DOI:
10.1039/C5NJ02124C
(Paper)
New J. Chem., 2016,
40, 402-412
Feasible energy level tuning in polymer solar cells based on broad band-gap polytriphenylamine derivatives
Received
(in Montpellier, France)
10th August 2015
, Accepted 23rd October 2015
First published on 27th October 2015
Abstract
A series of versatile broad band-gap alternating copolymers (P1, P2, P3 and P4) based on triphenylamine (TPA) and benzofurazan derivatives, differing in the substituted groups [–OC8H17, –C8H17, –CF3, –(CF3)2] in their triphenylamine units, were designed and synthesized by Suzuki polycondensation. The relationships between the substituted groups in TPA and the highest occupied molecular orbital (HOMO) energy levels, as well as the open circuit voltages (Vocs), were investigated in detail. The HOMO levels of these four polymers decreased sequentially when the substituted groups shifted from electron-donating groups [–OC8H17, –C8H17] to electron-withdrawing groups [–CF3, –(CF3)2], which led to the successive increase in Vocs of the polymer solar cells (PSCs) based on these polymers. Through the characterization of photovoltaic performance, the highest Voc, which reached up to 1.00 V, was achieved by the polymer with bis(trifluoromenthyl) substituted group (P4), which is one of the highest Voc values based on polytriphenylamine derived polymers for PSCs. Among these polymers, the one with octyl side chain (P2) showed the best photovoltaic performance with the highest short circuit current density (Jsc) and fill factor (FF), giving a Jsc of 4.84 mA cm−2, FF of 50%, Voc of 0.80 V and power conversion efficiency (PCE) of 2.22%.
Introduction
Bulk-heterojunction polymer solar cells (BHJ-PSCs), due to their versatile merits of low cost, large-scale fabrication, flexibility and diversity of polymer donors, have attracted considerable interest, both academically and industrially. To date, some novel PSCs with high photovoltaic performance and power conversion efficiencies (PCEs) exceeding 10% have been developed.1–3 Potentially, this success can lead to the application of PSCs in generating electricity in the future.
Generally, two components that contain narrow bandgap polymers and work as electron donators and electron-accepting units (acceptors) are utilized widely in BHJ-PSCs. For acceptors, fullerene derivatives are used comprehensively due to their advanced properties of high electron-accepting capability and electron mobility.4–6 On the other hand, narrow bandgap polymeric donors are another significant part of BHJ-PSCs. To obtain high photovoltaic performance, these donors should meet specific requirements: (1) high stability, (2) good solubility, (3) narrow band gap, (4) high molar extinction coefficient, (5) deep highest occupied molecular orbital (HOMO), and (6) good miscibility with acceptors.7
Among these requirements for polymer donors, a deep HOMO energy level is an indispensable parameter for evaluating polymers, because the open circuit voltage (Voc) is proportional to the energy level difference between the HOMO energy level of donor and lowest unoccupied molecular orbital (LUMO) energy level of acceptor.8 If the HOMOs of polymer donors are much deeper, higher Vocs in PSCs will be facilely accessible. Therefore, researchers should select molecular units with deep HOMOs, such as fluorene,9 indenofluorene,10 benzene,11 carbazole,12–15 indolocarbazole,16 diindenothieno[2,3-b]thiophene ladder-type hexacyclic arene17 and pyrrolo[3,4-g]quinoxaline-6,8-dione, to construct polymers.18 However, some of these units are not suitable for the construction of highly efficient narrow bandgap polymers due to their disadvantages of large band gaps and low hole mobility, for example, fluorene and benzene. Alternatively, another efficient strategy to achieve deep HOMO energy levels in polymer donors is the introduction of electron-withdrawing groups on the side chains of aromatic moieties. Liang et al. obtained high-efficiency PSCs with a higher Voc of 0.76 V by introducing the electron-withdrawing fluorine group on thieno[3,4-b]thiophene as compared to that of 0.68 V based on the non-fluorine substituted polymer.19,20 You's group, through the introduction of fluorinated benzothiadiazole21 and benzotriazole22 into the backbones of polymers, acquired a series of versatile polymers with enhanced Voc values. In addition, Huang et al. developed a new series of novel two-dimensional (2D) narrow bandgap polymers containing strong electron-withdrawing groups in the side chains of the polymers, such as malononitrile and 1,3-diethyl-2-thiobarbituric acid. Through the introduction of these electron-withdrawing groups, Voc values can not only be readily increased, but the band gaps of polymers are also decreased simultaneously, which is ascribed to the strong internal charge-transfer (ICT) effect between electron-rich polymer main chains and side groups. On the basis of the strategy for achieving 2D narrow bandgap polymers, the Voc value can reach 0.99 V, which is an excellent result in BHJ-PSCs.23,24 However, due to the process of post-linking reaction between polymers and formyl groups and strong electron-withdrawing groups, it is not a certain guarantee that every formyl group is reacted completely, and ultimately this possibly affects the batch-to-batch repeatable production of polymers.
Triphenylamine (TPA), which is a versatile unit, is utilized widely in organic light-emitting diodes (OLEDs),25–27 small molecular organic solar cells (OSCs)28–30 and organic field-effect transistors (OFETs)31,32 due to its excellent merits of low cost, feasible chemical functionalization, thermal stability, and high hole mobility. Normally, TPA, which is a strong electron-donating moiety, acts as a donor in the preparation of narrow bandgap polymers. However, very reactive points exist in the TPA backbone, which can be used to decorate it, and different substituting groups can be readily introduced to modify the chemical and physical properties of TPA. When electron-donating groups are introduced, the HOMOs of polymers are promoted; in contrast, the HOMOs decrease by the introduction of electron-withdrawing groups into the polymer side chains. Herein, we developed a series of TPA based broad bandgap polymers with different substituting groups ranging from the electron-donating groups of octyloxy [–OC8H17] and octyl [–C8H17] to the electron-withdrawing groups of trifluoromethyl [–CF3] and bis(trifluoromethyl) [–(CF3)2], and four polymers P1, P2, P3 and P4, were prepared accordingly. Through the introduction of these groups, the HOMO energy levels of the corresponding polymers can be tuned readily from −5.15, −5.23, −5.41, to −5.46 eV, respectively, and ultimately the Voc values in PSCs based on these polymers were simultaneously adjusted from 0.70, 0.80, 0.95 to 1.00 V. Therefore, this is a facile strategy to tune the energy levels in polymer donors by substituting different groups (electron-donating or electron-withdrawing) into the polymer side chains.
Experimental section
Characterization and instrumentation
1H nuclear magnetic resonance (NMR) and 13C NMR characterization were carried out on a Bruker 300 MHz or 600 MHz DRX spectrometer with tetramethylsilane (TMS) as the internal reference. The weight-averaged molecular weight (Mw) and number-averaged molecular weight (Mn) were determined on a waters gel permeation chromatography (GPC) system using linear polystyrene as standard and tetrahydrofuran (THF) as eluent. Cyclic voltammetry (CV) was carried out on a CHI600D electrochemical workstation with a standard three electrodes cell using a platinum (Pt) working electrode and Pt wire counter electrode, against a saturated calomel electrode (SCE) as the reference electrode at a scan rate of 50 mV s−1 in a nitrogen-saturated anhydrous solution of 0.1 mol L−1 tetrabutylammonium hexafluorophosphate (Bu4NPF6) in acetonitrile versus ferrocene/ferrocenium (Fc/Fc+) as the internal reference. UV-vis absorption spectra were obtained on an HP 8453 spectrophotometer. Tapping-mode atomic force microscopy (AFM) was performed on a Veeco Nanoscope V scanning probe microscope for the identification of topography and to obtain blend film phase images.
Device fabrication
All the devices were fabricated on ITO-coated glass substrates. A thin layer (40 nm) of PEDOT:PSS (Clevios 4083) was spin-casted onto UV-ozone treated ITO substrates at 4000 rpm for 40 s, and then baked at 140 °C for 15 min in air. The solutions containing the polymer and PC61BM with different weight ratios in o-dichlorobenzene (ODCB) were spin-casted inside a glove-box with nitrogen at 1200 rpm for 40 s to form active layers (∼80 nm). Under the device structure of ITO/PEDOT-PSS/active layer/poly[(9,9-dioctyl-2,7-fluorene)-alt-(9,9-bis(3′-(N,N-dimethylamino)propyl)-2,7-fluorene)] (PFN)/Al, the PFN layer (5 nm) was prepared by spin-coating a mixed solution (0.2 mg ml−1) in methanol with a small amount of acetic acid (volume ratio: 100![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) before cathode evaporation. The aluminum cathode (∼80 nm) was then thermally evaporated under vacuum (∼10−6 Torr) through a shadow mask defining an active area of 0.16 cm2. Current density–voltage (J–V) curves were obtained using a Keithley 2400 multimeter under AM 1.5 G solar illumination at 87 mW cm−2 and a Thermal-Oriel 150 W solar simulator. Incident photon-to-current conversion efficiency (IPCE) values were acquired with a monochromator and calibrated with a standard silicon photodiode. The hole mobility data of neat polymer films were characterized using the space charge limited current (SCLC) method with the device structure of ITO/PEDOT:PSS (40 nm)/active layer/MoO3 (10 nm)/Al (80 nm). Preliminarily, PEDOT:PSS was spin coated on an ITO covered glass substrate; then, active layers were prepared from an ODCB solution of 1% concentration by spin casting. Finally, MoO3 and Al layers were deposited via thermal evaporation under high vacuum.
:1) before cathode evaporation. The aluminum cathode (∼80 nm) was then thermally evaporated under vacuum (∼10−6 Torr) through a shadow mask defining an active area of 0.16 cm2. Current density–voltage (J–V) curves were obtained using a Keithley 2400 multimeter under AM 1.5 G solar illumination at 87 mW cm−2 and a Thermal-Oriel 150 W solar simulator. Incident photon-to-current conversion efficiency (IPCE) values were acquired with a monochromator and calibrated with a standard silicon photodiode. The hole mobility data of neat polymer films were characterized using the space charge limited current (SCLC) method with the device structure of ITO/PEDOT:PSS (40 nm)/active layer/MoO3 (10 nm)/Al (80 nm). Preliminarily, PEDOT:PSS was spin coated on an ITO covered glass substrate; then, active layers were prepared from an ODCB solution of 1% concentration by spin casting. Finally, MoO3 and Al layers were deposited via thermal evaporation under high vacuum.
