no tumor-specific signal enhancement or longer term imaging was shown to demonstrate the benefit of nanogels over small molecule contrast agents.
Without chelation, Lim et al. described the formulation of a nanogel MRI contrast agent through hybridization of poly(ethyleneimine) (PEI) with Gd3+ ions as the crosslinker.55 This design was intended to yield nanogels with increased elastic deformability to circumvent reticuloendothelial system (RES) sequestration and thus enhance tumor uptake. The nanogel surface was also modified with cyanine5.5 (Cy5.5) for NIR imaging. The 165 nm nanogels accumulated in SCC7 tumors in mice at a higher concentration than in the liver 12 h after injection. Unlike most other Gd-chelating particles, these nanogels exhibited a more significant transverse relaxivity (∼82.6 mM−1 s−1) than longitudinal relaxivity (2.1 mM−1 s−1), making them a better T2 contrast agent. T2-weighed MR imaging revealed negative contrast enhancement in the tumor 2 h after injection. Alternatively, Ahmed et al. recently published a nanogel system with chitosan and PAA as constituents.56 Negatively charged Gd–DTPA was adsorbed to the nanogels through electrostatic interaction with the positively charged chitosan. As the nanogels in these two papers carry Gd chelates or ions through physical interaction only, long-term Gd chelating stability, a critical safety concern for all clinical MRI contrast agents, may be an issue.
To address the concern of Gd-chelating instability, our group described a different formulation of a nanogel MRI contrast agent.20 Chan and Lux et al. synthesized three Gd-chelate crosslinkers that both held together polyacrylamide (PAAm) nanogels and stably retained Gd3+ ions. Incorporation of the Gd-chelating crosslinker into nanogels enhanced relaxivity by 4 to 6 times (18 mM−1 s−1 at 1.5 T) and essentially prevented transmetallation by Zn2+, the ions most likely to displace Gd3+in vivo.
3.2 T 2 agents
Most superparamagnetic contrast agents incorporate water-insoluble iron oxide crystals consisting of magnetite (Fe3O4) or maghemite (γ-Fe2O3) with a core diameter in the range of 4 to 180 nm.57–59 Iron oxide particles are of considerable interest as contrast agents because of their low toxicity. Due to their hydrophobicity, they are often encapsulated in nanoparticles to enhance their solubility in aqueous solution. In addition, the clustering of iron oxide particles inside nanoparticles allows them to work synergistically in enhancing T2 relaxation of water, leading to a higher relaxivity.Multiple studies have employed similar strategy in formulating Fe3O4-encapsulated nanogels by utilizing Fe3O4 as a core to build the hydrogel layer (Table 1a and b). Amphiphilic polymers, such as poly(butyl methacrylate) (PBMA) grafted with 1-dodecylamine (C12)60 and hydrophobized dextrin (a carbohydrate polymer),61 can self-assemble into nanogels encapsulating Fe3O4 nanoparticles in the hydrophobic core. This strategy is convenient, as Fe3O4 templates the one-step assembly of nanoparticles through hydrophobic–hydrophobic interactions between Fe3O4 and the hydrophobic moieties of the polymer. Alternatively, emulsion polymerization of monomers and crosslinkers, which adsorb onto Fe3O4 through inverse emulsion, has also been used to formulate nanogels.62 For example, Sun et al. and Liu et al. employed a commonly used stealth material, PEG methacrylate, and N,N′-methylenebisacrylamide (MBA, also called bisacrylamide in this review) to form their Fe3O4-carrying nanogel system.63,64 Likewise, Gong et al. encapsulated Fe3O4 in amine-containing nanogels prepared by photopolymerizing N-(2-aminoethyl) methacrylamide hydrochloride monomers.65 In this method, Fe3O4 assists the formation of a nanogel shell: the high surface area of Fe3O4 allows adsorption of monomers and crosslinkers at high concentrations, while strong UV absorption of Fe3O4 initiates the formation of radicals for the photopolymerization. All of these Fe3O4-encapsulating nanogels ranged in size from 68 nm to 250 nm, with T2 relaxivities ranging from 100 to 440 mM−1 s−1. As shown with these examples, encapsulation of Fe3O4 inside nanogels offers a simple way to formulate T2 contrast agents with the flexibility of using various materials as coating. However, the clinical potential of a T2 agent is limited. They offer negative contrast, darkening areas in which they accumulate; this makes interpretation and identification of lesions more difficult than with a T1 agent. In addition, Fe3O4, such as Feridex® or Resovist®, are readily taken up by reticuloendothelial cells in the liver and spleen. This limits their use to only liver lesion imaging. Expansion of their applications would require substantial materials engineering and innovative designs.
3.3 Dual MR and fluorescence imaging agents
While MRI provides excellent soft tissue contrast, it suffers from low sensitivity. The combination of MRI with other imaging modalities, such as PET or fluorescence, which have higher sensitivities, can compensate for this disadvantage of MRI and provide better information about disease location and progression and therapeutic efficacy. Various groups have reported nanogels with dual MR and fluorescence imaging properties, often by encapsulating Fe3O4 into nanogels and conjugating (or encapsulating) fluorescent dyes/quantum dots.67Jiang et al. formulated a pH- and thermo-sensitive nanogel as a dual MRI and fluorescence agent for medical imaging of brain glioma (Fig. 3).66 Fe3O4 was encapsulated in p(NIPAM-co-AA) (PNA), and the resulting nanogels were further conjugated with Cy5.5-labeled lactoferrin for fluorescence and tumor targeting. The pH sensitivity of the polymer translated to a lowered lower critical solution temperature (LCST) at slightly acidic pH (6.4), which caused nanogels to shrink at physiological temperature, enhancing uptake into tumor cells. MRI imaging revealed that uptake of lactoferrin-labeled nanogels into brain glioma of rats 48 h after injection was significantly higher than that of unlabeled nanogels. Park et al. utilized the same polymer to formulate a theranostic nanogel for both MRI and fluorescence imaging to visualize gene delivery.68 p(NIPAM-co-AA) self-assembled into nanogels around the amine-functionalized Fe3O4. Instead of chemically conjugating a dye, Dil was encapsulated. The nanogels were further coated with polyethyleneimine (PEI), which carries positively charged amines, for complexation with negatively charged DNA for gene delivery. Nanogels complexed with green fluorescent protein (GFP) plasmid DNA were internalized in human mesenchymal stem cells (hMSCs), leading to GFP expression. The transplantation of these hMSCs into mice was then monitored using MRI and fluorescence. Of these two studies, only the second demonstrated imaging by both modalities in vivo, and neither compare the data obtained from the two modalities. Thus, they do not fully demonstrate how MRI and fluorescence complement each other in providing morphological and functional information.
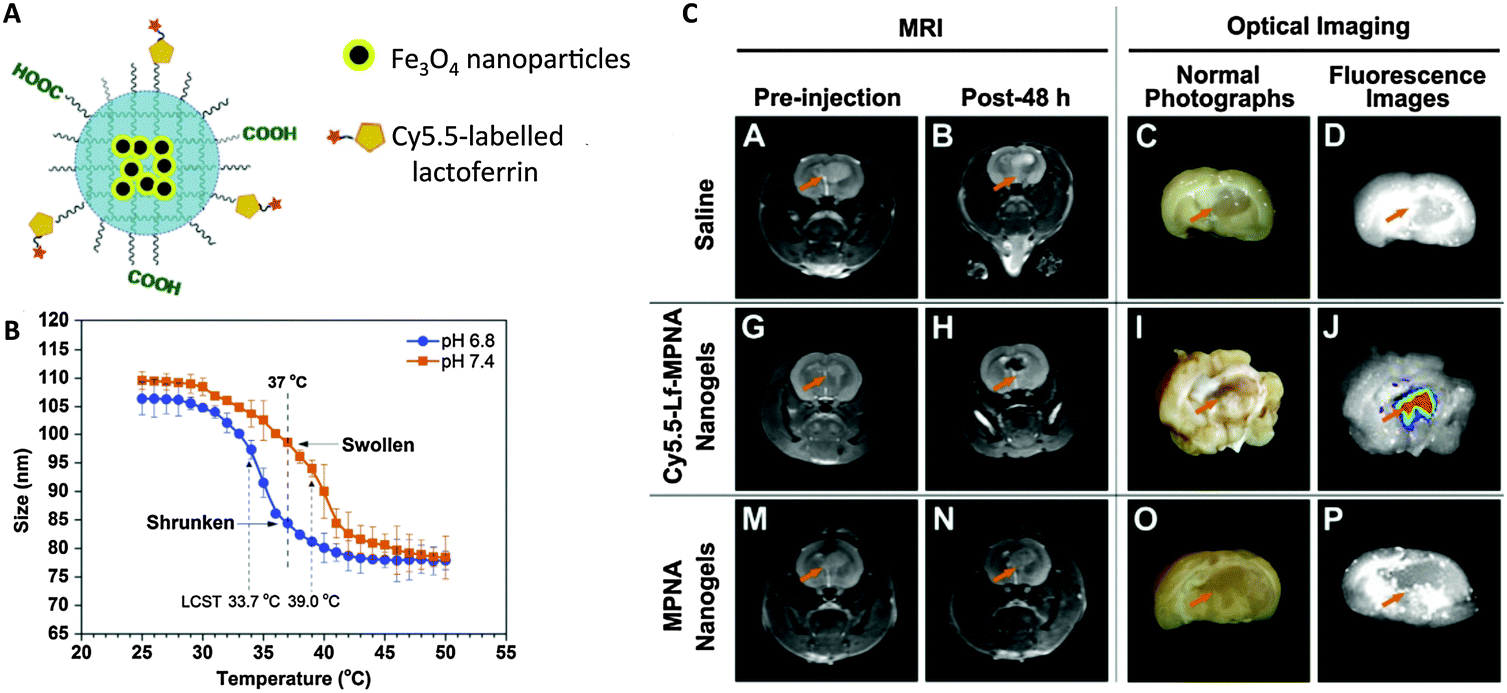 | ||
| Fig. 3 pH- and temperature-sensitive nanogels as dual T2 and optical imaging agents. (A) Cy5.5-Lf-MPNA: P(NIPAM-co-AA) nanogels encapsulating Fe3O4 nanoparticles and conjugated with Cy5.5-labeled lactoferrin as a glioma-targeting ligand. (B) Thermo- and pH-responsive change in nanogel size (due to change in hydrophobicity). (C) In vivo MR and ex vivo NIR fluorescence imaging showed higher uptake of Cy5.5-Lf-MPNA than MPNA (without Cy5.5-labeled lactoferrin) in rat glioma.66 | ||
Kim et al. employed a different approach for the development of dual MRI and fluorescent nanogels. Instead of encapsulating or conjugating a dye, these researchers used an autofluorescent nanogel matrix for tracking dendritic cell therapy.69 Positively charged poly(L-lysine) (PLL) was added to negatively charged poly(γ-glutamic acid) (γ-PGA)-coated manganese/iron oxide nanoparticles (MnFe2O4) to form ionic nanogels; PLL amines were crosslinked by the addition of glutaraldehyde, which was responsible for its fluorescence at around 553 nm. Incubation of dendritic cells with the nanogels allowed MR and fluorescence visualization of their accumulation in lymph nodes for dendritic cell therapy. It should be noted that fluorescence is very sensitive, making it well suited to complement the low sensitivity of MR imaging. However, its limited penetration depth in the UV to visible range may not be effective in imaging of tumors/lymph nodes deeper than a few millimeters inside the body.70,71 The systems covered in this section are good proofs-of-concept, showcasing the versatility of modifying nanogels as bimodal agents, yet likely have limited clinical utility.
