DOI:
10.1039/C5NR06734K
(Paper)
Nanoscale, 2016,
8, 283-291
Theranostic reduction-sensitive gemcitabine prodrug micelles for near-infrared imaging and pancreatic cancer therapy†
Received
29th September 2015
, Accepted 7th November 2015
First published on 9th November 2015
Abstract
A biodegradable and reduction-cleavable gemcitabine (GEM) polymeric prodrug with in vivo near-infrared (NIR) imaging ability was reported. This theranostic GEM prodrug PEG-b-[PLA-co-PMAC-graft-(IR820-co-GEM)] was synthesized by ring-opening polymerization and “click” reaction. The as-prepared reduction-sensitive prodrug could self-assemble into prodrug micelles in aqueous solution confirmed by dynamic light scattering (DLS) and transmission electron microscopy (TEM). In vitro drug release studies showed that these prodrug micelles were able to release GEM in an intracellular-mimicking reductive environment. These prodrug micelles could be effectively internalized by BxPC-3 pancreatic cancer cells, which were observed by confocal laser scanning microscopy (CLSM). Meanwhile, a methyl thiazolyl tetrazolium (MTT) assay demonstrated that this prodrug exhibited high cytotoxicity against BxPC-3 cells. The in vivo whole-animal near-infrared (NIR) imaging results showed that these prodrug micelles could be effectively accumulated in tumor tissue and had a longer blood circulation time than IR820-COOH. The endogenous reduction-sensitive gemcitabine prodrug micelles with the in vivo NIR imaging ability might have great potential in image-guided pancreatic cancer therapy.
Introduction
Pancreatic cancer is one of the most lethal human cancers, with only a 5% 5-year survival rate and less than 6 months median survival.1,2 80–85% of pancreatic cancer patients cannot be surgically resected, so chemotherapy is always the only choice for these patients.3 Gemcitabine (GEM) is currently the first line drug available for treatment of pancreatic cancer in clinics. GEM, a cell cycle-dependent (S-phase-specific) analogue of deoxycytidine, acts against a wide range of solid tumors, such as pancreatic, non-small lung, breast, and ovarian cancers. However, its clinical application is limited by high resistance towards tumor tissue because of insufficient activation or rapid inactivation of GEM. It can be rapidly deaminated in blood to the inactive metabolite 2′,2′-difluorodeoxyuridine to be rapidly excreted in urine.4,5 To address these limitations, gemcitabine prodrugs were well developed to protect drugs from deactivation.6–9 However, it remains a big challenge to release the active drugs from gemcitabine prodrugs after cellular internalization. It will be highly desirable to develop new smart drug delivery systems so that GEM can be released in response to biological stimuli, such as pH, redox environment, and enzyme levels.10–14 Reduction-responsive drug delivery systems have attracted considerable interest because of the large concentration gradient of glutathione (GSH) between the intracellular (≈10.0 mM) and the extracellular environment (≈2.0 μM) and at least 4-fold higher concentrations of GSH in tumor tissues than normal tissues.15–18 Small molecular reduction-responsive GEM prodrugs were reported by Kim et al.11,12 However, it is always a big challenge to achieve effective accumulation of such GEM prodrugs in tumor tissue by the enhanced permeability and retention (EPR) effect. Nanomedicine for GEM delivery might be one of the most effective strategies.
In recent years, polymeric prodrug micelles, which combine the advantages of prodrug technology (e.g., structural stability and minimal burst release) and micellar technology (e.g., high drug loading and efficient cell uptake) for drug formulation, have attracted extensive research interest, since they can increase the drug solubility, prolong blood circulation and reduce adverse effects. Therefore, various polymeric prodrug micelles have been developed for efficient delivery of chemotherapeutic agents, such as doxorubicin, cisplatin, and camptothecin. However, GEM prodrug micelles have rarely been reported. How to develop sophisticated multifunctional GEM prodrug micelles for efficient cancer therapy is a very important research subject. It has already been demonstrated that GEM encapsulated nanoparticles with stimuli responsiveness resulted in a better therapeutic effect and a lower drug resistance compared with free GEM.2,14,19
For ideal drug nanocarriers, it is very necessary to monitor the cellular uptake of nanocarriers, the accumulation of nanocarriers in tissues and the interactions between the nanocarriers and their microenvironments by a real-time and non-invasive way. However, it is very difficult to track the delivery of GEM in vitro and in vivo since GEM is a non-fluorescent chemotherapeutic agent. It would be of great importance to develop new theranostic GEM drug nanocarriers which are able to realize real-time monitoring of drug delivery and on-demand drug release. Fluorescence techniques have become one of the most effective diagnostic strategies for real-time and non-invasive monitoring of biological processes in cells and organisms. However, most of the fluorescent bioprobes may not be applicable for in vivo applications since the tissue penetration depth of UV light and visible light is extremely limited. Near-infrared (NIR) fluorescence dyes are very advantageous for in vivo imaging since the optical absorbance of NIR dyes in the NIR region (700–900 nm) is a transparency window for biological tissues.20–22 Cyanine-based NIR dyes, such as indocyanine green, IR780 and IR820, are the most widely used NIR imaging agents with commercial availability.23–26 However, there are still some limitations about cyanine dyes, including poor stability, short half-life in blood, quick clearance from the body, and lack of the targeting ability. Some attempts have been made to encapsulate cyanine dyes in micelles, nanoparticles and dendrimers.23,27–29 Compared to physical encapsulation, chemical conjugation of cyanine dyes to nanoparticles may be more advantageous since it prevents the dyes from leakage during blood circulation. In order to covalently conjugate cyanine dyes to nanoparticles, it is highly desirable to synthesize functionalized cyanine dyes by introduction of suitable functional groups.26,30–34
In this research, we developed biodegradable reduction-sensitive theranostic GEM prodrug micelles based on PEG-b-[PLA-co-PMAC-graft-(IR820-co-GEM)] containing IR820 for NIR imaging. GEM was conjugated to the polymer backbone via a reduction-sensitive disulfide bond. It was expected that the GEM prodrug micelles could be internalized by cancer cells. The disulfide bond was expected to be cleaved in the intracellular reductive environment with rapid release of GEM from prodrug micelles, resulting in enhanced inhibition of the cellular proliferation. Moreover, the IR820 conjugated prodrug micelles can be used for real-time monitoring of the in vivo distribution of the prodrug micelles with a tumor xenograft model. As far as we know, it is the first example of reduction-sensitive GEM prodrug micelles. Such theranostic GEM prodrug micelles might open an avenue for image-guided pancreatic cancer therapy.