Synthesis of monomers and polymers
All the starting materials utilized were purchased commercially and used without further purification. [6,6]-Phenyl-C61-butyric acid methyl ester (PC61BM) was purchased from Lumtec Corp. 4-Bromo-N-(4-bromophenyl)-N-(4-methoxyphenyl) aniline, 1,33 4-(bis(4-bromophenyl)amino)phenol, 2,34 4-bromo-N,N-diphenylaniline, 5,35 and 4,7-bis(5-bromothiophen-2-yl)-5,6-bis(octyloxy)benzofurazan, 15,15 were synthesized according to the published literature. Other monomers and polymers were synthesized via the following synthetic methodologies.
4-Bromo-N-(4-bromophenyl)-N-(4-(octyloxy)phenyl)aniline, 3.
To a solution of 4-(bis(4-bromophenyl)amino)phenol 2 (16.5 g, 39.4 mmol) in dry methanol (170 ml) under nitrogen, sodium methoxide (3.2 g, 59.1 mmol) was added in one portion. Then, the mixture was heated to reflux for one hour and 1-bromooctane (14 ml, 78.8 mmol) was added dropwise. The reaction mixture was refluxed overnight. Then, it was cooled to room temperature, poured into water, extracted with ethyl acetate three times and washed with an aqueous solution of sodium chloride and water. The organic extract was dried using MgSO4, and the solvent was removed by evaporation. The resulting product was purified by column chromatography using petroleum as the eluent to obtain 18.0 g of a viscous liquid, yield 86%. 1H NMR (CDCl3, 300 MHz, δ) 7.31 (d, 4H), 7.03 (d, 2H), 6.92–6.85 (m, 6H), 3.96 (t, 2H), 1.88–1.77 (m, 2H), 1.50–1.33 (m, 10H), 0.93 (t, 3H). 13C NMR (CDCl3, 75 MHz, δ) 152.37, 146.76, 136.12, 130.87, 124.34, 123.86, 117.71, 112.03, 31.81, 29.43, 29.32, 29.47, 26.13, 24.75, 22.57, 14.14.
4-(Octyloxy)-N,N-bis(4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)phenyl)aniline 4.
4-Bromo-N-(4-bromophenyl)-N-(4-(octyloxy)phenyl)aniline 3 (17.0 g, 32 mmol) was dissolved in 300 ml of dry tetrahydrofuran (THF) under the protection of dry nitrogen gas. The mixture was cooled to −78 °C, then BuLi (2.5 M, 51 ml, 128 mmol) was slowly added dropwise. When BuLi was added completely, it was left to react for another 1 hour at this temperature. 2-Isopropoxy-4,4,5,5-tetramethyl-1,3,2-dioxaborolane (53 ml, 256 mmol) was added slowly, and the reaction was allowed to continue for 1 hour at −78 °C. Then, the reaction mixture was warmed to room temperature spontaneously, and allowed to react overnight. It was poured into water, and extracted with dichloromethane (DCM) three times, then washed with sodium chloride aqueous solution and water, and dried over MgSO4. The solvent was removed by evaporation, and the crude product was purified by recrystallization in THF and methanol to obtain 14.3 g of target compound as a white crystal, yield 72%. 1H NMR (CDCl3, 300 MHz, δ) 7.65 (d, 4H), 7.05–7.01 (m, 6H), 6.83 (d, 2H), 3.94 (t, 2H), 1.80–1.73 (m, 2H), 1.46–1.29 (m, 34H), 0.89 (t, 3H). 13C NMR (CDCl3, 75 MHz, δ) 156.41, 150.34, 139.62, 135.81, 128.06, 127.75, 121.73, 115.40, 31.82, 29.38, 29.35, 29.25, 26.09, 24.87, 24.60, 22.66, 14.11.
4-Octyl-N,N-diphenylaniline 6.
4-Bromo-N,N-diphenylaniline 5 (6.48 g, 20 mmol) was dissolved in 100 ml of dry THF under the protection of dry nitrogen gas. The mixture was cooled to −78 °C, and then BuLi (2.5 M, 12 ml, 30 mmol) was slowly added dropwise. When BuLi was added completely, it was allowed to react for another 1 hour at this temperature. 1-Bromooctane (14 ml, 78.8 mmol) was added dropwise and 1-iodooctane (12 g, 50 mmol) was added in one portion. Then, the reaction mixture was warmed to room temperature spontaneously, and allowed to react overnight. The mixture was then poured into water, and extracted with DCM three times, then washed with sodium chloride aqueous solution and water, and dried over MgSO4. The solvent was removed by evaporation, and the crude product was purified by column chromatography using petroleum as the eluent to obtain 5.3 g of a viscous liquid, yield 74%. 1H NMR (CDCl3, 300 MH, δ) 7.31–7.27 (m, 4H), 7.24–6.94 (m, 10H), 2.55 (t, 2H), 1.62–1.55 (m, 2H), 1.31–1.26 (m, 10H), 0.88 (t, 3H). 13C NMR (CDCl3, 75 MHz, δ) 148.07, 145.35, 137.84, 129.19, 124.72,124.31, 124.18, 123.70, 123.10, 122.68, 122.24, 35.41, 31.93, 31.55, 29.73, 29.69, 29.52, 22.71, 14.16.
4-Bromo-N-(4-bromophenyl)-N-(4-octylphenyl)aniline 7.
4-Octyl-N,N-diphenylaniline 6 (5.19 g, 14.5 mmol) was dissolved in N,N-dimethylformamide (DMF) (50 ml) at 0 °C. Then, N-bromosuccinimide (NBS) (5.16 g, 29 mmol) was slowly added in portions in the absence of light. When NBS was added completely, the reaction mixture was warmed to room temperature spontaneously, and allowed to react overnight. The mixture was then poured into water, and extracted with DCM three times, then washed with sodium chloride aqueous solution and water, and dried over MgSO4. The solvent was removed by evaporation, and the crude product was purified by column chromatography in petroleum as the eluent to obtain 6.3 g of a viscous liquid, yield 84%. 1H NMR (CDCl3, 300 MHz, δ) 7.25–7.19 (m, 4H), 7.11–7.05 (m, 2H), 6.98–6.89 (m, 6H), 2.58 (t, 2H), 1.65–1.55 (m, 2H), 1.33–1.28 (m, 10H), 0.93 (t, 3H). 13C NMR (CDCl3, 75 MHz, δ) 146.70, 146.06, 144.37, 138.92, 132.52, 132.32, 129.47, 125.60, 124.96, 124.52, 116.10, 114.98, 35.37, 31.87, 31.42, 29.68, 29.63, 29.45, 22.64, 14.09.
4-Octyl-N,N-bis(4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)phenyl)aniline 8.
4-Bromo-N-(4-bromophenyl)-N-(4-octylphenyl)aniline 7 (23.4 g, 45.4 mmol) was dissolved in 300 ml dry THF under the protection of dry nitrogen gas. The mixture was cooled to −78 °C, then BuLi (2.5 M, 73 ml, 181.4 mmol) was slowly added dropwise. When the BuLi was added completely, it was allowed to react for another hour at this temperature. 2-Isopropoxy-4,4,5,5-tetramethyl-1,3,2-dioxaborolane (75.5 ml, 362.8 mmol) was added slowly, and the reaction was continued for 1 hour at −78 °C. Then, the reaction mixture was warmed to room temperature spontaneously, and allowed to react overnight. The mixture was then poured into water, and extracted with DCM three times, then washed with sodium chloride aqueous solution and water, and dried over MgSO4. The solvent was removed by evaporation, and the crude product was purified by recrystallization in THF and methanol to obtain 18.7 g of a white solid, yield 68%.1H NMR (CDCl3, 300 MHz, δ) 7.67–7.64 (m, 4H), 7.09–7.00 (m, 8H), 2.57 (t, 2H), 1.64–1.53 (m, 2H), 1.33–1.27 (m, 34H), 0.89 (t, 3H). 13C NMR (CDCl3, 75 MHz, δ) 150.26, 144.48, 138.98, 135.88, 129.32, 125.83, 123.88, 122.39, 35.44, 31.89, 31.42, 29.48, 29.41, 29.27, 24.86, 22.67, 14.12.
N,N-Diphenyl-4-(trifluoromethyl)aniline 9.
Diphenylamine (6.1 g, 36 mmol), 4-iodobenzotrifluoride (5 g, 18 mmol), palladium acetate (202 mg, 0.9 mmol), tris(2-methylphenyl)phosphine (548 mg, 1.8 mmol) and sodium tert-butoxide (3.57 g, 36 mmol) were dissolved in dry toluene (60 ml). The mixture was refluxed overnight. Then, it was cooled to room temperature and filtered to remove any insoluble solids. The filtrate was concentrated to remove the solvent, and the crude product was purified by column chromatography using petroleum as the eluent to obtain 7.8 g of a viscous liquid, yield 69%. 1H NMR (CDCl3, 300 MHz, δ) 7.55 (d, 2H), 7.37 (t, 4H), 7.19–7.11 (m, 6H), 6.96 (d, 2H). 13C NMR (CDCl3, 75 MHz, δ) 151.30, 146.51, 130.38, 126.94, 126.89, 125.24, 123.28, 120.20, 40.01.
4-Bromo-N-(4-bromophenyl)-N-(4-(trifluoromethyl) phenyl)aniline 10.
