
Redox stimuli-responsive hollow mesoporous silica nanocarriers for targeted drug delivery in cancer therapy†
Ye
Tian
a,
Ranran
Guo
a,
Yunfeng
Jiao
a,
Yangfei
Sun
b,
Shun
Shen
c,
Yajun
Wang
*d,
Daru
Lu
b,
Xingguo
Jiang
c and
Wuli
Yang
 *a
*a
aState Key Laboratory of Molecular Engineering of Polymers and Department of Macromolecular Science, Fudan University, Shanghai 200433, P. R. China. E-mail: wlyang@fudan.edu.cn; Tel: +86-21-65642385
bState Key Laboratory of Genetic Engineering and School of Life Sciences, Fudan University, Shanghai, 200433, P. R. China
cKey Laboratory of Smart Drug Delivery, Ministry of Education & PLA, School of Pharmacy, Fudan University, Shanghai 201203, P. R. China
dDepartment of Chemistry, Fudan University, Shanghai 200433, P. R. China. E-mail: yajunwang@fudan.edu.cn; Tel: +86-21-65642975
First published on 26th August 2016
Abstract
In order to specifically deliver drugs into cancer cells with targeted recognition and controlled release, biocompatible hollow mesoporous silica nanocarriers with tumor-targeting and glutathione-responsive release dual properties were developed. These multifunctional nanocarriers were fabricated by anchoring transferrin on the surface of hollow mesoporous silica nanoparticles through disulfide bond conjugation, which could be cleaved in the presence of glutathione. In this case, transferrin acted as the gatekeeper to control the drug release, and as a tumor-targeting agent to improve drug accumulation at the tumor site simultaneously. The detailed investigations indicate that the anticancer drug (doxorubicin) release from the nanocarriers was strongly dependent on the concentration of glutathione. The capacity of the nanocarriers to selectively deliver doxorubicin to the tumor cells was demonstrated in vitro and in vivo. The doxorubicin-loaded nanocarriers showed enhanced inhibition of tumor growth and minimal side-effects in vivo compared to free doxorubicin. These redox stimuli-responsive nanocarriers that achieved a combination of tumor targeting and controlled drug release provide a promising platform for efficient cancer therapies.
Conceptual insightsVarious nanocarrier drug delivery systems (DDSs) have been exploited to improve the treatment effect of chemotherapeutics. However, most of these DDSs also have severe side effects on normal tissues due to the premature release and a nonspecific targeting ability, which largely restrict their further development in clinical applications. Here, we report a hollow mesoporous silica nanoparticle based drug delivery system which could achieve controlled drug release and specific cancer cell targeting. In this study, transferrin is designed as a gatekeeper and tumor-targeting unit simultaneously through disulfide linkage, which could be removed by intracellular glutathione of cancer cells. The subsequent experiments indicate that more drug is released from the nanocarriers as the glutathione concentration increases. Moreover, in vitro and in vivo experiments demonstrate that the nanocarriers could greatly increase the amount of drug at the tumor site and improve the therapeutic efficacy of chemotherapy with minimal side effects. Therefore, the glutathione-responsive nanocarriers are expected to be an effective chemotherapeutic platform in oncotherapy. |
Cancer has become a leading cause of death around the world, with both the incidence and mortality of cancers continuously increasing. Recently, various chemotherapeutics have been exploited for effective cancer treatments, however, the cytotoxicity of these drugs to normal tissues is also prominent due to the random distribution of them in the body. Therefore, the development of elegant drug delivery systems (DDSs) that could specifically transport drugs into the target sites with minimal premature release has been widely studied to improve the efficacy of chemotherapeutics as well as to minimize the severe side effects on normal tissues.1–4
Among the various DDSs, mesoporous silica nanoparticles (MSNs) are considered to be promising candidates due to their high surface area, tunable pore size, controlled mesoporous structure, good biocompatibility and well-defined surface properties.5–9 The unique mesopore structures and facile surface functionalization properties of MSNs make it feasible to design various stimuli-responsive gatekeepers to control the opening and closing of the mesopores for the triggered release of the drugs that are loaded inside the porous particles. To date, several materials, such as magnetic nanoparticles,10 gold nanoparticles,11 quantum dots,12 rotaxanes,13 cyclodextrins,14,15 DNA16 and polymers,17 have been applied as gatekeepers to cap the mesoporous openings on the surface of the MSNs. They block the pores, avoiding the leakage of the loaded drug in the bloodstream, and can be removed by specific internal or external stimuli such as redox potential,13,18,19 pH,20,21 temperature,22 enzymes,23,24 and photo irradiation.11,25–27 However, the side effects of most of these DDSs still cannot be ignored in practice since these particles are often short of a specific cancer cell targeting capacity and can be efficiently internalized by normal cells as well,28 which may be the reason why few MSN-based stimuli-responsive drug delivery systems have been involved in the in vivo investigation. Therefore, there is an urgent need to develop effecient stimuli-responsive nanocarriers that could achieve improved drug release control and specific drug enrichment in cancer tissue simultaneously.19,29
Here, we report a versatile drug delivery system for tumor cell-targeted, glutathione (GSH)-triggered release based on transferrin (Tf)-capped hollow mesoporous silica nanoparticles (HMSNs) (Scheme 1). HMSNs were used as they can effectively enhance the drug loading and reduce the nanoparticles administrated in treatment due to their unique hollow cores.30–32 The mesopores in the HMSN shells were further covalently capped with Tf through disulfide linkage to prevent anticancer drug (i.e. doxorubicin, DOX) leakage during circulation. As an iron-binding blood plasma glycoprotein, Tf is also considered as a particularly effective tumor-targeting agent due to the significantly higher expression of Tf receptors on tumor cells.33 Through the recognition of the Tf target site, the nanocarriers could preferentially enter the target tumor cells. As the concentration of GSH in the intracellular matrix of cells is 102–103 times higher than that in the extracellular environment,34 the Tf gatekeeper would be removed from the surface of a mesopore by GSH to trigger the release of DOX once the nanocarriers were internalized into tumor cells. We demonstrated that Tf-capped HMSNs could realize the specific accumulation of drugs in tumor tissues to improve the therapeutic efficacy and minimize the side effects in vivo.