Experimental
Materials
2,2-Dimethylol propionic acid, 3-bromopropene, 4-dimethylaminopyridine (DMAP), 1,8-diazabicyclo[5,4,0]undec-7-ene (DBU), methoxypolyethylene glycol (PEG, Mw 5000), 6-aminohexanoic acid, cysteamine hydrochloride and L-buthionine-sulfoximine (BSO) were purchased from Aladdin Industrial Corporation. N,N-Diisopropylethylamine (DIPEA), 4-nitrophenyl chloroformate, 2-hydroxyethyl disulfide, acryloyl chloride, new indocyanine green (IR820), 3-(4,5-dimethyl-thiazol-2-yl)-2,5-diphenyl tetrazolium bromide (MTT), 2-(4-amidinophenyl)-6-indolecarbamidine dihydrochloride (DAPI) and glutathione reduced ethyl ester (GSH-OEt) were purchased from Sigma-Aldrich. DL-Lactide was received from Beijing Glaco Co., Ltd. Gemcitabine was obtained from Beijing HWRK Chem Co., Ltd. 5-Methyl-5-allyloxycarbonyl-1,3-dioxan-2-one (MAC) and 2-((2-hydroxyethyl)disulfanyl) ethyl acrylate (HSEA) were synthesized according to the previously published procedures.35,36 Carboxylated IR820 (IR820-COOH) was synthesized according to a published article.26 MilliQ Water (18.2 MΩ cm−1) was obtained using a Millipore MilliQ Academic Water Purification System. Chloroform was dried over CaH2. Other reagents not listed here were used as received.
Instrumentation
1H NMR spectra were recorded on a Bruker DMX500 instrument. The mass spectra were recorded on a Bruker Esquire 3000 plus ion trap mass spectrometer equipped with an ESI source. Molecular weights and molecular weight distributions of the polymers were studied by using a gel permeation chromatograph (GPC) equipped with PL gel 10 mm MIXED-B columns using a series of narrow polystyrene as standards for the calibration. The eluent used was THF, and the flow rate was 1.0 mL min−1 at 40 °C. Dynamic light scattering (DLS) measurements were performed using a Zetasizer Nano-ZS from Malvern Instruments equipped with a He–Ne laser at a wavelength of 633 nm at 25 °C. The intensity-average hydrodynamic diameter (Dh) was adopted in this research. The samples were cleaned using a 0.45 μm Millipore filter before the measurements. Transmission electron microscopy (TEM) was performed on a HT7700 TEM (HITACHI, Japan) operated at an accelerating voltage of 100 kV. UV-vis spectra were recorded using a Shimadzu UV-2550 UV-vis spectrometer and the absorption was recorded from 400 to 900 nm. The fluorometric measurements were carried out using a Shimadzu RF-530 spectrometer. The excitation wavelength was set at 650 nm with a slit width of 5 nm and the emission spectra were recorded from 690 to 900 nm. In vivo fluorescence images were obtained with a CRI Maestro in vivo imaging system equipped with a tunable liquid-crystal filter and a cooled scientific-grade monochrome CCD camera. The filter was set to 665 nm/785 nm for excitation (narrow band) and emission (long-pass), and used to excite and obtain the fluorescence signals of the prodrug micelles, respectively. The liquid-crystal tunable filter was automatically tuned with 10 nm increments, and all image cubes were captured with the same exposure time.
Synthesis of reduction-responsive and clickable gemcitabine derivative 2-((2-hydroxyethyl)disulfanyl) ethyl acrylate (HSEA-GEM)
To a stirred solution of HSEA (200 mg, 0.962 mmol), DIPEA (497 mg, 3.846 mmol) and a catalytic amount of pyridine in dry dichloromethane (DCM) (5.0 mL), 4-nitrophenyl chloroformate (582 mg, 2.886 mmol) in dry DCM was added dropwise at 0 °C and stirred for 4 h at room temperature. The reaction mixture was concentrated in vacuo and then dissolved in 3 mL DMF. To this solution, GEM (760 mg, 2.889 mmol) in DMF (3 mL) and TEA (0.8 mL) were added and continued to be stirred for 24 h. After completion of the reaction, the reaction mixture was diluted in DCM and washed with water three times. Finally, the crude product was purified by column chromatography on silica gel using DCM/MeOH (15![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) as the eluent to afford a yellow solid.
:1) as the eluent to afford a yellow solid.
Synthesis of the degradable copolymer poly(ethylene glycol)-block-[poly(DL-lactide)-co-poly(5-methyl-5-allyloxycarbonyl-1,3-dioxan-2-one)] (PEG-b-(PLA-co-PMAC))
PEG-b-(PLA-co-PMAC) was synthesized by the ring-opening copolymerization of DL-lactide and MAC using PEG as the initiator. In a typical experiment, PEG (500 mg, 0.10 mmol), MAC (1000 mg, 5.0 mmol), DL-lactide (650 mg, 3.0 mmol), DBU (30 μL, 0.20 mmol) and dry chloroform (8 mL) were added into a 25 mL flask under a nitrogen atmosphere. The mixture solution was degassed by three freeze–pump–thaw cycles and stirred at 30 °C. After 48 h, the reaction mixture was precipitated directly into methanol and diethyl ether three times, respectively.
Synthesis of amino functionalized PEG-b-(PLA-co-PMAC)
PEG-b-(PLA-co-PMAC) was functionalized using cysteamine hydrochloride by thiol–ene “click” reaction. Briefly, PEG-b-(PLA-co-PMAC) (200 mg), DMAP (50 mg, 0.41 mmol) in dry THF (8 mL) and cysteamine hydrochloride (3000 mg, 26.4 mmol) in methanol (8 mL) were added into a 25 mL round flask. The solution was degassed by three freeze–pump–thaw cycles and was irradiated at room temperature for 3 h. Finally, the crude product was isolated by extensive dialysis against distilled water (MWCO 3500) for 48 h and freeze-drying.
Synthesis of PEG-b-[PLA-co-PMAC-graft-(IR820-co-GEM)]
PEG-b-(PLA-co-PMAC-graft-GEM) was first synthesized by the Michael addition reaction. Briefly, PEG-b-(PLA-co-PMAC) (160 mg) and HSEA-GEM (80 mg) in DMF (10 ml) were added in a 25 mL round flask and continued to be stirred for 24 h at 50 °C. After completion of the reaction, the reaction mixture was isolated by extensive dialysis against DMF (MWCO 3500), and distilled water (MWCO 3500) for 48 h, respectively, and then freeze-drying. Next, the obtained PEG-b-(PLA-co-PMAC-graft-GEM) (100 mg), IR820-COOH (80 mg), EDC (30 mg) and NHS (30 mg) were added in DMSO (5 ml) and stirred for 12 h at room temperature. After completion of the reaction, the reaction mixture was extensively dialysed against DMSO (MWCO 3500) for 48 h and distilled water (MWCO 3500) for another 48 h. The solution was then freeze-dried.
Preparation of prodrug micelles
PEG-b-[PLA-co-PMAC-graft-(IR820-co-GEM)] micelles were prepared by dialysis. 10 mg of the prodrug was dissolved in 2 mL DMSO and stirred for 4 h. 2 mL water was added dropwise and stirred for 4 h. Then, the solution was transferred into a dialysis tube, followed by dialyzing against water for 48 h to remove the organic solution.
Photophysical characterization
Absorption spectra and fluorescence emission spectra of IR820-COOH and IR820 conjugated prodrug micelles at 10 μg mL−1 dye content were recorded in deionized water at room temperature. The excitation wavelengths of IR820-COOH and IR820 conjugated prodrug micelles were both 650 nm.
In vitro drug release
The release profile of gemcitabine from prodrug micelles was followed by using a UV spectrometer in PBS with or without 10.0 mM GSH at 37 °C. In a typical experiment, 2 mL of prodrug micellar solution (1.6 mg mL−1) was added to two dialysis bags and then immersed in 8 mL PBS with or without 10.0 mM GSH media in a prepared tube, respectively. The tubes were kept at 37 °C in a thermostated incubator with constant shaking (100 rpm). At regular time points, 2 mL of media were taken out and replaced with fresh medium. The samples were subjected to UV measurements.
