
One-pot synthesis of carbazoles via tandem C–C cross-coupling and reductive amination†
Deuk-Young
Goo
and
Sang Kook
Woo
*
Department of Chemistry, University of Ulsan, 93 Daehak-Ro, Nam-Gu, Ulsan 44610, Korea. E-mail: woosk@ulsan.ac.kr; Fax: +82-52-259-2348; Tel: +82-52-259-2338
First published on 28th October 2015
Abstract
We have developed a highly efficient synthetic route to carbazoles that employs sequential C–C/C–N bond formation via Suzuki cross-coupling and Cadogan cyclization using commercially available or easily preparable starting materials. The developed method is compatible with electron neutral, rich or deficient substrates. The synthetic utility of this method was demonstrated by the concise syntheses of four natural products (glycozoline, glycozolicine, glycozolidine and clausenalene).
Introduction
Carbazole is a ubiquitous structural motif in a large number of biologically active natural products and pharmaceuticals.1 In addition, carbazole derivatives have been widely used as functional organic materials.2 Thus, the development of efficient synthetic routes to structurally diverse carbazoles represents an important objective in organic synthesis. Many methods have been developed for the preparation of carbazole.3 The most widely used approaches for the synthesis of the carbazole framework are C–N or C–C bond forming cyclizations from substituted biaryls or N,N-biaryl amines. Representative C–N bond forming reactions for the synthesis of carbazoles are summarized in Scheme 1(a): (A) transition metal free4 or palladium catalyzed Buchwald–Hartwig5 amination from 2-amino-2′-halo-biphenyls; (B) dehydrogenative C–H/N–H coupling of 2-amino-biphenyls;6 (C) iron-mediated oxidative amination of an iron complex;7 (D) transition metal mediated double amination of 2,2′-dihalobiphenyls or cyclic diphenyleneiodoniums;8 (E) transition metal free9 or rhodium catalyzed10 nitrene insertion of 2-azido-biphenyls; and (F) reductive amination of 2-nitro-biphenyls.11 Similarly, dehydrogenative C–C bond formation of N,N-biaryl amines leads to carbazoles (Scheme 1(b)).12 Although these methods proceed in good yields and are tolerant of most organic functional groups, they require the preparation of complex starting materials, such as substituted biaryls or N,N-biaryl amines, for the synthesis of diverse carbazoles. Therefore, we investigated the development of a one-pot C–C and C–N bond coupling reaction that utilizes commercially available or easily preparable starting materials.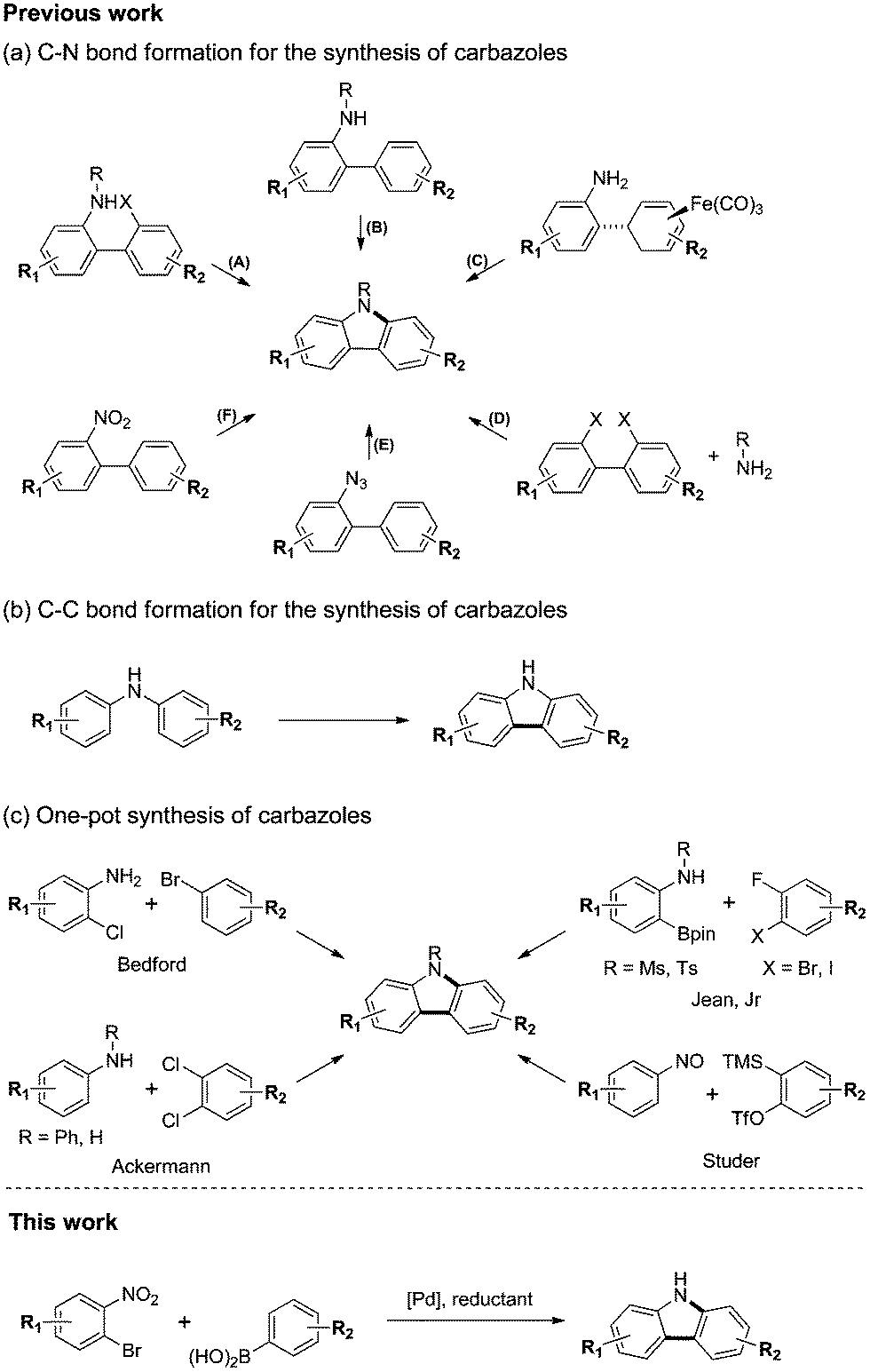 | ||
| Scheme 1 Various synthetic approaches to carbazoles. | ||
More recently, one-pot syntheses of carbazoles have been developed (Scheme 1(c)).13 Bedford13a and Ackermann13b,d reported a palladium catalyzed domino Buchwald–Hartwig amination and C–H activation of 2-chloroanilines with aryl bromides or anilines with dichloroarenes. Jean Jr13c reported the Pd-catalyzed tandem Suzuki cross-coupling and SNAr reaction of aniline-derived boronic esters with 2-fluoro-3-halobenzenes. Studer13f reported a transition metal-free cascade reaction of nitrosoarenes with in situ generated arynes.