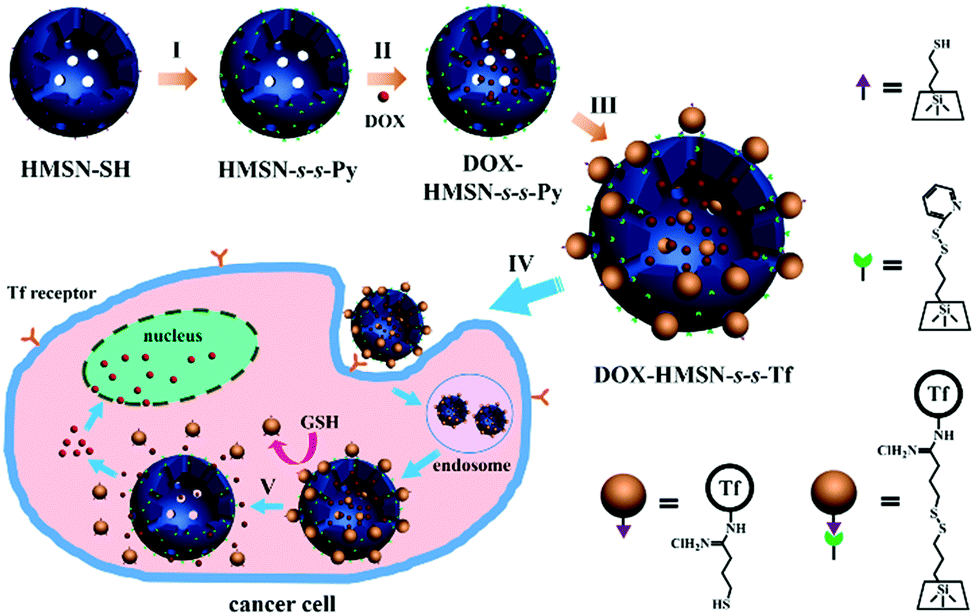 | ||
| Scheme 1 Schematic representation of the preparation and cancer cell internalization of the Tf-capped HMSN nanocarriers with both tumor-targeting and GSH-stimuli responsive properties. Stage I: functionalization of the mercaptopropyl-derivatized HMSNs (HMSN-SH) with 2,2′-dipyridyl disulfide to yield 2-propyldisulfanyl pyridine-functionalized HMSNs (HMSN-s-s-Py); stage II: loading DOX into the hollow core of HMSN-s-s-Py to obtain DOX-HMSN-s-s-Py; stage III: conjugation of thiol groups functionalized transferrin (Tf–SH) to the surface of the HMSN-s-s-Py nanocarriers by disulfide linkage to yield Tf-capped HMSNs (HMSN-s-s-Tf); stage IV: Tf-mediated cancer cell recognition and endocytosis of the nanocarriers; and stage V: GSH-triggered release of DOX by uncapping the Tf gatekeeper. | ||
Results and discussion
Preparation of Tf-capped HMSNs
The thiolated HMSN (HMSN-SH) nanocarriers were first prepared using poly(tert-butylacrylate) (PTBA) microspheres as the templates,35 through sol–gel co-condensation of TEOS and 3-mercaptopropyl-trimethoxysilane. After the removal of the templates, HMSN-SH was treated with 2,2′-dipyridyl disulfide to yield HMSN-s-s-Py. TEM images reveal that both HMSN-SH and HMSN-s-s-Py are monodisperse and show a similar hollow structure (highlighted by the dotted circles in the TEM images) with an average particle diameter of 100 nm and a shell thickness of 15 nm (Fig. 1a and b). These particles have a similar particle distribution with an average hydrodynamic diameter of 108 nm as measured using dynamic light scattering (DLS) (Fig. 2a). The N2 sorption results indicated that both HMSN-SH and HMSN-s-s-Py possessed a type IV isotherm with a high surface area of 1083 m2 g−1 and 746 m2 g−1 for HMSN-SH and HMSN-s-s-Py, respectively (Fig. 1d). The average pore diameter of HMSN-s-s-Py is slightly decreased from 3.2 nm (in HMSN-SH) to 3.1 nm (Fig. 1e), which can be attributed to a layer of 2-propyldisulfanyl pyridine-modification on the surface of the mesopores.36 The success of the surface functionalization can also be confirmed using thermal gravimetric analysis (TGA) data (Fig. S1, ESI†), X-ray photoelectron spectroscopy (XPS) data (Fig. S2, ESI†) and Fourier transform infared (FTIR) spectra (Fig. S3, ESI†). The presence of the peak at 505 cm−1 and the absence of the peak at 2583 cm−1 in the Raman spectrum of HMSN-s-s-Py, as compared to that of HMSN-SH, further demonstrated the formation of the disulfide bonds through the thiol-disulfide exchange reaction (Fig. S4, ESI†).37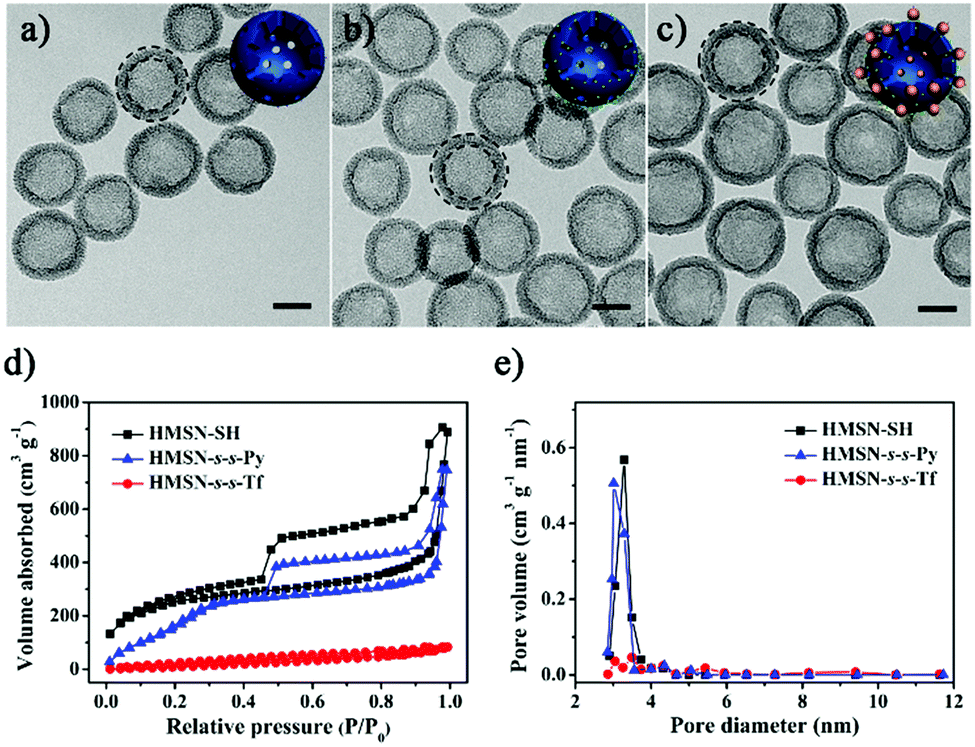 | ||
| Fig. 1 TEM images of (a) HMSN-SH, (b) HMSN-s-s-Py and (c) HMSN-s-s-Tf. Scale bars: 50 nm. Insets are the cartoons used in Scheme 1 to show different states of the nanocarriers. (d) The N2 adsorption–desorption isotherms and (e) pore distributions of HMSN-SH, HMSN-s-s-Py and HMSN-s-s-Tf. | ||
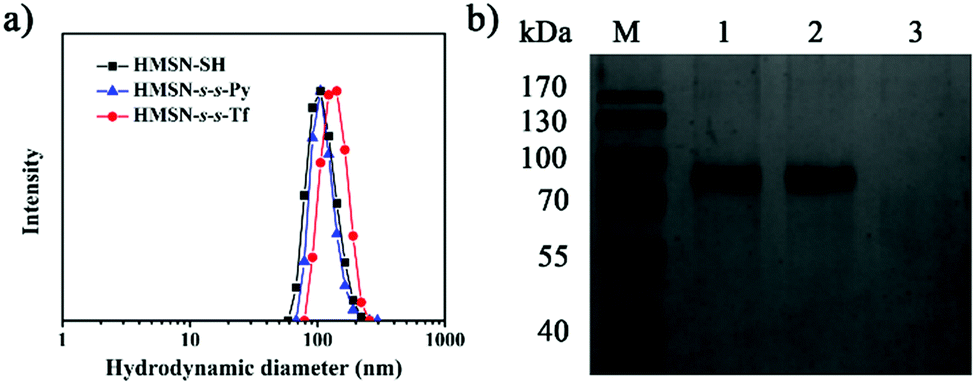 | ||
| Fig. 2 (a) Hydrodynamic diameters of HMSN-SH, HMSN-s-s-Py and HMSN-s-s-Tf determined using DLS in PBS (pH 7.4). (b) The SDS-PAGE analysis of pure Tf (lane 1) and HMSN-s-s-Tf incubated with 10 mM GSH (lane 2) and 0 mM GSH (lane 3), respectively. | ||