3.4 Fluorine-19 (19F) MRI probes
In addition to Gd and iron oxide-based contrast agents, 19F-based agents have recently gained increasing attention. As the abundance of water in the body leads to a high background signal in 1H MRI, magnetic species that are less abundant allow a higher signal-to-noise ratio. One such species is 19F, whose signal has a similar sensitivity to that of 1H, and is relatively safe. Most 19F agents developed so far have been perfluorocarbon-based polymeric nanoparticles or micelles.Oishi et al. translated this concept to nanogels by formulating a pH-activated 19F MRI nanogel system through co-polymerization of 19F-bearing 2,2,2-trifluoroethyl methacrylate (TFEMA), pH-sensitive 2-(N,N-diethylamino)ethyl methacrylate (DEAMA), and ethylene glycol dimethacrylate crosslinkers.72,73 At pH 7.4, nanogels were hydrophobic and shrunken due to deprotonation of amines, reducing molecular motion of 19F compounds in the hydrophobic gel core and thus broadening signal (off state). The signal was turned on at pH 6.5 as amines were protonated and nanogels became hydrophilic and swollen. Though 19F MR imaging was done only in vitro, the elegant design of the nanogels and high pH sensitivity of their system provides new insight into development of 19F-containing nanogels as tumor-responsive imaging agents.
3.5 MR theranostic imaging agents
Considerable effort has been made toward the development of MRI theranostic agents, which have both MR imaging and drug delivery capabilities, using micelles or polymeric nanoparticles.74 Characteristic properties of diseases, for example, low extracellular pH, hypoxia, and the reducing environment of tumors, can be exploited in the design of disease-specific theranostic agents. However, reports of nanogel MRI theranostic systems are still scarce.As a proof of concept Zhang et al. formulated a reduction-sensitive nanogel, with a disulfide-containing crosslinker, encapsulating Fe3O4 using non-fouling carboxybetaine methacrylate (CBMA) as a monomer.75 Dithiothreitol (DTT)-induced degradation of nanogels resulted in the release of model drugs (fluorescein isothiocyanate (FITC)-labeled dextran) and a reduction in relaxivity which translated to higher MR signal intensity. Since DTT is a harsh reducing agent, glutathione (GSH), an abundant thiol reducing agent that maintains the redox state in cells, would have allowed a more biologically relevant proof of principle; this species is upregulated in many diseases, including tumors.76 while the study shows the potential of nanogels as reduction-responsive theranostics, high sensitivity to the low GSH concentrations found in vivo (millimolar range) remains a major challenge.
Chiang et al., on the other hand, utilized another property of tumors, low extracellular pH, to develop a tumor-specific theranostic system. Fe3O4 and Dox were encapsulated into self-assembled pH-sensitive poly(acrylic acid-co-distearin acrylate) polymersomes, stabilized by an electrostatically assembled nanogel shell of positively charged chitosan and a negatively charged folic acid (FA)-tagged block copolymer for active targeting.77 Similar to the other nanogel theranostic study, in vivo imaging and drug delivery was not evaluated. Though achieving sensitive disease-responsive MR imaging with optimal release kinetics for drug delivery remains a challenge, the studies mentioned here suggest how stimuli-responsive materials together with proper system design would allow potential realization of nanogels as MRI theranostic agents.
3.6 CEST agents
In addition to the previously mentioned MRI contrast agent chemical exchange saturation transfer (CEST) agents have gained substantial interest in the last decades.78,79 CEST offer multiple advantages over other conventional contrast agents as it provides frequency-dependent signal, therefore signal can be switched “off” and “on”. This unique property also allows potential visualization of multiple imaging agents and quantitative characterization of in vivo environment with ratiometric method. Both diamagnetic and paramagnetic CEST agents have been incorporated into nanocarriers, specifically liposomes, to increase the number of exchangeable protons for contrast sensitivity enhancement.80–82 Though there are currently no report on nanogels-based CEST agents, the capability of nanogels to encapsulate large amount of molecules and their high water content make nanogels an attractive platform CEST agents.4. PET imaging
In addition to MRI, positron emission tomography (PET) is also commonly used for clinical molecular imaging, especially in oncology, neurology and cardiology. PET uses gamma rays, which have the highest energy among imaging modalities (wavelength in order of 10−12 m) compared to MRI, its sensitivity is very high, requiring only nanomolar or even lower concentrations of the imaging agent,1,83,84 allowing visualization and quantification of disease markers. PET signal arises from correlated gamma photons generated upon annihilation of electrons by tracer-emitted positrons. These photons are generated some distance away from the site of emission, as annihilation occurs when positrons lose enough kinetic energy to collide with an electron.85,86 Commonly used tracers in organic molecules include 18F and 11C. Among these, 18F fluorodeoxyglucose, a glucose analog, is often used as a tracer for oncological imaging because many tumors consume more glucose than surrounding tissues.87 Other studies mainly focus on metal radionuclei with longer half-lives, such as copper-64 (64Cu), yttrium-86 (86Y), and zirconium-89 (89Zr).88,89 gallium-68 (68Ga) has gained increasing attention since the development of modern 68Ge/68Ga generators, as their generation does not require an on-site cyclotron.904.1 PET nanogels
Although many polymers, micelles and polymeric nanoparticles have been used in experimental macromolecular radiopharmaceutics, the use of nanogels as PET imaging tracers has rarely been explored (Table 2). Soni et al. published the first radiolabeled nanogels in 2006, encapsulating N-hexylcarbamoyl-5-fluorouracil (HCFU), a prodrug of 5-fluorouracil (FU), into nanogels for drug delivery to the brain.91 A 68Ga-labeled polysorbate 80 coating granted the nanogels with gamma imaging properties. Instead of using a coating, Singh et al. labeled the nanogel matrix directly by attaching 68Ga-chelating 1,4,7-triazacyclononane-1-glutaric acid-4,7-diacetic acid (NODAGA) to the arms of a hydroxy- and thiol-terminated star-shaped poly(ethylene oxide-stat-polypropylene oxide) prepolymer.92 Nanogels were formed by self-assembly, followed by crosslinking of thiols on the polymer, which were sensitive to reducing environments. Although radiochemical yield was measured in vitro in a competition experiment, the in vivo stability of the system should be further investigated.Recently, Lux et al. altered the chemistry of the metal-chelating crosslinkers used in our MRI nanogel contrast agent to formulate a PET nanogel tracer (Fig. 4).9364Cu was selected because of its advantageous long half-life (12.7 h). To enhance Cu-chelating stability, a 1,4,7-triazacyclononane-1,4,7-triacetic acid (NOTA) crosslinker was synthesized and used in the PAAm nanogel formulation, allowing retention of 94% of the copper in the nanogels after 48 h of incubation in serum. In vivo PET imaging revealed high nanogel uptake in 4T1 mouse breast tumors. The tumor/muscle intensity ratio was greater than nine at 48 h after injection, which clearly delineated the tumor. In addition to primary tumors, the nanogels were also able to image metastasis, highlighting this system's clinical potential as a tumor-imaging PET agent. While PET has superior sensitivity for functional imaging, MRI offers unmatched soft tissue resolution for anatomical imaging. Combined PET and MRI therefore provides information about both structure and function, which can significantly improve disease identification, localization and thus the diagnostic evaluation. This new imaging field also opens up the opportunity to develop bimodal PET imaging agents, particularly after the commercialization of fully-integrated whole-body PET–MRI scanner. Simultaneous PET and MRI scanning can not only shorten imaging time, but also reduce motion artifacts. The above 64Cu-chelating and Gd-chelating crosslinkers could thus be combined in one nanogel system for simultaneous PET and MRI.
