
Bioinspired nanophotosensitizers: synthesis and characterization of porphyrin–noble metal nanoparticle conjugates†
Jayeeta
Bhaumik
*a,
Gitanjali
Gogia
b,
Seema
Kirar
a,
Lekshmi
Vijay
b,
Neeraj S.
Thakur
a,
Uttam C.
Banerjee
a and
Joydev K.
Laha
*b
aDepartment of Pharmaceutical Technology (Biotechnology), National Institute of Pharmaceutical Education and Research, Sector-67, S.A.S. Nagar – 160062, Punjab, India. E-mail: jbhaumi@gmail.com
bDepartment of Pharmaceutical Technology (Process Chemistry), National Institute of Pharmaceutical Education and Research, Sector-67, S.A.S. Nagar – 160062, Punjab, India. E-mail: jlaha@niper.ac.in
First published on 10th November 2015
Abstract
A methodology to enhance biological delivery of photosensitizers by incorporating them into nanomaterials has been developed. In order to prepare photosensitizer nanoconjugates as biocompatible and selective probes, initially, bioconjugatable porphyrinic photosensitizers were prepared through rational routes. The porphyrins with carboxyl groups (as conjugatable handles) were successfully attached on the surface of the bioinspired nanoparticles (through a stable ester bond formation) affording hydrophilic and biocompatible nanophotosensitizers. The loading efficiency of the photosensitizer into nanomaterials was found to be 12–16%. Given their biocompatibility and efficient loading on nanoparticles, the photosensitizers prepared in this study could find use in photodynamic therapy and dual photodynamic–photothermal therapy.
Introduction
Photodynamic therapy (PDT) has become an important therapeutic tool for the treatment of many diseases including cancer, microbial infection and cardiovascular diseases.1–3 Photosensitizers (PSs) are essential components of PDT, which upon absorption of light are activated, and generate singlet oxygen that causes tumor cell destruction.4,5 Among other photosensitizers, tetrapyrrolic macrocycles have found an extensive use as photosensitizers due to their unique absorption properties in the visible and/or near infrared region of the electromagnetic spectrum.6,7 Porphyrins or their hydrogenated derivatives such as chlorins are effective tetrapyrrolic photosensitizers due to their high stability and ease of synthesis.8 For example, commercially available Photofrin, a porphyrin, or Foscan, a chlorin has found large applications for the treatment of certain cancer.9 However, limitations of tetrapyrrolic macrocycles as photosensitizers may include their hydrophobicity, biocompatibility, and non-selective delivery to biological systems.10Nanoparticles have largely been used for efficient drug delivery in tumors due to the enhanced permeability and retention (EPR) effect.1,11,12 Nowadays, biocompatible metal nanoparticles (MNPs) are gaining popularity as delivery vehicles for hydrophobic drugs and/or imaging agents. The incorporation of hydrophobic photosensitizers into NPs (through encapsulation or conjugation) facilitates their delivery into biological systems.21 A photosensitizer-nanoconjugate has been demonstrated to possess enhanced hydrophilicity, selectivity and biocompatibility compared to the free photosensitizer. In this regard, gold nanoparticles (AuNPs) have widely been used in photothermal therapy (PTT) of cancer treatment.13–17 Upon photosensitizer incorporation into AuNPs, a laser source of desired wavelengths is applied to simultaneously activate AuNPs and PSs resulting in dual PDT–PTT effects.18–20 Furthermore, the incorporation of a diagnostic agent into nanomaterials forming phototheranostic nanoagents has also been realized.22
Porphyrins are hydrophobic due to the presence of a large tetrapyrrolic macrocycle.23–25 This prevents their applications in PDT.22 Their biocompatibility can be tuned by incorporating them into nanoparticles. Porphyrinic macrocycles can serve as bioconjugatable photosensitizers when appropriate functional groups are present on their periphery. Previously we demonstrated that a rationally synthesized porphyrin with a conjugatable handle (e.g. carboxylic acid) is capable of attaching to nanoparticles or other biomaterials containing counter functionalities such as an amine or a hydroxyl group.26 We recently demonstrated that the bioinspired noble metal (gold and silver) nanoparticles rich in hydroxyl groups can incorporate imaging (rhodamine B) and therapeutic agents (rose bengal) containing carboxylic acid groups through simple ester bond formation.27,28 These findings motivated us to conjugate the porphyrins on the surface of bioinspired noble metal NPs to construct dual PDT–PTT agents.
Herein we report the development of porphyrin-nanoconjugates as biocompatible photosensitizers for potential applications in PDT and/or dual PDT–PTT. Initially, several conjugatable heterocycles containing compact trans-AB porphyrinic photosensitizers (PSs) were synthesized via rational routes. Those compact porphyrins with a carboxyl tether were then successfully attached to the surface of the bioinspired metal nanoparticles (rich in –OH group) via covalent (ester) bond formation. These nanophotosensitizer conjugates were further characterized (by absorption spectroscopy, FTIR spectroscopy and HPLC) and the photosensitizer loading efficiency on nanoscaffolds was also determined. Overall, these bioinspired photosensitizer nanoconjugates with enhanced biocompatibility are promising candidates for photomedical applications.
Results and discussion
Design plan
The concept of porphyrin conjugation on metal nanoparticles largely stems from our recent success in preparing bioinspired noble metal nanoparticles from various plant extracts.27 To explore further novel applications of these nanoparticles, we aimed to develop new photosensitizers that could largely address limitations of photosensitizers currently available in the literature or in market. Despite being an ambitious objective, we embarked on this challenging task and initially designed new photosensitizers as shown in Fig. 1. A rational synthesis of conjugatable trans-AB porphyrins could be achieved by condensing two building blocks, dipyrromethane (DPM) and 1,9-diformyldipyrromethane together using an improved procedure previously developed by us.23 The conjugatable porphyrin (containing carboxyl moieties) can further be attached to bioinspired metal nanomaterials (containing hydroxyl groups) through ester bond formation as shown in Fig. 1. Polar (due to the presence of quaternized nitrogen in the pyridyl moiety) and conjugatable (due to the presence of carboxylic acid) porphyrins thus obtained were used in the preparation of nanophotosensitizer conjugates.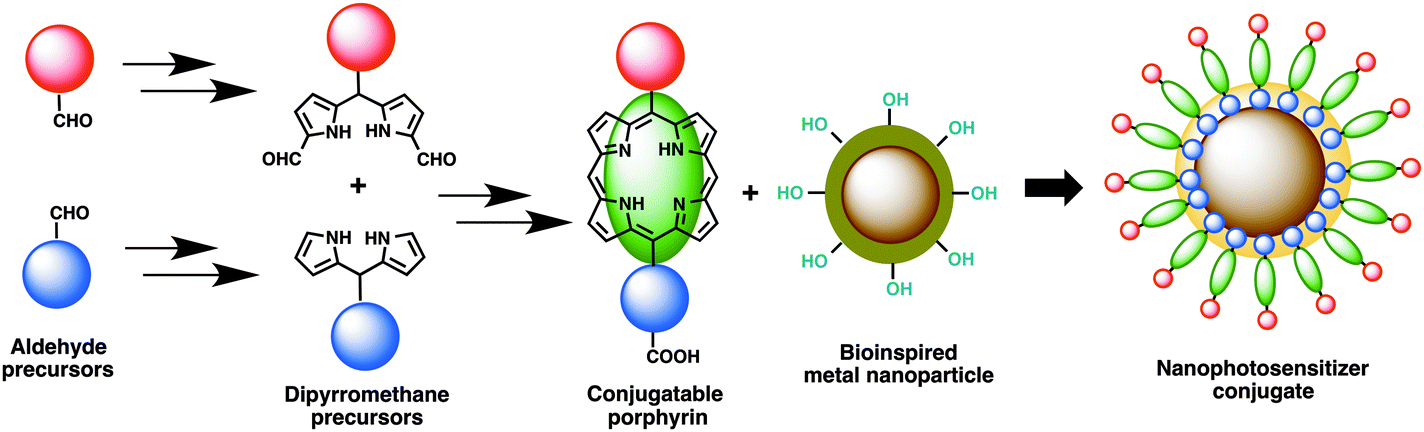 | ||
| Fig. 1 Synthetic design towards the development of nanophotosensitizer conjugates. | ||
The preparation of building blocks for the synthesis of dipyrromethanes is shown in Scheme 1. Treatment of 4-hydroxy benzaldehyde with ethyl bromoacetate or methyl glutaryl chloride at room temperature (or heating) gave ethyl 2-(4-formylphenoxy)acetate (1) or 4-formylphenyl methyl glutarate (2) in 66 and 80% yields, respectively.23 Following reported methods, N-alkylation of imidazole-2-carboxaldehyde (3) and indole-3-carboxaldehyde (4) with excess n-butyl iodide afforded N-butyl aldehydes 3a29 and 4a30 in 82 and 83% isolated yields, respectively.