We envision a one-pot reaction sequence of C–C cross-coupling and C–N bond forming cyclization to develop a step-economical method to prepare carbazoles. We considered the Suzuki reaction14 and the Cadogan cyclization15 for one-pot reaction. Both reactions are the widely used methods for C–C cross-coupling or reductive amination and use similar phosphine reagents for the ligand or reductant.15d Herein, we present a highly efficient synthetic method to carbazoles that consists of palladium-catalyzed tandem C–C/C–N bond formation via the Suzuki reaction and reductive Cadogan cyclization (Scheme 1).
Results and discussion
Initially, we explored the domino reaction of 2-iodonitrobenzene 1a with phenylboronic acid 2a in the presence of Pd(OAc)2 and triphenylphosphine in o-DCB at 150 °C, which delivered the carbazole 3a and 2-nitrobiphenyl in 24% and 20% yields (Table 1, entry 1). Based on this result, other 2-halonitrobenzenes were assayed, and it was found that 2-bromonitrobenzene 1b was the most efficient (Table 1, entry 2).|
|
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Entry | 1, X | Catalyst (mol%) | Reductant | T (°C) | Time (h) | Yb (%) | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| a Reaction conditions: 1 (0.5 mmol), 2a (0.6 mmol), catalyst (quantity noted), reductant (250 mol%), K2CO3 (200 mol%), o-DCB (1.0 mL) under air in pressure tubes. b Isolated yield by flash column chromatography. c Using a reflux condenser. d 2a (130 mol%). | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 1 | 1a, I | Pd(OAc) (2) | PPh3 | 150 | 16 | 24 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 2 | 1b, Br | Pd(OAc) (2) | PPh3 | 150 | 16 | 56 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 3 | 1c, Cl | Pd(OAc) (2) | PPh3 | 150 | 16 | 31 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 4 | 1b, Br | Pd(OAc) (2) | PEt3 | 150 | 16 | 12 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 5 | 1b, Br | Pd(OAc) (2) | P(OEt)3 | 150 | 16 | 21 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 6 | 1b, Br | Pd2(dba)3 (2) | PPh3 | 150 | 16 | 41 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 7 | 1b, Br | Pd(PPh3)4 (2) | PPh3 | 150 | 16 | 29 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 8 | 1b, Br | Pd(OAc) (1) | PPh3 | 150 | 16 | 17 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 9 | 1b, Br | Pd(OAc) (4) | PPh3 | 150 | 16 | 13 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 10 | 1b, Br | Pd(OAc) (2) | PPh3 | 180 | 16 | 72 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 11 | 1b, Br | Pd(OAc) (2) | PPh3 | 180 | 24 | 78 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 12c | 1b, Br | Pd(OAc) (2) | PPh3 | Reflux | 24 | 66 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| ⇒13d | 1b , Br | Pd(OAc) (2) | PPh 3 | 180 | 24 | 86 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
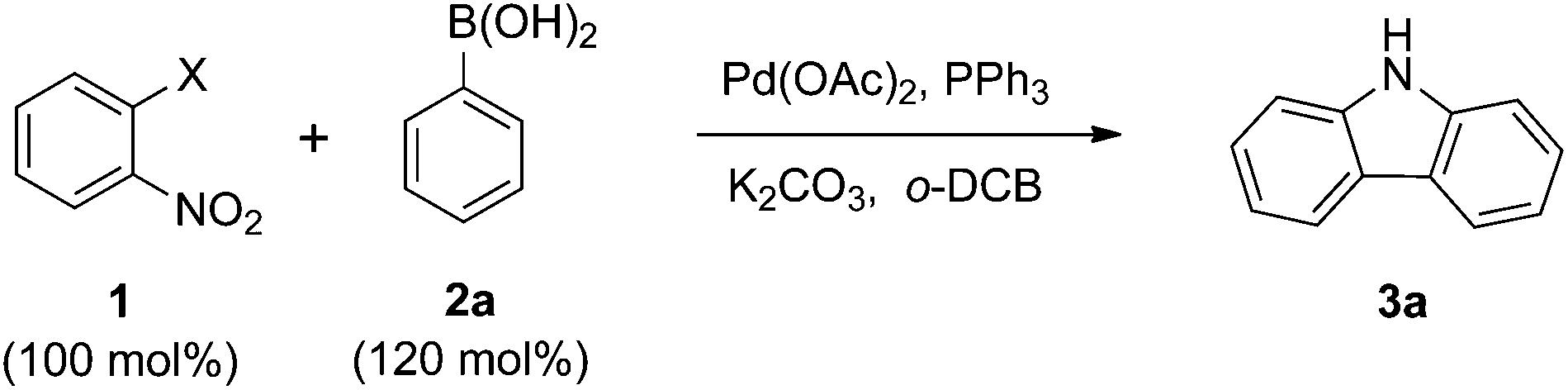
Thus they were chosen for further studies. When other reductants such as PEt3 and P(OEt)3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 15e were used, the yield of the reaction did not improve (Table 1, entries 4 and 5). Moreover, other catalysts such as Pd2(dba)3 and Pd(PPh3)4 were not effective in this transformation (Table 1, entries 6 and 7). Reductant and catalyst screening indicated that Pd(OAc)2 and PPh3 were the best choices. To improve the yield, the catalyst loading, as well as the reaction temperature and time were surveyed. Increasing and decreasing the catalyst loading were not efficient (Table 1, entries 8 and 9). However, the yield did increase with a higher temperature and longer reaction time (Table 1, entries 10 and 11). The coupling reaction was not efficient at atmospheric pressure (Table 1, entry 12). Using 130 mol% phenylboronic acid 2a gave the best result (Table 1, entry 13).
15e were used, the yield of the reaction did not improve (Table 1, entries 4 and 5). Moreover, other catalysts such as Pd2(dba)3 and Pd(PPh3)4 were not effective in this transformation (Table 1, entries 6 and 7). Reductant and catalyst screening indicated that Pd(OAc)2 and PPh3 were the best choices. To improve the yield, the catalyst loading, as well as the reaction temperature and time were surveyed. Increasing and decreasing the catalyst loading were not efficient (Table 1, entries 8 and 9). However, the yield did increase with a higher temperature and longer reaction time (Table 1, entries 10 and 11). The coupling reaction was not efficient at atmospheric pressure (Table 1, entry 12). Using 130 mol% phenylboronic acid 2a gave the best result (Table 1, entry 13).