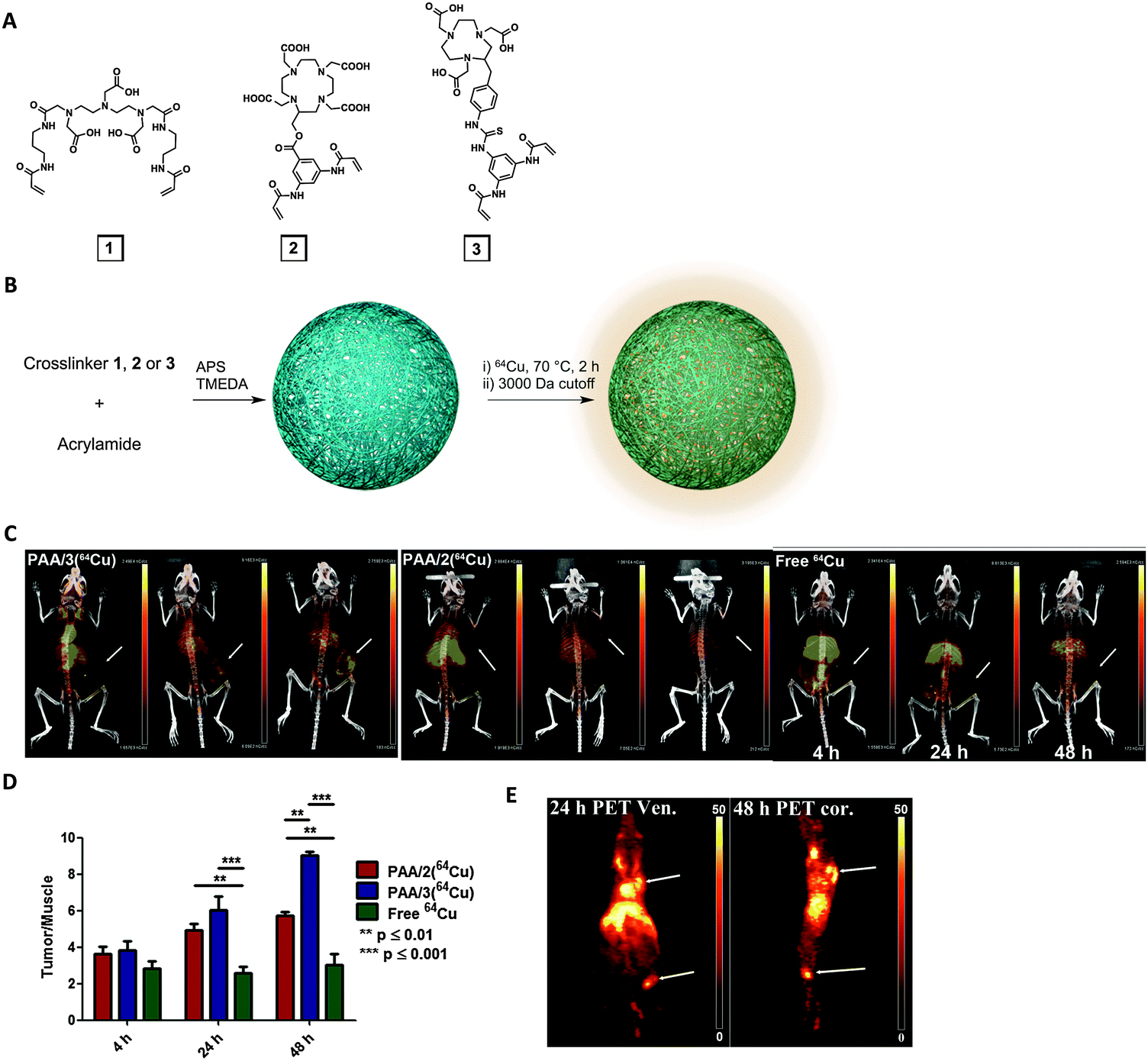 | ||
| Fig. 4 64Cu-bearing polyacrylamide (PAAm) nanogels as PET imaging nanogels. (A) Structures of three metal-chelating crosslinkers: DTPA (1), DOTA (2) and NOTA (3)-based. (B) Acrylamide was used as monomer and either 1, 2, or 3 was used as crosslinker to formulate nanogels (PAA/1, PAA/2 or PAA/3). (C) PET-CT imaging of mice with 4T1 tumors at 4 h, 24 h and 48 h after injection with PAA/2, PAA/3 or free 64Cu2+. Arrows indicate tumors. (D) Tumor/muscle ratio of PET signal. (E) PET imaging (arrows indicate popliteal lymph node in leg and primary tumor on shoulder) at 24 or 48 h post-injection.93 | ||
4.2 Dual PET imaging
Other groups have developed bimodal PET nanogel imaging agents. Gallo et al. developed a dual PET/fluorescence imaging nanogel, in which fluorescent upconverting nanoparticles, NaYF4:Yb/Er/Tm (18![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1.5:0.5 mol%), were encapsulated in a PEI shell.94 The amines on PEI allowed conjugation with 68Ga-chelating 1,4,7,10-tetraazacyclododecane-1,4,7,10-tetraacetic acid (DOTA) and iRGD, a tumor-cell-targeting peptide. The 136 nm nanogels absorbed NIR light and emitted visible light; whole-body PET imaging of mice with M21 melanoma tumors allowed quantification of nanogels in various organs. Uptake of iRGD-labeled nanogels in tumors was approximately 30% greater than that of unlabeled nanogels. Both fluorescence and PET imaging offer great sensitivity for functional imaging but have low three-dimensional spatial resolution. Therefore, MRI or CT would be better choices for coregistration with PET to provide complementary anatomical and morphological information.
:1.5:0.5 mol%), were encapsulated in a PEI shell.94 The amines on PEI allowed conjugation with 68Ga-chelating 1,4,7,10-tetraazacyclododecane-1,4,7,10-tetraacetic acid (DOTA) and iRGD, a tumor-cell-targeting peptide. The 136 nm nanogels absorbed NIR light and emitted visible light; whole-body PET imaging of mice with M21 melanoma tumors allowed quantification of nanogels in various organs. Uptake of iRGD-labeled nanogels in tumors was approximately 30% greater than that of unlabeled nanogels. Both fluorescence and PET imaging offer great sensitivity for functional imaging but have low three-dimensional spatial resolution. Therefore, MRI or CT would be better choices for coregistration with PET to provide complementary anatomical and morphological information.
Recently, Lee et al. published a dual PET and metalloprotease-activable optical imaging probe prepared from glycol chitosan nanogels (CNP).95 Glycol chitosan modified with 5β-cholanic acid and azide groups self-assembled into nanogels via hydrophobic interactions among the 5β-cholanic acid moieties. Using bio-orthogonal click chemistry with surface azides, 64Cu-chelating DOTA and an activable matrix metalloproteinase (MMP)-specific peptide probe, bearing Cy5.5 NIR dye and black hole quencher 3 (BHQ3) on opposite ends, were conjugated onto the nanogels' surfaces. MMP is overexpressed in many tumors and is responsible for tumor progression and metastasis. Quenched fluorescence was recovered only upon peptide cleavage by the target MMP. While the always “on” PET imaging signal can overcome the shortcomings of fluorescence imaging, such as limited tissue penetration depth, MMP-triggered “off-to-on” NIR fluorescence provides tumor-imaging specificity, which would improve tumor diagnosis accuracy.
Gupta et al. employed a different approach, instead loading PAA nanogels with 124I-labeled porphyrin and conjugating fluorescent cyanine dyes to surface amines to prepare dual PET-NIR fluorescence theranostic agents.96 The inclusion of porphyrin confers possible theranostic applications as a photodynamic therapy agent. In contrast to the previous system, in which the radioisotopes are chemically conjugated to nanogels, this system may suffer from leakage of encapsulated 124I-labeled porphyrin.
As mentioned above, PET is an essential tool in clinical imaging given its superior sensitivity. However, this modality alone does not provide any structural and morphological information. Combination with CT or MRI is required to identify where the PET signal originates in the body. In particular, MRI, because it does not use strong ionizing radiation, is highly preferable to CT. This encouraged the recent rapid development of novel and effective dual PET–MRI imaging agents for simultaneous PET and MR imaging with a new level of precision and resolution.