Transferrin functionalized with thiol groups (Tf–SH) was prepared according to a reported procedure.38 The degree of thiolation of Tf (–SH/Tf) was measured to be 3.0 from the UV/Vis data (Fig. S5, ESI†).39 DLS results showed that the hydrodynamic diameter of Tf–SH was ca. 5 nm (Fig. S6, ESI†), which is large enough to block the 3.1 nm pores on the mesoporous shells. HMSN-s-s-Py was treated with Tf–SH to yield Tf-capped HMSNs (HMSN-s-s-Tf) via the thiol-disulfide exchange reaction.40 The TEM image revealed that HMSN-s-s-Tf remained a clear core–shell structure and had a similar particle size of ca. 100 nm (Fig. 1c). However, the hydrodynamic diameter of HMSN-s-s-Tf was increased from 108 nm (HMSN-s-s-Py) to 131 nm (Fig. 2a), suggesting a successful conjugation of a layer of soft Tf–SH moieties on the particle surface. In contrast to the HMSN-s-s-Py that had a high surface area, HMSN-s-s-Tf showed a type I sorption isotherm (typical of nonporous materials) with a surface area of 283 m2 g−1 (Fig. 1d), and no obvious pore distribution could be observed (Fig. 1e), suggesting that most of the mesopores in the nanocarrier shells were blocked by Tf. The success of the Tf–SH functionalization can also be indicated from the significantly increased (ca. 25 wt%) weight loss of the HMSN-s-s-Py particles in the TGA curves (Fig. S1, ESI†). Functionalization of Tf–SH to the surface of HMSN-s-s-Py through the formation of disulfide bonds was evidenced from the decrease of the absorbance of Tf–SH at 412 nm in the supernatant after the reaction of Tf–SH with HMSN-s-s-Py (Fig. S7, ESI†). As a control, the UV/Vis absorbance spectrum of the supernatant showed no variation when Tf–SH was treated with HMSN-SH (Fig. S7, ESI†), suggesting that Tf could not be physically adsorbed on the surface of HMSN-SH.
The mechanism of the GSH-triggered uncapping of the mesopores was investigated by sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) analysis (Fig. 2b). When the concentration of GSH was 10 mM, the resulting solution showed the same band (lane 2) as pure Tf solution (lane 1), indicating that Tf was shed from HMSN-s-s-Tf as a result of cleavage of the disulfide linkages by GSH.41,42 In the absence of GSH, the resulting solution had no band of Tf (lane 3). These results confirmed that the Tf gatekeeper could be removed efficiently by GSH.
Glutathione-triggered drug release
To evaluate the GSH-triggered release properties of HMSN-s-s-Tf, DOX was used as a model anticancer drug. The DOX-loaded HMSN-s-s-Py (DOX-HMSN-s-s-Py) retained a type IV sorption isotherm with a high surface area of 680 m2 g−1 (Fig. S8, ESI†), suggesting that DOX was mainly loaded in the hollow core and the mesoporous structure in the shell was hardly influenced.43 After capping the mesoporous shell with Tf, an ultimate DOX loading of 265 mg g−1 was obtained, which is higher than that in conventional MSNs (189 mg g−1, as reported in our previous work),44 and the resulting DOX-HMSN-s-s-Tf showed good colloidal stability in PBS buffer (pH 7.4) (Fig. S9, ESI†). The release of DOX from the nanocarriers was also studied (Fig. 3a). In the absence of GSH, a small leakage of DOX (ca. 5.2%) was observed from DOX-HMSN-s-s-Tf in PBS buffer (pH 6.5) in 28 h. Considering the variations of pH from extracellular to intracellular,45,46 the stability of DOX-HMSN-s-s-Tf was further evaluated at different pH values (Fig. S10, ESI†). In the absence of GSH, the release of DOX was prevented by Tf in all the pH ranges (pH 5–7.4) studied. In contrast, DOX reached ca. 78% release from DOX-HMSN-SH under the same release conditions (Fig. S11, ESI†). Once GSH was added to the suspension, the release of DOX from DOX-HMSN-s-s-Tf could be triggered, and the release rates were largely governed by GSH concentration (Fig. 3b). The release amount of DOX could reach ca. 70% in 24 h when the GSH concentration was increased to 10 mM. These results clearly demonstrate that the Tf gatekeeper could effectively prevent the leakage of DOX during the circulation of the nanocarriers and that the encapsulated drug could be trigger-released once the Tf-cap was removed by GSH (Fig. 3c).