To a solution of N,N-diphenyl-4-(trifluoromethyl)aniline 9 (21.2 g, 67.7 mmol) dissolved in DMF (210 ml) at room temperature, NBS (24.7 g, 138.8 mmol) dissolved in DMF (210 ml) was added dropwise slowly in the absence of light. When NBS was added completely, the reaction was continued at room temperature for one hour. The mixture was then poured into water and extracted with DCM three times, then washed with sodium chloride aqueous solution and water, and dried over MgSO4. The solvent was removed by evaporation, and the crude product was purified by recrystallization in ethanol to obtain 31.5 g of white crystals, yield 99%. 1H NMR (CDCl3, 300 MHz, δ) 7.47–7.32 (m, 6H), 7.06 (d, 2H), 6.97 (d, 4H). 13C NMR (CDCl3, 75 MHz, δ) 150.00, 145.61, 132.77, 126.61, 126.54, 126.49, 121.92, 117.17.
N,N-Bis(4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)phenyl)-(4-(trifluoromethyl)phenyl)aniline 11.
4-Bromo-N-(4-bromophenyl)-N-(4-(trifluoromethyl)phenyl)aniline 10 (15.0 g, 31.8 mmol) was dissolved in 300 ml of dry THF under the protection of dry nitrogen gas. The mixture was cooled to −78 °C, and then BuLi (2.5 M, 45 ml, 111.4 mmol) was slowly added dropwise. When the BuLi was added completely, it was left to react for another hour at this temperature. 2-Isopropoxy-4,4,5,5-tetramethyl-1,3,2-dioxaborolane (40 ml, 191 mmol) was added slowly, and the reaction was continued for 1 hour at −78 °C. Then, the reaction mixture was warmed to room temperature spontaneously, and allowed to react overnight. It was then poured into water, extracted with DCM three times, washed with sodium chloride aqueous solution and water, and dried over MgSO4. The solvent was removed by evaporation, and the crude product was purified by recrystallization in THF and methanol to obtain 12.4 g of a white crystal, yield 69%. 1H NMR (CDCl3, 300 MHz, δ) 7.72 (d, 4H), 7.44 (d, 2H), 7.14–7.07 (m, 6H), 1.35 (s, 24H). 13C NMR (CDCl3, 75 MHz, δ) 150.24, 149.31, 136.14, 126.39, 126.33, 126.11, 124.40, 122.99, 83.77, 25.60.
N,N-Diphenyl-3,5-bis(trifluoromethyl)aniline 12.
Diphenylamine (3.7 g, 22.1 mmol), 1-iodo-3,5-bis(trifluoromethyl)benzene (5 g, 14.7 mmol), palladium acetate (166 mg, 0.74 mmol), tris(2-methylphenyl)phosphine (447 mg, 1.47 mmol) and sodium tert-butoxide (2.9 g, 29.4 mmol) were dissolved in dry toluene (50 ml). The mixture was refluxed overnight. Then, it was cooled to room temperature, and filtered to remove any insoluble solid. The filtrate was concentrated to remove the solvent, and the crude product was purified by column chromatography using petroleum as the eluent to obtain 4.3 g of a viscous liquid, yield 77%. 1H NMR (CDCl3, 300 MHz, δ) 7.42–7.34 (m, 7H), 7.22–7.14 (m, 6H). 13C NMR (CDCl3, 75 MHz, δ) 150.14, 146.31, 133.14, 125.39, 125.33, 125.11, 124.40, 113.99.
N,N-Bis(4-bromophenyl)-3,5-bis(trifluoromethyl)aniline 13.
To a solution of N,N-bis(4-bromophenyl)-3,5-bis(trifluoromethyl)aniline 13 (4.8 g, 12.6 mmol) dissolved in DMF (50 ml) at room temperature, NBS (4.5 g, 25.2 mmol) dissolved in DMF (50 ml) was slowly added dropwise in the absence of light. When NBS was added completely, the reaction was continued at room temperature for one hour. Then, the mixture was poured into water and extracted with DCM three times, then washed with sodium chloride aqueous solution and water, and dried over MgSO4. The solvent was removed by evaporation, and the crude product was purified by recrystallization in ethanol to obtain 5.6 g of a white crystal, yield 82%. 1H NMR (CDCl3, 300 MHz, δ) 7.48–7.37 (m, 7H), 6.96 (d, 4H). 13C NMR (CDCl3, 75 MHz, δ) 148.46, 144.92, 133.53, 126.62, 121.22, 118.08, 115.35, 115.25.
N,N-Bis(4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)phenyl)-3,5-bis(trifluoromethyl)aniline 14.
N,N-Bis(4-bromophenyl)-3,5-bis(trifluoromethyl)aniline 13 (15.0 g, 27.8 mmol) was dissolved in 350 ml of dry THF under the protection of dry nitrogen gas. The mixture was cooled to −78 °C, and then BuLi (2.5 M, 33 ml, 83.5 mmol) was slowly added dropwise. When the BuLi was added completely, it was left to react for another hour at this temperature. 2-Isopropoxy-4,4,5,5-tetramethyl-1,3,2-dioxaborolane (35 ml, 167 mmol) was added slowly, and the reaction was continued for 1 hour at −78 °C. Then, the reaction mixture was warmed to room temperature spontaneously, and allowed to react overnight. It was then poured into water and extracted with DCM three times, then washed with sodium chloride aqueous solution and water, and dried over MgSO4. The solvent was removed by evaporation, and the crude product was purified by recrystallization in THF and methanol to obtain 13.2 g of a white crystal, yield 75%. 1H NMR (CDCl3, 300 MHz, δ) 7.75 (d, 4H), 7.44–7.41 (m, 3H), 7.08 (d, 4H), 1.35 (s, 24H). 13C NMR (CDCl3, 75 MHz, δ) 148.69, 148.61, 136.43, 132.84, 132.40, 123.89, 122.35, 115.40, 83.88, 24.87.
Poly(4-(octyloxy)-N,N-diphenylaniline-alt-4,7di(thiophen-2-yl)-5,6-bis(octyloxy) benzofurazan) P1.
4-(Octyloxy)-N,N-bis(4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)phenyl)aniline 4 (313 mg, 0.5 mmol), 4,7-bis(5-bromothiophen-2-yl)-5,6-bis(octyloxy)benzofurazan 5 (349 mg, 0.5 mmol) and Pd(PPh3)4 (11.6 mg, 0.01 mmol) were dissolved in 15 ml toluene under nitrogen protection. Then, a tetraethyl ammonium hydroxide aqueous solution (2 ml, 20 wt/wt%) was added in one portion and the mixture was heated to 85 °C for 48 h. Phenylboronic acid (0.609 g, 5 mmol) was added and the mixture was left to react for 12 h, then bromobenzene (1.57 g, 10 mmol) was added in one portion and allowed to react for an additional 12 h. The reaction mixture was cooled to room temperature and precipitated in methanol. The crude polymer was purified by Soxhlet extraction with methanol, acetone, hexane and chloroform. The final chloroform solution was precipitated in methanol, filtered and dried at 80 °C in a vacuum oven for 10 hours to yield 247 mg of the target polymer as a red solid. 1H NMR (CDCl3, 600 MHz, δ): 7.71–7.31 (br, 8H, Ar-H), 7.22–7.00 (br, 6H, Ar-H), 6.91–6.86 (m, 2H, Ar-H), 4.20–4.14 (m, 4H, CH2), 3.73–3.72 (m, 2H, CH2), 2.04–2.03 (m, 4H, CH2), 1.83–1.79 (m, 2H, CH2), 1.29–1.27 (br, 30H, CH2), 0.90–0.86 (m, 9H, CH3). GPC (THF): Mn = 12400, Mw = 26800, PDI = 2.16.
Poly(4-octyl-N,N-diphenylaniline-alt-4,7di(thiophen-2-yl)-5,6-bis(octyloxy)benzofurazan) P2.
4-Octyl-N,N-bis(4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)phenyl)aniline 8 (305 mg, 0.5 mmol), 4,7-bis(5-bromothiophen-2-yl)-5,6-bis(octyloxy)benzofurazan 5 (349 mg, 0.5 mmol) and Pd(PPh3)4 (11.6 mg, 0.01 mmol) were dissolved in 15 ml toluene under nitrogen protection. Then, tetraethyl ammonium hydroxide aqueous solution (2 ml, 20 wt/wt%) was added in one portion and the mixture was heated to 85 °C for 48 h. Phenylboronic acid (0.609 g, 5 mmol) was added and reacted for 12 h. Then, bromobenzene (1.57 g, 10 mmol) was added in one portion and reacted for an additional 12 h. The reaction mixture was cooled to room temperature and precipitated in methanol. The crude polymer was purified by Soxhlet extraction in methanol, acetone, hexane and chloroform. The final chloroform solution was precipitated in methanol, filtered and dried at 80 °C in a vacuum oven for 10 hours to yield 189 mg of the target polymer as a red solid. 1H NMR (CDCl3, 600 MHz, δ): 7.70–7.58 (br, 4H, Ar-H), 7.49–7.30 (br, 4H, Ar-H), 7.18–7.06 (br, 8H, Ar-H), 4.20–4.15 (m, 4H, CH2), 2.62–2.59 (m, 2H, CH2), 2.05–1.99 (m, 4H, CH2), 1.38–1.28 (br, 32H, CH2), 0.90–0.85 (m, 9H, CH3). GPC (THF): Mn = 11700, Mw = 20600, PDI = 1.76.
Poly(N,N-diphenyl-4-(trifluoromethyl)aniline-alt-4,7di(thiophen-2-yl)-5,6-bis(octyl oxy)benzofurazan) P3.