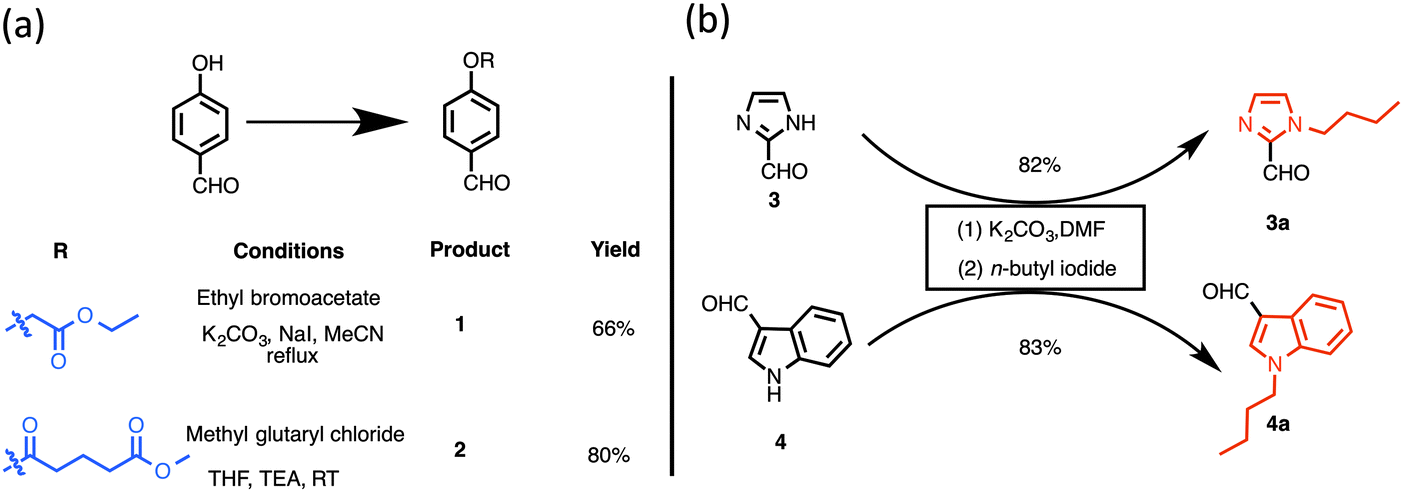 | ||
| Scheme 1 Synthetic precursors to dipyrromethanes: (a) synthesis of aldehydes containing masked conjugatable groups; (b) synthesis of butylated heterocyclic aldehydes. | ||
Rational synthesis of conjugatable trans-AB porphyrins
The preparation of synthetic precursors to porphyrins is shown in Scheme 1. The synthesis of dipyrromethanes was carried out using one of the following approaches developed earlier by us: (a) TFA-catalyzed reaction at room temperature, (b) InCl3-catalyzed reaction at room temperature or an elevated temperature in the presence of a large excess pyrrole,31 or (c) heating a neat mixture at an elevated temperature. Thus, aldehyde 1 was treated with excess pyrrole in the presence of TFA (10 mol%), which upon chromatographic purification produced dipyrromethane 5 in 66% yield (Scheme 2).32 Reaction of a mixture of benzaldehyde 2 and excess pyrrole at room temperature (25 °C) in the presence of a catalytic amount of InCl3 (20 mol%) followed by chromatography gave dipyrromethane 6 in 73% yield. A neat mixture of 4-pyridine carboxaldehyde and excess pyrrole was heated at 80 °C for 16 h, which upon chromatographic separation gave the desired 5-pyridyldipyrromethane (7) in 75% yield.33 Likewise, compounds 3a and 4a afforded 5-[1-butyl-imidazol-2-yl]dipyrromethane (8) and 5-[1-butyl-indol-3-yl]dipyrromethane (9) in 58 and 40% yields, respectively. Diformylation of dipyrromethanes 5, 6 or 7 at 1,9-positions, following a protocol previously demonstrated by us,23 yielded 1,9-diformyldipyrromethanes (10–12) in 20–40% yield.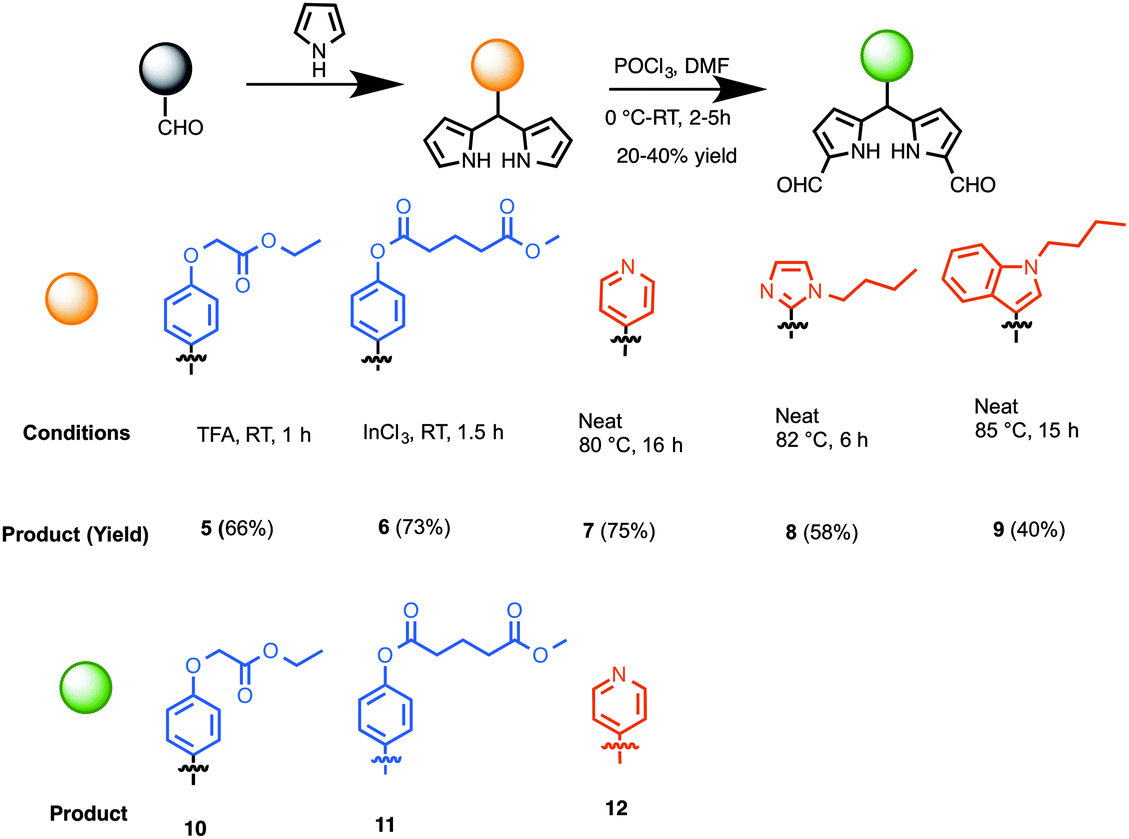 | ||
| Scheme 2 Synthesis of conjugatable and heterocyclic dipyrromethanes, and 1,9-diformyldipyrromethanes as porphyrin precursors. | ||
Using a protocol developed by us earlier,23 the synthesis of trans-AB porphyrins was achieved by the condensation of 1,9-diformyldipyrromethanes and dipyrromethanes in the presence of excess n-propylamine in THF and subsequent metalation with Zn(OAc)2. Thus, reaction of 1,9-diformyldipyrromethane 12 and dipyrromethane 5 delivered porphyrin 13 as a purple solid (Scheme 3, 20% yield). However, acquisition of spectroscopic data for the characterization of porphyrin 13 was largely impeded probably because of self-aggregation, encountered due to the co-ordination between pyridyl nitrogen and zinc metal. We then sought to remove the central zinc atom from the porphyrin macrocycle. Demetalation of trans-AB porphyrin 13 was achieved by treating a solution of 13 in CH2Cl2 with excess TFA at room temperature (25 °C) for 2 h, which upon subsequent purification afforded porphyrin 14. To our delight, the free-base porphyrin 14 provided satisfactory characterization data. Next, the ester group in porphyrin 14 was subjected to base mediated hydrolysis to engender carboxylic acid tether required for conjugation to nanoparticles. Thus, treating a solution of 14 in THF with LiOH·H2O at room temperature and subsequent acidification with 10% HCl afforded porphyrin 15 containing a carboxylic acid handle. The pyridyl nitrogen in porphyrin 15 was methylated with excess methyl iodide to obtain 16 (80% yield in two steps).