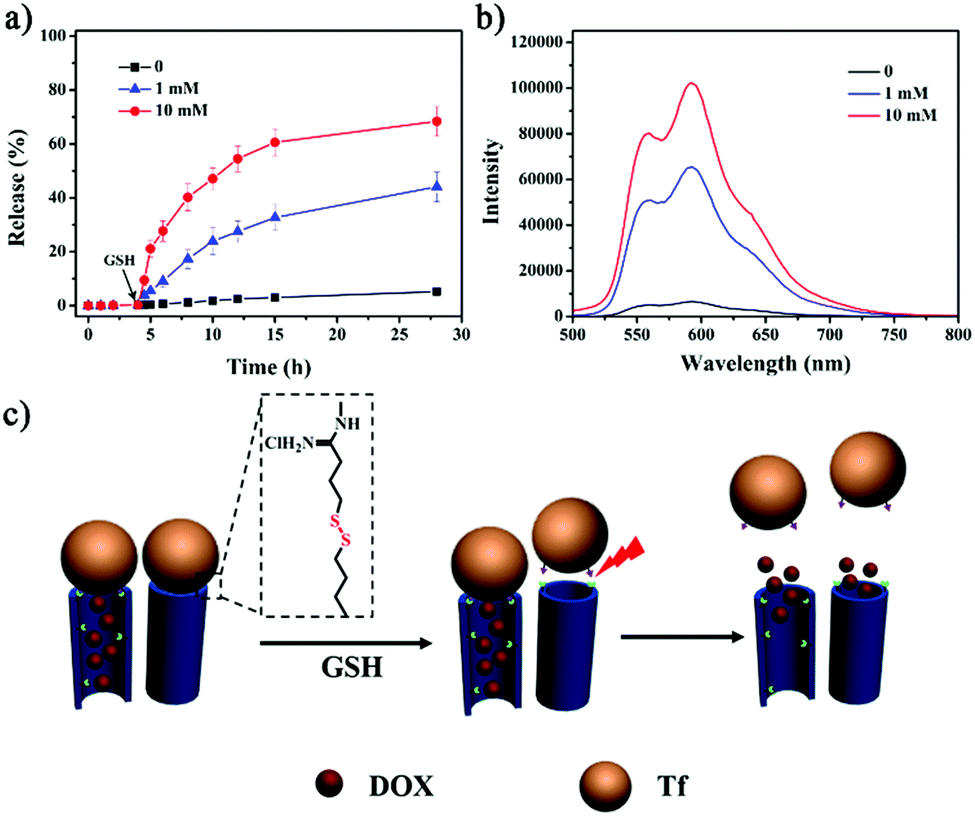 | ||
| Fig. 3 (a) The release profiles of DOX after the addition of GSH with different concentrations in PBS (pH 6.5). (b) The fluorescence spectra of the solution measured at 24 h after the addition of GSH. (c) Schematic illustration of the GSH-triggered DOX release through uncapping of the Tf from the mesopores. | ||
Interactions of DOX-HMSN-s-s-Tf with tumor cells
In vitro cell experiments were performed to evaluate the targeting efficiency and specificity of DOX-HMSN-s-s-Tf. The human adenocarcinoma alveolar basal epithelial cell line (A549 cells) was chosen for the research because of their overexpression of Tf-receptors on the cell membrane. Fig. 4a shows the confocal laser scanning microscope (CLSM) images of the A549 cells incubated with DOX-HMSN-s-s-Tf for 2, 4 and 8 h, respectively. The observation of the red fluorescence (from DOX) inside the cells confirmed that DOX-HMSN-s-s-Tf could be endocytosed by the A549 cells and then DOX was trigger-released into the cytoplasm by intracellular GSH. In addition, with the incubation time increased from 2 to 8 h, the fluorescence intensity of DOX in the cell was significantly increased, which could be attributed to the improved endocytosis of the nanoparticles and enhanced DOX release with the incubation time increase. In the control group, A549 cells incubated with DOX-HMSN-SH showed a much weaker DOX fluorescence than that of the cells incubated with DOX-HMSN-s-s-Tf in the same incubation time (Fig. 4b). For quantitative analysis, the fluorescence intensity (MFI) per cell was measured by flow cytometry analysis. The MFI per cell with DOX-HMSN-s-s-Tf was 1.7-fold and 2.0-fold higher than that of DOX-HMSN-SH after incubation with the cells for 2 and 4 h, respectively (Fig. 4e). It could be interpreted that Tf units on the surface of DOX-HMSN-s-s-Tf could enhance the internalization efficiency of the nanoparticles through specific receptor-mediated endocytosis.39,47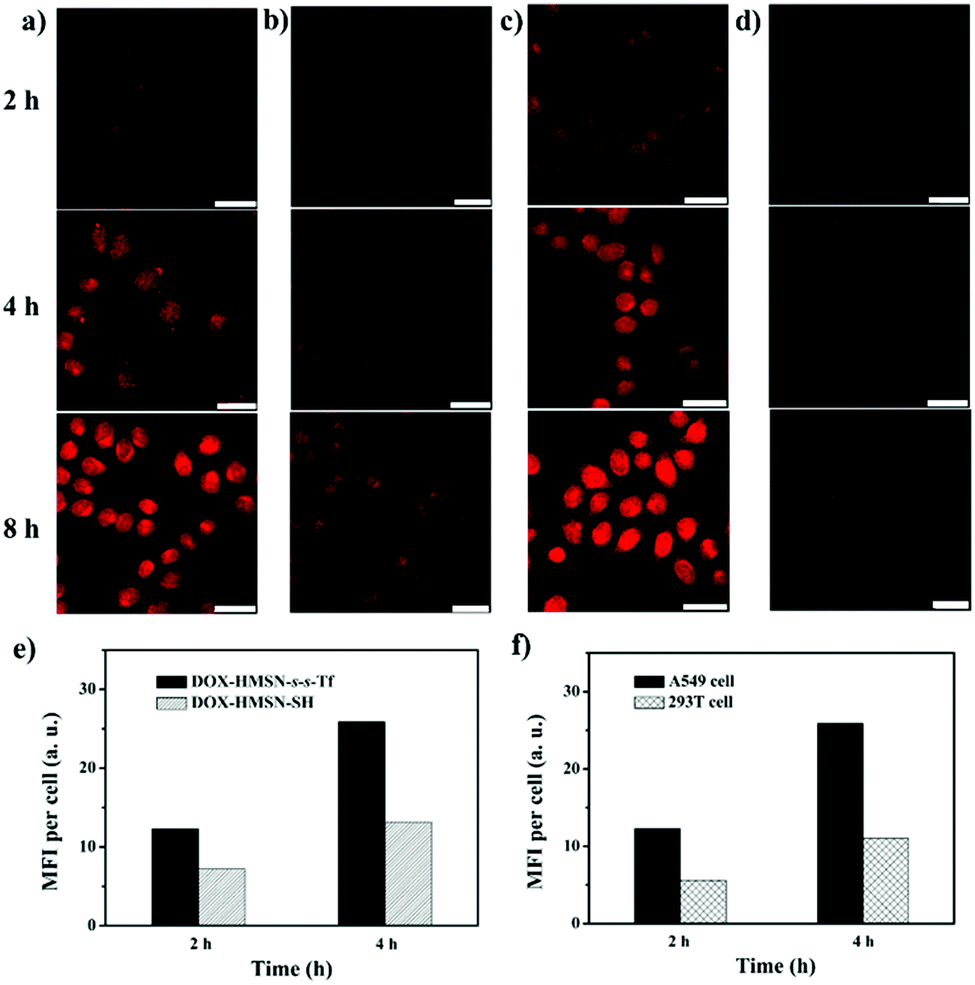 | ||
| Fig. 4 CLSM images of A549 cells incubated with (a) DOX-HMSN-s-s-Tf and (b) DOX-HMSN-SH. (c) A549 cells incubated with DOX-HMSN-s-s-Tf containing 5 mM GSH-OEt and (d) 293T cells incubated with DOX-HMSN-s-s-Tf for 2, 4, and 8 h. Scale bars: 50 μm. (e) MFI per cell of the A549 cells incubated with DOX-HMSN-s-s-Tf and DOX-HMSN-SH for 2 and 4 h. (f) MFI per cell of the A549 cells and 293T cells incubated with DOX-HMSN-s-s-Tf for 2 and 4 h. | ||
In addition, after the A549 cells were treated with free Tf, the internalization efficiency of DOX-HMSN-s-s-Tf decreased as the concentration of free Tf increased from 0 to 1 mg mL−1 (Fig. S12, ESI†). It could be explained by the fact that the free Tf would occupy part of the Tf-receptors on the cells, resulting in a decrease of the binding sites with DOX-HMSN-s-s-Tf, which confirmed that the uptake process by the A549 cells was indeed Tf-mediated endocytosis.