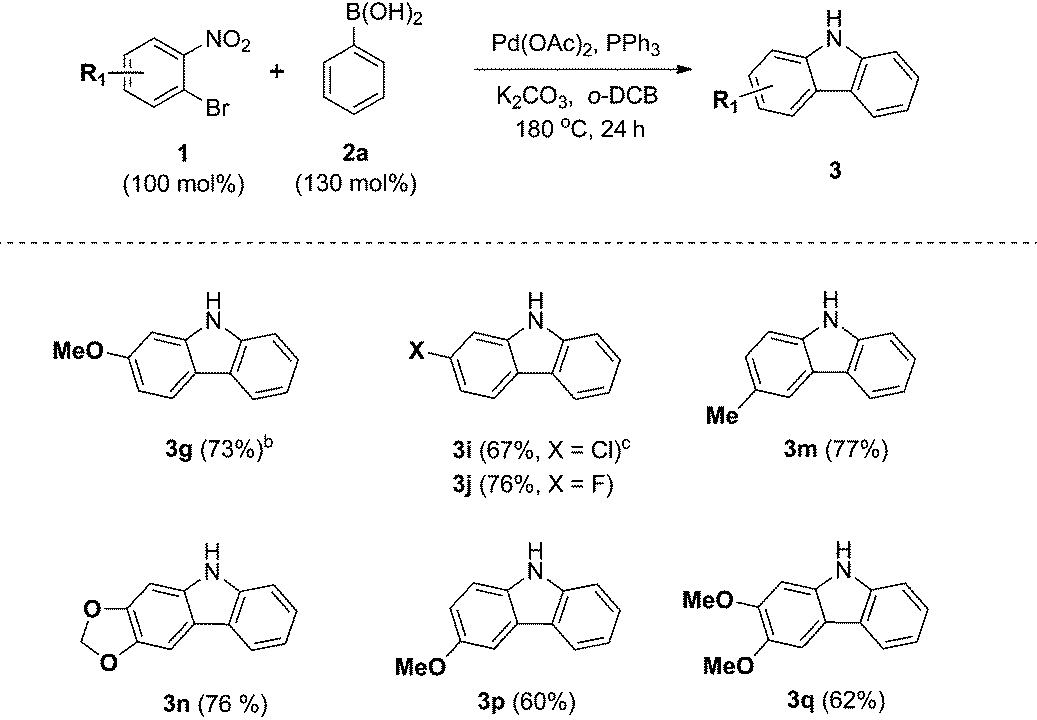
We next examined the substrate scope with respect to the substituted arylboronic acid (Table 2). Under the optimized conditions, o-bromonitrobenzene 1b was coupled to a diverse range of arylboronic acids 2 to form carbazoles 3a–3o in moderate to excellent yields.
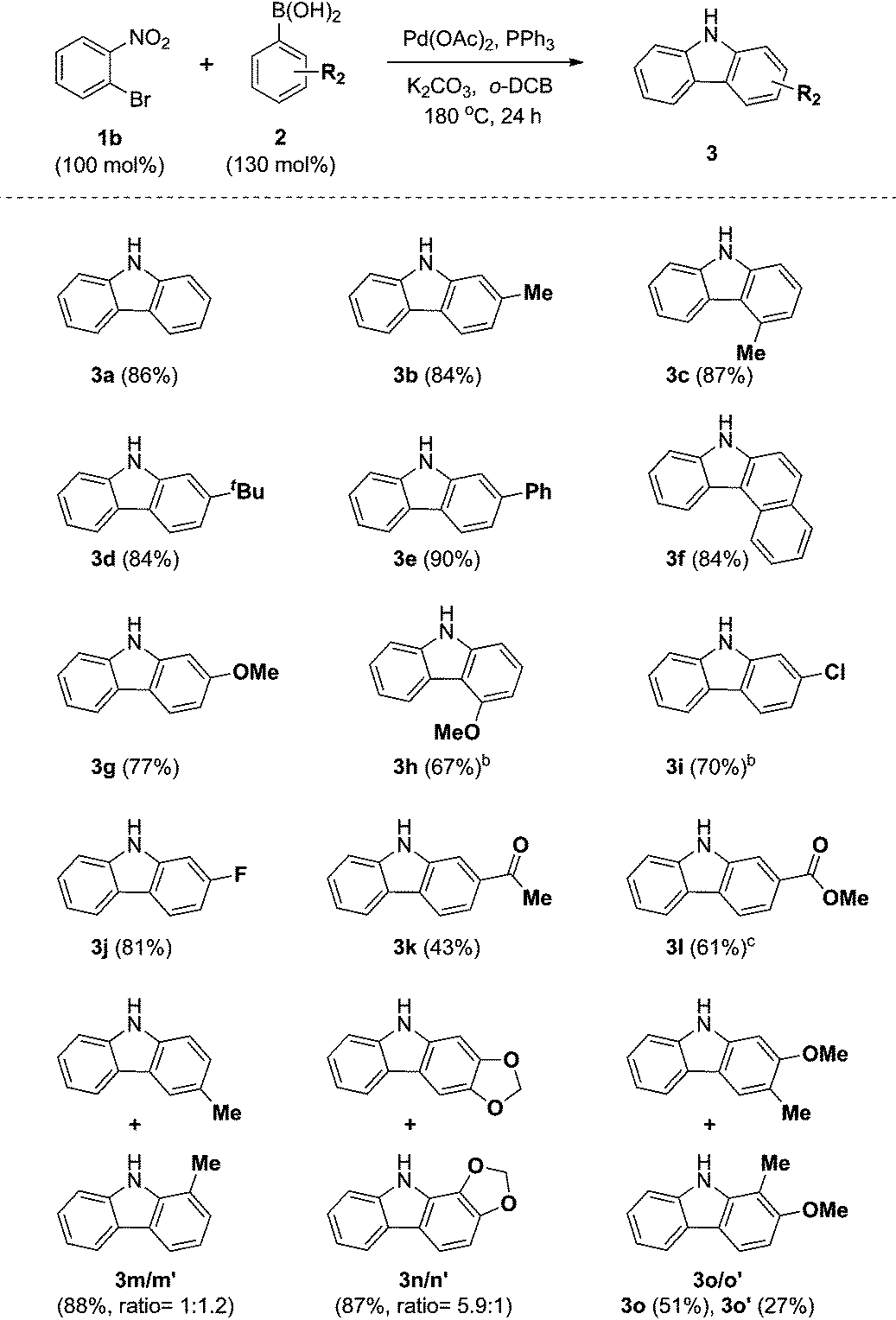
Notably, electron neutral and electron rich arylboronic acids possessing alkyl, aryl, and alkoxy substituents were tolerated in the one-pot reaction (3a–3h, 3m–o), as o-, m- and p-substituted arylboronic acids and 3,4-disubstituted arylboronic acids afforded the corresponding products (3a–3h, 3m–o) in good to high yields. Aryl boronic acids possessing bulky groups at the para position such as t-butyl and phenyl were also converted to carbazoles in excellent yields. As expected, coupling of m-substituted arylboronic acids with o-bromonitrobenzene 1b gave a mixture of corresponding 1- and 3-substituted carbazoles as regioisomers. In these experiments, 3-substituted carbazoles were obtained as the major regioisomer except with 3-tolylboronic acid. In particular, 3,4-(methylenedioxy)phenylboronic acid gave a 5.9:1 mixture of 3n and 3n′, demonstrating moderate regioselectivity, while 3-tolylboronic acid and 4-methoxy-3-methylphenylboronic acid gave mixtures with low regioselectivity (3m16/m′, 3o17/o′).