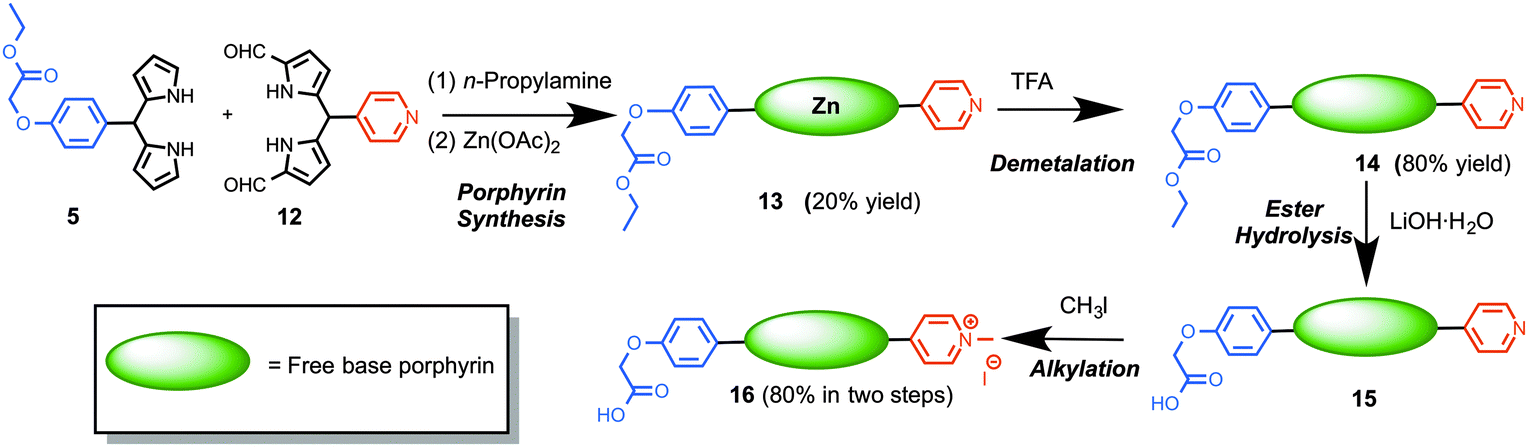 | ||
| Scheme 3 Preparation of a trans-AB porphyrin with a cationic and a conjugatable handle. | ||
Nanophotosensitizer preparation through the surface modification of metal nanomaterials with conjugatable porphyrins
Central to this study was to demonstrate the conjugation of porphyrin 16 with metal NPs, prepared by us previously.27 Silver and gold nanoparticles (AgNPs and AuNPs) were prepared from Camellia sinensis (CS, green tea) and Potentilla fulgens (PF, vajradanti) in high yields. Three types of bioinspired nanoparticles (CS-AgNPs, CS-AuNPs and PF-AuNPs) were developed where plant materials acted as reducing, stabilizing and capping agents. The biomaterials present (from plant extracts) on the surface of the metal nanoparticles are a class of flavonoids that are rich in hydroxyl functionalities.27 The carboxylic acid moiety in the porphyrin 16 could be easily coupled with the hydroxyl groups present in metal NPs via an ester linkage (Scheme 2, top panel).The conjugation of porphyrin 16 and metal NPs was carried out via EDC mediated coupling reaction. The analytical data (FTIR, UV-vis, and HPLC) obtained for the samples prepared from these reactions strongly support the formation of conjugated nanophotosensitizers from porphyrin 16. For example, FTIR data showed a peak at ∼1732 nm as a characteristic of ester functionality present in porphyrin-nanoconjugates (see ESI,† Fig. S1–S3). UV-visible spectra of porphyrin-nanoconjugates are distinct from those of their unconjugated metal NPs or porphyrin 16 (Fig. 2a–g). For example, the UV-visible spectrum of porphyrin 16 shows a strong intense band at 410 nm, while UV-visible spectra of silver nanoparticles in our study showed a peak at 450 nm, and UV-visible spectra of gold nanoparticles appeared at 535 nm. For example, the porphyrin-nanoconjugates of silver nanoparticles (CS-AgNPs) showed broad absorption peaks at 411 and 450 nm (Fig. 2a). Similarly, porphyrin conjugated gold nanoparticles (both CS-AuNPs and PF-AuNPs) also showed two peaks at 411 and 535 nm (Fig. 2b and c). Further confirmation of porphyrin-nanoconjugates is apparent from the fact that treatment with NaOH resulted in the hydrolysis of the ester bond formed between porphyrin and metal nanoparticles. Centrifugation of the supernatant revealed the appearance of only one peak at 411 nm upon absorption spectroscopy indicating release of the porphyrin from the nanomaterial surface.