Cell culture
The human pancreatic cancer cell line BxPC-3 cells were purchased from KeyGen BioTECH (Nanjing, China) and cultured in RPMI 1640 containing 10% fetal bovine serum (FBS) in 5% CO2 at 37 °C.
In vitro cell cytotoxicity assay
The cytotoxicities of PEG-b-[PLA-co-PMAC-graft-(IR820-co-GEM)] prodrug micelles against BxPC-3 cells were evaluated in vitro by MTT assays. Briefly, cells were seeded in 96-well plates at 6 × 103 cells per well in 200 μL of culture medium for 12 h. Then, cells were treated with 0.5 mM BSO for 12 h or 10.0 mM GSH-OEt for 2 h. Cells without pretreatment were used as a control. After washing cells with PBS, 180 μL of prodrug and free gemcitabine in fresh culture medium (final concentration from 1 to 40 μg mL−1 equivalent to gemcitabine) were added and incubated for another 48 h. Then, 20 μL of MTT (5 mg mL−1) was added. After 4 h, the medium was replaced by 150 μL DMSO to dissolve the obtained crystals, and the absorbance was measured at a wavelength of 490 nm using a microplate Bio-Rad reader (Thermo Fisher Scientific, Waltham, MA). Data were expressed as average ± SD (n = 3).
Cellular uptake and in vitro imaging
BxPC-3 cells were seeded in a confocal dish at 3 × 104 cells per well and incubated for 12 h at 37 °C. Then, the medium was replaced with 0.1 mg mL−1 prodrug micelles in fresh culture. After being incubated for 0.5 h, 2 h and 6 h, the medium was removed and the confocal dishes were washed with PBS three times. The cells were fixed with 4% formaldehyde for 15 min and the cell nuclei were dyed with DAPI for 15 min. The fluorescence images of IR-820 of prodrug micelles in cells were observed by confocal laser scanning microscopy (CLSM).
In vivo NIR imaging and biodistribution
All animal experiments were performed according to the “Principles of Laboratory Animal Care” (NIH publication NO. 86–23, revised 1985) and the guidelines for Animal Care and Use Committee, Zhejiang University. Healthy male BALB/c nude mice (5 week old, weight around 15 g), which were purchased from the animal center of Zhejiang Academy of Medical Sciences, were used as a tumor model. BxPC-3 cells (2 × 106) in 0.1 mL of PBS were injected subcutaneously into the alar area of male nude BALB/c mice of weight 20 g. When the tumor volumes reached about 100 mm3, the nude mice were divided into two groups. 200 μL of free IR820-COOH (75 μg mL−1) or 200 μL of nanoparticles (75 μg mL−1 equivalent to IR820-COOH) were injected into nude mice via a tail vein and images of IR820 were obtained at 3, 8 and 24 h after injection using a Maestro in vivo imaging system.
Results and discussion
Synthesis of the polymeric prodrug PEG-b-[PLA-co-PMAC-graft-(IR820-co-GEM)]
Synthesis of the polymeric prodrug PEG-b-[PLA-co-PMAC-graft-(IR820-co-GEM)] was performed according to the synthetic route shown in Scheme 1: (A) preparation of a double bond-functionalized reduction-responsive GEM derivative (HSEA-GEM); (B) preparation of IR820-COOH and (C) synthesis of a polycarbonate-based triblock copolymer PEG-b-(PLA-co-PMAC) by ring opening polymerization followed by amination of the prepared polymer by “click” reaction and synthesis of the final polymeric prodrug PEG-b-[PLA-co-PMAC-graft-(IR820-co-GEM)] by chemical conjugation of HSEA-GEM and IR820-COOH.