In addition, we explored the scope of electron deficient arylboronic acids possessing fluorine, chlorine, ketone, and ester moieties and found that they also gave the desired products (3i–3l). Slightly lower yields were observed for electron deficient arylboronic acids except for 4-fluorophenylboronic acid.
In particular, 4-fluoro and 4-chloroboronic acids were tolerated under these coupling conditions. Halogen containing carbazoles are useful for the introduction of additional functionalization. Thus, we examined the further functionalization of 3-chlorocarbazole 3i (Scheme 2). Under modified Suzuki cross-coupling conditions18 for unprotected N-heterocycles, 3-chlorocarbazole 3i was coupled to a diverse range of arylboronic acids to form functionalized carbazoles (3e, 3ia–3id) in good to excellent yields.
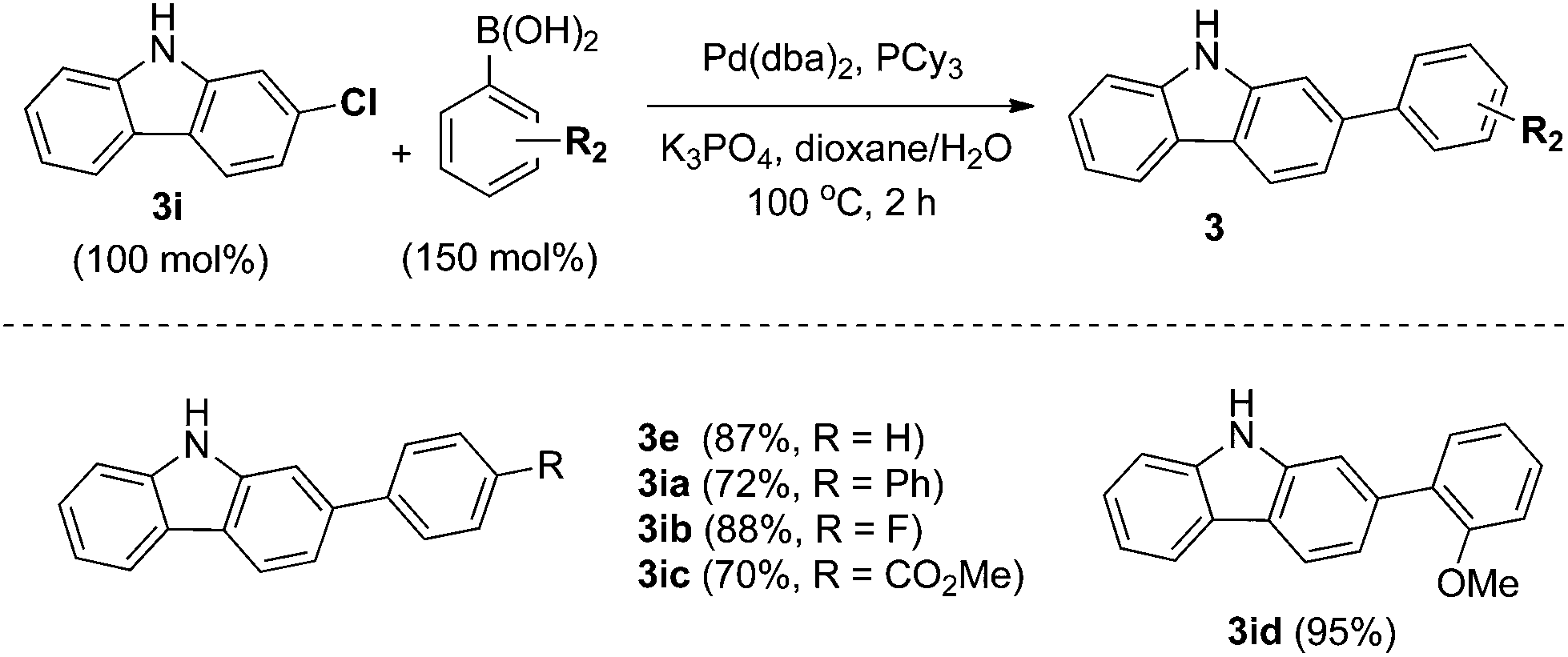 | ||
| Scheme 2 Further functionalization of 3-chlorocarbazole (3i). | ||
To demonstrate the efficiency of this method, we conducted the gram scale synthesis of carbazole 3a. The coupling reaction of 2-bromonitrobenzene 1b (1 g, 5 mmol) with phenylboronic acid 2a (750 mg, 6.5 mmol) afforded the carbazole 3a in 75% yield.
Next, we turned our attention to the domino reaction of phenylboronic acid with a broad range of substituted o-bromonitrobenzenes (Table 3). Under the optimized conditions, the one-pot reaction of substituted o-bromonitrobenzenes 1 with phenylboronic acid 2a was converted to the corresponding carbazoles in good yields, and similar results with respect to substrate scope of various aryl boronic acids were observed (Table 3, 3g, 3i, 3j, 3m and 3n). These approaches constitute an alternative route that avoids the regioselectivity issue in the synthesis of 1- or 3-substituted carbazoles (3m–3q).
The scope and limitations of the domino reaction with respect to various substituted o-bromonitrobenzenes and arylboronic acids were next investigated under the optimized conditions (Table 4). Coupling reactions of 1-bromo-4-methoxy-2-nitrobenzene with 3-tolylboronic acid and 4-fluorophenylboronic acid afforded the corresponding carbazoles 3s and 3t in 71% and 66% yields. Likewise, o-bromonitrobenzenes possessing methyl and fluoro substituents were coupled to arylboronic acids to form carbazoles 3u and 3v in 70% and 53% yields. This tandem reaction was applied to the syntheses of four bioactive carbazole alkaloids, glycozoline 3w,19 glycozolicine 3w′,20 glycozolidine 3x13a,21 and clausenalene 3y,22 in good yields and moderate regioselectivity.