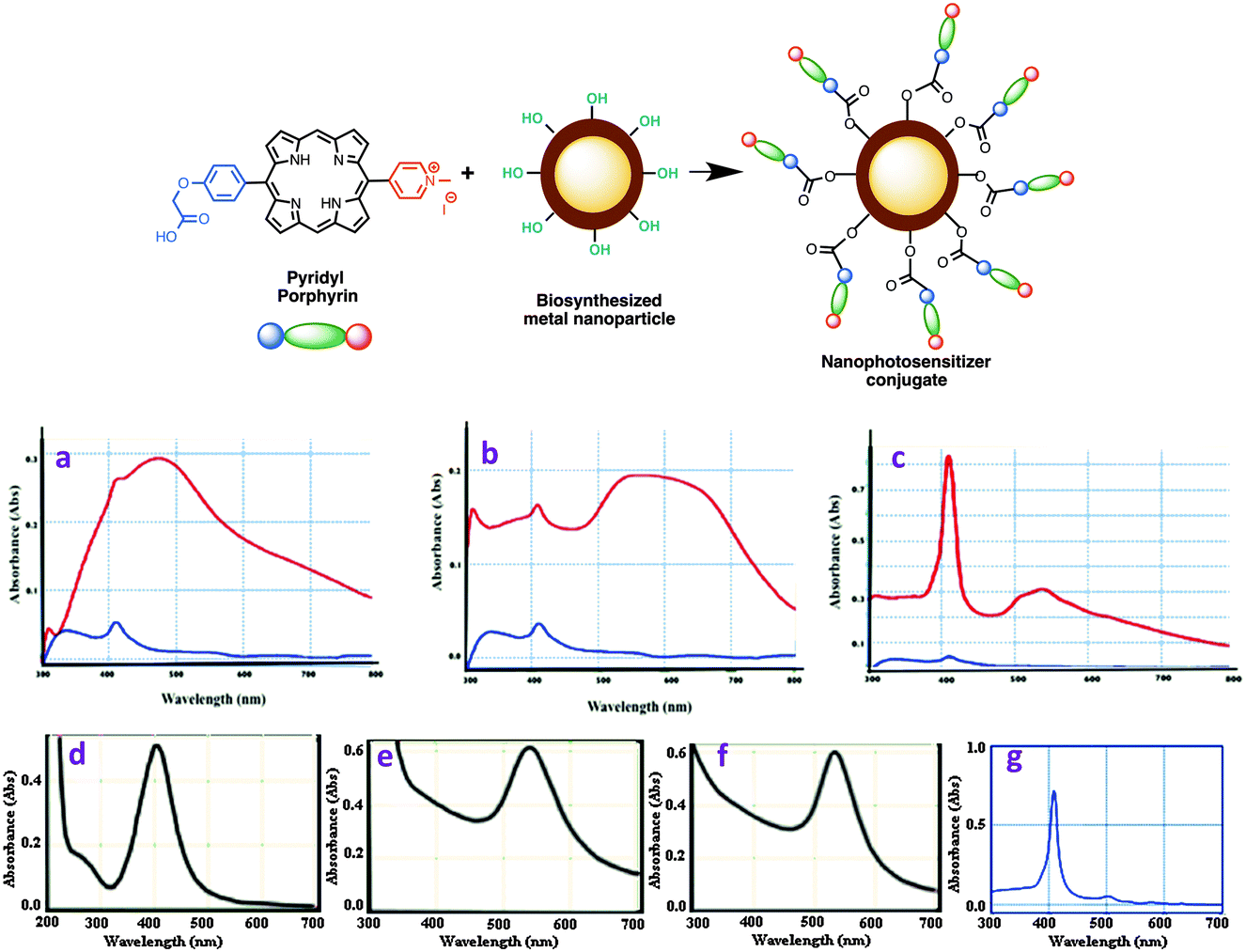 | ||
| Fig. 2 (top panel) Development of nanophotosensitizer conjugates via the condensation of a conjugatable porphyrin on the surface of biosynthesized metal nanoparticles. (bottom panel) Conjugation of porphyrin with metallic NPs: (a) conjugation with CS-AgNPs; (b) conjugation with CS-AuNPs, and (c) conjugation with PF-AuNPs. Red line: UV-vis spectra before NaOH treatment; blue line: UV-vis spectra after NaOH treatment; absorption spectra of (d) CS-AgNPs, (e) CS-AuNPs (f) PF-AuNPs and (g) free pyridyl porphyrin 16. | ||
As photosensitizers show therapeutic efficacy at lower concentration,2 photosensitizers loaded with nanoparticles at the 12–16% level have successfully been delivered. The ability of porphyrin 16 loaded on CS-AgNPs, CS-AuNPs, and PF-AuNPs was estimated to be 16, 12, and 16%, respectively (by the HPLC method, Table 1). The observed loading efficiency of 16 is proportional to the number of hydroxyl functionalities present on metal nanoparticles. Notably, the loading efficiency of porphyrin 16 is found to be significant for drug delivery and also leaves room to develop phototheranostic nanoagents by the incorporation of diagnostic and targeting agents on the nanoparticles. Overall these bioinspired photosensitizers are substantial precursors for phototheranostic nanoagents for finding better treatment using PDT or dual PDT–PTT.