N,N-Bis(4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)phenyl)-(4-(trifluoromethyl)phenyl)aniline 11 (283 mg, 0.5 mmol), 4,7-bis(5-bromothiophen-2-yl)-5,6-bis(octyloxy)benzofurazan 5 (349 mg, 0.5 mmol) and Pd(PPh3)4 (11.6 mg, 0.01 mmol) were dissolved in 15 ml toluene under nitrogen protection. Then, a tetraethyl ammonium hydroxide aqueous solution (2 ml, 20 wt/wt%) was added in one portion and the mixture was heated to 85 °C for 48 h. Phenylboronic acid (0.609 g, 5 mmol) was added and allowed to react for 12 h. Then, bromobenzene (1.57 g, 10 mmol) was added in one portion and reacted for an additional 12 h. The reaction mixture was cooled to room temperature and precipitated in methanol. The crude polymer was purified by Soxhlet extraction in methanol, acetone, hexane and chloroform. The final chloroform solution was precipitated in methanol, filtered and dried at 80 °C in a vacuum oven for 10 hours to yield 207 mg of the target polymer as a red solid. 1H NMR (CDCl3, 600 MHz, δ): 8.37–8.17 (m, 2H, Ar-H), 7.64–7.31 (br, 8H, Ar-H), 7.19–7.01 (br, 6H, Ar-H), 4.14–4.09 (m, 4H, CH2), 1.98–1.91 (m, 4H, CH2), 1.40–1.16 (br, 20H, CH2), 0.87–0.78 (m, 6H, CH3). GPC (THF): Mn = 8700, Mw = 12400, PDI = 1.43.
Poly(N,N-diphenyl-3,5-bis(trifluoromethyl)aniline-alt-4,7di(thiophen-2-yl)-5,6-bis(octyloxy)benzofurazan) P4.
N,N-Bis(4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)phenyl)-3,5-bis(trifluoromethyl)aniline 14 (317 mg, 0.5 mmol), 4,7-bis(5-bromothiophen-2-yl)-5,6-bis(octyloxy) benzofurazan 5 (349 mg, 0.5 mmol) and Pd(PPh3)4 (11.6 mg, 0.01 mmol) were dissolved in 15 ml toluene under nitrogen protection. Then, a tetraethyl ammonium hydroxide aqueous solution (2 ml, 20 wt/wt%) was added in one portion and the mixture was heated to 85 °C for 48 h. Phenylboronic acid (0.609 g, 5 mmol) was added and reacted for 12 h. Then bromobenzene (1.57 g, 10 mmol) was added in one portion and reacted for an additional 12 h. The reaction mixture was cooled to room temperature and precipitated in methanol. The crude polymer was purified by Soxhlet extraction in methanol, acetone, hexane and chloroform. The final chloroform solution was precipitated in methanol, filtered and dried at 80 °C a the vacuum oven for 10 hours to yield 162 mg of the target polymer as a red solid. 1H NMR (CDCl3, 600 MHz, δ): 8.64 (s, 1H, Ar-H), 7.68–7.33 (br, 10H, Ar-H), 7.20–7.14 (br, 4H, Ar-H), 4.22–4.18 (m, 4H, CH2), 2.05–2.04 (m, 4H, CH2), 1.53–1.26 (br, 20H, CH2), 0.91–0.84 (m, 6H, CH3). GPC (THF): Mn = 7600, Mw = 10700, PDI = 1.41.
Results and discussion
Synthesis
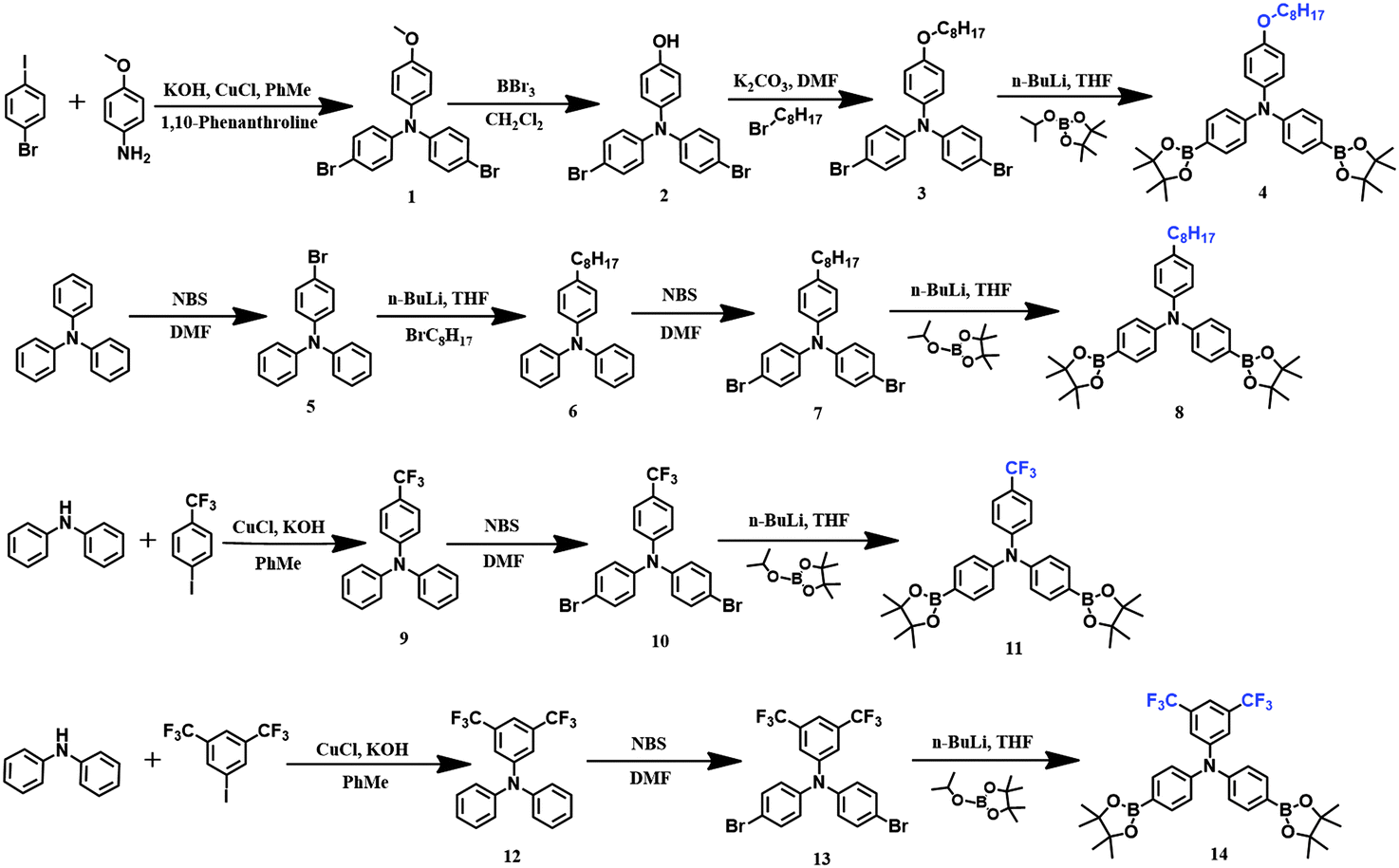
The synthetic routes of the monomers are presented in Scheme 1. For the synthesis of 4-(octyloxy)-N,N-bis(4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)phenyl)aniline 4, 4-bromo-N-(4-bromophenyl)-N-(4-methoxyphenyl) aniline 1 was prepared preliminarily from 1-bromo-4-iodobenzene and p-methoxylaniline via the Ullmann reaction, and then compound 1 was demethylated to afford 4-(bis(4-bromophenyl)amino)phenol 2 with the assistance of boron tribromide as the catalyst. By the alkylation of monomer 2 in the presence of 1-bromooctane, 4-bromo-N-(4-bromophenyl)-N-(4-(octyloxy)phenyl)aniline 3 was obtained with good yield, and finally target compound 4 was prepared from monomer 3 by a general low temperature reaction (−78 °C) in the presence of n-butyllithium and 2-isopropoxy-4,4,5,5-tetramethyl-1,3,2-dioxaborolane. 4-Octyl-N,N-diphenylaniline 6 was prepared feasibly from 4-bromo-N,N-diphenylaniline 5via the reaction with n-butyllithium and 1-bromooctane at −78 °C sequentially; then, through general bromination in DMF, 4-bromo-N-(4-bromophenyl)-N-(4-octylphenyl)aniline 7 was obtained in 84% yield, and ultimately 4-octyl-N,N-bis(4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)phenyl)aniline 8 was prepared through the same procedure used for the synthesis of compound 4. The synthetic procedures of N,N-bis(4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)phenyl)-(4-(trifluoromethyl)phenyl)aniline 11 and N,N-bis(4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)phenyl)-3,5-bis (trifluoromethyl)aniline 14 are very similar, where N,N-diphenyl-4-(trifluoromethyl)aniline 9 and N,N-diphenyl-3,5-bis(trifluoromethyl)aniline 12 were synthesized first via the Buchwald–Hartwig reaction; then, through general bromination in DMF, 4-bromo-N-(4-bromophenyl)-N-(4-(trifluoromethyl)phenyl)aniline 10 and N,N-bis(4-bromophenyl)-3,5-bis(trifluoromethyl)aniline 13 were obtained, respectively, and finally using the same synthetic procedure for the preparation of compound 4 and 8, we acquired target compound 11 and 14.