To evaluate the role of GSH in cargo release, the A549 cells were treated with 5 mM glutathione reduced ethyl ester (GSH-OEt) to enhance the GSH level in the cell.48,49 Stronger DOX fluorescence could be observed in the GSH-OEt-treated cells after incubating them with DOX-HMSN-s-s-Tf for 2, 4 and 8 h (Fig. 4c). These results indicate that the release rate of DOX from DOX-HMSN-s-s-Tf was dependent on the intracellular GSH level, suggesting that the GSH-triggered DOX release mechanism in solution is also applicable in the cell. We further investigated the DOX fluorescence in normal cells (293T cells) incubated with DOX-HMSN-s-s-Tf for 2, 4 and 8 h by CLSM (Fig. 4d). The DOX fluorescence in the 293T cells was significantly lower than that in the A549 cells incubated with DOX-HMSN-s-s-Tf for the same period. The flow cytometry analysis results indicate that the MFI per 293T cell was 2.2-fold and 2.4-fold lower than the MFI per A549 cell for the samples with an incubation time of 2 and 4 h, respectively (Fig. 4f). These results could be attributed to the cooperative effects of Tf-mediated targeting and GSH-triggered release because of the overexpression of Tf receptors on the surface of the A549 cells and the higher intracellular GSH level in cancer cells compared with normal cells.50,51 Therefore, DOX-HMSN-s-s-Tf with tumor-targeting and GSH-triggered release properties could effectively and preferentially deliver DOX into cancer cells.
In vitro cytotoxicity assays
In cancer therapy, non-cytotoxic nanocarriers are desirable to avoid unwanted side effects. Therefore, the cell viability incubated with HMSN-s-s-Tf was evaluated using MTT assays in both A549 and 293T cells (Fig. S13, ESI†). The results revealed that there was almost no cytotoxicity of HMSN-s-s-Tf to the cells under a measured particle concentration (100 μg mL−1). To evaluate the therapeutic efficacy of DOX-HMSN-s-s-Tf, the capability of free DOX, DOX-HMSN-SH, and DOX-HMSN-s-s-Tf to kill cancer cells was investigated (Fig. 5a). The half maximal inhibitory concentration (IC50) of DOX-HMSN-s-s-Tf was 1.93 μg mL−1, which was lower than that of DOX-HMSN-SH (4.20 μg mL−1) and even free DOX (2.84 μg mL−1). The higher cytotoxicity of DOX-HMSN-s-s-Tf compared with free DOX and DOX-HMSN-SH indicated that DOX-HMSN-s-s-Tf could be more effectively internalized by A549 cells through Tf receptor-mediated endocytosis, resulting in a higher intracellular concentration of DOX. To evaluate the selectivity of DOX-HMSN-s-s-Tf for cancer cells over normal cells, the cytotoxicity of free DOX, DOX-HMSN-SH, and DOX-HMSN-s-s-Tf to 293T cells was also investigated (Fig. 5b). The IC50 of free DOX and DOX-HMSN-SH was 2.05 μg mL−1 and 3.16 μg mL−1, indicating that free DOX and DOX-HMSN-SH had high cytotoxicity to 293T cells. Interestingly, the IC50 of DOX-HMSN-s-s-Tf was increased to 12.2 μg mL−1, indicating a much lower cytotoxicity to 293T cells in contrast to the high cytotoxicity for A549 cells (1.93 μg mL−1). These results undoubtedly demonstrated that DOX-HMSN-s-s-Tf had much higher cytotoxicity for cancer cells and could reduce the side-effects for normal cells due to the cooperative effects of Tf-mediated targeting and GSH-triggered DOX release. | ||
| Fig. 5 The cytotoxicity of DOX-HMSN-s-s-Tf, DOX-HMSN-SH and free DOX to the A549 cells (a) and 293T cells (b) for an incubation time of 24 h. | ||
The tumor-targeted delivery in vivo
To confirm the potential of using HMSN-s-s-Tf as a drug delivery system in vivo, A549 tumor-bearing mice were established and indocyanine green (ICG), a near-infrared fluorescent (NIRF) dye, was loaded into HMSN-s-s-Tf (ICG-HMSN-s-s-Tf) instead of DOX for in vivo tracking (Fig. S14, ESI†). All animal experiments were performed according to the guidelines evaluated and approved by the Ethics Committee of Fudan University. At 1 h post injection, the tumor area showed the maximal NIRF intensity (Fig. 6a). As a comparison, at 1 h post injection of ICG-HMSN-SH or free ICG, the maximal NIRF signal presented at the liver area. At 12 h post injection of ICG-HMSN-s-s-Tf, a clear tumor delineation was observed. Importantly, the NIRF images revealed a clearly delineated tumor for up to 48 h (Fig. 6a), and the contrast index (tumor/normal tissues) improved over time from 3.0 at 1 h to 4.7 at 48 h (Fig. S15, ESI†). These data demonstrated that the ICG-HMSN-s-s-Tf was preferentially retained in the tumor tissue and experienced a slower clearance (Fig. S16, ESI†).52,53 In mice treated with free ICG, the NIRF intensity decreased sharply at 12 h post injection and little NIRF signal could be detected at 48 h due to the rapid clearance of the dye from the body. At 12 h post injection of ICG-HMSN-SH, the tumor with a weak NIFR signal could be observed, but the contrast index was only 0.83 (Fig. S15, ESI†), which was much lower than that of ICG-HMSN-s-s-Tf. The substantially higher contrast index in the mice treated with ICG-HMSN-s-s-Tf indicated that ICG-HMSN-s-s-Tf preferentially accumulated in the tumor tissue. | ||