| Parameters | Pyridyl porphyrin (16) [y = 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 687347x, R2 = 0.999] 687347x, R2 = 0.999] |
|||
|---|---|---|---|---|
| Nanoparticle | Blank porphyrin | CS-AgNPs | CS-AuNPs | PF-AuNPs |
| a Area under the curve in the HPLC chromatogram of supernatant solutions containing pyridyl porphyrin after conjugation reaction and centrifugation. b Concentration of pyridyl porphyrin in the supernatant solution, calculated using linear regression equation y = mx + c. c Initial concentrations of pyridyl porphyrin used in the conjugation reaction. d Percent conjugation of pyridyl porphyrin. This was calculated using the equation, Conj(%) = {[Ci − Co]/[Ci]} × 100. e Percent loading of pyridyl porphyrin on the surface of nanoparticles. This was calculated using the equation, Ld(%) = Conj(%) − Conj(%)blank. | ||||
| AUCa | 40251 |
34011 |
35212 |
34152 |
| C o (μg mL−1) | 24 | 20 | 21 | 20 |
| C i (μg mL−1) | 25 | 25 | 25 | 25 |
| Conjd (%) | 4 | 20 | 16 | 20 |
| L d (%) | 0 | 16 | 12 | 16 |
Stability studies of porphyrin-nanoconjugates under physiological conditions
After successful synthesis of porphyrin-nanoconjugates we investigated their stability under physiological conditions.5 For this purpose, each of the nanoconjugates were dispersed in MES buffer (pH 7.4) and incubated at 37 °C. The nanoconjugate stability was monitored at various time points (0–48 h) by absorption spectroscopy. The colloidal solution of the nanoconjugates (CSAuNP-POR, CSAgNP-POR and PFAuNP-POR) showed similar absorbance values (at 410 nm) at various time points (Fig. 3a). The absorbance (at 410 nm) of the centrifuged supernatant of each sample slightly increased due to the presence of porphyrin in the solution (Fig. 3b). This confirms the stability of porphyrin-nanoconjugates. All the nanoconjugates showed negligible decomposition under physiological conditions. This stability study under physiological conditions indicates that the nanoconjugates can be applied in biological systems for further studies involving photodynamic therapy.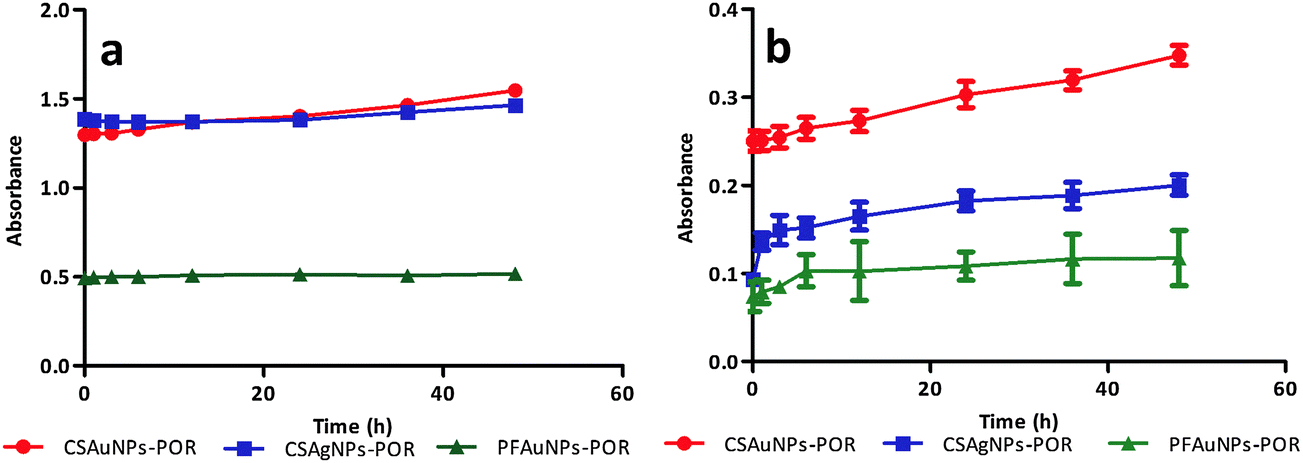 | ||
| Fig. 3 Stability study of porphyrin-nanoconjugates under physiological conditions. Graph (a) showing the absorbance of the colloidal solution of nanoconjugates at various incubation time intervals. Graph (b) showing the gradual increment in absorbance of the supernatant solution of nanoconjugates after the centrifugation due to the release of porphyrin in the solution under physiological conditions with time. | ||
Conclusions
In summary, photosensitizer metal nanoconjugates were developed as better alternatives to conventional photosensitizers for PDT or combined PDT–PTT applications. First, various building blocks (aldehydes, dipyrromethanes and diformyldipyrromethanes) containing heterocyclic (pyridyl, imidazole, and indole) and conjugatable moieties as precursors to porphyrins were synthesized. Later, rational syntheses of versatile cationic (positively charged) and conjugatable tetrapyrrolic macrocycles were accomplished. Unlike the statistical route, which generates a mixture of porphyrinic derivatives, the rational route resulted in single porphyrinic species in highly pure form. Afterwards, newly constructed conjugatable tetrapyrrolic photosensitizers were characterized by different analytical techniques including absorption spectroscopy, NMR and mass spectrometry. Taking the advantage of hydroxyl functionalities present on the periphery of bioinspired noble metal nanoparticles (silver and gold), porphyrins containing carboxyl tethers were attached on their surface via ester bond formation (through EDC mediated coupling). The loading efficiency of the photosensitizers on nanoscaffolds was determined by both absorption spectroscopy and HPLC methods. These nanophotosensitizers were designed in order to possess enhanced biocompatibility as observed by their stability under physiological conditions. These photosensitizer nanoconjugates can serve as PDT or dual PDT–PTT agents with improved efficacy compared to native PSs or NPs. Taken together, these bioengineered nanophotosensitizer probes are potential candidates for photomedicines.Experimental section
The known compounds were prepared by following the reported methods: ethyl 2-(4-formylphenoxy)acetate (1),23 1-butyl-imidazole-2-carbaldehyde (3a),29 1-butyl-indole-3-carbaldehyde (4a),30 5-[4-(ethoxycarbonylmethoxy)phenyl]dipyrromethane (5),23 biosynthesized silver and gold nanoparticles (CS-AgNPs, CS-AuNPs and PF-AuNPs).274-Formylphenyl methyl glutarate (2)
A sample of 4-hydroxybenzaldehyde (0.12 g, 1.0 mmol) was taken in THF (3 mL) at room temperature. The reaction mixture was cooled to 0 °C and TEA (0.15 g, 1.5 mmol) was added followed by the addition of methyl glutaryl chloride (0.25 g, 1.5 mmol). The reaction mixture was warmed to room temperature and stirred for 45 min. Upon reaction completion, the reaction mixture was diluted with ethyl acetate and aq. NH4Cl. The aqueous layer was extracted with ethyl acetate and the combined organic layer was separated and dried (Na2SO4). Evaporation of the solvent and preparative TLC [silica, hexane/ethyl acetate (3:1)] afforded a white sticky solid (0.20 g, 80%): 1H NMR (400 MHz) δ 1.93–2.00 (m, J = 16 Hz, 12 Hz, 2H), 2.49 (t, J = 12 Hz, 2H), 2.86 (t, J = 12 Hz, 2H), 3.76 (s, 3H), 7.30 (d, J = 8 Hz, 2H), 7.93 (d, J = 8 Hz, 2H), 10.00 (s, 1H); 13C NMR δ 20.0, 33.0, 51.1, 122.3, 131.2, 155.2, 170.7, 173.3, 190.9; HRMS obsd 252.1567, calcd 250.0841 [(M + 2H)+, for M = C13H14O5].
5-(Phenylmethylglutarate)dipyrromethane (6)
Following a general procedure for dipyrromethane synthesis,31 a solution of 4-formylphenyl methyl glutarate 2 (0.10 g, 0.40 mmol) in pyrrole (2.0 mL, 29 mmol) was flushed with argon for 10 min. A sample of InCl3 (22 mg, 0.12 mmol) was added and the reaction was allowed to proceed for 1.5 h. Upon completion, the reaction was quenched by the addition of powdered NaHCO3 (0.10 g, 1.2 mmol). The mixture was stirred for 45 min. The mixture was then filtered. The filtrate was concentrated at reduced pressure. Preparative TLC [silica, hexane/ethyl acetate, (3:1)] afforded the desired product as a dark brown sticky solid (107 mg, 73%): 1H NMR (400 Hz) δ 2.00–2.07 (m, 3H), 2.48 (t, J = 12 Hz, 2H), 2.64 (t, J = 12 Hz, 2H), 3.71 (s, 2H), 5.49 (s, 1H), 5.92 (s, 2H), 6.17 (m, 2H), 6.71 (t, J = 8 Hz, 2H), 7.04 (td, J = 8 Hz, 4 Hz, 2H), 7.22 (d, J = 8 Hz, 2H), 7.98 (br, 2H); 13C NMR 20.0, 32.9, 33.3, 43.37, 51.7, 107.3, 108.4, 117.4, 121.6, 129.41, 132.2, 139.7, 149.4, 171.6, 173.3; HRMS obsd 365.1508, calcd 366.1580 [(M − H)+, M = C21H22N2O4].