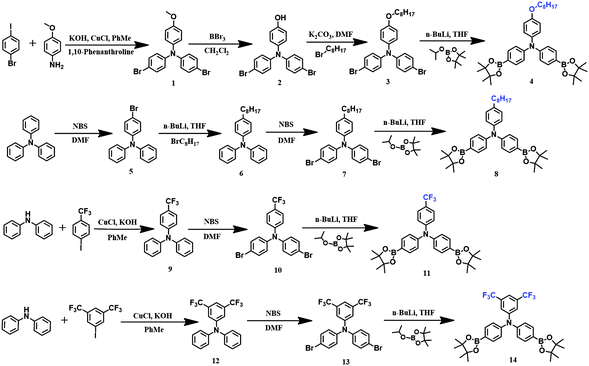 |
| | Scheme 1 Synthetic routes for the monomers. | |
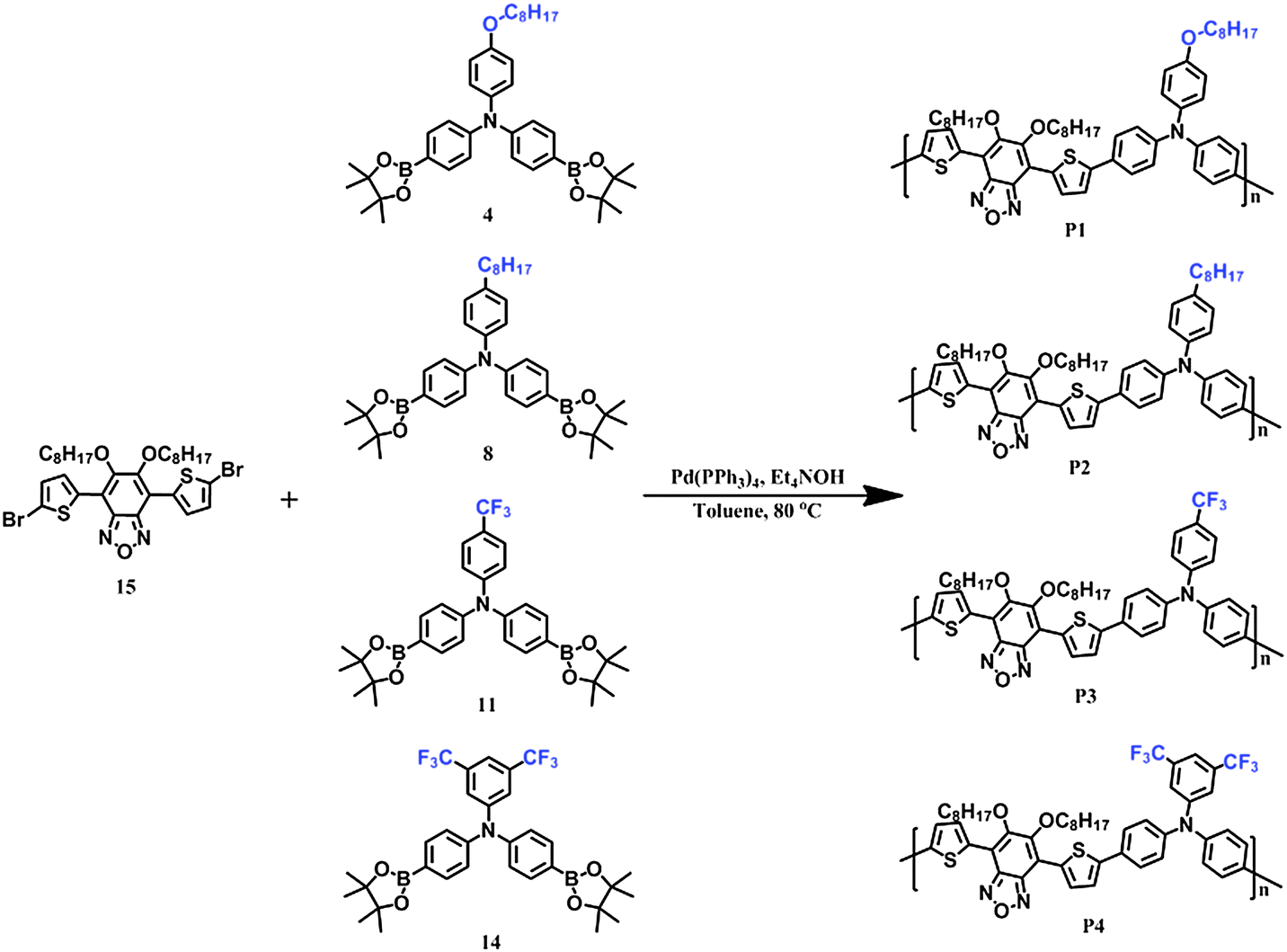
The synthetic routes of polymers are shown in Scheme 2. All the polymers, P1, P2, P3 and P4, were synthesized by Suzuki polycondensation in toluene under catalysis of tetrakis(triphenylphosphine)palladium, and then purified by Soxhlet extraction in methanol, acetone, hexane and chloroform, respectively. The final polymer solution in chloroform was precipitated in methanol, filtered and dried at 60 °C under vacuum for 24 hours. These four polymers have excellent solubility in general organic solvents such as toluene, tetrahydrofuran, chloroform, and chlorobenzene. Molecular weights were characterized by gel permission chromatography (GPC), which shows that the number-averaged molecular weights (Mn) and weight-averaged molecular weights (Mw) are 12400, 11700, 8700, 7600, and 26800, 20600, 12400 and 10700 with the polydispersity index (PDI) of 2.16, 1.76, 1.43 and 1.41 for P1, P2, P3 and P4, respectively (Table 1). In comparison with P3 and P4, P1 and P2 have higher molecular weights due to the longer octyl side chains in P1 and octoxyl in P2, which may facilitate the good solubility of the polymers and realize the increment of molecular weights when polymerization occurs.
Table 1 Molecular weights and thermal properties
| Polymers |
M
n
|
M
w
|
PDI |
T
d (°C) |
|
P1
|
12400 |
26800 |
2.16 |
324 |
|
P2
|
11700 |
20600 |
1.76 |
321 |
|
P3
|
8700 |
12400 |
1.43 |
321 |
|
P4
|
7600 |
10700 |
1.41 |
323 |
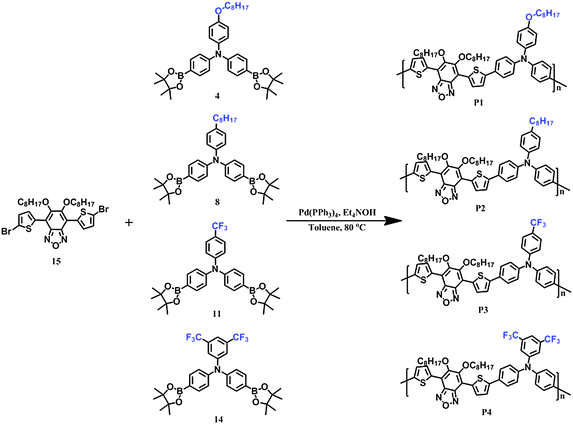 |
| | Scheme 2 Synthetic routes for the polymers. | |
Thermal stability
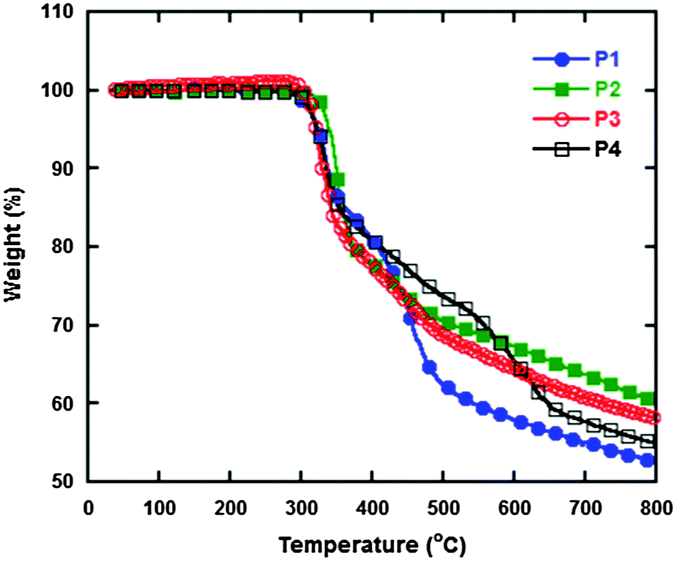
The thermal stability of polymers is an indispensable parameter for evaluating the performance of PSCs. Therefore, thermal gravimetric analysis (TGA) was used to characterize the polymers and the resulting curves are displayed in Fig. 1, and detailed data are summarized in Table 1. The degradation temperatures at 5% weight loss of P1, P2, P3 and P4 are 324, 321, 321 and 321 °C, respectively. All of them are thermally stable up to 300 °C, which indicates that they potentially possess high stability for the construction of PSCs.
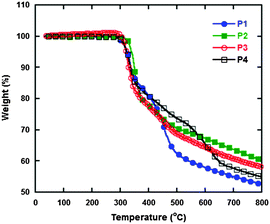 |
| | Fig. 1 Thermal gravimetric analysis curves of the polymers. | |
UV-vis absorption
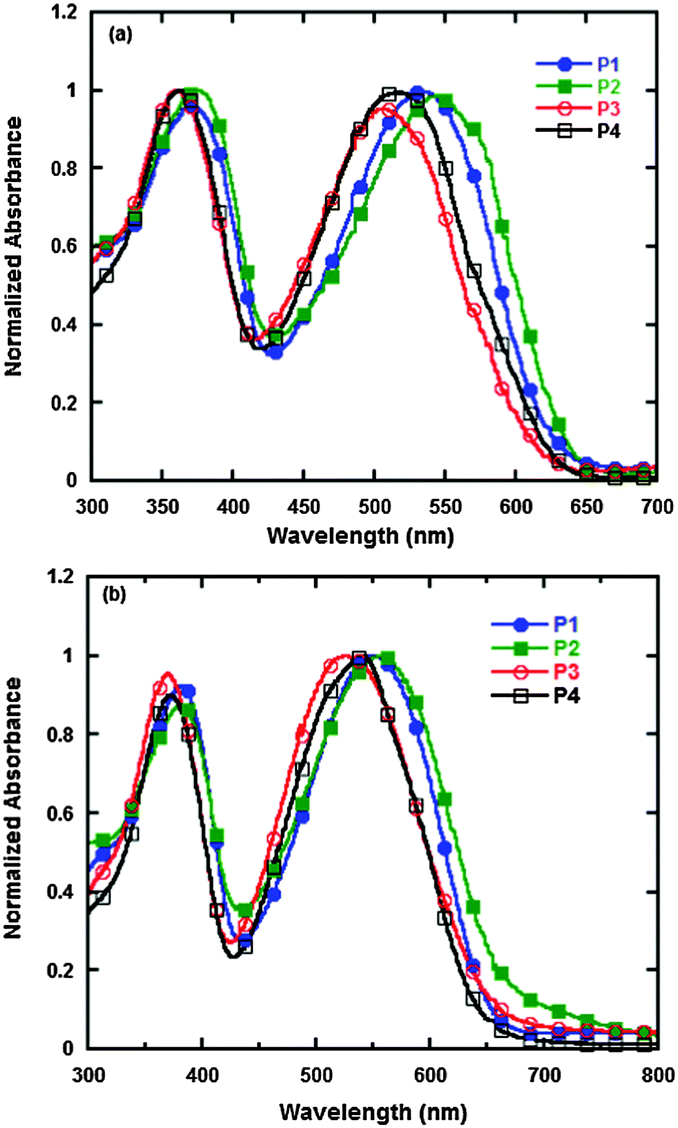
The UV-vis absorption spectra of P1, P2, P3 and P4 in chloroform and in film are shown in Fig. 2, and the data in detail are summarized in Table 2. In Fig. 2, we can see that all the polymers, both in chloroform and in film, show two distinct absorption peaks. The peaks at short wavelengths, which range from 360 to 380 nm, are attributed to the localized π–π* transition, and the other peaks at longer wavelengths, which range from 500 to 560 nm, are due to the internal charge transfer (ICT) interaction between the donor and acceptor units in the polymer main chains.36 The onsets of absorption spectra of P1, P2, P3 and P4 in films are 649, 656, 643 and 640 nm, which correspond to the optical band gaps of 1.91, 1.89, 1.93 and 1.94 eV, respectively. The absorption spectra in film are clearly red-shifted by about 20 nm in comparison to that in chloroform, which indicates that strong intermolecular π–π stacking exists between the polymer backbones. In comparison with the absorption spectra of P3, which has trifluoromethyl, and P4, which has bis(trifluoromethyl), P1, which has octyloxy, and P2, which has octyl, display much more red-shifted absorptions and much lower optical band gaps. This result is possibly due to the fact that octyloxy and octyl belong to strong electron-donating groups that enhance the ICT effect and decrease the band gaps of polymers, whereas the electron-withdrawing groups of trifluoromethyl or bis(trifluoromethyl) weaken the ICT effect and cause the absorption peaks of P3 and P4 to be blue-shifted.