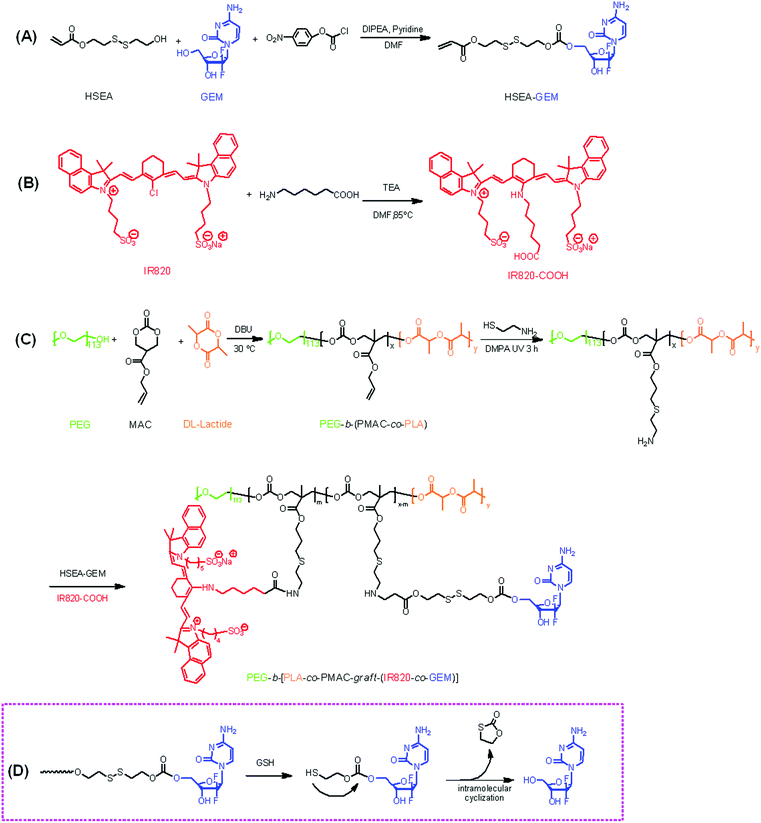 |
| | Scheme 1 Synthetic routes of (A) clickable GEM derivative HSEA-GEM, (B) IR820-COOH and (C) reduction-responsive polymeric prodrug PEG-b-[PLA-co-PMAC-graft-(IR820-co-GEM)]; (D) mechanism of GEM release from polymeric prodrug in the presence of GSH. | |
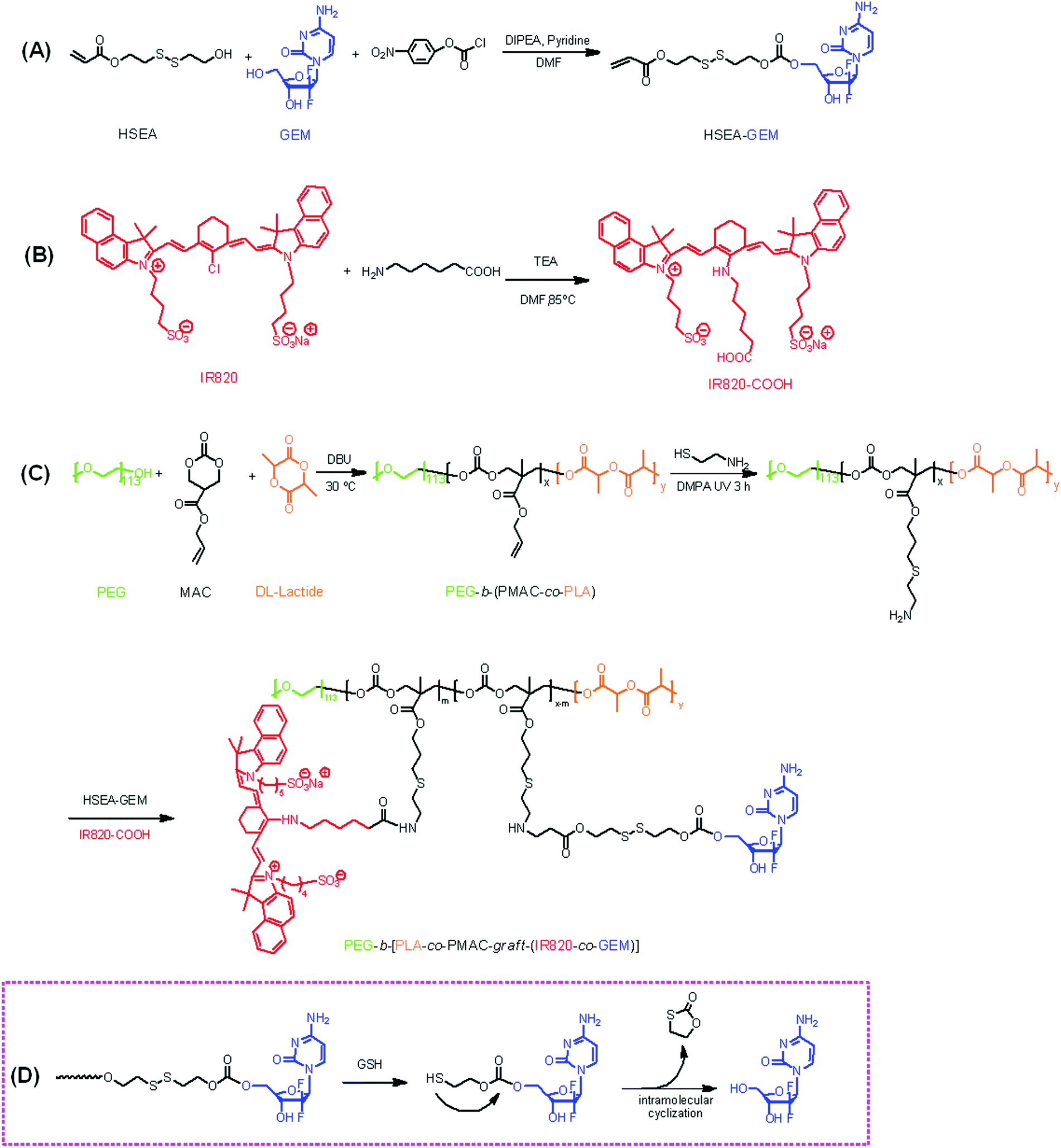
The chemical structures of HSEA-GEM and IR820-COOH were verified by 1H NMR and MS analyses (Fig. S1–S4†). The block copolymer PEG-b-(PLA-co-PMAC) was synthesized using PEG as the initiator and DBU as the catalyst. The typical 1H NMR spectrum of PEG-b-(PLA-co-PMAC) is shown in Fig. 1. The characteristic peaks of PEG at δ 3.6, PMAC at δ 5.8, 5.2, and PLA at δ 1.5 can be observed in Fig. 1. These results confirmed the successful synthesis of PEG-b-(PLA-co-PMAC).
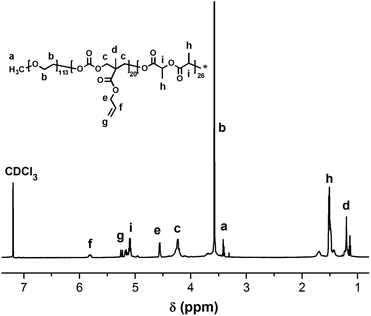 |
| | Fig. 1
1H NMR spectrum of PEG-b-(PLA-co-PMAC) (CDCl3). | |
By comparing the well-defined peak integrals of the initiator PEG with PMAC and PLA, the polymerization degree of PMAC and PLA was calculated as 20 and 26, respectively. Thus, the calculated molecular weight of PEG-b-(PLA-co-PMAC) is 12700. The number-average molecular weight and molecular weight distribution (Mw/Mn) of PEG-b-(PLA-co-PMAC) were measured by GPC analysis (Fig. S5†). As indicated by GPC, the resulting PEG-b-(PLA-co-PMAC) had relatively low polydispersity.
Almost 80% of the side chain from PEG-b-(PLA-co-PMAC) was functionalized with the amino group, which was verified by 1H NMR (Fig. S6†). Its molecular weight is calculated as 14000 accordingly. Finally, the obtained polymeric prodrug PEG-b-[PLA-co-PMAC-graft-(IR820-co-GEM)] was confirmed by 1H NMR as characteristic peaks of IR820-COOH and GEM labeled in Fig. 2. With eight GEM molecules and five IR820-COOH molecules conjugated to one polymer chain, the molecular weight of this polymeric prodrug is 22600. Therefore, the loading content of GEM and IR820-COOH was 9.3% and 20.7%, respectively.
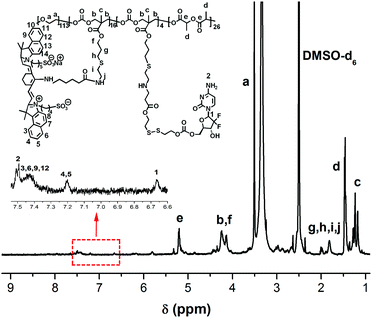 |
| | Fig. 2
1H NMR spectrum of PEG-b-[PLA-co-PMAC-graft-(IR820-co-GEM)] (DMSO-d6). | |
Photophysical characterization
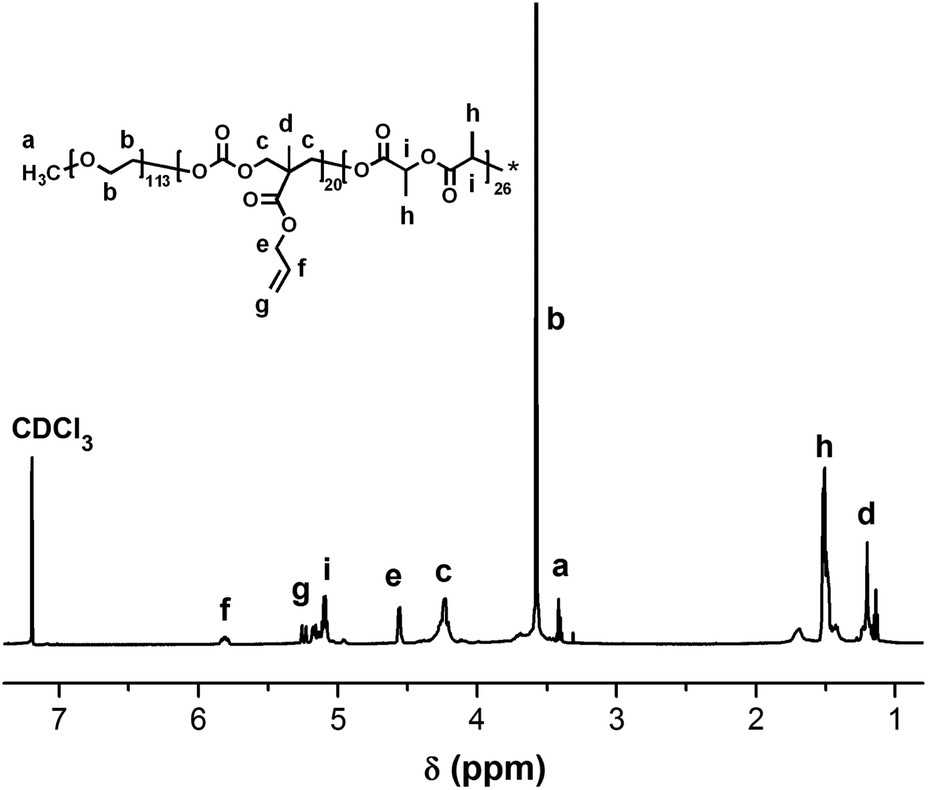
The UV-vis-NIR spectra and fluorescence emission spectra of PEG-b-[PLA-co-PMAC-graft-(IR820-co-GEM)] prodrug and IR820-COOH are presented in Fig. 3. IR820-COOH showed a characteristic peak located at 629 nm in aqueous solution, whereas IR820 conjugated prodrug micelles showed a peak at 675 nm with a red shift in water. This red shift can be attributed to an increase in conjugation length and the formation of an extended π system, which further proved the successful conjugation of IR820 to prodrug micelles.32 IR820 conjugated prodrug micelles and IR820-COOH showed a similar emission peak at about 810 nm.