5. UV-visible/near-infra-red optical imaging
The popularity of optical imaging as an in vitro analytical tool is driven by its ease of use and high sensitivity (similar to PET) without ionizing radiation.97,98 These advantageous properties also enable their use in imaged-guide surgeries.2 Optical fluorescence can be categorized into three ranges according to the emission wavelength: near-infra-red (NIR) from 750 nm to 1000 nm, visible light from 450 nm to 750 nm and ultraviolet from 320 nm to 450 nm. In the last decade, advancements in hardware, imaging probes designs and mathematical models have made in vivo whole-body fluorescence imaging possible. This imaging modality is especially attractive given its sensitivity, allowing for the detection of fluorescent dyes in the picomolar range. In addition, quantum dots, gold/silver nanoparticles, and fluorescent dyes with various excitation and emission wavelengths, photostabilities, quantum yields, and functional groups are commercially available. Delivering these fluorescent entities using nanogels can improve their solubility, in vivo stability, and pharmacokinetics.5.1 Quantum dots
Quantum dots (QDs), usually nanocrystals of the semiconductor cadmium selenide (CdSe), have attracted attention as imaging probes due to their unique properties: high resistance to photobleaching, high quantum yield, narrow emission peak, and commercial availability at various emission wavelengths ranging from UV to NIR.Instead of being loaded into nanogels like other small dyes or drugs, QD can act as a core to template nanogels formation (Table 3). Surface ligand exchange of QD allows modification of QD with surface charges to electrostatically interact with oppositely charged polymers or monomers that form the nanogel matrix.99–102 The physical attraction between the QD and polymer matrix can minimize unwanted QD release. For example, QD was modified with a surface ligand carrying thiol groups at one end for QD surface adsorption, and amine groups on the other end for positive charges (Fig. 5).99 The positively charged QD electrostatically attracted the negatively charged hyaluronic acid (HA) polymer during formation of nanogels. Since HA has been shown to bind to lymphatic vessel endothelial receptor 1 (LYVE-1), this group employed the fluorescent HA nanogels to visualize lymphatic vessels, which may be used in lymphangiogenesis imaging for better understanding of cancer progression. Subcutaneous injection of the nanogels into mouse ears showed their accumulation in lymphatic vessels with strong fluorescence signal for up to a day. Yang et al. did not rely on electrostatic interactions, instead relying on the affinity between CdSe–ZnS QD and polyhistidine tag to build a nanogel for imaging cellular drug delivery.103 The polypeptide also included two hydrophobic and one hydrophilic domain to form a sandwiched layer for encapsulating both hydrophobic and hydrophilic drugs.
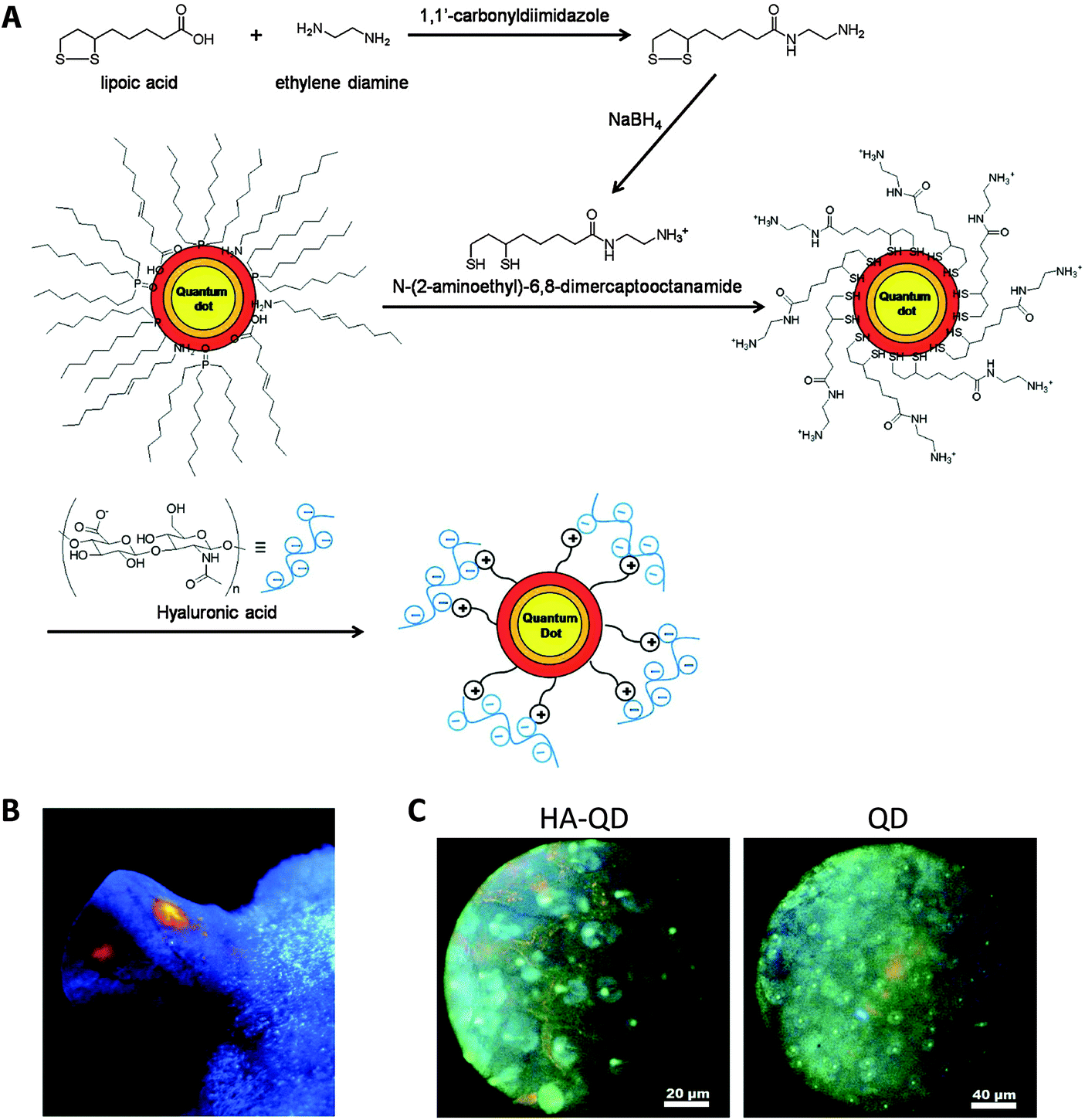 | ||
| Fig. 5 QD-encapsulating nanogels as lymph node imaging agents. (A) Synthesis and formulation scheme of HA-QD nanogels. (B) Image of a mouse ear under UV lamp 30 minutes after subcutaneous injection of HA-QD. (C) Fluorescence images of lymphatic vessels under microscope 30 minuets after injection of either HA-QD or QD. HA-QD provides a clear delineation of lymphatic vessel.99 | ||
Another strategy to formulate QD nanogels is through in situ synthesis of QD.104,105 Wu et al. utilized chitosan interpenetrated with polymethacrylic acid (PMAA) as the nanogel matrix.105 Chitosan's abundant hydroxyl groups sequestered Cd3+ and stabilized CdSe QDs formed in situ inside the nanogels; chemical crosslinking and hydrophobic interactions enhanced stability. Chitosan's amines (–NH2) and carboxylates (COO−) from PMAA gave the nanogels a pH-dependent volume phase transition. At approximately pH 5, the electrostatic interactions between deprotonated carboxylates (COO−) and protonated amines (NH3+) caused maximal shrinking; as pH increased (>pH 5.5) amines were deprotonated decreasing interactions with COO− and causing repulsion among polymers, swelling the nanogels. Not only did this enhance release of the encapsulated anticancer drug, temozolomide (TMZ), but changes in the protonation state of the counterions COO− and NH3+ also caused changes in the local dielectric environment of the QDs. This yielded pH-dependent optical properties: quenched NIR photoluminescence became unquenched as pH increased. This study demonstrated the application of nanogels as NIR theranostic agents for tumor drug delivery and pH sensing.
Carbon dots, with even higher biocompatibility and lower toxicity, have recently been investigated as an alternative to QD. Wang et al. encapsulated magnetic iron oxide nanocrystal cores and carbon dots in a carbon shell, which were further encapsulated into a poly(NIPAM-co-AA) nanogel.106 The temperature-dependent emission of carbon dots at 377 nm allowed sensing of the environmental temperature. In addition, NIR irradiation of the carbon shell or application of an alternating magnetic field induced localized heating and triggered drug release.
While QD-encapsulated nanogels have the advantage of direct and easy fabrication, the high toxicity of QD associated with the release of free Cd3+ remains a problem. In addition, though QD are widely commercially available at various emission wavelengths, their absorption lies in UV-blue ranges, which further limits their application as in vivo agents.