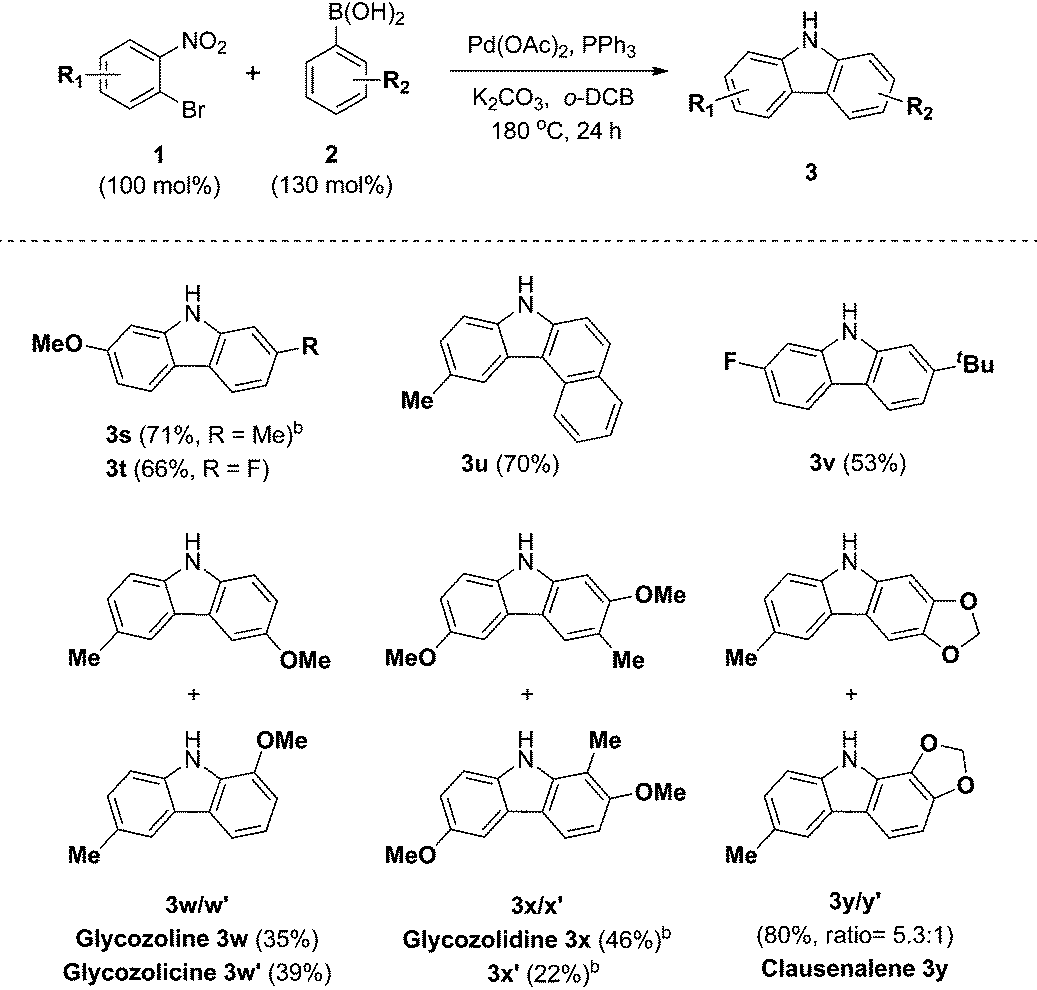
Conclusions
In conclusion, we have developed highly efficient synthetic methods for carbazoles that are palladium-catalyzed sequential C–C/C–N bond formations via Suzuki cross-coupling and reductive Cadogan cyclization. Electron neutral, rich and deficient substrates were tolerated in this method. The synthetic utility of this method was demonstrated by the concise syntheses of four naturally occurring bioactive carbazole alkaloids, glycozoline 3w, glycozolicine 3w′, glycozolidine 3x and clausenalene 3y, from commercially available simple starting materials. Future studies will focus on further applications of this one-pot strategy to the syntheses of complex natural products.Experimental
General information
All reactions were run under an atmosphere of air under anhydrous conditions unless otherwise indicated. Dichloromethane (CH2Cl2), tetrahydrofuran (THF), dimethylformamide (DMF) and toluene (PhMe) were obtained from the Pure-Solv MD-5 Solvent Purification System (Innovative Technology). Pressure tubes (13 × 100 mm, PYREXPLUS) were dried in an oven overnight and cooled under a stream of nitrogen prior to use. All commercial reagents were used directly without further purification. The progress of the reaction was checked on TLC plates (Merck 5554 Kiesel gel 60 F254). Column chromatography was performed on silica gel (Merck 9385 Kiesel gel 60) using hexanes–EtOAc (v/v) or hexanes–acetone (v/v). Infrared spectra were recorded on a Varian 2000 FT-IR. High-resolution mass spectra (EI) were obtained on a Jeol JMS700 HRMS at the Korea Basic Science Center (KBSI), Daegu, Korea. Low-resolution mass spectra (EI) were obtained on a Varian 450-GC/Varian 220-MS and are reported as m/z (relative intensity). Accurate masses are reported for the molecular ion ([M + H]+ or [M + Na]+). Nuclear magnetic resonance spectra (1H NMR and 13C NMR) were recorded by using a Bruker (300 MHz) spectrometer. Chemical shift values were recorded as parts per million relative to tetramethylsilane as an internal standard unless otherwise indicated, and coupling constants in hertz. The following abbreviations are used: m (multiplet), s (singlet), d (doublet), t (triplet), q (quartet), dd (doublet of doublets), etc.General procedure for carbazole synthesis
To a re-sealable pressure tube (13 × 100 mm) equipped with a magnetic stir bar were added o-bromonitrobenzene 1 (0.5 mmol, 100 mol%), aryl boronic acid 2 (0.65 mmol, 130 mol%), Pd(OAc) (0.01 mmol, 2 mol%), PPh3 (1.25 mmol, 250 mol%), K2CO3 (1 mmol, 200 mol%) and o-DCB (1 mL, 0.5 M concentration with respect to o-bromonitrobenzene 1). The mixture was heated at 180 °C (oil bath temperature) for 24–48 h, at which point the reaction mixture was allowed to cool to ambient temperature. The reaction mixture was filtered through a pad of celite and the resulting liquor was concentrated in vacuo and purified by flash column chromatography (SiO2) under the conditions noted to furnish the corresponding product.:3m′ = 1:1.2 (determined by using 1H NMR and a GC-mass spectrometer)). GC (Varian 450-GC, SPB™-680 (30 m × 0.25 mm × 0.25 μm), T0: 60 °C (hold 4 min), T1: ramp, 250 °C at 10 °C min−1, carrier: He (1.0 mL min−1), total time 60 min), t3m′ = 21.95 min, t3m = 22.32 min, ratio (3m:3m′ = 1:1.2).