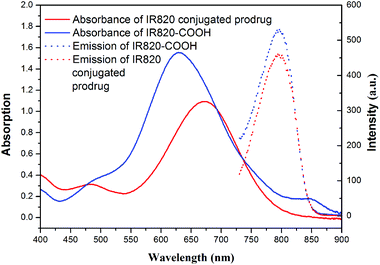 |
| | Fig. 3 Absorbance (solid line) and emission (dashed line) spectra of free IR820-COOH (blue) and IR820 conjugated prodrug micelles (red) in water. | |
Self-assembly of the polymeric prodrug micelles
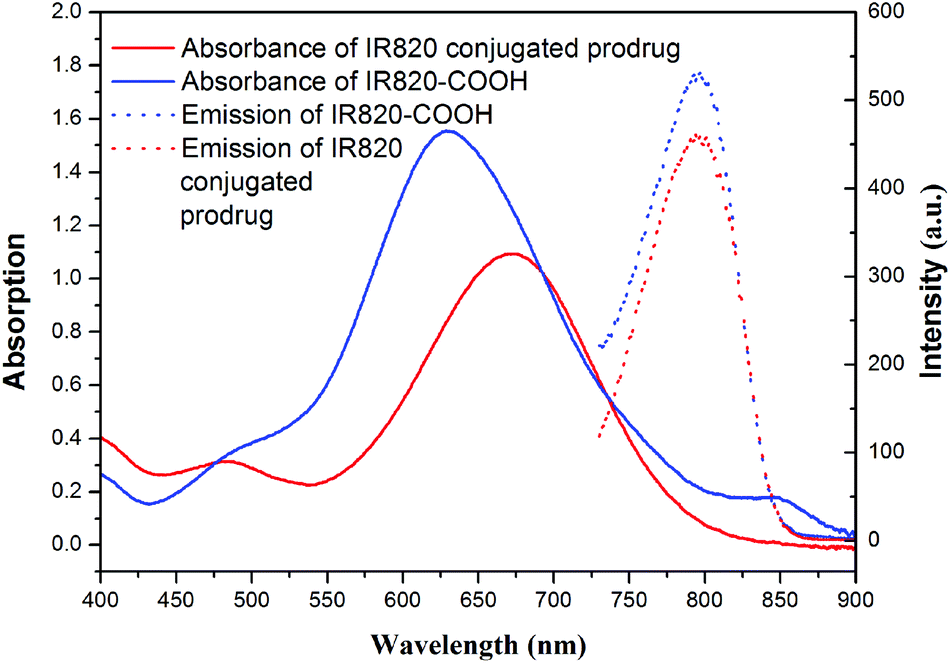
As is known, the size and size distribution of nanoparticles are important factors for drug delivery. Nanoparticles of 50–200 nm may preferentially be accumulated in the tumor tissues because of the enhanced permeability and retention (EPR) effect.37 The self-assembly and reduction-sensitive properties of the GEM prodrug micelles were evaluated by DLS and TEM analyses. The DLS results indicated that the intensity-average hydrodynamic diameter (Dh) of the prodrug micelles was 175.4 nm with a relatively narrow size distribution (PDI: 0.174) as shown in
Fig. 4. TEM images of the prodrug micelles revealed the presence of presumably spherical morphologies with an average micellar diameter of 160 nm as shown in Fig. 5a. The DLS and TEM results indicated that such GEM prodrug micelles had an ideal size for drug delivery.
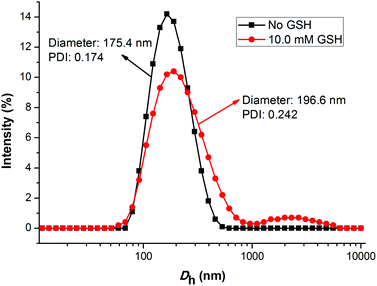 |
| | Fig. 4 The change of the size for PEG-b-[PLA-co-PMAC-graft-(IR820-co-GEM)] prodrug micelles with 10.0 mM GSH treated for 6 h and without GSH treated determined by DLS. | |
 |
| | Fig. 5 TEM images of PEG-b-[PLA-co-PMAC-graft-(IR820-co-GEM)] prodrug micelles with 10.0 mM GSH treated for 6 h (b) and without GSH treated (a). | |
The disulfide bonds caused the reduction-sensitive release of GEM from GEM prodrug micelles. Compared with GEM prodrug micelles, the Dh of GEM prodrug micelles treated with 10.0 mM GSH was a little larger and the particle dispersion index (PDI) was relatively broader. What's more, large aggregates with an average diameter of more than 1000 nm could be observed. TEM images showed that the diameter of prodrug micelles treated with GSH is larger than the untreated, which is coincident with the DLS results. The increase of the size might be ascribed to the cleavage of the disulfide bonds and release of GEM. When GEM was cleaved from the polymeric backbone, the structure of the micelles became looser, and some looser micelles would further form large aggregates due to the inferior stability.38
In vitro drug release
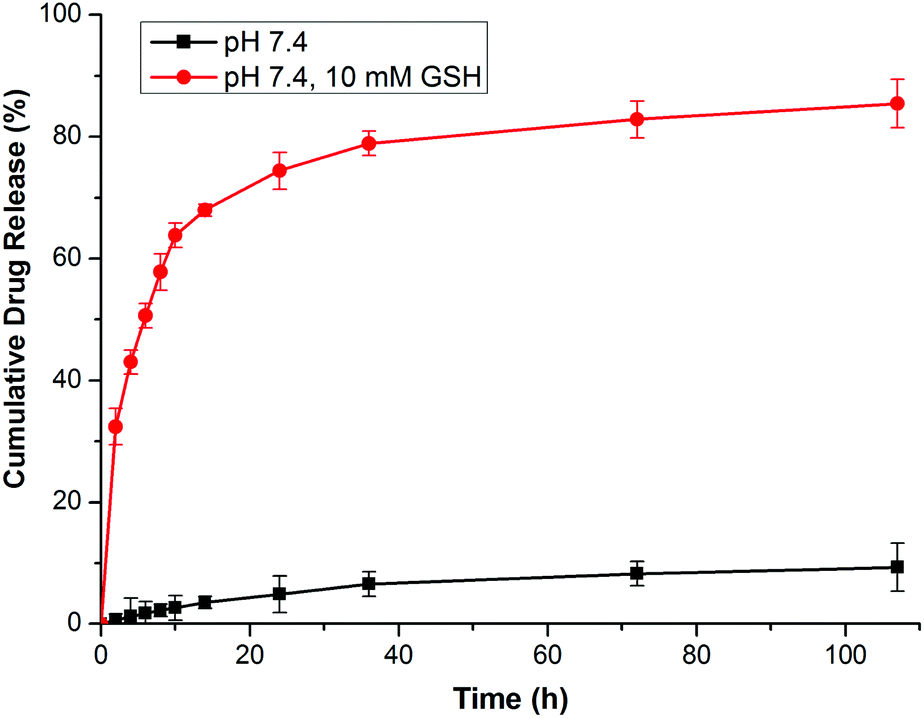
In this research, GEM was conjugated to the polymer backbone by a disulfide carbonate linker, which caused a reduction-sensitive release of GEM from GEM prodrug micelles. It has been demonstrated that the reduction of the vulnerable disulfide carbonate bond by GSH generates a thiol intermediate, followed by intramolecular cyclization and the cleavage of the neighboring carbonate bridge, thus releasing drug molecules in a pharmaceutically active form12,38,39 (Scheme 1D). The GEM release behaviors of the prodrug micelles were investigated with or without 10.0 mM GSH in PBS at pH 7.4. In the absence of GSH, only a minimal GEM was released from the micelles in 108 h. However, GEM release was accelerated with 10.0 mM GSH in PBS, analogous to the intracellular reductive environment. Nearly 77% of the GEM was released in 24 h and 83% of GEM was released over 108 h (Fig. 6). Therefore, it is anticipated that these prodrug micelles can realize triggered drug release in the intracellular reductive environment.