5.2 Au/Ag nanoparticles or nanorods
Gold (Au) or silver (Ag) nanoparticles with sizes larger than 3 nm exhibit strong surface plasmon resonance (SPR) absorption in the visible spectrum.107,108 The SPR originates from the oscillation of conductive electrons on the surface upon irradiation at resonant wavelengths. The surface plasmon band depends on various factors, including nanoparticle size, shape, and the surrounding environment. These metallic nanoparticles have the advantage of being less susceptible to photobleaching, and with absorption and emission orders of magnitude greater than those of small fluorescent dyes, making them better suited for imaging. Finally, because these nanoparticles can generate heat by absorbing NIR light, they can also provide photothermal treatment, and be formulated as theranostic agents.Similar to QD, surface modified Au/Ag nanoparticles can act as a core for a hydrogel shell to be built on,109–113 or could be synthesized in situ within nanogels (Table 3).114,115 Wu et al. developed multiple thermo or pH responsive nanogels with metallic nanoparticle cores for cellular imaging, chemo- and NIR-induced photothermal therapy. In one study, they encapsulated bimetallic Ag–Au nanoparticles into thermo-responsive PEG-based nanogels. In addition to fluorescence imaging, Ag–Au nanoparticles allowed conversion of NIR light to heat for photothermal therapy. Thermo-responsive PEG polymeric shells switched from hydrophilic to hydrophobic as the temperature increased, shrinking the nanogels, increasing photoluminescence, and releasing the hydrophilic model anticancer drug (temozolomide). The temperature responsiveness could be tuned by changing the thickness of the polymer shell. HA has been shown to bind CD44 receptors, which are overexpressed in various cancers and contribute to cell proliferation and migration,116,117 The HA conjugated-PEG nanogels enabled uptake by CD44-overexpressing B16F10 murine melanoma cells through endocytosis; these cells emitted fluorescence when excited at 405 nm. The therapeutic efficacy, as measured by cytotoxicity, of the combined chemo and photothermal therapy was higher than the additive effect of either alone. The group subsequently improved the system to include a hydrophobic polystyrene gel layer inside the PEG shell to encapsulate hydrophobic drugs.112 Their studies demonstrated the advantage of Ag/Au nanoparticles over other fluorescent dyes or QD, as they can provide photothermal therapy in addition to optical imaging, which allows them to be developed into multifunctional theranostics.
5.3 Small fluorescent dyes
A wide range of fluorescent dyes in the UV-vis to NIR range have been synthesized and studied as they are frequently used in microscopy. Conjugation or encapsulation of small dyes to nanogels provides a simple and direct route to formulate fluorescent nanogel imaging probes (Table 4). One strategy is to conjugate dyes to the nanogel surface post-formulation.118,119 Fluorescent dyes can also be modified as monomers or crosslinkers to be used in nanogels formulation.120–122 Incorporation of dyes in the particle matrix can minimize spontaneous release of labels due to instability in vivo, which complicates image interpretation,120 and avoids the need for excessive washing to remove dyes adsorbed to nanogels. Chen et al. utilized this strategy and modified autofluorescent abietane-based acid as a methacrylic monomer which was crosslinked with PEG diacrylate to yield a nanogel for both drug delivery and imaging. FA was conjugated to the surface as a targeting ligand, and doxorubicin was loaded and stabilized by the hydrophobic abietane. Fluorescent imaging showed uptake into MCF-7 human breast cancer cell cytoplasm within 1 h, illustrating their potential as cellular drug delivery tracking agents. Encapsulation of dyes into nanogels without covalent attachment has also been reported.19,123 Cao et al. encapsulated a pH-sensitive dye, 8-hydroxypyrene-1-carbaldehyde (HPC), into polyurethane nanogels as an intracellular fluorescence pH indicator.19 The nanogels' pH sensitivity allowed imaging of H2O2-induced cytosolic acidosis.While the small size and resulting high cellular uptake make UV-visible fluorescent nanogels good for sensing and detecting intracellular states, their use as in vivo imaging agents is limited. As shown in the above examples, almost all of these optical nanogels system were examined as cell imaging agents only. This choice may relate to the overlap of these nanogels' absorption and emission with the absorption of hemoglobin, melanin and other proteins in the body (from 200 to 650 nm),70,98,124,125 and interference by autofluorescence from tissues in this wavelength range.
To achieve deep tissue penetration and accurate detection, research on optical imaging agents has focused on those with absorption and emission wavelengths in the NIR region (from 750 nm to 1000 nm), in which absorption and autofluorescence from biological tissue is substantially lower. Generally, NIR probes can be categorized as either inorganic, such as quantum dots, gold (Au) nanorods/nanoclusters/nanoparticles and upconverting phosphor (UCP), or organic, such as cyanine, squaraine, BODIPY® and Alexa Fluor dyes® etc.124 These probes have been thoroughly compared elsewhere.124 Most of them suffer from low water solubility, a drawback for in vivo applications. In addition, the rapid degradation of organic NIR dyes in vivo prevents their wide use in clinical applications. Encapsulation or conjugation to hydrophilic nanogels can enhance their stability in aqueous solution, making them suitable for in vivo imaging and potentially enabling NIR imaging in humans.
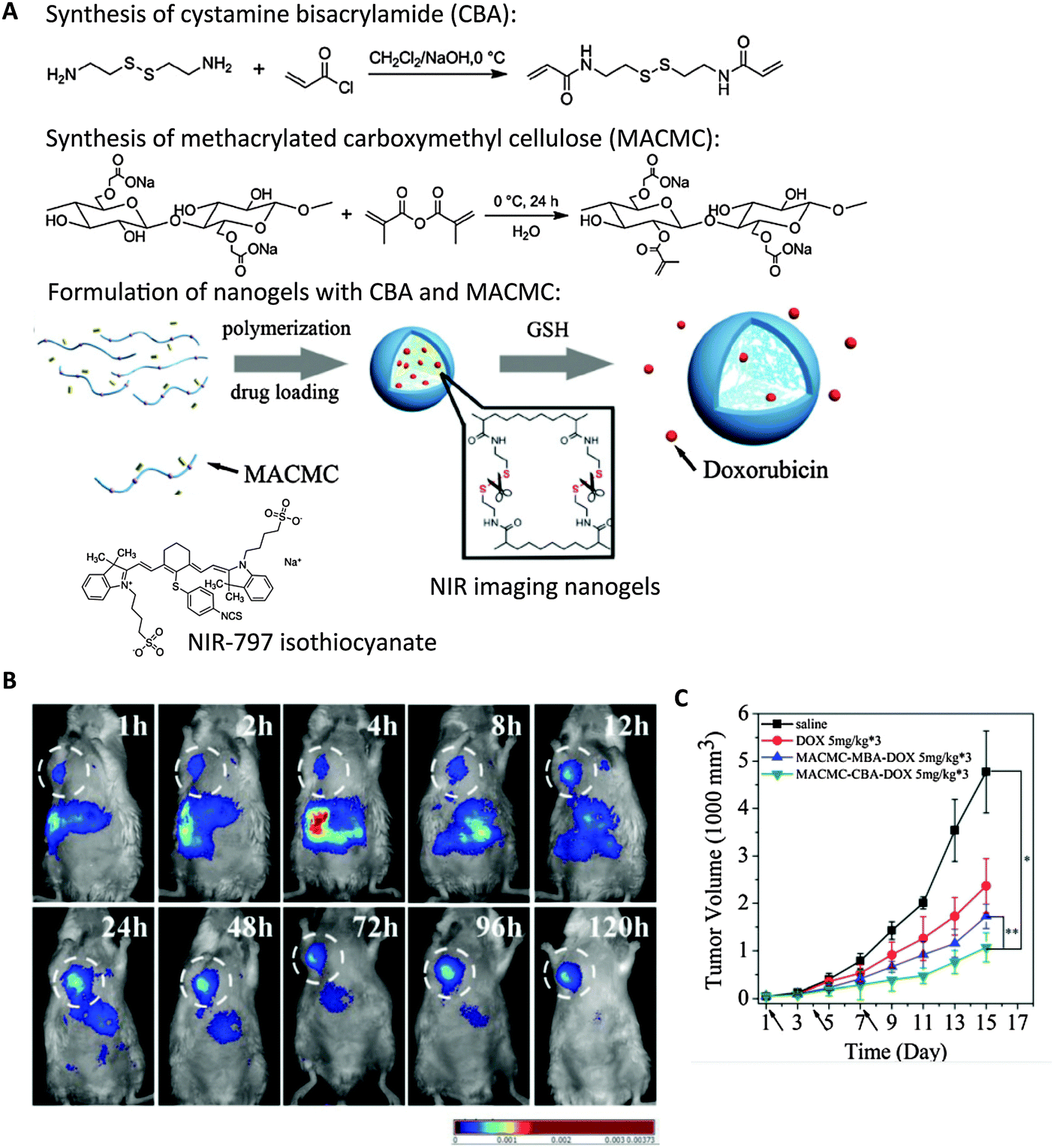 | ||
| Fig. 6 Cellulose-based nanogels for NIR tumor imaging. (A) Synthesis of disulfide crosslinkers (CBA), methacrylated cellulose (MACMC) and formulation of nanogels encapsulating doxorubicin (Dox) through radical polymerization. NIR-797 isothiocyanate was conjugated to render nanogels with NIR emitting properties. (B) Real-time NIR imaging of subcutaneous hepatic H22 tumor-bearing mice injected with NIR nanogels at 120 h post-injection. White circle highlights the tumor. (C) Tumor size was substantially decreased upon multiple dosage of Dox-containing nanogels compared with controls. (dosage time indicated by arrows). Saline, free Dox, and nanogels formulated with bisacrylamide (MACMC–MBA–Dox) without reduction-sensitive drug release were injected as controls.126 | ||
5.4 NIR tumor imaging
NIR nanogels have also been examined as tumor imaging agents. The enzyme hyaluronidase (HAdase), which is responsible for the degradation of hyaluronic acid (HA), is upregulated in tumor cells and contributes to tumor progression, angiogenesis, and metastasis.132,133 Overexpression of HAdase can thus serve as a biomarker for cancer progression. Mok et al. designed a hyaluronidase-activable NIR tumor-imaging nanogel using indocyanine green (ICG) dye-conjugated HA.134,135 ICG-conjugated HA self-assembled into nanogels through the hydrophobic interaction among ICG dyes. Park et al. utilized another tumor biomarker, low extracellular pH, for tumor-specific imaging in their system with ICG dyes.136 ICG dyes were encapsulated inside the nanogels composed of HA polymer and pH-sensitive poly(beta-amino)ester (PBAE) through electrostatic and hydrophobic interaction. In all three cases, embedding the dyes inside nanogels quenched the NIR signal; presence of HAdase or low pH liberated ICG and turned on the NIR signal, allowing tumor-specific imaging. Mok et al. showed that their nanogel system allowed longitudinal NIR imaging of tumors for up to three days post-injection. Instead of conjugation to a dye, Fu et al. formulated label-free NIR nanogels by incorporating a Ga-porphyrin complex as a crosslinker.137 Ga-porphyrin was modified with tetra-alkyne functionalities and reacted with difunctionalized azide-PEG through Cu2+-catalyzed click chemistry, to form nanogels of 30 or 120 nm. Though these nanogels exhibit maximum emission at approximately 725 nm, in vivo imaging was not shown to illustrate their feasibility as in vivo NIR tumor imaging agents.Conclusion
In this review, we discussed nanogels as imaging agents in various imaging modalities spanning the electromagnetic spectrum. Imaging agents for each imaging modality have different requirements. They need to be able to carry molecules at different concentrations, of different sizes, and different chemical properties, ranging from small fluorescent/NIR dyes, metal-chelates, quantum dots to iron oxide nanoparticles. Because of the availability of numerous formulation methods and building materials, nanogels can be utilized as imaging agents in many imaging modalities, making them very versatile. Specifically, due to flexibility in their constituents, more biocompatible and less immunogenic materials, such as natural polymers (chitosan, dextran, and pullulan), can be used. This ideal characteristic of nanogels gives them great potential as a platform for clinical contrast agents. However, the development of nanogels as imaging agents is in its infancy. As reviewed herein, while a few systems have been tested as in vivo whole body imaging/theranostic agents for imaging tumors, most have only been used in vitro. Increasing structural stability, in vivo contrast, photostability, and disease-specific accumulation, and reducing toxicity and overcoming background signal, still remain major challenges for nanogels' full realization as in vivo imaging agents. Despite these problems, fast advances in imaging hardware and the constant discovery and design of newer and safer materials suggest that these obstacles could be overcome in the near future for applications in oncological, neuropsychiatric and cardiac imaging.References
- L. Fass, Mol. Oncol., 2008, 2, 115–152 CrossRef PubMed.