:3n′ = 5.9:1 (determined by using 1H NMR and a GC-mass spectrometer)). GC (Varian 450-GC, SPB™-680 (30 m × 0.25 mm × 0.25 μm), T0: 60 °C (hold 4 min), T1: ramp, 250 °C at 10 °C min−1, carrier: He (1.0 mL min−1), total time 60 min), t3n′ = 25.38 min, t3n = 26.90 min, ratio (3n:3n′ = 5.9:1).
:3y′ = 5.3:1 (determined by using 1H NMR and a GC-mass spectrometer)). GC (Varian 450-GC, SPB™-680 (30 m × 0.25 mm × 0.25 μm), T0: 60 °C (hold 4 min), T1: ramp, 250 °C at 10 °C min−1, carrier: He (1.0 mL min−1), total time 60 min), t3y′ = 26.98 min, t3y = 28.69 min, ratio (3y:3y′ = 5.3:1).
General procedure for Suzuki coupling of 3-chlorocarbazole (3i)
To a re-sealable pressure tube (13 × 100 mm) equipped with a magnetic stir bar were added 2-chloro-9H-carbazole 3i (0.1 mmol, 100 mol%), aryl boronic acid 2 (0.15 mmol, 150 mol%), Pd2(dba)3 (0.01 mmol, 10 mol%), PCy3 (0.25 mmol, 25 mol%), K3PO4 (1.27 M in water, 0.34 mmol) and dioxane (2 mL, 0.05 M concentration with respect to 2-chloro-9H-carbazole 3i). The slurry was sealed and purged with argon gas for 5 minutes. The mixture was heated at 100 °C (oil bath temperature) for 2 h, at which point the reaction mixture was allowed to cool to ambient temperature. The reaction mixture was treated with saturated sodium bicarbonate solution and extracted with dichloromethane (3x). The combined organic layers were dried with magnesium sulfate, filtered, and concentrated in vacuo and purified by flash column chromatography (SiO2) and recrystallization from dichloromethane and hexanes under the conditions noted to furnish the corresponding product.Acknowledgements
This work was supported by the 2013 Research Fund of University of Ulsan.Notes and references
- (a) J. Cheng, K. Kamiya and I. Kodama, Cardiovasc. Drug Rev., 2001, 19, 152–171 CrossRef CAS PubMed; (b) M.-J. Hsu, Y. Chao, Y.-H. Chang, F.-M. Ho, L.-J. Huang, Y.-L. Huang, T.-Y. Luh, C.-P. Chen and W.-W. Lin, Biochem. Pharmacol., 2005, 70, 102–112 CrossRef CAS PubMed; (c) M. K. Roy, V. N. Thalang, G. Trakoontivakorn and K. Nakahara, Br. J. Pharmacol., 2005, 145, 145–155 CrossRef CAS PubMed; (d) S. Oishi, T. Watanabe, J.-i. Sawada, A. Asai, H. Ohno and N. Fujii, J. Med. Chem., 2010, 53, 5054–5058 CrossRef CAS PubMed; (e) T. Takeuchi, S. Oishi, T. Watanabe, H. Ohno, J.-i. Sawada, K. Matsuno, A. Asai, N. Asada, K. Kitaura and N. Fujii, J. Med. Chem., 2011, 54, 4839–4846 CrossRef CAS PubMed.
- (a) Y. Zhang, T. Wada and H. Sasabe, J. Mater. Chem., 1998, 8, 809–828 RSC; (b) J. L. Díaz, A. Dobarro, B. Villacampa and D. Velasco, Chem. Mater., 2001, 13, 2528–2536 CrossRef; (c) K. R. Justin Thomas, J. T. Lin, Y.-T. Tao and C.-W. Ko, J. Am. Chem. Soc., 2001, 123, 9404–9411 CrossRef; (d) J. V. Grazulevicius, P. Strohriegl, J. Pielichowski and K. Pielichowski, Prog. Polym. Sci., 2003, 28, 1297–1353 CrossRef CAS; (e) J. Li and A. C. Grimsdale, Chem. Soc. Rev., 2010, 39, 2399–2410 RSC; (f) A. D. Finke, D. E. Gross, A. Han and J. S. Moore, J. Am. Chem. Soc., 2011, 133, 14063–14070 CrossRef CAS PubMed; (g) D. Kim, V. Coropceanu and J.-L. Brédas, J. Am. Chem. Soc., 2011, 133, 17895–17900 CrossRef CAS PubMed.
- (a) H.-J. Knölker and K. R. Reddy, Chem. Rev., 2002, 102, 4303–4428 CrossRef; (b) I. Bauer and H.-J. Knölker, Top. Curr. Chem., 2012, 309, 203–253 CrossRef CAS PubMed; (c) A. W. Schmidt, K. R. Reddy and H.-J. Knölker, Chem. Rev., 2012, 112, 3193–3328 CrossRef CAS PubMed; (d) N. Yoshikai and Y. Wei, Asian J. Org. Chem., 2013, 2, 466–478 CrossRef CAS.
- (a) T. Noji, H. Fujiwara, K. Okano and H. Tokuyama, Org. Lett., 2013, 15, 1946–1949 CrossRef CAS PubMed; (b) W. D. Guerra, R. A. Rossi, A. B. Pierini and S. M. Barolo, J. Org. Chem., 2015, 80, 928–941 CrossRef CAS PubMed.
- C. P. Leslie, R. D. Fabio, F. Bonetti, M. Borriello, S. Braggio, G. D. Forno, D. Donati, A. Falchi, D. Ghirlanda, R. Giovannini, F. Pavone, A. Pecunioso, G. Pentassuglia, D. A. Pizzi, G. Rumboldt and L. Stasi, Bioorg. Med. Chem. Lett., 2007, 17, 1043–1046 CrossRef CAS PubMed.