| Fig. 6 (a) In vivo NIRF images of ICG-HMSN-s-s-Tf, ICG-HMSN-SH and ICG in tumor-bearing mice at 1, 12, 24 and 48 h post injection (the arrows show the tumor location). (b) NIRF images of dissected organs and tumor tissues obtained from the mice treated with ICG-HMSN-s-s-Tf and ICG-HMSN-SH. The mean NIRF intensity of the organs and tumor tissues from the mice treated with ICG-HMSN-s-s-Tf and ICG-HMSN-SH at 1 h (c) and 48 h (d) post injection. | ||
The ex vivo evaluation of excised tissues was further performed to investigate the biodistribution of ICG-HMSN-s-s-Tf and ICG-HMSN-SH in mice (Fig. 6b). At 1 h post injection of ICG-HMSN-s-s-Tf, the tumor tissue showed the strongest NIRF signal compared with the other major organs including heart, liver, spleen, lung and kidney, indicating that ICG-HMSN-s-s-Tf could preferentially accumulate in the tumor tissue. As a control, at 1 h post injection of ICG-HMSN-SH, the strongest NIRF signal was observed in the liver. When the time was prolonged to 48 h, the mice treated with ICG-HMSN-s-s-Tf still showed the strongest NIRF signal in the tumor tissue, but in contrast, the NIRF signals in the organs and tumor from the mice treated with ICG-HMSN-SH became negligible. For the quantitative analysis, the mean NIRF intensity of all the organs and tumor were measured (Fig. 6c and d). In the mice treated with ICG-HMSN-s-s-Tf, the tumor/liver ratio increased from 1.73 at 1 h to 6.26 at 48 h, which was much higher than that for ICG-HMSN-SH (0.18 at 1 h and 0.89 at 48 h). These data provided decisive evidence that ICG-HMSN-s-s-Tf could effectively target tumor tissues in vivo.
Therapeutic efficacy of DOX-HMSN-s-s-Tf on the tumor in vivo
To investigate the therapeutic efficacy on the tumor in vivo, the A549 tumor-bearing mice weights and tumor sizes were periodically measured after injection of saline, DOX, DOX-HMSN-SH and DOX-HMSN-s-s-T. Every group had six duplicate A549 tumor-bearing mice (n = 6), and the injection was operated once every two days and lasted for 8 days. The mice treated with saline, DOX-HMSN-SH and DOX-HMSN-s-s-Tf maintained their weight around 25 g for 14 days. However, the average weights of the mice treated with free DOX decreased from 24.5 to 17.2 g (Fig. 7a). These results indicated that the functionalized HMSNs as drug carriers could effectively reduce the side effects of DOX in mice.54,55 Meanwhile, the quantitative analysis showed that in vivo tumor sizes increased upon feeding time after injection of saline, DOX and DOX-HMSN-SH (Fig. 7b). Compared with the control groups of saline, the growth of tumor tissue was partly inhibited when injected with DOX and DOX-HMSN-SH. Most importantly, the mice injected with DOX-HMSN-s-s-Tf showed much better therapeutic efficacy than the other groups. The growth of the tumor tissue was effectively inhibited by DOX-HMSN-s-s-Tf, and the tumor size got smaller after 14 days (Fig. 7b). The therapeutic efficacy was also confirmed by the images and weights of the tumor tissues measured at the final stage of the in vivo studies (Fig. 7c and d). The mice treated with DOX-HMSN-s-s-Tf showed the smallest tumor tissues. All these results indicated that DOX-HMSN-s-s-Tf could effectively accumulate in tumor tissues following DOX release into the cytoplasm triggered by intracellular GSH, accomplishing the maximum inhibition of tumor growth.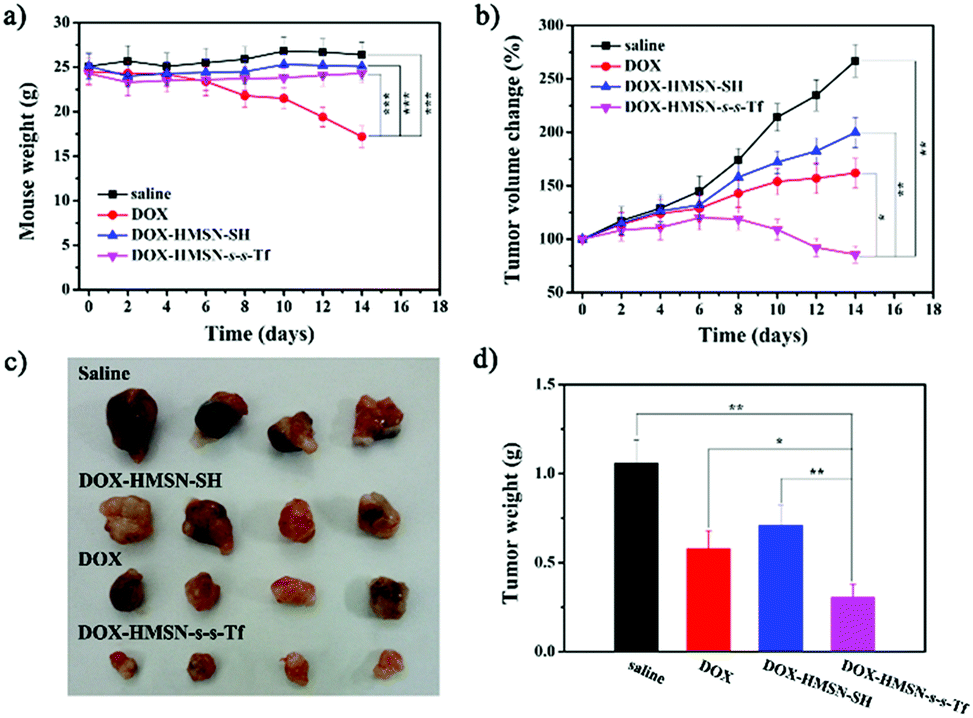 | ||
| Fig. 7 (a) Real-time weight analysis of mice injected with saline, DOX, DOX-HMSN-SH and DOX-HMSN-s-s-Tf, respectively. Error bars represent means ± standard deviation (SD) for n = 6, ***p < 0.001. (b) Real-time observation of tumor sizes in vivo after being injected with these samples. Error bars represent means ± SD for n = 6, *p < 0.05, **p < 0.01. (c) Representative images of excised tumor tissues injected with these samples for 14 days. (d) The final tumor weight of each group after treatment for 14 days. Error bars represent means ± SD for n = 6, *p < 0.05, **p < 0.01. | ||
Conclusions
In conclusion, we have demonstrated that the Tf linked on the surface of the HMSNs through biologically cleavable disulfide bonds could effectively prevent drug leakage from the nanocarrier during circulation, and be removed by the high concentration of intracellular GSH. In view of the overexpression of Tf-receptors on the surface of tumor cells, tumor-targeted delivery and GSH-controlled release could be achieved simultaneously, which resulted in maximum therapeutic efficacy and minimized side effects of the drugs. In vivo experiments have confirmed that DOX-HMSN-s-s-Tf could specifically deliver DOX to tumor cells and effectively inhibit the growth of tumor tissues. As this Tf-capped HMSN system could achieve two things in one stroke, we anticipate that this facile yet efficient DDS will present a new strategy to develop a controlled drug release system with targeting capacity for cancer treatment.Acknowledgements
We are grateful for the financial support of the National Science Foundation of China (Grant No. 51273047 and 51473037), the State Key Project of Research and Development (Grant No. 2016YFC1100300) and the “Shu Guang Project” (12SG07) from supported by the Shanghai Municipal Education Commission and Shanghai Education Development Foundation.Notes and references
- Y. H. Bae and K. Park, J. Controlled Release, 2011, 153, 198–205 CrossRef CAS PubMed.