- J. V. Frangioni, J. Clin. Oncol., 2008, 26, 4012–4021 CrossRef PubMed.
- G. Themelis, L. M. A. Crane, N. J. Harlaar, R. G. Pleijhuis, W. Kelder, A. Sarantopoulos, J. S. de Jong, H. J. G. Arts, A. G. J. van der Zee, J. Bart, P. S. Low, G. M. van Dam and V. Ntziachristos, Nat. Med., 2011, 17, 1315–1319 CrossRef PubMed.
- B. P. Joshi and T. D. Wang, Cancers, 2010, 2, 1251–1288 CrossRef CAS PubMed.
- V. S. Khoo and D. L. Joon, Br. J. Radiol., 2006, 79, S2–S15 CrossRef PubMed.
- C. Oerlemans, W. Bult, M. Bos, G. Storm, J. F. W. Nijsen and W. E. Hennink, Pharm. Res., 2010, 27, 2569–2589 CrossRef CAS PubMed.
- V. P. Torchilin, Adv. Drug Delivery Rev., 2002, 54, 235–252 CrossRef CAS.
- B. Dubertret, P. Skourides, D. J. Norris, V. Noireaux, A. H. Brivanlou and A. Libchaber, Science, 2002, 298, 1759–1762 CrossRef CAS PubMed.
- Y. Shimizu, T. Temma, I. Hara, A. Makino, R. Yamahara, E.-I. Ozeki, M. Ono and H. Saji, Nanomedicine, 2014, 10, 187–195 CrossRef CAS PubMed.
- W. Wang, D. Cheng, F. Gong, X. Miao and X. Shuai, Adv. Mater., 2011, 24, 115–120 CrossRef PubMed.
- L. K. Bogart, G. Pourroy, C. J. Murphy, V. Puntes, T. Pellegrino, D. Rosenblum, D. Peer and R. Lévy, ACS Nano, 2014, 8, 3107–3122 CrossRef CAS PubMed.
- M. K. Yu, J. Park and S. Jon, Theranostics, 2012, 2, 3–44 CrossRef CAS PubMed.
- Y. Wang, Nat. Mater., 2013, 13, 1–9 CrossRef PubMed.
- M. Longmire, P. L. Choyke and H. Kobayashi, Curr. Top. Med. Chem., 2008, 8, 1180–1186 CrossRef CAS.
- A. R. Menjoge, R. M. Kannan and D. A. Tomalia, Drug Discovery Today, 2010, 15, 171–185 CrossRef CAS PubMed.
- H. Ow, D. R. Larson, M. Srivastava, B. A. Baird, W. W. Webb and U. Wiesner, Nano Lett., 2005, 5, 113–117 CrossRef CAS PubMed.
- J. Yan, M. C. Estevez, J. Smith, K. Wang, X. He, L. Wang and W. Tan, Nano Today, 2007, 2, 44–50 CrossRef.
- A. Burns, H. Ow and U. Wiesner, Chem. Soc. Rev., 2006, 35, 1028–1042 RSC.
- L. Cao, X. Li, S. Wang, S. Li, Y. Li and G. Yang, Chem. Commun., 2014, 50, 8787–8790 RSC.
- M. Chan, J. Lux, L. Vander Elst, E. Schopf, E. Mahmoud, S. Laurent and A. Almutairi, J. Mater. Chem. B, 2013, 1, 6359–6364 RSC.
- J. R. I. Kopeček and J. Yang, Polym. Int., 2007, 56, 1078–1098 CrossRef PubMed.
- N. A. Peppas, J. Z. Hilt, A. Khademhosseini and R. Langer, Adv. Mater., 2006, 18, 1345–1360 CrossRef CAS PubMed.
- N. A. Peppas, P. Bures, W. Leobandung and H. Ichikawa, Eur. J. Pharm. Biopharm., 2000, 50, 27–46 CrossRef CAS.
- N. B. Graham and A. Cameron, Pure Appl. Chem., 1998, 70, 1271–1275 CrossRef CAS.
- A. V. Kabanov and S. V. Vinogradov, Angew. Chem., Int. Ed., 2009, 48, 5418–5429 CrossRef CAS PubMed.
- F. Sultana, M. Manirujjaman, M. Imran-Ul-Haque, M. Arafat and S. Sharmin, J. Appl. Pharm. Sci., 2013, 3, S95–S105 Search PubMed.
- S. Maya, B. Sarmentob, A. Naira, N. S. Rejinold, S. V. Nair and R. Jayakumar, Curr. Pharm. Des., 2013, 19, 7203–7218 CrossRef CAS.
- M. A. C. Stuart, W. T. S. Huck, J. Genzer, M. Müller, C. Ober, M. Stamm, G. B. Sukhorukov, I. Szleifer, V. V. Tsukruk, M. Urban, F. Winnik, S. Zauscher, I. Luzinov and S. Minko, Nat. Mater., 2010, 9, 101–113 CrossRef PubMed.
- S.-I. Sawada, Y. Sasaki, Y. Nomura and K. Akiyoshi, Colloid Polym. Sci., 2010, 289, 685–691 Search PubMed.
- Y. Sasaki, Y. Nomura, S.-I. Sawada and K. Akiyoshi, Polym. J., 2010, 42, 823–828 CrossRef CAS PubMed.
- N. Singh, Nisha, V. Gill and P. Gill, Am. J. Adv. Drug Delivery, 2013, 1, 271–276 Search PubMed.
- A. J. Sivaram, P. Rajitha, S. Maya, R. Jayakumar and M. Sabitha, Wiley Interdiscip. Rev.: Nanomed. Nanobiotechnol., 2015, 7, 509–533 CrossRef CAS PubMed.
- X. Wang, D. Niu, P. Li, Q. Wu, X. Bo, B. Liu, S. Bao, T. Su, H. Xu and Q. Wang, ACS Nano, 2015, 9, 5646–5656 CrossRef CAS PubMed.
- W. Wu and S. Zhou, Nano Rev., 2010, 1, 1718 Search PubMed.
- Y. Sasaki and K. Akiyoshi, Chem. Rec., 2010, 366–376 CAS.
- S. S. Pedrosa, C. Gonçalves, L. David and M. Gama, Macromol. Biosci., 2014, 14, 1556–1568 CrossRef CAS PubMed.
- W. Wu, W. Yao, X. Wang, C. Xie, J. Zhang and X. Jiang, Biomaterials, 2015, 39, 260–268 CrossRef CAS PubMed.
- E. M. Ahmed, J. Adv. Res., 2013, 6, 105–121 CrossRef PubMed.
- N. Das, Int. J. Pharm. Pharm. Sci., 2013, 5, 112–117 CAS.
- M. Baker, Nature, 2010, 463, 977–980 CrossRef CAS PubMed.