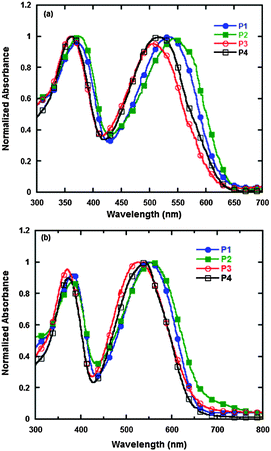 |
| | Fig. 2 UV-vis absorption spectra of the polymers in chloroform (a) and in film (b). | |
Table 2 UV-vis absorption properties of the polymers
| Polymer |
λ
max,inchloroform (nm) |
λ
max,infilm (nm) |
λ
onset (nm) |
E
g,optical (eV) |
|
P1
|
371, 533 |
381, 550 |
649 |
1.91 |
|
P2
|
377, 547 |
381, 558 |
656 |
1.89 |
|
P3
|
360, 505 |
370, 525 |
643 |
1.93 |
|
P4
|
361, 516 |
372, 542 |
640 |
1.94 |
Electrochemical properties
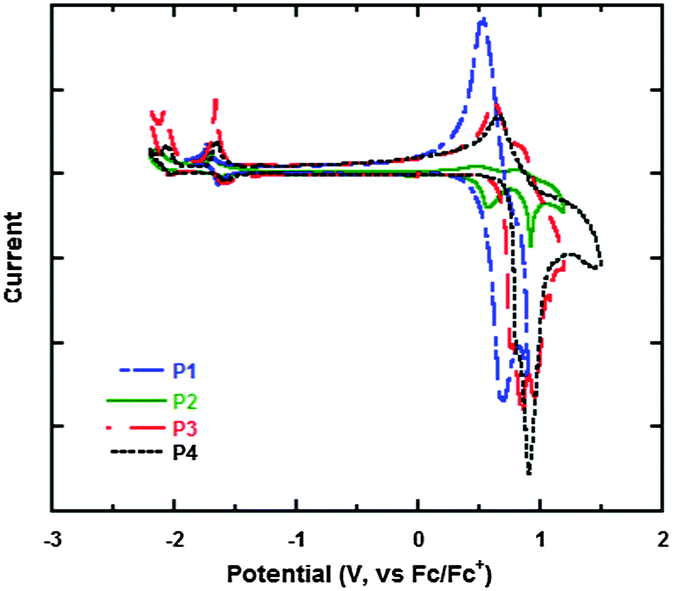
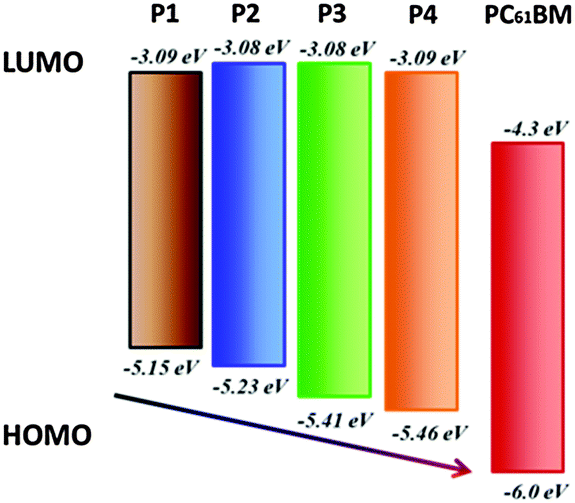
To further investigate the energy levels of the polymers, the highest occupied molecular orbit (HOMO) and the lowest unoccupied molecular orbit (LUMO) energy levels of P1, P2, P3 and P4 were acquired from the cyclic voltammogram (CV) characterization. Fig. 3 gives the CV curves of P1, P2, P3 and P4 in films and the detailed data are summarized in Table 3. The CV curves present reversibly oxidative and reductive curves for all the polymers, in which the onsets of the oxidation and reduction potentials for P1, P2, P3 and P4 are located at 0.45, 0.53, 0.71, 0.76 and −1.61, −1.62, −1.62, −1.61 V versus SCE, respectively. The redox potential of the Fc/Fc+ internal reference is 0.1 V vs. SCE. The HOMO and LUMO energy levels of P1, P2, P3 and P4, which were determined using the empirical formula EHOMO = −e(Eox + 4.8 − E1/2,(Fc/Fc+)) and ELUMO = −e(Ere + 4.8 − E1/2,(Fc/Fc+)), are −5.15, −5.23, −5.41, −5.46 and −3.09, −3.08, −3.08, −3.09 eV, respectively. It is found that the HOMO energy levels decreased obviously from P1, P2, P3 to P4, which is because of the electron-withdrawing ability of the substituted groups of octoxy, octyl, trifluoromethyl and bis(trifluoromenthyl) in P1, P2, P3 and P4, which increase sequentially. Octoxy and octyl are electron-donating groups and they promote the HOMO energy levels, while trifluoromethyl and bis(trifluoromenthyl) are attributed to strong electron-withdrawing groups and they reduce the HOMO energy levels. Therefore, it is a feasible method to tune the energy levels of semiconducting polymers by adjusting the properties of substituted groups in the construction units (Fig. 4). On the other hand, the reduction potentials and LUMO energy levels of P1, P2, P3 and P4 are very similar, because they have the same accepting unit of bis(octyloxy)benzofurazan in their polymer main chains, thus they show similar reductive properties.
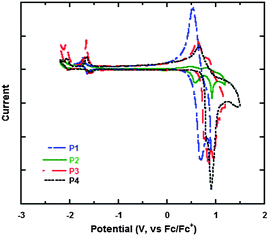 |
| | Fig. 3 Cyclic voltammogram curves of the polymers in films. | |
Table 3 Electrochemical properties of the polymers
| Polymer |
E
ox (V) |
E
re (V) |
E
HOMO (eV) |
E
LUMO (eV) |
E
g,CV (eV) |
|
P1
|
0.45 |
−1.61 |
−5.15 |
−3.09 |
2.06 |
|
P2
|
0.53 |
−1.62 |
−5.23 |
−3.08 |
2.15 |
|
P3
|
0.71 |
−1.62 |
−5.41 |
−3.08 |
2.33 |
|
P4
|
0.76 |
−1.61 |
−5.46 |
−3.09 |
2.37 |
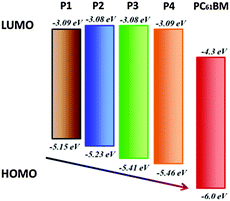 |
| | Fig. 4 Energy level diagram of the polymers. | |
Hole mobility
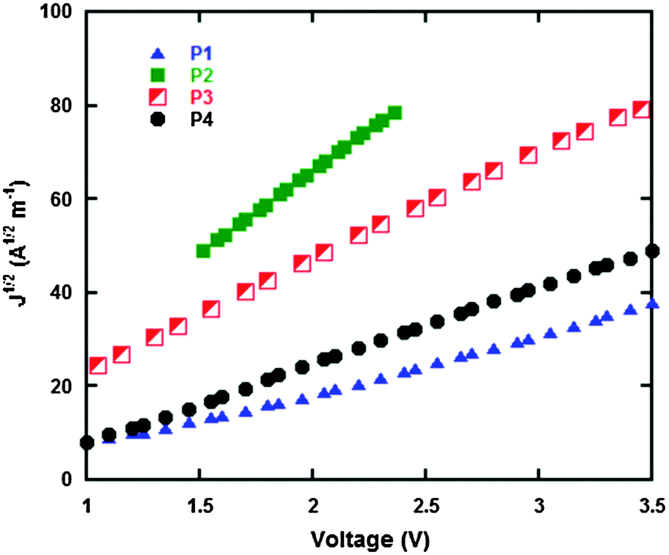
Hole mobility is one of the indispensable parameters to evaluate donor polymers in BHJ PSCs. In this study, the hole mobility of neat polymers were measured using the space charge limited current (SCLC) method with the device configuration of ITO/PEDOT:PSS/active layer/MoO3/Al. The thickness of the active layers is very similar, in which for P1 it is 79 nm, P2 it is 73 nm, P3 it is 77 nm and P4 it is 78 nm. Fig. 5 shows the current density–voltage (J–V) curves of the neat polymer films from SCLC devices. The calculated hole mobilities of P1, P2, P3 and P4 are 2.7 × 10−5, 1.6 × 10−4, 1.7 × 10−5 and 2.7 × 10−5, respectively. In comparison to P1, P3 and P4, the hole mobility of P2 is an order of magnitude higher, which is possible because the octyl substituted TPA unit in P2 is straighter than the other group substituted TPA derivatives in P1, P3 and P4, and this effect can improve the polymer main chain stacking and ultimately strengthen the hole transporting ability in P2. Due to the higher hole mobility of P2, it is more beneficial for the improvement of the photovoltaic performance of the P2 based PSC (Table 4).