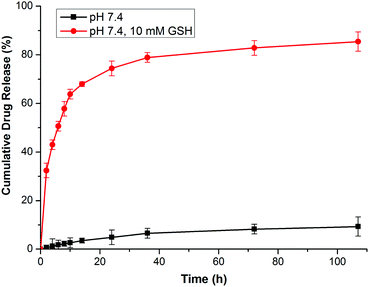 |
| | Fig. 6
In vitro GEM release from prodrug micelles in PBS with 10.0 mM GSH and without GSH. | |
In vitro cytotoxicity assay
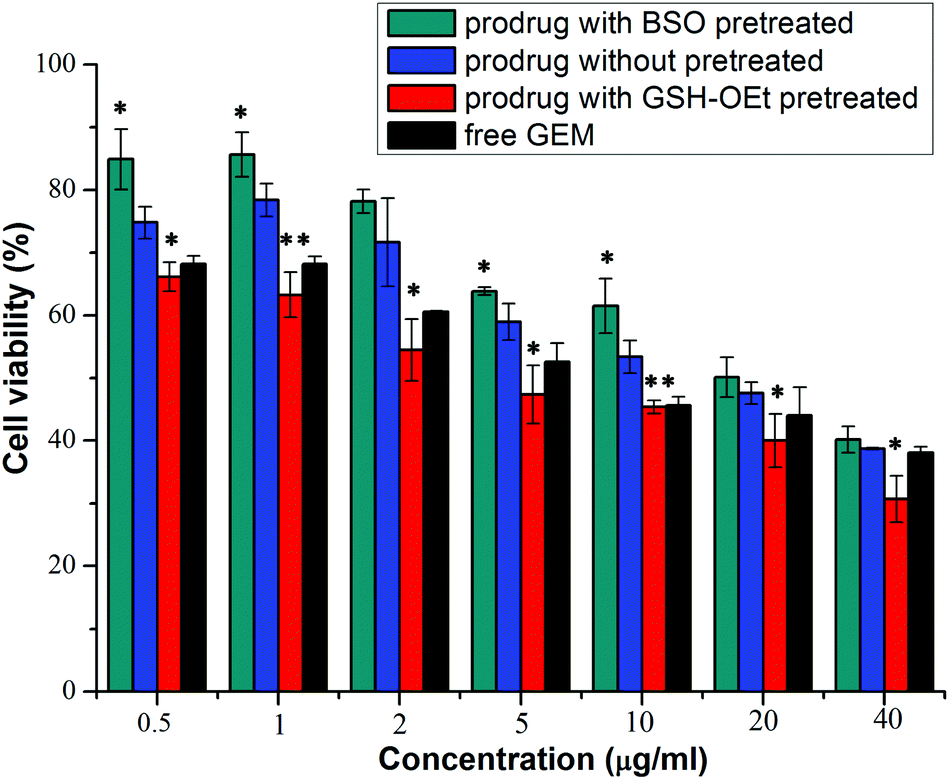
The in vitro cellular proliferation inhibitions of GEM prodrug micelles against BxPC-3 cells were evaluated by the MTT assay. Cells were first pretreated with 0.5 mM BSO for 12 h to inhibit the intracellular synthesis of GSH or with 10.0 M GSH-OEt for 2 h to increase the intracellular GSH concentration and then incubated with the GEM prodrug nanoparticles for 48 h.40,41 Cells without pretreatment and incubated with free GEM were both used as control. As is shown in Fig. 7, the BxPC-3 cells pretreated with BSO exhibited the lowest inhibition efficiency. While the cells pretreated with GSH-OEt showed even higher inhibition efficiency than free GEM. These results indicated that the faster GEM release from GEM prodrug micelles was triggered by higher intracellular GSH concentration in the tumor cell environment, which enhanced the inhibition of the cellular proliferation.