5-(4-Pyridyl)dipyrromethane (7)
Following a general procedure33 with modification, pyrrole (4.0 mL, 58 mmol) was flushed with nitrogen, 4-pyridine carboxaldehyde (0.54 g, 5.0 mmol) was added and the reaction mixture was heated for 22 h at 82 °C. Upon reaction completion, ethyl acetate was added and the entire mixture was concentrated at reduced pressure. Column chromatography [alumina, hexane/ethyl acetate (9:1)] afforded a grey-white low melting solid (0.84 g, 75%): 1H NMR (400 Hz) δ 5.47 (s, 1H), 5.91 (s, 2H), 6.10 (s, 2H), 6.74 (s, 2H), 7.13 (d, J = 12 Hz, 2H), 8.11 (br, 2H), 8.56 (d, J = 8 Hz, 2H); 13C NMR 43.4, 107.7, 108.7, 111.7, 117.9, 120.3, 122.4, 123.6, 126.5, 130.6, 149.9, 150.3, 151.2; HRMS obsd 224.1184 calcd 223.1109 [(M + H)+, M = C14H13N3].
5-(1-Butyl-imidazol-2-yl)dipyrromethane (8)
Following a general procedure for dipyrromethane synthesis31 with modification, pyrrole (1.0 mL, 14 mmol) was flushed with nitrogen, 1-butyl-2-imidazolecarboxaldehyde (3a) (0.16 g, 1.0 mmol) was then added and the reaction mixture was heated for 12 h at 82 °C. Upon reaction completion ethyl acetate was added to the reaction mixture and concentrated under reduced pressure. Column chromatography [alumina, DCM/methanol (99:1)] afforded the desired product as dark brown liquid (155 mg, 58%): 1H NMR (400 MHz) δ 0.93 (t, J = 7.2 Hz, 3H), 1.28–1.35 (m, 2H), 1.61–1.65 (m, 2H), 3.94 (t, J = 7.2 Hz, 2H), 6.69–6.71 (m, 2H), 6.85 (s, 1H), 7.05 (s, 1H), 9.24 (br, 2H); 13C NMR δ 13.6, 19.8, 32.9, 35.1, 46.1, 105.9, 108.1, 117.6, 126.9, 129.7, 146.8, 176.1; HRMS obsd 268.1655, calcd 268.1688 (C16H20N4).
5-(1-Butyl-indol-3-yl)dipyrromethane (9)
Following a general procedure for dipyrromethane synthesis31 with modification, pyrrole (1.0 mL, 14 mmol) was flushed with nitrogen, 1-butyl-3-indolecarboxaldehyde (4a) (0.20 g, 1.0 mmol) was added and the reaction mixture was heated for 13 h at 85 °C. Upon reaction completion ethyl acetate was added to the reaction mixture and concentrated under reduced pressure. Column chromatography (hexanes/ethyl acetate, 7:3) afforded the desired product as a dark brown sticky solid (127 mg, 40%): 1H NMR (400 Hz) δ 0.89–0.99 (m, 3H), 1.32–1.41 (m, 2H), 1.78–1.86 (m, 2H), 4.08 (t, J = 7.16 Hz, 2H), 5.79 (s, 1H), 6.07 (s, 2H), 6.19 (d, J = 4 Hz, 2H), 6.66 (d, J = 4 Hz, 2H), 6.90 (s, 1H), 7.04 (t, J = 16 Hz, 1H), 7.22 (t, J = 16 Hz, 1H), 7.36 (m, 2H), 8.02 (br, s, 2H); 13C NMR δ 13.8, 20.3, 32.4, 35.3, 46.2, 106.4, 108.4, 109.9, 115.3, 116.7, 119.2, 119.9, 121.7, 126.8, 132.8, 137.0; HRMS obsd 316.30, calcd 317.18 [(M − H)+ due to the presence of an acidic proton at the methyne bridge, M = C21H23N3].
1,9-Diformyl-5-4-(phenylmethylglutarate)dipyrromethane (11)
Following a general procedure of formylation,23 a solution of dipyrromethane 6 (0.16 g, 0.50 mmol) in DMF (0.5 mL) at 0 °C under argon was treated with POCl3 (0.190 g, 1.25 mmol). The mixture was allowed to warm to room temperature. Stirring was continued for 2 h. The mixture was then poured into aqueous NaCO3 (10% solution) to make it basic, ethyl acetate was added and an organic layer was extracted. The organic layer was washed (water and brine), dried (Na2SO4), and concentrated. Preparative TLC [silica, hexane/ethyl acetate (7:3)] was performed to isolate the desired compound as orange-brown colored oil (50 mg, 24%): 1H NMR (400 Hz) δ 2.00–2.10 (m, 2H), 2.49 (t, J = 16 Hz, 2H), 2.66 (t, J = 12 Hz, 2H), 3.72 (s, 3H), 5.59 (s, 1H), 6.08–6.09 (m, 2H), 6.89–6.91 (m, 2H), 7.10 (d, J = 9 Hz, 2H), 7.22–7.35 (m, 2H), 9.20 (s, 2H), 10.32–10.48 (br, 2H); HRMS obsd 419.27, calcd 422.15 [(M − 3H)+, M = C23H22N2O6].
1,9-Diformyl-5-(4-pyridyl)dipyrromethane (12)
Following a general procedure of formylation,23 a solution of pyridyl dipyrromethane 7 (45 mg, 0.20 mmol) in DMF (0.2 mL) at 0 °C was treated with POCl3 (68 mg, 0.44 mmol) under argon atmosphere. The mixture was allowed to warm to room temperature. Stirring was continued for 2 h. The mixture was then poured into aqueous Na2CO3 (10% solution) to make it basic, ethyl acetate was added and an organic layer was extracted. The organic layer was washed (water and brine), dried (Na2SO4) and concentrated. Preparative TLC [alumina, hexanes/ethyl acetate (7:3)] was performed to isolate the desired compound as dark orange oil (13 mg, 23%): 1H NMR (400 Hz) δ 5.60 (s, 1H), 6.07–6.09 (m, 2H), 6.90–6.91 (m, 2H), 7.26–7.28 (m, 2H), 8.58–8.60 (m, 2H), 9.20 (s, 2H), 10.4 (br, 2H); 13C NMR δ 14.2, 22.7, 29.7, 31.9, 43.8, 60.4, 77.0, 111.5, 111.9, 122.4, 123.6, 132.9, 139.9, 148.38, 150.5, 179.3.HRMS obsd 280.1078 calcd 279.1008 [(M − H)+, M = C16H13N3O2].