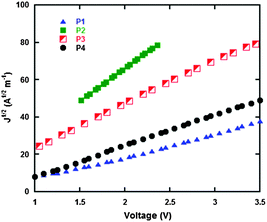 |
| | Fig. 5
J–V characteristics of neat films of the polymers from SCLC devices. | |
Table 4 Hole mobility of neat polymers
| Polymer |
Thickness (nm) |
μ (× 10−4, cm2 V−1 s−1) |
|
P1
|
79 |
0.27 |
|
P2
|
73 |
1.60 |
|
P3
|
77 |
0.17 |
|
P4
|
78 |
0.27 |
Photovoltaic properties
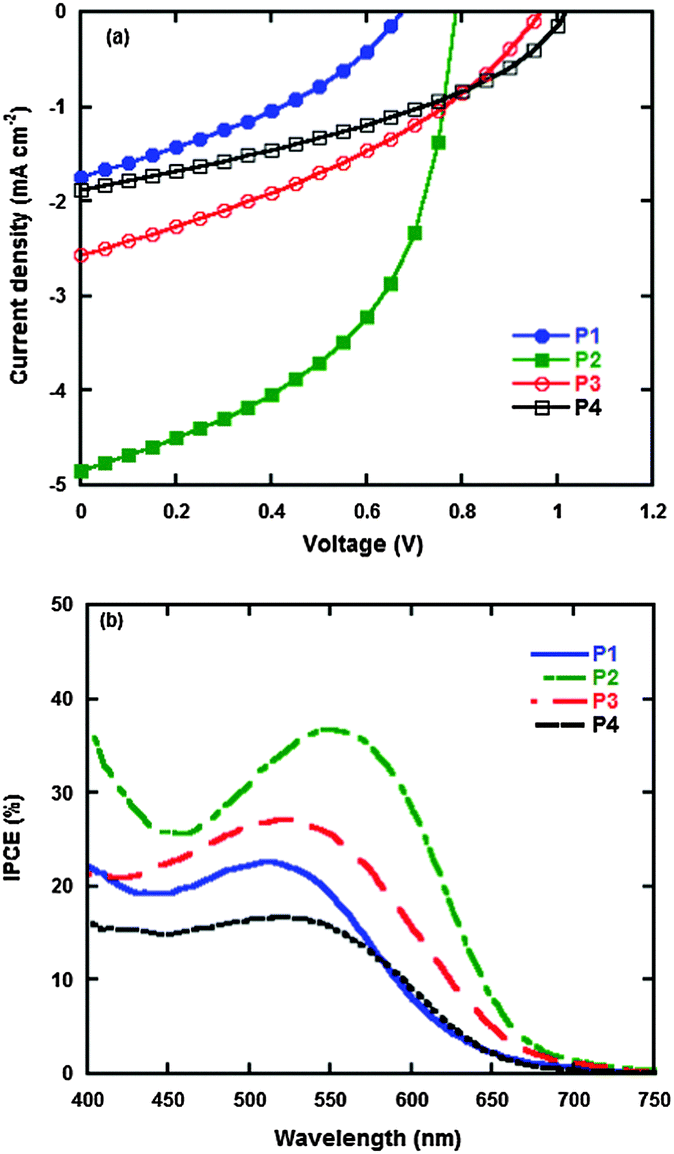
PSCs were fabricated under the device structure of ITO/PEDOT:PSS/polymer:PC61BM/PFN/Al. Herein, PFN was utilized as a cathode interfacial layer due to its versatile functions as a protecting active layer during cathode evaporation and the possible existence of built-in electric fields between the active layer and the cathode.37,38 The current density–voltage (J–V) curves are shown in Fig. 6a, and the photovoltaic properties in detail are summarized in Table 5. The P2 based PSC exhibits a higher short circuit current density (Jsc), which is ascribed to its better light absorption in the visible region and higher hole mobility than P1, P3 and P4. The open circuit voltages (Voc) of the P1, P2, P3 and P4 based PSCs increase from 0.70, 0.80, 0.95 to 1.00 V, respectively, which result from the fact that the HOMO energy levels of P1, P2, P3 to P4 decrease successively from −5.15, −5.23, −5.41 to −5.46 eV, where the value of Voc is proportional to the energy level difference between the HOMO energy level of the donor and LUMO energy level of the acceptor.8 Herein, the P4 based PSC gives the highest Voc of 1.00 V which is, to our best knowledge, one of the highest Voc values based on polytriphenylamine derived broad band-gap polymers for PSCs. On the other hand, although the P2 based PSC gives the lower Voc of 0.80 V, it also exhibits the highest Jsc and FF among all four polymers, which is ascribed to its higher absorption and hole mobility. Comprehensively, the P2 based PSC has the best photovoltaic performance with the Jsc of 4.84 mA cm−2, FF of 50%, Voc of 0.80 V and PCE of 2.22%.
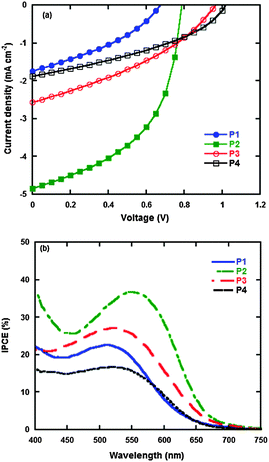 |
| | Fig. 6 Current density–voltage (J–V) characteristics of polymers:PC61BM under AM 1.5 G, 87 mW cm−2 (a); incident photon-to-current conversion efficiency (IPCE) curves of polymers:PC61BM blend films (b). | |
Table 5 Photovoltaic performances with the device structure of ITO/PEDOT:PSS/polymer:PC61BM/PFN/Al under AM 1.5 G, 87 mW cm−2
| Polymer |
Ratio |
J
sc (mA cm−2) |
FF (%) |
V
oc (V) |
PCE (%) |
|
P1
|
1:3 |
2.06 |
43 |
0.70 |
0.86 |
|
P2
|
1:2 |
4.84 |
50 |
0.80 |
2.22 |
|
P3
|
1:2 |
2.57 |
36 |
0.95 |
1.28 |
|
P4
|
1:2 |
1.87 |
38 |
1.00 |
1.05 |
The incident photon-to-current conversion efficiency (IPCE) curves of the PSCs are shown in Fig. 6b. All the polymers display the corresponding photon-to-current conversion effect, which is in agreement with the results of the absorption spectra. Among these polymers, the P2 based PSC gives the best result with the broadened and intensified IPCE in the visible region, which is ascribed to its natural characteristic of better light absorption than P1, P3 and P4.
AFM morphologies
To study the interaction of the blend films between the polymers and PC61BM, AFM images of the blend films were obtained, which are shown in Fig. 7. In the topography of Fig. 7a–d, the root-mean-square (RMS) roughness of the blend films are 0.476, 0.23, 2.488 and 2.550 nm for P1, P2, P3 and P4, respectively, which means that the P1 and P2 based blend films are much smoother than the P3 and P4 based films. Furthermore, the phase images (Fig. 7e–h) display that P1 and P2 based blend films give better nano-scale phase separation, while the P3 and P4 based blend films show worse phase separation and bigger aggregates, which inhibit holes and electrons transport and ultimately decrease their FF values and photovoltaic properties. According to the photovoltaic performances displayed in Table 5, P1 and P2 exhibit higher FF values than P3 and P4, which are ascribed to the better nano-scale phase separation in P1:PC61BM and P2:PC61BM blend films.39
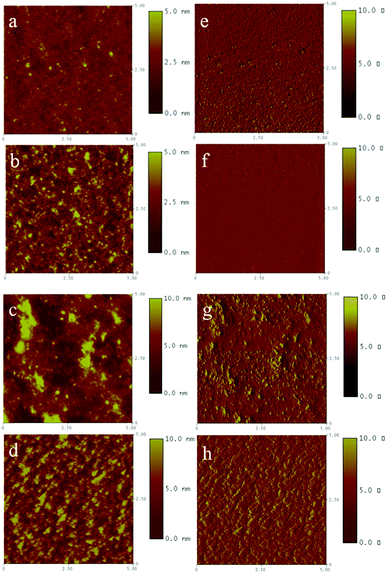 |
| | Fig. 7 AFM images of blend films (1:3 weight ratio for P1 and 1:2 weight ratio for P2, P3 and P4); topography of P1: PC61BM (a), P2: PC61BM (b), P3: PC61BM (c), and P4: PC61BM (d); phase images of P1: PC61BM (e), P2: PC61BM (f), P3: PC61BM (g), and P4: PC61BM (h). | |
Conclusions
In summary, a series of novel broad band-gap D–A copolymers (P1, P2, P3, and P4) based on triphenylamine and benzofurazan derivatives were prepared. The HOMO energy levels of these polymers can be tuned feasibly by changing the side chains [–OC8H17, –C8H17, –CF3, –(CF3)2] in the triphenylamine unit. Their HOMO energy levels are decreased successively from −5.15, −5.23, −5.41 to −5.46 eV, as the electron-withdrawing ability of the octoxy, octyl, trifluoromethyl and bis(trifluoromenthyl) substituted groups are increased sequentially. Octoxy and octyl are electron-donating groups that promote the HOMO energy levels, while the trifluoromethyl and bistrifluoromenthyl are the strong electron-withdrawing groups that should reduce the HOMO energy levels. As a result, the Vocs of the PSCs with the device structure of ITO/PEDOT:PSS/polymer:PC61BM/PFN/Al can be adjusted from 0.70, 0.80, 0.95 to 1.00 V. To the best of our knowledge, the Voc of 1.00 V (P4) is one of the highest Voc values based on polytriphenylamine derived narrow band-gap polymers for PSCs. Therefore, it is a feasible way to achieve high Vocs and thus high PCE values by optimizing the substituted groups of the polymers. Among these four polymers, the one with the octyl side chain shows the best photovoltaic performance with the highest Jsc (due to the best light absorption in visible region and highest hole mobility) and FF (resulting from its better nano-scale phase separation), giving a Jsc of 4.84 mA cm−2, FF of 50%, Voc of 0.80 V and PCE of 2.22%.