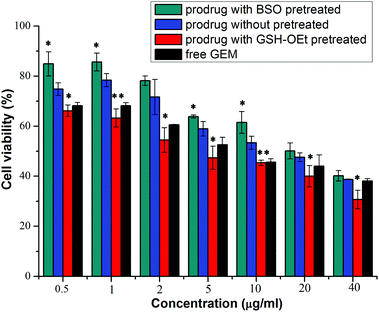 |
| | Fig. 7 Proliferation inhibitions toward BxPC-3 cells incubated with prodrug micelles which cells pretreated with 0.5 mM BSO and 10.0 mM GSH-OEt or untreated, and with free gemcitabine with various gemcitabine concentrations for 48 h. *P < 0.05, **P < 0.01, compared with the untreated group. | |
Cellular uptake and imaging
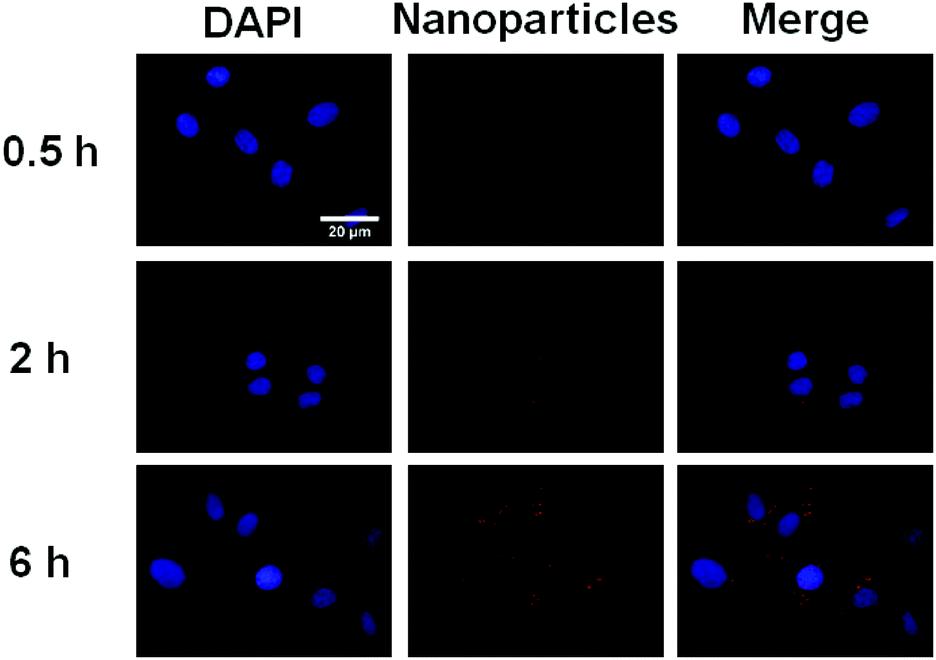
In order to demonstrate the potential of IR820 conjugated prodrug micelles in NIR bioimaging applications, the cellular uptake of prodrug micelles against BxPC-3 cells was studied by CLSM. As shown in Fig. 8, little fluorescence was observed in BxPC-3 cells after 0.5 h incubation, while strong fluorescence of IR820 can be observed after 2 h incubation. The fluorescence intensity of IR820 became much stronger with a longer incubation time. The IR820 conjugated prodrug micelles can be effectively internalized by BxPC-3 cells and intensively distributed in the cytoplasm.
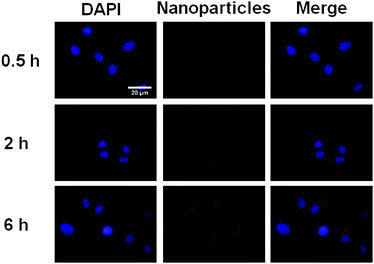 |
| | Fig. 8 CLSM images of BxPC-3 incubated with the IR820 conjugated prodrug micelles. From left to right: DAPI (blue), IR820 of prodrug micelles (red) and a merge of the two images. | |
Prodrug micelles for in vivo imaging
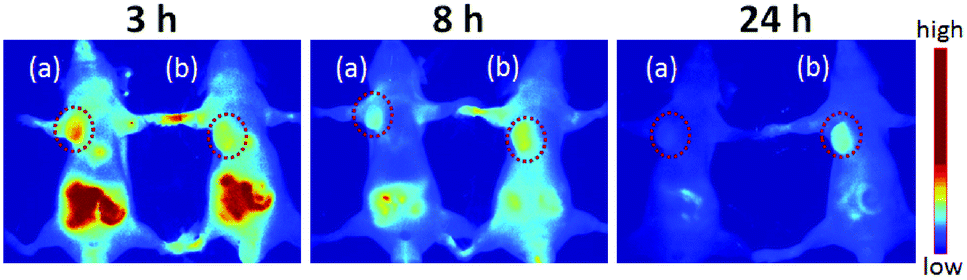
In order to visualize the prodrug micelles in vivo, biodistribution studies were performed on BxPC-3 tumor xenografted mice intravenously injected with IR820 conjugated prodrug micelles and free IR820-COOH, respectively. Fig. 9 shows the fluorescence signals and intensity distributions after 3 h, 8 h and 24 h injection. As shown in Fig. 9, the fluorescence signals of IR820 conjugated prodrug micelles and free IR820-COOH were both whole body distributed after 3 h injection. The signals of IR820 on the whole mouse body gradually decreased from 3 h to 24 h post injection. At first, the signal of free IR820-COOH was stronger than that of prodrug nanoparticles in tumor tissues at 3 h after injection. After 8 h and 24 h post-injection, the tumor signal intensity of prodrug nanoparticles remained high. However, the signal intensity of free IR820 almost disappeared. It indicated that covalently bonded IR820 with micelles was more stable and had a longer blood circulation time in vivo.
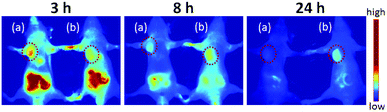 |
| | Fig. 9
In vivo NIR imaging of nude mice bearing tumors after tail vein injection of (a) free IR820-COOH and (b) IR820 conjugated prodrug nanoparticles. The tumors were circled with a red dotted line. | |
Conclusions
In summary, a novel theranostic reduction-sensitive GEM prodrug was successfully synthesized by chemical conjugation of GEM and IR820 to a biodegradable polycarbonate backbone. The IR820 conjugated GEM prodrug can self-assemble into micelles as verified by DLS and TEM. The MTT assay demonstrated that the released GEM from prodrug micelles can inhibit cell proliferation against BxPC-3 cells, which was achieved by the cleavage of the disulfide linkage in the intracellular reductive environment of tumor cells. Moreover, the xenograft tumor model indicated that these prodrug micelles could be accumulated in tumor tissue by the EPR effect and had a longer blood circulation time than IR820-COOH. Overall, such theranostic GEM prodrug micelles with favorable reduction sensitivity would be a promising candidate for enhancing the therapeutic efficacy of gemcitabine and simultaneously being used as in vivo bioprobes for pancreatic cancer therapy.
Acknowledgements
Financial support from the National Natural Science Foundation of China (21174126, 51303154, 51573160, 21574114), the Key Science Technology Innovation Team of Zhejiang Province (no. 2013TD02), the Research Fund for the Doctoral Program of Higher Education of China (20130101120177), and the Scientific Research Foundation for the Returned Overseas Chinese Scholars, State Education Ministry ([2015]311) is gratefully acknowledged.
Notes and references
- N. Bardeesy and R. A. DePinho, Nat. Rev. Cancer, 2002, 2, 897–909 CrossRef CAS PubMed.
- H. Meng, M. Wang, H. Liu, X. Liu, A. Situ, B. Wu, Z. Ji, C. H. Chang and A. E. Nel, ACS Nano, 2015, 9, 3540–3557 CrossRef CAS PubMed.
- D. Li, K. Xie, R. Wolff and J. L. Abbruzzese, Lancet, 2004, 363, 1049–1057 CrossRef CAS.
- E. Moysan, G. Bastiat and J. P. Benoit, Mol. Pharmaceutics, 2013, 10, 430–444 CrossRef CAS PubMed.
- M. Erkan, S. Hausmann, C. W. Michalski, A. A. Fingerle, M. Dobritz, J. Kleeff and H. Friess, Nat. Rev. Gastroenterol. Hepatol., 2012, 9, 454–467 CrossRef CAS PubMed.
- M. Vandana and S. K. Sahoo, Biomaterials, 2010, 31, 9340–9356 CrossRef CAS PubMed.
- L. H. Reddy, C. Dubernet, S. L. Mouelhi, P. E. Marque, D. Desmaele and P. Couvreur, J. Controlled Release, 2007, 124, 20–27 CrossRef CAS PubMed.