Zn(II)-5-[4-(ethoxycarbonylmethoxy)phenyl]-15-(4-pyridyl)porphyrin (13)
Following a general procedure23 with modification, a solution of diformyldipyrromethane 10 (28 mg, 0.10 mmol) and n-propylamine (15 mg) in THF (1 mL) was stirred at room temperature for 1 h, and then TLC was checked. After removal of excess n-propylamine and THF under reduced pressure, the residue and dipyrromethane 5 (32 mg, 0.10 mmol) were dissolved in ethanol (8 mL). The mixture was then treated with Zn(OAc)2 (0.18 g, 1.0 mmol)) and refluxed open to air for 18 h. Since TLC indicated non-completion of the reaction, it was continued for an additional 5 h. Afterwards TLC was checked using a DCM/MeOH (49:1) solvent system, which showed red fluorescence as an indication of porphyrin formation. The reaction mixture was first filtered through celite to remove polar impurities. The resulting material was purified by column chromatography (alumina, loading and packing in DCM, then DCM:MeOH (49:1) to give dark purple porphyrin 13 (13 mg, 20%). Though a single compound was isolated from column purification, even after repeated attempts (using various solvents) clear NMR could not be obtained due to extensive aggregation resulting from the co-ordination of zinc metal and a lone pair of pyridyl nitrogen present in the porphyrin: MALDI obsd 627.1, calcd 627.4 (C35H25N5O3Zn). Due to severe aggregation caused by the presence of pyridyl nitrogen and zinc metal, clear NMR data could not be obtained. Hence, this porphyrin was directly used in the next step without further characterization.
Demetalation of zinc(II)-trans AB-porphyrin: 5-[4-(ethoxycarbonylmethoxy)phenyl]-15-(4-pyridyl)porphyrin (14)
Following a method of porphyrin demetalation,32 the reaction of a solution of zinc porphyrin 13 (19 mg, 0.030 mmol) in DCM (3 mL) and TFA (0.23 mL) was allowed to proceed for 4 h at room temperature (green color appeared). The mixture was poured into aqueous solution of sodium bicarbonate. The mixture was extracted into DCM, dried (Na2SO4), and concentrated to afford a dark purple solid (15 mg, 89%). After demetalation clear NMR was obtained. 1H NMR (400 Hz) δ −3.18 (s, 2H), 1.35 (t, J = 16 Hz, 3H), 4.35 (q, J = 8 Hz, 2H), 5.08 (s, 2H), 7.43 (d, J = 12 Hz, 2H), 8.15–8.27 (m, 4H), 9.00–9.12 (m, 6H), 9.65 (q, J = 8 Hz, 4H), 10.61 (s, 2H); MALDI obsd 565.6, calcd 565.2 [C35H27N5O3]; HPLC tR = 8.40 min (using isocratic solvent system 90% acetonitrile–10% H2O–0.1% TFA ran over 40 min, purity > 95%).N-Alkylation followed by ester hydrolysis of trans AB-pyridyl porphyrin: 5-[4-(carbonylmethoxy)phenyl]-15-(1-methylpyridinium-4-yl)porphyrin (16)
Following a general procedure26 with modification, a solution of trans-AB porphyrin 14 (11 mg, 0.020 mmol) was dissolved in THF (2 mL). A sample of LiOH·H2O (8 mg in 1 mL of water) was added and the reaction was allowed to proceed for 2 h at room temperature. When reaction was completed, the reaction mixture was cooled to 0 °C. The pH was then adjusted to slightly acidic (∼pH 3–4) by adding 10% HCl. Ethyl acetate was then added to the reaction mixture and the organic layer was separated and concentrated affording porphyrin 16 as a dark green solid. This porphyrin was used directly in the next step without further characterization. Porphyrin 15 was suspended in DCM (1 mL) and treated with excess methyl iodide (0.1 mL). The mixture was stirred at room temperature for 15 h. After completion of the reaction, the mixture was washed with ethyl acetate and dried to afford porphyrin 16 (9.0 mg, 80% yield in two steps). MALDI obsd 552.5 calcd 552.2 [C34H26N5O3]; HPLC tR = 5.71 min (using a isocratic solvent system 90% acetonitrile–10% H2O–0.1% TFA ran over 40 min, purity >95%).Surface functionalization of Camellia sinensis mediated synthesized silver nanoparticles with pyridyl porphyrin
Following a general procedure for direct conjugation on the NP surface,27 a stock solution of conjugatable pyridyl porphyrin 16 was prepared in DMSO (2 mg mL−1). Purified CS-AgNPs (250 μg) were interacted with pyridyl porphyrin (100 μg) and 13 mM solution of 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide (EDC, coupling agent) in the presence of 50 mM HEPES buffer, and the final volume of reaction mixture was 2 mL, which was incubated at 30 °C and 120 rpm for 18 h in the dark. After incubation, the reaction mixture was centrifuged at 14600 rpm for 60 min, the supernatant was discarded and the pellet was dispersed in water containing 5% DMSO. This procedure was repeated until a clear supernatant was observed. The porphyrin-nanoconjugate was characterized by absorption spectroscopy, HPLC and FTIR spectroscopy (see ESI† for details).
Surface functionalization of Camellia sinensis mediated synthesized gold nanoparticles with pyridyl porphyrin
Following a general procedure,27 purified CS-AuNPs (250 μg) were interacted with conjugatable pyridyl porphyrin 16 (100 μg) and 13 mM solution of EDC in the presence of 50 mM HEPES buffer, and the final volume of reaction mixture was 2 mL, which was incubated at 30 °C and 120 rpm for 18 h in the dark. After incubation, the reaction mixture was centrifuged at 14600 rpm for 60 min, the supernatant was discarded and the pellet was dispersed in water containing 5% DMSO. This procedure was repeated until a clear supernatant was observed. The porphyrin-nanoconjugate was characterized by absorption spectroscopy, HPLC and FTIR spectroscopy (see ESI† for details).
Surface functionalization of Potentilla fulgens mediated synthesized gold nanoparticles with pyridyl porphyrins
Following a general procedure with modification,27 a sample of purified PF-AuNPs (500 μg) was interacted with pyridyl porphyrin 16 (200 μg) along with 12 mM solution of EDC and the final volume was made up to 5 mL using 50 mM HEPES buffer (pKa 7.4). The reaction mixture was then incubated in a shaker at 30 °C and 200 rpm for 18 h (in the dark). After the incubation time the reaction mixture was centrifuged at 14800 × g for 30 min, the supernatant was discarded and the pellet was re-dispersed in DMSO. This procedure was repeated until a clear supernatant was observed. The porphyrin-nanoconjugate was characterized by absorption spectroscopy, HPLC and FTIR spectroscopy (see ESI† for details).
Stability studies of porphyrin-nanoconjugates under physiological conditions
The porphyrin-nanoconjugates (50 μL, 50 μg mL−1, in DMSO) were dispersed in 950 μL of MES buffer:DMSO (99.5:0.5), pH 7.4, and incubated at 37 °C. The sample of each porphyrin-nanoconjugate was collected at various time points (0, 1, 3, 6, 12, 24, 36 and 48 h). The absorbance of each collected sample was recorded at 410 nm and the values were plotted against time. Each sample was then individually centrifuged at 14000 rpm for 1 h. The absorbance of the supernatant solutions was recorded at 410 nm and the values were plotted against time. Both plots were carefully analyzed and investigated whether the porphyrin has been released from the nanoconjugate or not.