Acknowledgements
This study was financially supported by the National Nature Science Foundation of China (No. 51273069, 91333206), the Research Fund for the Doctoral Program of Higher Education of China (No. 20130172110005) and the Fundamental Research Funds for the Central Universities (No. 2014ZB0017).
References
- Y. H. Liu, J. B. Zhao, Z. K. Li, C. Mu, M. Ma, H. W. Hu, K. Jiang, H. R. Lin, H. Ade and H. Yan, Nat. Commun., 2014, 5, 5293–5300 CrossRef CAS PubMed.
- Z. Zheng, S. Q. Zhang, M. J. Zhang, K. Zhao, L. Ye, Y. Chen, B. Yang and J. H. Hou, Adv. Mater., 2015, 27, 1189–1194 CrossRef CAS PubMed.
- Z. C. He, B. Xiao, F. Liu, H. B. Wu, Y. L. Yan, S. Xiao, C. Wang, T. P. Russell and Y. Cao, Nat. Photonics, 2015, 9, 174–179 CrossRef CAS.
- H. Choi, S. J. Ko, T. Kim, P. O. Morin, B. Walker, B. H. Lee, M. Leclerc, J. Y. Kim and A. J. Heeger, Adv. Mater., 2015, 27, 3318–3324 CrossRef CAS PubMed.
- C. Cabanetos, A. E. Labban, J. A. Bartelt, J. D. Douglas, W. R. Mateker, J. M. J. Fréchet, M. D. McGehee and P. M. Beaujuge, J. Am. Chem. Soc., 2013, 135, 4656–4659 CrossRef CAS PubMed.
- C. J. Brabec, A. Cravino, D. Meissner, N. S. Sariciftci, T. Fromherz, M. T. Rispens, L. Sanchez and J. C. Hummelen, Adv. Funct. Mater., 2001, 11, 374–380 CrossRef CAS.
- Y. F. Li, Acc. Chem. Res., 2012, 45, 723–733 CrossRef CAS PubMed.
- M. C. Scharber, D. Muhlbacher, M. Koppe, P. Denk, C. Waldorf, A. J. Heeger and C. J. Brabec, Adv. Mater., 2006, 18, 789–794 CrossRef CAS.
- L. Q. Liu, G. C. Zhang, P. Liu, J. Zhang, S. Dong, M. Wang, Y. G. Ma, H. L. Yip and F. Huang, Chem. – Asian J., 2014, 9, 2104–2112 CrossRef CAS PubMed.
- J. F. Li, J. F. Tong, P. Zhang, C. Y. Yang, D. J. Chen, Y. C. Zhu, Y. J. Xia and D. W. Fan, Bull. Korean Chem. Soc., 2014, 35, 505–512 CrossRef CAS.
- L. Wang, M. Xu, L. Ying, F. Liu and Y. Cao, Acta Polym. Sin., 2008, 10, 993–997 Search PubMed.
- N. Blouin, A. Michaud and M. Leclerc, Adv. Mater., 2007, 19, 2295–2300 CrossRef CAS.
- S. H. Park, A. Roy, S. Beaupré, S. Cho, N. Coates, J. S. Moon, D. Moses, M. Leclerc, K. Lee and A. J. Heeger, Nat. Photonics, 2009, 3, 297–303 CrossRef CAS.
- R. P. Qin, W. W. Li, C. H. Li, C. Du, C. Veit, H. F. Schleiermacher, M. Andersson, Z. S. Bo, Z. P. Liu, O. Inganäs, U. Wuerfel and F. L. Zhang, J. Am. Chem. Soc., 2009, 131, 14612–14613 CrossRef CAS PubMed.
- B. Zhang, X. W. Hu, M. Q. Wang, H. P. Xiao, X. Gong, W. Yang and Y. Cao, New J. Chem., 2012, 36, 2042–2047 RSC.
- B. Zhang, L. Yu, L. Fan, N. Wang, L. W. Hu and W. Yang, New J. Chem., 2014, 38, 4587–4593 RSC.
- Y. J. Cheng, S. W. Cheng, C. Y. Chang, W. S. Kao, M. H. Liao and C. S. Hsu, Chem. Commun., 2012, 48, 3203–3205 RSC.
- X. F. Xu, C. F. Wang, O. Bäcke, D. I. James, K. Bini, E. Olsson, M. R. Andersson, M. Fahlman and E. G. Wang, Polym. Chem., 2015, 6, 4624–4633 RSC.
- Y. Y. Liang, D. Q. Feng, Y. Wu, S. T. Tsai, G. Li, C. Ray and L. P. Yu, J. Am. Chem. Soc., 2009, 131, 7792–7799 CrossRef CAS PubMed.
- Y. Y. Liang, Z. Xu, J. B. Xia, S. T. Tsai, Y. Wu, G. Li, C. Ray and L. P. Yu, Adv. Mater., 2010, 22, E135–E138 CrossRef CAS PubMed.
- H. X. Zhou, L. Q. Yang, A. C. Stuart, S. C. Price, S. B. Liu and W. You, Angew. Chem., Int. Ed., 2011, 50, 2995–2998 CrossRef CAS PubMed.
- S. C. Price, A. C. Stuart, L. Q. Yang, H. X. Zhou and W. You, J. Am. Chem. Soc., 2011, 133, 4625–4631 CrossRef CAS PubMed.
- F. Huang, K. S. Chen, H. L. Yip, S. K. Hau, O. Acton, Y. Zhang, J. D. Luo and A. K. Y. Jen, J. Am. Chem. Soc., 2009, 131, 13886–13887 CrossRef CAS PubMed.
- C. H. Duan, K. S. Chen, F. Huang, H. L. Yip, S. J. Liu, J. Zhang, A. K. Y. Jen and Y. Cao, Chem. Mater., 2010, 22, 6444–6452 CrossRef CAS.
- T. Guo, L. Yu, B. F. Zhao, L. Ying, H. B. Wu, W. Yang and Y. Cao, J. Polym. Sci., Part A: Polym. Chem., 2015, 53, 1043–1051 CrossRef CAS.
- X. Y. Du, Y. Huang, S. L. Tao, X. X. Yang, X. L. Ding and X. H. Zhang, Dyes Pigm., 2015, 115, 149–153 CrossRef CAS.
- M. R. Zhu, J. H. Zou, S. J. Hu, C. Q. Li, C. L. Yang, H. B. Wu, J. Q. Qin and Y. Cao, J. Mater. Chem., 2012, 22, 361–366 RSC.
- O. Vybornyi, Y. Jiang, F. Baert, D. Demeter, J. Roncali, P. Blanchard and C. Cabanetos, Dyes Pigm., 2015, 115, 17–22 CrossRef CAS.
- Y. M. Zhang, M. J. Xiao, N. Su, J. Zhong, H. Tan, Y. F. Wang, Y. Liu, Y. Pei, R. Q. Yang and W. G. Zhu, Org. Electron., 2015, 17, 198–207 CrossRef CAS.
- H. F. Feng, W. F. Fu, L. J. Li, Q. C. Yu, H. Lu, J. H. Wan, M. M. Shi, H. Z. Chen, Z. A. Tan and Y. F. Li, Org. Electron., 2014, 15, 2575–2586 CrossRef CAS.
- S. S. Dharmapurikar, A. Arulkashmir, C. Das, P. Muddellu and K. Krishnamoorthy, ACS Appl. Mater. Interfaces, 2013, 5, 7086–7093 CAS.
- E. J. Wren, K. Mutkins, M. Aljada, P. L. Burn, P. Meredith and G. Vamvounis, Polym. Chem., 2010, 1, 1117–1126 RSC.
- S. G. Javad, A. Zeinab and Z. Abolfazl, RSC Adv., 2014, 4, 16385–16390 RSC.
- Q. Cassandre, A. R. Valérie, D. V. Cécile, C. Gilles, M. Fabien and A. Pierre, Eur. J. Org. Chem., 2012, 1394–1403 Search PubMed.
- L. Q. Shi, C. He, D. F. Zhu, Q. G. He, Y. Li, Y. Chen, Y. X. Sun, Y. Y. Fu, D. Wen, H. M. Cao and J. G. Cheng, J. Mater. Chem., 2012, 22, 11629–11635 RSC.
- J. M. Jiang, P. A. Yang, H. C. Chen and K. H. Wei, Chem. Commun., 2011, 47, 8877–8879 RSC.
- C. He, C. M. Zhong, H. B. Wu, R. Q. Yang, W. Yang, F. Huang, G. C. Bazan and Y. Cao, J. Mater. Chem., 2010, 20, 2617–2622 RSC.
- Z. C. He, C. M. Zhong, X. Huang, W. Y. Wong, H. B. Wu, L. W. Chen, S. J. Su and Y. Cao, Adv. Mater., 2011, 23, 4636–4643 CrossRef CAS PubMed.
- G. J. Zhao, Y. J. He and Y. F. Li, Adv. Mater., 2010, 22, 4355–4358 CrossRef CAS PubMed.
|
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2016 |
Click here to see how this site uses Cookies. View our privacy policy here.