- G. K. Such, A. P. R. Johnston and F. Caruso, Chem. Soc. Rev., 2011, 40, 19–29 RSC.
- Y. Yan, M. Bjornmalm and F. Caruso, Chem. Mater., 2014, 26, 452–460 CrossRef CAS.
- D. P. Ferris, J. Lu, C. Gothard, R. Yanes, C. R. Thomas, J. Olsen, J. F. Stoddart, F. Tamanoi and J. I. Zink, Small, 2011, 7, 1816–1826 CrossRef CAS PubMed.
- M. Vallet-Regi, F. Balas and D. Arcos, Angew. Chem., Int. Ed., 2007, 46, 7548–7558 CrossRef CAS PubMed.
- Y. J. Wang, A. D. Price and F. Caruso, J. Mater. Chem., 2009, 19, 6451–6464 RSC.
- Z. X. Li, J. C. Barnes, A. Bosoy, J. F. Stoddart and J. I. Zink, Chem. Soc. Rev., 2012, 41, 2590–2605 RSC.
- Y. J. Wang, A. K. Wise, J. Tan, J. Maina, R. K. Shepherd and F. Caruso, Small, 2014, 21, 4244–4248 Search PubMed.
- Q. J. He and J. L. Shi, Adv. Mater., 2014, 26, 391–411 CrossRef CAS PubMed.
- S. Giri, B. G. Trewyn, M. P. Stellmaker and V. S. Y. Lin, Angew. Chem., Int. Ed., 2005, 44, 5038–5044 CrossRef CAS PubMed.
- J. L. Vivero-Escoto, I. I. Slowing, C. W. Wu and V. S. Y. Lin, J. Am. Chem. Soc., 2009, 131, 3462–3463 CrossRef CAS PubMed.
- F. Muhammad, M. Y. Guo, W. X. Qi, F. X. Sun, A. F. Wang, Y. J. Guo and G. S. Zhu, J. Am. Chem. Soc., 2011, 133, 8778–8781 CrossRef CAS PubMed.
- T. D. Nguyen, Y. Liu, S. Saha, K. C. F. Leung, J. F. Stoddart and J. I. Zink, J. Am. Chem. Soc., 2007, 129, 626–634 CrossRef CAS PubMed.
- H. Kin, S. Kim, C. Park, H. Lee, H. J. Park and C. Kim, Adv. Mater., 2010, 22, 4280–4281 CrossRef PubMed.
- B. Y. Lee, Z. Li, D. L. Clemens, B. J. Dillon, A. A. Hwang, J. I. Zink and M. A. Horwitz, Small, 2016, 12, 3690–3702 CrossRef CAS PubMed.
- P. H. Zhang, F. F. Cheng, R. Zhou, J. T. Cao, J. J. Li, C. Burda, Q. H. Min and J. J. Zhu, Angew. Chem., Int. Ed., 2014, 53, 2371–2375 CrossRef CAS PubMed.
- T. A. Xia, M. Kovochich, M. Liong, H. Meng, S. Kabehie, S. George, J. I. Zink and A. E. Nel, ACS Nano, 2009, 3, 3273–3286 CrossRef CAS PubMed.
- R. Liu, X. Zhao, T. Wu and P. Y. Feng, J. Am. Chem. Soc., 2008, 130, 14418–14419 CrossRef CAS PubMed.
- Z. Luo, X. W. Ding, Y. Hu, S. J. Wu, Y. Xiang, Y. F. Zeng, H. Yan, H. C. Zhang, L. L. Zhu, J. J. Liu, J. H. Li, K. Y. Cai and Y. L. Zhao, ACS Nano, 2013, 7, 10271–10284 CrossRef CAS PubMed.
- M. Xue, X. Zhong, Z. Shaposhnik, Y. Q. Qu, F. Tamanoi, X. F. Duan and J. I. Zink, J. Am. Chem. Soc., 2011, 133, 8798–8801 CrossRef CAS PubMed.
- C. H. Lee, S. H. Cheng, I. P. Huang, J. S. Souris, C. S. Yang, C. Y. Mou and L. W. Lo, Angew. Chem., Int. Ed., 2010, 49, 8214–8219 CrossRef CAS PubMed.
- X. J. Yang, X. Liu, Z. Liu, F. Pu, J. S. Ren and X. G. Qu, Adv. Mater., 2012, 24, 2890–2895 CrossRef CAS PubMed.
- C. Park, H. Kim, S. Kim and C. Kim, J. Am. Chem. Soc., 2009, 131, 16614–16615 CrossRef CAS PubMed.
- Z. Zhang, D. Balogh, F. Wang and I. Willner, J. Am. Chem. Soc., 2013, 135, 1934–1940 CrossRef CAS PubMed.
- C. Chen, L. Zhou, J. Geng, J. Ren and X. Qu, Small, 2013, 9, 2793–2800 CrossRef CAS PubMed.