- D.-E. Lee, H. Koo, I.-C. Sun, J. H. Ryu, K. Kim and I. C. Kwon, Chem. Soc. Rev., 2012, 41, 2656–2672 RSC.
- P. Caravan, J. J. Ellison, T. J. McMurry and R. B. Lauffer, Chem. Rev., 1999, 99, 2293–2352 CrossRef CAS PubMed.
- C. Corot, P. Robert, J. M. Idee and M. Port, Adv. Drug Delivery Rev., 2006, 58, 1471–1504 CrossRef CAS PubMed.
- E. Toth, L. Helm and A. E. Merbach, Topics in Current Chemistry, 2002, vol. 221, pp. 62–98 Search PubMed.
- E. Boros, E. M. Gale and P. Caravan, Dalton Trans., 2014, 44, 4805–4818 Search PubMed.
- S. Aime and P. Caravan, J. Magn. Reson. Imaging, 2009, 30, 1259–1267 CrossRef PubMed.
- P. Caravan, Chem. Soc. Rev., 2006, 35, 512 RSC.
- P. Caravan, G. Parigi, J. M. Chasse, N. J. Cloutier, J. J. Ellison, R. B. Lauffer, C. Luchinat, S. A. McDermid, M. Spiller and T. J. McMurry, Inorg. Chem., 2007, 46, 6632–6639 CrossRef CAS PubMed.
- Y. Li, M. Beija, S. Laurent, L. V. Elst, R. N. Muller, H. T. T. Duong, A. B. Lowe, T. P. Davis and C. Boyer, Macromolecules, 2012, 45, 4196–4204 CrossRef CAS.
- Y. Liu, Z. Chen, C. Liu, D. Yu, Z. Lu and N. Zhang, Biomaterials, 2011, 32, 5167–5176 CrossRef CAS PubMed.
- P. Mi, H. Cabral, D. Kokuryo, M. Rafi, Y. Terada, I. Aoki, T. Saga, I. Takehiko, N. Nishiyama and K. Kataoka, Biomaterials, 2013, 34, 492–500 CrossRef CAS PubMed.
- H. Kobayashi and M. W. Brechbiel, Adv. Drug Delivery Rev., 2005, 57, 2271–2286 CrossRef CAS PubMed.
- E. Schopf, J. Sankaranarayanan, M. Chan, R. Mattrey and A. Almutairi, Mol. Pharmaceutics, 2012, 9, 1911–1918 CrossRef CAS PubMed.
- A. Soleimani, F. Martínez, V. Economopoulos, P. J. Foster, T. J. Scholl and E. R. Gillies, J. Mater. Chem. B, 2013, 1, 1027–1034 RSC.
- C. K. Lim, A. Singh, J. Heo, D. Kim, K. E. Lee, H. Jeon, J. Koh, I. C. Kwon and S. Kim, Biomaterials, 2013, 34, 6846–6852 CrossRef CAS PubMed.
- A. Ahmed, C. Zhang, J. Guo, Y. Hu and X. Jiang, Macromol. Biosci., 2015, 15, 1105–1114 CrossRef CAS PubMed.
- C. Prashant, M. Dipak, C. T. Yang, K. H. Chuang, D. Jun and S. S. Feng, Biomaterials, 2010, 31, 5588–5597 CrossRef CAS PubMed.
- A. K. Gupta and M. Gupta, Biomaterials, 2005, 26, 3995–4021 CrossRef CAS PubMed.
- R. Qiao, C. Yang and M. Gao, J. Mater. Chem., 2009, 19, 6274 RSC.
- E. S. Guang Choo, X. Tang, Y. Sheng, B. Shuter and J. Xue, J. Mater. Chem., 2011, 21, 2310 RSC.
- C. Gonçalves, Y. Lalatonne, L. Melro, G. Badino, M. F. M. Ferreira, L. David, C. F. G. C. Geraldes, L. Motte, J. A. Martins and F. M. Gama, J. Mater. Chem. B, 2013, 1, 5853 RSC.
- T. Fan, M. Li, X. Wu, M. Li and Y. Wu, Colloids Surf., B, 2011, 88, 593–600 CrossRef CAS PubMed.
- H. Sun, L. Zhang, X. Zhu, C. Kong, C. Zhang and S. Yao, Sci. China, Ser. B: Chem., 2009, 52, 69–75 CrossRef CAS.
- H. Liu and J. Li, Iran. Polym. J., 2008, 17, 721–727 CAS.
- Y. Gong, M. Fan, F. Gao, J. Hong, S. Liu, S. Luo, J. Yu and J. Huang, Colloids Surf., B, 2009, 71, 243–247 CrossRef CAS PubMed.
- L. Jiang, Q. Zhou, K. Mu, H. Xie, Y. Zhu, W. Zhu, Y. Zhao, H. Xu and X. Yang, Biomaterials, 2013, 34, 7418–7428 CrossRef CAS PubMed.
- J.-M. Shen, L. Xu, Y. Lu, H.-M. Cao, Z.-G. Xu, T. Chen and H.-X. Zhang, Int. J. Pharm., 2012, 427, 400–409 CrossRef CAS PubMed.
- J. S. Park, H. N. Yang, D. G. Woo, S. Y. Jeon and K.-H. Park, Biomaterials, 2013, 34, 8819–8834 CrossRef CAS PubMed.
- H. M. Kim, Y.-W. Noh, H. S. Park, M. Y. Cho, K. S. Hong, H. Lee, D. H. Shin, J. Kang, M.-H. Sung, H. Poo and Y. T. Lim, Small, 2012, 8, 666–670 CrossRef CAS PubMed.
- V. J. Pansare, S. Hejazi, W. J. Faenza and R. K. Prud'homme, Chem. Mater., 2012, 24, 812–827 CrossRef CAS PubMed.
- P. Juzenas, A. Juzeniene, O. Kaalhus, V. Iani and J. Moan, Photochem. Photobiol. Sci., 2002, 1, 745–748 CAS.
- M. Oishi, S. Sumitani and Y. Nagasaki, Bioconjugate Chem., 2007, 18, 1379–1382 CrossRef CAS PubMed.
- M. Oishi and Y. Nagasaki, Nanomedicine, 2010, 5, 451–468 CrossRef CAS PubMed.
- M. L. Viger, J. Sankaranarayanan, C. de Gracia Lux, M. Chan and A. Almutairi, J. Am. Chem. Soc., 2013, 135, 7847–7850 CrossRef CAS PubMed.
- L. Zhang, H. Xue, Z. Cao, A. Keefe, J. Wang and S. Jiang, Biomaterials, 2011, 32, 4604–4608 CrossRef CAS PubMed.
- J. M. Estrela, A. Ortega and E. Obrador, Crit. Rev. Clin. Lab. Sci., 2006, 43, 143–181 CrossRef CAS PubMed.
- W.-H. Chiang, W.-C. Huang, C.-W. Chang, M.-Y. Shen, Z.-F. Shih, Y.-F. Huang, S.-C. Lin and H.-C. Chiu, J. Controlled Release, 2013, 168, 280–288 CrossRef CAS PubMed.
- A. D. Sherry and M. Woods, Annu. Rev. Biomed. Eng., 2008, 10, 391–411 CrossRef CAS PubMed.
- E. Terreno, D. D. Castelli and S. Aime, Contrast Media Mol. Imaging, 2010, 5, 78–98 CAS.
- K. W. Y. Chan, T. Yu, Y. Qiao, Q. Liu, M. Yang, H. Patel, G. Liu, K. W. Kinzler, B. Vogelstein, J. W. M. Bulte, P. C. M. van Zijl, J. Hanes, S. Zhou and M. T. McMahon, J. Controlled Release, 2014, 180, 51–59 CrossRef CAS PubMed.
- D. D. Castelli, C. Boffa, P. Giustetto, E. Terreno and S. Aime, J. Biol. Inorg. Chem., 2014, 19, 207–214 CrossRef CAS PubMed.
- K. W. Y. Chan, G. Liu, X. Song, H. Kim, T. Yu, D. R. Arifin, A. A. Gilad, J. Hanes, P. Walczak, P. C. M. van Zijl, J. W. M. Bulte and M. T. McMahon, Nat. Mater., 2013, 12, 268–275 CrossRef CAS PubMed.
- S. L. Pimlott and A. Sutherland, Chem. Soc. Rev., 2010, 40, 149 RSC.
- Y. Li, W. Zhang, H. Wu and G. Liu, BioMed Res. Int., 2014, 2014, 1–13 CAS.
- M. D. Farwell, D. A. Pryma and D. A. Mankoff, Cancer, 2014, 120, 3433–3445 CrossRef CAS PubMed.
- V. Kapoor, B. M. McCook and F. S. Torok, RadioGraphics, 2004, 24, 523–543 CrossRef PubMed.
- J. G. Rajendran, D. A. Mankoff, F. O'Sullivan, L. M. Peterson, D. L. Schwartz, E. U. Conrad, A. M. Spence, M. Muzi, D. G. Farwell and K. Krohn, Clin. Cancer Res., 2004, 10, 2245–2252 CrossRef CAS.
- Z. Cai and C. J. Anderson, J. Labelled Compd. Radiopharm., 2014, 57, 224–230 CrossRef CAS PubMed.