- G. Arya, M. Vandana, S. Acharya and S. K. Sahoo, Nanomed. Nanotechnol. Biol. Med., 2011, 7, 859–870 CrossRef CAS PubMed.
- W. Wu, W. Driessen and X. Jiang, J. Am. Chem. Soc., 2014, 136, 3145–3155 CrossRef CAS PubMed.
- C. Ren, C. Xu, D. Li, H. Ren, J. Hao and Z. Yang, RSC Adv., 2014, 4, 34729–34732 RSC.
- Z. Yang, J. H. Lee, H. M. Jeon, J. H. Han, N. Park, Y. He, H. Lee, K. S. Hong, C. Kang and J. S. Kim, J. Am. Chem. Soc., 2013, 135, 11657–11662 CrossRef CAS PubMed.
- S. Maiti, N. Park, J. H. Han, H. M. Jeon, J. H. Lee, S. Bhuniya, C. Kang and J. S. Kim, J. Am. Chem. Soc., 2013, 135, 4567–4572 CrossRef CAS PubMed.
- R. Zhang, J. Yang, M. Sima, Y. Zhou and J. Kopecek, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 12181–12186 CrossRef CAS PubMed.
- G. Y. Lee, W. P. Qian, L. Wang, Y. A. Wang, C. A. Staley, M. Satpathy, S. Nie, H. Mao and L. Yang, ACS Nano, 2013, 7, 2078–2089 CrossRef CAS PubMed.
- M. H. Lee, Z. Yang, C. W. Lim, Y. H. Lee, S. Dongbang, C. Kang and J. S. Kim, Chem. Rev., 2013, 113, 5071–5109 CrossRef CAS PubMed.
- P. Kuppusamy, H. Li, G. Ilangovan, A. J. Cardounel, J. L. Zweier, K. Yamada, M. C. Krishna and J. B. Mitchell, Cancer Res., 2002, 62, 307–312 CAS.
- B. Deng, P. Ma and Y. Xie, Nanoscale, 2015, 7, 12773–12795 RSC.
- H. Sun, F. Meng, R. Cheng, C. Deng and Z. Zhong, Expert Opin. Drug Delivery, 2013, 10, 1109–1122 CrossRef CAS PubMed.
- W. Wang, C. Li, J. Zhang, A. Dong and D. Kong, J. Mater. Chem. B, 2014, 2, 1891–1901 RSC.
- S. Luo, E. Zhang, Y. Su, T. Cheng and C. Shi, Biomaterials, 2011, 32, 7127–7138 CrossRef CAS PubMed.
- E. Cassette, M. Helle, L. Bezdetnaya, F. Marchal, B. Dubertret and T. Pons, Adv. Drug Delivery Rev., 2013, 65, 719–731 CrossRef CAS PubMed.
- R. Wang and F. Zhang, J. Mater. Chem. B, 2014, 2, 2422–2443 RSC.
- L. Wu, S. Fang, S. Shi, J. Deng, B. Liu and L. Cai, Biomacromolecules, 2013, 14, 3027–3033 CrossRef CAS PubMed.
- Z. Sheng, D. Hu, M. Zheng, P. Zhao, H. Liu, D. Gao, P. Gong, G. Gao, P. Zhang, Y. Ma and L. Cai, ACS Nano, 2014, 8, 12310–12322 CrossRef CAS PubMed.
- C. H. Kao, J. Y. Wang, K. H. Chuang, C. H. Chuang, T. C. Cheng, Y. C. Hsieh, Y. L. Tseng, B. M. Chen, S. R. Roffler and T. L. Cheng, Biomaterials, 2014, 35, 9930–9940 CrossRef CAS PubMed.
- A. Masotti, P. Vicennati, F. Boschi, L. Calderan, A. Sbarbati and G. Ortaggi, Bioconjugate Chem., 2008, 19, 983–987 CrossRef CAS PubMed.
- X. Zheng, F. Zhou, B. Wu, W. R. Chen and D. Xing, Mol. Pharmaceutics, 2012, 9, 514–522 CrossRef CAS PubMed.
- O. Taratula, C. Schumann, T. Duong, K. L. Taylor and O. Taratula, Nanoscale, 2015, 7, 3888–3902 RSC.
- C. Yue, P. Liu, M. Zheng, P. Zhao, Y. Wang, Y. Ma and L. Cai, Biomaterials, 2013, 34, 6853–6861 CrossRef CAS PubMed.
- R. P. Johnson, Y. I. Jeong, J. V. John, C. W. Chung, S. H. Choi, S. Y. Song, D. H. Kang, H. Suh and I. Kim, Macromol. Rapid Commun., 2014, 35, 888–894 CrossRef CAS PubMed.
- C. Ornelas, R. Lodescar, A. Durandin, J. W. Canary, R. Pennell, L. F. Liebes and M. Weck, Polym. Chem., 2011, 17, 3619–3629 CAS.
- S. Srinivasan, R. Manchanda, A. Fernandez-Fernandez, T. Lei and A. J. McGoron, J. Photochem. Photobiol., B, 2013, 119, 52–59 CrossRef CAS PubMed.
- K. Miki, T. Inoue, Y. Kobayashi, K. Nakano, H. Matsuoka, F. Yamauchi, T. Yano and K. Ohe, Biomacromolecules, 2015, 16, 219–227 CrossRef CAS PubMed.
- S. K. Yen, D. Jańczewski, J. L. Lakshmi, S. B. Dolmanan, S. Tripathy, V. H. B. Ho, V. Vijayaragavan, A. Hariharan, P. Padmanabhan, K. K. Bhakoo, T. Sudhaharan, S. Ahmed, Y. Zhang and S. Tamil Selvan, ACS Nano, 2013, 7, 6796–6805 CrossRef CAS PubMed.
- H. Wang, Y. Wang, Y. Chen, Q. Jin and J. Ji, Polym. Chem., 2014, 5, 854–861 RSC.
- S. S. Dunn, S. Tian, S. Blake, J. Wang, A. L. Galloway, A. Murphy, P. D. Pohlhaus, J. P. Rolland, M. E. Napier and J. M. DeSimone, J. Am. Chem. Soc., 2012, 134, 7423–7430 CrossRef CAS PubMed.
- V. Torchilin, Adv. Drug Delivery Rev., 2011, 63, 131–135 CrossRef CAS PubMed.
- Q. Zhang, J. He, M. Zhang and P. Ni, J. Mater. Chem. B, 2015, 3, 4922–4932 RSC.
- X. Hu, J. Hu, J. Tian, Z. Ge, G. Zhang, K. Luo and S. Liu, J. Am. Chem. Soc., 2013, 135, 17617–17629 CrossRef CAS PubMed.
- A. Zhang, Z. Zhang, F. Shi, C. Xiao, J. Ding, X. Zhuang, C. He, L. Chen and X. Chen, Macromol. Biosci., 2013, 13, 1249–1258 CrossRef CAS PubMed.
- J. Liu, Y. Pang, W. Huang, X. Huang, L. Meng, X. Zhu, Y. Zhou and D. Yan, Biomacromolecules, 2011, 12, 1567–1577 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5nr06734k |
|
| This journal is © The Royal Society of Chemistry 2016 |
Click here to see how this site uses Cookies. View our privacy policy here.