Competing financial interest
The authors declare no competing financial interest.Acknowledgements
J. B. is supported by the Department of Science and Technology (DST) Women Scientists Scheme A (WOS-A), Govt. of India. S. K. would like to acknowledge the receipt of the fellowship from Department of Biotechnology (DBT). The receipt of the DST Inspire PhD fellowship is acknowledged by N. T. The authors would like to thank P. K. Aili, B. S. Rathore and R. Bhimpuria for their technical assistance in carrying out this research.References
- J. Bhaumik, A. K. Mittal, A. Banerjee, Y. Chisti and U. C. Banerjee, Nano Res., 2015, 8, 1373–1394 CrossRef CAS.
- P. Mroz, J. Bhaumik, D. K. Dogutan, Z. Aly, Z. Kamal, L. Khalid, H. L. Kee, D. F. Bocian, D. Holten, J. S. Lindsey and M. R. Hamblin, Cancer Lett., 2009, 282, 63–76 CrossRef CAS PubMed.
- J. R. McCarthy, E. Korngold, R. Weissleder and F. A. Jaffer, Small, 2010, 6, 2041–2049 CrossRef CAS PubMed.
- D. E. Dolmans, D. Fukumura and R. K. Jain, Nat. Rev. Cancer, 2003, 3, 380–387 CrossRef CAS PubMed.
- J. Bhaumik, J. R. McCarthy and R. Weissleder, J. Org. Chem., 2009, 74, 5894–5901 CrossRef CAS PubMed.
- H. L. Kee, J. Bhaumik, J. R. Diers, P. Mroz, M. R. Hamblin, D. F. Bocian, J. S. Lindsey and D. Holten, J. Photochem. Photobiol., A, 2008, 200, 346–355 CrossRef CAS PubMed.
- C. Muthiah, J. Bhaumik and J. S. Lindsey, J. Org. Chem., 2007, 72, 5839–5842 CrossRef CAS PubMed.
- M. Ethirajan, Y. Chen, P. Joshi and R. K. Pandey, Chem. Soc. Rev., 2011, 40, 340–362 RSC.
- L. B. Josefsen and R. W. Boyle, Theranostics, 2012, 2, 916–966 CrossRef CAS PubMed.
- M. Ethirajan, Y. Chen, P. Joshi and R. K. Pandey, Chem. Soc. Rev., 2011, 40, 340–362 RSC.
- J. R. McCarthy, J. Bhaumik, M. R. Karver, S. S. Erdem and R. Weissleder, Mol. Oncol., 2010, 4, 511–528 CrossRef CAS PubMed.
- L. Dennany, P. Sherrell, J. Chen, P. C. Innis, G. G. Wallace and A. I. Minett, Phys. Chem. Chem. Phys., 2010, 12, 4135–4141 RSC.
- B. Jang, J. Park, C. Tung, I. Kim and Y. Choi, ACS Nano, 2011, 5, 1086–1094 CrossRef CAS PubMed.
- T. Curry, R. Kopelman, M. Shilo and R. Popovtzer, Contrast Media Mol. Imaging, 2014, 9, 53–61 CrossRef CAS PubMed.
- L. Vigderman and E. R. Zubarev, Adv. Drug Delivery Rev., 2012, 65, 663–676 CrossRef PubMed.
- T. T. Zhao, K. Yu, L. Li, T. S. Zhang, Z. P. Guan, N. Y. Gao, P. Y. Yuan, S. Li and S. Q. Yao, et al. , ACS Appl. Mater. Interfaces, 2014, 6, 2700–2708 CAS.
- K. Záruba, J. Králová, P. Rezanka, P. Poucková, L. Veverková and V. Král, Org. Biomol. Chem., 2010, 8, 3202–3206 Search PubMed.
- W. S. Kuo, C. N. Chang, Y. T. Chang and C. S. Yeh, Chem. Commun., 2009, 4853–4855 RSC.
- M. Guo, H. J. Mao, Y. L. Li, A. J. Zhu, H. He, H. Yang, Y. Y. Wang and X. Tian, et al. , Biomaterials, 2014, 35, 4656–4666 CrossRef CAS PubMed.
- Q. Chen, C. Wang, L. Cheng, W. W. He, Z. P. Cheng and Z. Liu, Biomaterials, 2014, 35, 2915–2923 CrossRef CAS PubMed.
- S. Wang, G. Kim, Y. E. K. Lee, H. J. Hah, M. Ethirajan, R. K. Pandey and R. Kopelman, ACS Nano, 2012, 6, 6843–6851 CrossRef CAS PubMed.
- S. S. Kelkar and T. M. Reineke, Bioconjugate Chem., 2011, 22, 1879–1903 CrossRef CAS PubMed.
- J. Bhaumik, Z. Yao, E. K. Borbas, M. Taniguchi and J. S. Lindsey, J. Org. Chem., 2006, 71, 8807–8817 CrossRef CAS PubMed.
- M. Liu, M. Ptaszek, O. Mass, D. F. Minkler, R. D. Sommer, J. Bhaumik and J. S. Lindsey, New J. Chem., 2014, 38, 1717–1730 RSC.
- M. Ptaszek, J. Bhaumik, H.-J. Kim, M. Taniguchi and J. S. Lindsey, Org. Process Res. Dev., 2005, 9, 651–658 CrossRef CAS PubMed.
- J. R. McCarthy, J. Bhaumik, N. Merbouh and R. Weissleder, Org. Biomol. Chem., 2009, 7, 3430–3436 CAS.
- J. Bhaumik, N. S. Thakur, P. K. Aili, A. Ghanghoriya, A. K. Mittal and U. C. Banerjee, ACS Biomater. Sci. Eng., 2015, 6, 382–392 CrossRef.
- A. K. Mittal, J. Bhaumik, S. Kumar and U. C. Banerjee, J. Colloid Interface Sci., 2014, 415, 39–47 CrossRef CAS PubMed.
- M. Seredyuk, M. C. Muñoz, M. Castro, T. Romero-Morcillo, A. B. Gaspar and J. A. Real, et al. , Chem. – Eur. J., 2013, 19, 6591–6596 CrossRef CAS PubMed.
- L. R. Marcin, A. Mendi, F. Higgins, C. Zusi, Y. Zhang and M. F. Dee, et al. , Bioorg. Med. Chem. Lett., 2011, 21, 537–541 CrossRef CAS PubMed.
- J. K. Laha, S. Dhanalekshmi, M. Taniguchi, A. Ambroise and J. S. Lindsey, Org. Process Res. Dev., 2003, 7, 799–812 CrossRef CAS.
- Z. Yao, J. Bhaumik, S. Dhanalekshmi, M. Ptaszek, P. R. Rodriguez and J. S. Lindsey, Tetrahedron, 2007, 63, 10657–10670 CrossRef CAS PubMed.
- D. Gryko and J. S. Lindsey, J. Org. Chem., 2000, 65, 2249–2252 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Materials and methods; FTIR spectra; HPLC data and NMR spectra of selected compounds. See DOI: 10.1039/c5nj02056e |
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2016 |