- Y. Lu, K. Yang, K. Zhou, B. Pang, G. Wang, Y. Ding, Q. Zhang, H. Han, J. Tian, C. Li and Q. Ren, J. Nucl. Med., 2014, 55, 1375–1379 CrossRef CAS PubMed.
- I. Velikyan, Theranostics, 2014, 4, 47–80 CrossRef CAS PubMed.
- S. Soni, A. K. Babbar, R. K. Sharma and A. Maitra, J. Drug Targeting, 2006, 14, 87–95 CrossRef CAS PubMed.
- S. Singh, B. Bingöl, A. Morgenroth, F. M. Mottaghy, M. Möller and J. Schmaljohann, Macromol. Rapid Commun., 2013, 34, 562–567 CrossRef CAS PubMed.
- J. Lux, A. G. White, M. Chan, C. J. Anderson and A. Almutairi, Theranostics, 2014, 5, 277–288 CrossRef PubMed.
- J. Gallo, I. S. Alam, J. Jin, Y.-J. Gu, E. O. Aboagye, W.-T. Wong and N. J. Long, Dalton Trans., 2014, 43, 5535–5545 RSC.
- S. Lee, S.-W. Kang, J. H. Ryu, J. H. Na, D.-E. Lee, S. J. Han, C. M. Kang, Y. S. Choe, K. C. Lee, J. F. Leary, K. Choi, K.-H. Lee and K. Kim, Bioconjugate Chem., 2014, 25, 601–610 CrossRef CAS PubMed.
- A. Gupta, S. Wang, A. Marko, P. Joshi, M. Ethirajan, Y. Chen, R. Yao, M. Sajjad, R. Kopelman and R. K. Pandey, Theranostics, 2014, 4, 614–628 CrossRef CAS PubMed.
- J. W. Lichtman and J.-A. Conchello, Nat. Methods, 2005, 2, 910–919 CrossRef CAS PubMed.
- J. Rao, A. Dragulescu-Andrasi and H. Yao, Curr. Opin. Biotechnol., 2007, 18, 17–25 CrossRef CAS PubMed.
- S. H. Bhang, N. Won, T.-J. Lee, H. Jin, J. Nam, J. Park, H. Chung, H.-S. Park, Y.-E. Sung, S. K. Hahn, B.-S. Kim and S. Kim, ACS Nano, 2009, 3, 1389–1398 CrossRef CAS PubMed.
- Y. Chen, T. Ji and Z. Rosenzweig, Nano Lett., 2003, 3, 581–584 CrossRef CAS.
- Z. Li, W. Xu, Y. Wang, B. R. Shah, C. Zhang, Y. Chen, Y. Li and B. Li, Carbohydr. Polym., 2015, 121, 477–485 CrossRef CAS PubMed.
- U. Hasegawa, S.-I. M. Nomura, S. C. Kaul, T. Hirano and K. Akiyoshi, Biochem. Biophys. Res. Commun., 2005, 331, 917–921 CrossRef CAS PubMed.
- J. Yang, M.-H. Yao, L. Wen, J.-T. Song, M.-Z. Zhang, Y.-D. Zhao and B. Liu, Nanoscale, 2014, 6, 11282–11292 RSC.
- W. Wu, M. Aiello, T. Zhou, A. Berliner, P. Banerjee and S. Zhou, Biomaterials, 2010, 31, 3023–3031 CrossRef CAS PubMed.
- W. Wu, J. Shen, P. Banerjee and S. Zhou, Biomaterials, 2010, 31, 8371–8381 CrossRef CAS PubMed.
- H. Wang, J. Yi, S. Mukherjee, P. Banerjee and S. Zhou, Nanoscale, 2014, 6, 13001–13011 RSC.
- S. Eustis and M. A. El-Sayed, Chem. Soc. Rev., 2006, 35, 209–217 RSC.
- T. A. El-Brolossy, T. Abdallah, M. B. Mohamed, S. Abdallah, K. Easawi, S. Negm and H. Talaat, Eur. Phys. J.: Spec. Top., 2008, 153, 361–364 CrossRef PubMed.
- W. Wu, T. Zhou, A. Berliner, P. Banerjee and S. Zhou, Chem. Mater., 2010, 22, 1966–1976 CrossRef CAS.
- X.-Q. Zhao, T.-X. Wang, W. Liu, C.-D. Wang, D. Wang, T. Shang, L.-H. Shen and L. Ren, J. Mater. Chem., 2011, 21, 7240–7247 RSC.
- W. Wu, J. Shen, P. Banerjee and S. Zhou, Biomaterials, 2010, 31, 7555–7566 CrossRef CAS PubMed.
- W. Wu, J. Shen, P. Banerjee and S. Zhou, Biomaterials, 2011, 32, 598–609 CrossRef CAS PubMed.
- Y. Ding, Q. Chen, H. Qian, Y. Chen, W. Wu, Y. Hu and X. Jiang, Macromol. Biosci., 2009, 9, 1272–1280 CrossRef CAS PubMed.
- H. Cai and P. Yao, Nanoscale, 2013, 5, 2892 RSC.
- R. Guo, L. Zhang, Z. Zhu and X. Jiang, Langmuir, 2008, 24, 3459–3464 CrossRef CAS PubMed.
- D. Naor, S. Nedvetzki, I. Golan, L. Melnik and Y. Faitelson, Crit. Rev. Clin. Lab. Sci., 2002, 39, 527–579 CrossRef CAS PubMed.
- R. Marhaba and M. Zoller, J. Mol. Histol., 2004, 35, 211–231 CrossRef CAS.
- T. Dai, S. Zhou, C. Yin, S. Li, W. Cao, W. Liu, K. Sun, H. Dou, Y. Cao and G. Zhou, Biomaterials, 2014, 35, 8227–8235 CrossRef CAS PubMed.
- M. Gonçalves, D. Maciel, D. Capelo, S. Xiao, W. Sun, X. Shi, J. Rodrigues, H. Tomás and Y. Li, Biomacromolecules, 2014, 15, 492–499 CrossRef PubMed.
- Y. Chen, P. A. Wilbon, J. Zhou, M. Nagarkatti, C. Wang, F. Chu and C. Tang, Chem. Commun., 2012, 49, 297 RSC.
- B. Shi, H. Zhang, S. Z. Qiao, J. Bi and S. Dai, Adv. Healthcare Mater., 2014, 3, 1839–1848 CrossRef CAS PubMed.
- X. Wan, H. Liu, S. Yao, T. Liu and Y. Yao, Macromol. Rapid Commun., 2013, 35, 323–329 CrossRef PubMed.
- X. Guo, X. Zhang, S. Wang, S. Li, R. Hu, Y. Li and G. Yang, Anal. Chim. Acta, 2015, 869, 81–88 CrossRef CAS PubMed.
- S. Luo, E. Zhang, Y. Su, T. Cheng and C. Shi, Biomaterials, 2011, 32, 7127–7138 CrossRef CAS PubMed.
- G. D. Luker and K. E. Luker, J. Nucl. Med., 2007, 49, 1–4 CrossRef PubMed.
- H. Qian, X. Wang, K. Yuan, C. Xie, W. Wu, X. Jiang and L. Hu, Biomater. Sci., 2013, 2, 220 RSC.
- T. Xing, B. Lai and L. Yan, Macromol. Chem. Phys., 2012, 214, 578–588 CrossRef PubMed.
- A. Cousins, S. K. Thompson, A. B. Wedding and B. Thierry, Biotechnol. Adv., 2014, 32, 269–279 CrossRef PubMed.
- J. Aarsvold and N. Alazraki, Semin. Nucl. Med., 2005, 35, 116–128 CrossRef PubMed.
- Y.-W. Noh, S.-H. Kong, D.-Y. Choi, H. S. Park, H.-K. Yang, H.-J. Lee, H. C. Kim, K. W. Kang, M.-H. Sung and Y. T. Lim, ACS Nano, 2012, 6, 7820–7831 CrossRef CAS PubMed.
- S.-H. Kong, Y.-W. Noh, Y.-S. Suh, H. S. Park, H.-J. Lee, K. W. Kang, H. C. Kim, Y. T. Lim and H.-K. Yang, Gastric Cancer, 2014, 18, 55–64 CrossRef PubMed.
- J.-X. Tan, X.-Y. Wang, H.-Y. Li, X.-L. Su, L. Wang, L. Ran, K. Zheng and G.-S. Ren, Int. J. Cancer, 2011, 128, 1303–1315 CrossRef CAS PubMed.
- A. Benitez, T. J. Yates, L. E. Lopez, W. H. Cerwinka, A. Bakkar and V. B. Lokeshwar, Cancer Res., 2011, 71, 4085–4095 CrossRef CAS PubMed.
- H. Mok, H. Jeong, S.-J. Kim and B. H. Chung, Chem. Commun., 2012, 48, 8628 RSC.
- J. Kim, Y. Chong and H. Mok, Macromol. Biosci., 2014, 14, 881–888 CrossRef CAS PubMed.
- H. S. Park, J. E. Lee, M. Y. Cho, J. H. Hong, S. H. Cho and Y. T. Lim, Macromol. Rapid Commun., 2012, 33, 1549–1555 CrossRef CAS PubMed.
- G.-D. Fu, H. Jiang, F. Yao, L.-Q. Xu, J. Ling and E.-T. Kang, Macromol. Rapid Commun., 2012, 33, 1523–1527 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2016 |