- (a) J. A. Jordan-Hore, C. C. C. Johansson, M. Gulias, E. M. Beck and M. J. Gaunt, J. Am. Chem. Soc., 2008, 130, 16184–16186 CrossRef CAS PubMed; (b) W. C. P. Tsang, R. H. Munday, G. Brasche, N. Zheng and S. L. Buchwald, J. Org. Chem., 2008, 73, 7603–7610 CrossRef CAS PubMed; (c) S. H. Cho, J. Yoon and S. Chang, J. Am. Chem. Soc., 2011, 133, 5996–6005 CrossRef CAS PubMed; (d) K. Takamatsu, K. Hirano, T. Satoh and M. Miura, Org. Lett., 2014, 16, 2892–2895 CrossRef CAS PubMed; (e) C. Suzuki, K. Hirano, T. Satoh and M. Miura, Org. Lett., 2015, 17, 1597–1600 CrossRef CAS PubMed.
- (a) H.-J. Knölker and M. Bauermeister, J. Chem. Soc., Chem. Commun., 1989, 1468–1470 RSC; (b) H.-J. Knölker and W. Fröhner, Tetrahedron Lett., 1997, 38, 1535–1538 CrossRef; (c) O. Kataeva, M. P. Krahl and H.-J. Knölker, Org. Biomol. Chem., 2005, 3, 3099–3101 RSC; (d) R. Czerwonka, K. R. Reddy, E. Baum and H.-J. Knölker, Chem. Commun., 2006, 711–713 RSC; (e) K. E. Knott, S. Auschill, A. Jager and H.-J. Knölker, Chem. Commun., 2009, 1467–1469 RSC; (f) K. K. Gruner, T. Hopfmann, K. Matsumoto, A. Jager, T. Katsuki and H.-J. Knölker, Org. Biomol. Chem., 2011, 9, 2057–2061 RSC.
- (a) K. Nozaki, K. Takahashi, K. Nakano, T. Hiyama, H.-Z. Tang, M. Fujiki, S. Yamaguchi and K. Tamao, Angew. Chem., Int. Ed., 2003, 42, 2051–2053 CrossRef CAS PubMed; (b) D. Zhu, Q. Liu, B. Luo, M. Chen, R. Pi, P. Huang and S. Wen, Adv. Synth. Catal., 2013, 355, 2172–2178 CrossRef CAS.
- M. Pudlo, D. Csányi, F. Moreau, G. Hajós, Z. Riedl and J. Sapi, Tetrahedron, 2007, 63, 10320–10329 CrossRef CAS.
- B. J. Stokes, B. Jovanović, H. Dong, K. J. Richert, R. D. Riell and T. G. Driver, J. Org. Chem., 2009, 74, 3225–3228 CrossRef CAS PubMed.
- (a) I. C. F. R. Ferreira, M.-J. R. P. Queiroz and G. Kirsch, Tetrahedron Lett., 2003, 44, 4327–4329 CrossRef CAS; (b) J. H. Smitrovich and I. W. Davies, Org. Lett., 2004, 6, 533–535 CrossRef CAS PubMed; (c) P. Appukkuttan, E. Van der Eycken and W. Dehaen, Synlett, 2005, 127–133 CAS; (d) A. W. Freeman, M. Urvoy and M. E. Criswell, J. Org. Chem., 2005, 70, 5014–5019 CrossRef CAS PubMed; (e) R. Sanz, J. Escribano, M. R. Pedrosa, R. Aguado and F. J. Arnáiz, Adv. Synth. Catal., 2007, 349, 713–718 CrossRef CAS; (f) J. T. Kuethe and K. G. Childers, Adv. Synth. Catal., 2008, 350, 1577–1586 CrossRef CAS; (g) H. Gao, Q.-L. Xu, M. Yousufuddin, D. H. Ess and L. Kürti, Angew. Chem., Int. Ed., 2014, 53, 2701–2705 CrossRef CAS PubMed.
- (a) B. Akermark, L. Eberson, E. Jonsson and E. Pettersson, J. Org. Chem., 1975, 40, 1365–1367 CrossRef CAS; (b) H.-J. Knölker and N. O'Sullivan, Tetrahedron, 1994, 50, 10893–10908 CrossRef; (c) H. J. Knölker and K. R. Reddy, Heterocycles, 2003, 60, 1049–1052 CrossRef; (d) R. Forke, M. P. Krahl, T. Krause, G. Schlechtingen and H. J. Knölker, Synlett, 2007, 268–272 CAS; (e) R. Forke, A. Jänger and H.-J. Knölker, Org. Biomol. Chem., 2008, 6, 2481–2483 RSC; (f) K. K. Gruner and H.-J. Knölker, Org. Biomol. Chem., 2008, 6, 3902–3904 RSC; (g) H.-J. Knölker, Chem. Lett., 2009, 38, 8–13 CrossRef; (h) M. Fuchsenberger, R. Forke and H. J. Knölker, Synlett, 2011, 2056–2058 CAS; (i) T. Gensch, M. Rönnefahrt, R. Czerwonka, A. Jäger, O. Kataeva, I. Bauer and H.-J. Knölker, Chem. – Eur. J., 2012, 18, 770–776 CrossRef CAS PubMed; (j) L. Huet, R. Forke, A. Jänger and H. J. Knölker, Synlett, 2012, 1230–1234 CAS; (k) R. Hesse, K. K. Gruner, O. Kataeva, A. W. Schmidt and H.-J. Knölker, Chem. – Eur. J., 2013, 19, 14098–14111 CrossRef CAS PubMed; (l) V. P. Kumar, K. K. Gruner, O. Kataeva and H.-J. Knölker, Angew. Chem., Int. Ed., 2013, 52, 11073–11077 CrossRef CAS PubMed.
- (a) R. B. Bedford and M. Betham, J. Org. Chem., 2006, 71, 9403–9410 CrossRef CAS PubMed; (b) L. Ackermann and A. Althammer, Angew. Chem., Int. Ed., 2007, 46, 1627–1629 CrossRef CAS PubMed; (c) D. J. St. Jean, S. F. Poon and J. L. Schwarzbach, Org. Lett., 2007, 9, 4893–4896 CrossRef PubMed; (d) L. Ackermann, A. Althammer and P. Mayer, Synthesis, 2009, 3493–3503 CrossRef CAS; (e) J. K. Laha, P. Petrou and G. D. Cuny, J. Org. Chem., 2009, 74, 3152–3155 CrossRef CAS PubMed; (f) S. Chakrabarty, I. Chatterjee, L. Tebben and A. Studer, Angew. Chem., Int. Ed., 2013, 52, 2968–2971 CrossRef CAS PubMed.
- (a) N. Miyaura and A. Suzuki, J. Chem. Soc., Chem. Commun., 1979, 866–867 RSC; (b) N. Miyaura, K. Yamada and A. Suzuki, Tetrahedron Lett., 1979, 20, 3437–3440 CrossRef; (c) N. Miyaura, T. Yanagi and A. Suzuki, Synth. Commun., 1981, 11, 513–519 CrossRef CAS; (d) N. Miyaura and A. Suzuki, Chem. Rev., 1995, 95, 2457–2483 CrossRef CAS; (e) A. Suzuki, J. Organomet. Chem., 1999, 576, 147–168 CrossRef CAS.