- M. Martinez-Carmona, A. Baeza, M. A. Rodriguez-Milla, J. Garcia-Castro and M. Vallet-Regi, J. Mater. Chem. B, 2015, 3, 5746–5752 RSC.
- J. G. Croissant, C. Qi, O. Mongin, V. Hugues, M. Blanchard-Desce, L. Raehm, X. Cattoen, M. W. C. Man, M. Maynadier, M. Gary-Bobo, M. Garcia, J. I. Zink and J. O. Durand, J. Mater. Chem. B, 2015, 3, 6456–6461 RSC.
- F. Q. Tang, L. L. Li and D. Chen, Adv. Mater., 2012, 24, 1504–1534 CrossRef CAS PubMed.
- O. Shimoni, A. Postma, Y. Yan, A. M. Scott, J. K. Heath, E. C. Nice, A. N. Zelikin and F. Caruso, ACS Nano, 2012, 6, 1463–1472 CrossRef CAS PubMed.
- Y. Chen, H. R. Chen, L. M. Guo, Q. J. He, F. Chen, J. Zhou, J. W. Feng and J. L. Shi, ACS Nano, 2010, 4, 529–539 CrossRef CAS PubMed.
- L. L. Li, F. Q. Tang, H. Y. Liu, T. L. Liu, N. J. Hao, D. Chen, X. Teng and J. Q. He, ACS Nano, 2010, 4, 6874–6882 CrossRef CAS PubMed.
- Y. Chen, P. F. Xu, H. R. Chen, Y. S. Li, W. B. Bu, Z. Shu, Y. P. Li, J. M. Zhang, L. X. Zhang, L. M. Pan, X. Z. Cui, Z. L. Hua, J. Wang, L. L. Zhang and J. L. Shi, Adv. Mater., 2013, 25, 3100–3105 CrossRef CAS PubMed.
- P. T. Gomme and K. B. McCann, Drug Discovery Today, 2005, 10, 267–273 CrossRef CAS PubMed.
- A. Verma, J. M. Simard, J. W. E. Worrall and V. M. Rotello, J. Am. Chem. Soc., 2004, 126, 13987–13991 CrossRef CAS PubMed.
- Y. F. Jiao, J. Guo, S. Shen, B. S. Chang, Y. H. Zhang, X. G. Jiang and W. L. Yang, J. Mater. Chem., 2012, 22, 17636–17643 RSC.
- C. Y. Lai, B. G. Trewyn, D. M. Jeftinjia, K. Jeftinija, S. Xu, S. Jeftinjia and V. S. Y. Lin, J. Am. Chem. Soc., 2003, 125, 4451–4459 CrossRef CAS PubMed.
- J. Dai, S. D. Lin, D. Cheng, S. Y. Zou and X. T. Shuai, Angew. Chem., Int. Ed., 2011, 50, 9404–9408 CrossRef CAS PubMed.
- Y. Y. Jiang, C. Liu, M. H. Hong, S. J. Zhu and Y. Y. Pei, Bioconjugate Chem., 2007, 18, 41–49 CrossRef CAS PubMed.
- G. L. Ellman, Arch. Biochem. Biophys., 1959, 82, 70–77 CrossRef CAS PubMed.
- S. F. Chong, R. Chandrawati, B. Stadler, J. Park, J. H. Cho, Y. J. Wang, Z. F. Jia, V. Bulmus, T. P. Davis, A. N. Zelikin and F. Caruso, Small, 2009, 5, 2601–2610 CrossRef CAS PubMed.
- Y. J. Wang, Y. Yan, J. W. Cui, L. Hosta-Rigau, J. K. Heath, E. C. Nice and F. Caruso, Adv. Mater., 2010, 22, 4293–4297 CrossRef CAS PubMed.
- S. S. Bian, J. Zheng, X. L. Tang, D. L. Yi, Y. J. Wang and W. L. Yang, Chem. Mater., 2015, 27, 1262–1268 CrossRef CAS.
- Y. F. Zhu, J. L. Shi, W. H. Shen, H. R. Chen, X. P. Dong and M. L. Ruan, Nanotechnology, 2005, 16, 2633–2638 CrossRef CAS.
- J. Zheng, X. Tian, Y. Sun, D. Lu and W. Yang, Int. J. Pharm., 2013, 450, 296–303 CrossRef CAS PubMed.
- S. Ganta, H. Devalapally, A. Shahiwala and M. Amiji, J. Controlled Release, 2008, 126, 187–204 CrossRef CAS PubMed.
- J. W. Cui, Y. Yan, Y. J. Wang and F. Caruso, Adv. Funct. Mater., 2012, 22, 4718–4723 CrossRef CAS.
- D. P. Ferris, J. Lu, C. Gothard, R. Yanes, C. R. Thomas, J. C. Olsen, J. F. Stoddart, F. Tamanoi and J. I. Zink, Small, 2011, 7, 1816–1826 CrossRef CAS PubMed.
- R. Hong, G. Han, J. M. Fernández, B. Kim, N. S. Forbes and V. M. Rotello, J. Am. Chem. Soc., 2006, 128, 1078–1079 CrossRef CAS PubMed.
- Q. Zhang, F. Liu, K. T. Nguyen, X. Ma, X. J. Wang, B. G. Xing and Y. L. Zhao, Adv. Funct. Mater., 2012, 22, 5144–5156 CrossRef CAS.
- G. Saito, J. A. Swanson and K. D. Lee, Adv. Drug Delivery Rev., 2003, 55, 199–215 CrossRef CAS PubMed.
- A. Gupte and R. J. Mumper, Cancer Treat. Rev., 2009, 35, 32–46 CrossRef CAS PubMed.
- Q. J. He, Z. W. Zhang, F. Gao, Y. P. Li and J. L. Shi, Small, 2011, 7, 271–280 CrossRef CAS PubMed.
- H. Meng, M. Xue, T. Xia, Z. Ji, D. Y. Tarn, J. I. Zink and A. E. Nel, ACS Nano, 2011, 5, 4131–4144 CrossRef CAS PubMed.
- N. Singh, A. Karambelkar, L. Gu, K. Lin, J. S. Miller, C. S. Chen, M. J. Sailor and S. N. Bhatia, J. Am. Chem. Soc., 2011, 133, 19582–19585 CrossRef CAS PubMed.
- H. Meng, W. X. Mai, H. Zhang, M. Xue, T. Xia, S. Lin, X. Wang, Y. Zhao, Z. Ji, J. I. Zink and A. E. Nel, ACS Nano, 2013, 7, 994–1005 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Details on experimental information. See DOI: 10.1039/c6nh00139d |
| This journal is © The Royal Society of Chemistry 2016 |