- (a) J. I. G. Cadogan, Q. Rev., Chem. Soc., 1962, 16, 208–239 RSC; (b) J. I. G. Cadogan and M. Cameron-Wood, Proc. Chem. Soc., 1962, 361 CAS; (c) J. I. G. Cadogan, M. Cameron-Wood, R. K. Mackie and R. J. G. Searle, J. Chem. Soc., 1965, 4831–4837 RSC; (d) J. I. G. Cadogan, Q. Rev., Chem. Soc., 1968, 22, 222–251 RSC; (e) J. I. G. Cadogan, Synthesis, 1969, 11–17 CrossRef CAS; (f) J. I. G. Cadogan and M. J. Todd, J. Chem. Soc. C, 1969, 2808–2813 RSC.
- (a) H. J. Knölker, M. Bauermeister and J. B. Pannek, Chem. Ber., 1992, 125, 2783–2793 CrossRef; (b) M. Chakrabarty, A. C. Nath, S. Khasnobis, M. Chakrabarty, Y. Konda, Y. Harigaya and K. Komiyama, Phytochemistry, 1997, 46, 751–755 CrossRef CAS PubMed; (c) T. Watanabe, S. Oishi, N. Fujii and H. Ohno, J. Org. Chem., 2009, 74, 4720–4726 CrossRef CAS PubMed.
- (a) R. Forke, M. P. Krahl, F. Däbritz, A. Jäger and H.-J. Knölker, Synlett, 2008, 1870–1876 CAS; (b) V. Sridharan, M. A. Martín and J. C. Menéndez, Eur. J. Org. Chem., 2009, 4614–4621 CrossRef CAS.
- J. Lim, B. Taoka, R. D. Otte, K. Spencer, C. J. Dinsmore, M. D. Altman, G. Chan, C. Rosenstein, S. Sharma, H.-P. Su, A. A. Szewczak, L. Xu, H. Yin, J. Zugay-Murphy, C. G. Marshall and J. R. Young, J. Med. Chem., 2011, 54, 7334–7349 CrossRef CAS PubMed.
- (a) D. P. Chakraborty, Phytochemistry, 1969, 8, 769–772 CrossRef CAS; (b) H. Peng, X. Chen, Y. Chen, Q. He, Y. Xie and C. Yang, Tetrahedron, 2011, 67, 5725–5731 CrossRef CAS; (c) S. W. Youn, J. H. Bihn and B. S. Kim, Org. Lett., 2011, 13, 3738–3741 CrossRef CAS PubMed; (d) T. Pirali, F. Zhang, A. H. Miller, J. L. Head, D. McAusland and M. F. Greaney, Angew. Chem., Int. Ed., 2012, 51, 1006–1009 CrossRef CAS PubMed; (e) R. Hesse, A. W. Schmidt and H.-J. Knölker, Tetrahedron, 2015, 71, 3485–3490 CrossRef CAS; (f) M. S. Naykode, V. T. Humne and P. D. Lokhande, J. Org. Chem., 2015, 80, 2392–2396 CrossRef CAS PubMed.
- (a) A. Chakraborty, B. K. Chowdhury, S. S. Jash, G. K. Biswas, S. K. Bhattacharyya and P. Bhattacharyya, Phytochemistry, 1992, 31, 2503–2505 CrossRef; (b) A. K. Chakravarty, T. Sarkar, K. Masuda, T. Takey, H. Doi, E. Kotani and K. Shiojima, Indian J., Chem. Sect B: Org. Chem. Incl. Med. Chem., 2001, 40, 484–489 Search PubMed; (c) D. Ding, X. Lv, J. Li, G. Xu, B. Ma and J. Sun, Chem. – Asian J., 2014, 9, 1539–1542 CrossRef CAS PubMed; (d) C. Schuster, C. Börger, K. K. Julich-Gruner, R. Hesse, A. Jäger, G. Kaufmann, A. W. Schmidt and H.-J. Knölker, Eur. J. Org. Chem., 2014, 4741–4752 CrossRef CAS.
- (a) M. Schmidt and H.-J. Knölker, Synlett, 2009, 2421–2424 CAS; (b) C. Ito, M. Itoigawa, A. Sato, C. M. Hasan, M. A. Rashid, H. Tokuda, T. Mukainaka, H. Nishino and H. Furukawa, J. Nat. Prod., 2004, 67, 1488–1491 CrossRef CAS PubMed; (c) M. Iwao, H. Takehara, S. Furukawa and M. Watanabe, Heterocycles, 1993, 36, 1483–1488 CrossRef CAS.
- (a) P. Bhattacharyya, G. K. Biswas, A. K. Barua, C. Saha, I. B. Roy and B. K. Chowdhury, Phytochemistry, 1993, 33, 248–250 CrossRef CAS; (b) T. L. Scott, X. Yu, S. P. Gorugantula, G. Carrero-Martínez and B. C. G. Söderberg, Tetrahedron, 2006, 62, 10835–10842 CrossRef CAS; (c) S. Rasheed, D. N. Rao, K. R. Reddy, S. Aravinda, R. A. Vishwakarma and P. Das, RSC Adv., 2014, 4, 4960–4969 RSC.
- (a) B. Alcaide, P. Almendros, J. M. Alonso, M. T. Quirós and P. Gadziński, Adv. Synth. Catal., 2011, 353, 1871–1876 CrossRef CAS; (b) C. Wang, W. Zhang, S. Lu, J. Wu and Z. Shi, Chem. Commun., 2008, 5176–5178 RSC; (c) S. Chakraborty, G. Chattopadhyay and C. Saha, J. Heterocycl. Chem., 2013, 50, 91–98 CrossRef CAS; (d) B. Liegault, D. Lee, M. P. Huestis, D. R. Stuart and K. Fagnou, J. Org. Chem., 2008, 73, 5022–5028 CrossRef CAS PubMed; (e) S. Kureel, R. Kapil and S. Popli, J. Chem. Soc. D, 1969, 1120–1121 RSC; (f) B.-Y. Lim, M.-K. Choi and C.-G. Cho, Tetrahedron Lett., 2011, 52, 6015–6017 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Details of experimental procedures and spectroscopic data for all new compounds (1H NMR, 13C NMR, IR, MS), including images of NMR spectra. See DOI: 10.1039/c5ob01952d |
| This journal is © The Royal Society of Chemistry 2016 |