
Characterization of structural elements in native autoinducing peptides and non-native analogues that permit the differential modulation of AgrC-type quorum sensing receptors in Staphylococcus aureus†
Yftah
Tal-Gan‡
a,
Monika
Ivancic§
a,
Gabriel
Cornilescu
b and
Helen E.
Blackwell
*a
aDepartment of Chemistry, University of Wisconsin-Madison, 1101 University Avenue, Madison, WI 53706, USA. E-mail: blackwell@chem.wisc.edu
bNational Magnetic Resonance Facility at Madison, University of Wisconsin-Madison, 433 Babcock Drive, Madison, WI 53706, USA
First published on 23rd September 2015
Abstract
Staphylococcus aureus uses short macrocyclic peptides (i.e., autoinducing peptides, or AIPs) to assess its local population density in a cell–cell signaling mechanism called quorum sensing (QS). At high cell numbers, this pathogen can initiate many virulent behaviors that allow for the establishment of infection. Binding of the AIP signal to its cognate transmembrane AgrC-type receptor is a critical event in the QS signaling cascade; consequently, interference of AIP:receptor interactions may have the potential to prevent and eradicate certain S. aureus infections. To date, four pairs of AIP:AgrC receptors have been identified in S. aureus, each pair being utilized by a specific S. aureus group (I–IV). Other staphylococcal species also use closely related, but distinct, AIP:AgrC pairs to control QS. We seek to develop non-native ligands capable of intercepting AIP:AgrC binding in each S. aureus group and in related species. As these bacteria may use their respective AIP signal to attenuate the QS systems of other groups/species, such ligands would provide valuable chemical tools to probe possible interference mechanisms in a range of contexts. In the current study, we used solution-phase NMR techniques to characterize the 3-D structures of a set of known native and non-native peptides that have differential modulatory activity in certain AgrC receptors. Analysis of these structures revealed several distinct structural motifs that belay differential activity in selected S. aureus AgrC receptors (i.e., AgrC-I, AgrC-II, and AgrC-III). The results of this study can be leveraged for the design of new synthetic ligands with enhanced selectivities and potencies for these AgrC receptors.
Introduction
Staphylococcus aureus is a common human pathogen that utilizes a cell-density sensing mechanism called quorum sensing (QS) to initiate virulence and establish infections.1–3 This bacterium uses the accessory gene regulator (agr) system for QS, which is regulated by short macrocyclic peptide signals, termed autoinducing peptides (AIPs), and their cognate transmembrane AgrC receptors.4S. aureus continuously produces the AIP signal at low levels, and the concentration of signal increases with cell density. Once a threshold AIP level is reached in a given environment, and thus a “quorate” population of bacteria has assembled, the AIP signal can bind and activate the extracellular sensor domain of the AgrC receptor, setting off a signalling cascade to activate the transcription of genes involved in group behaviours (Fig. 1).1,3,4 The majority of these QS genes control virulence phenotypes in S. aureus. Consequently, as AIP:AgrC receptor binding is integral to the activation of virulence, considerable research has focused on the development of strategies to intercept this binding event.4–8 Our laboratory9–12 and others13–18 have focused on the AIP ligand, and we have designed and synthesized a variety of AIP mimetics that can block native AIP binding and strongly attenuate QS virulence phenotypes in S. aureus.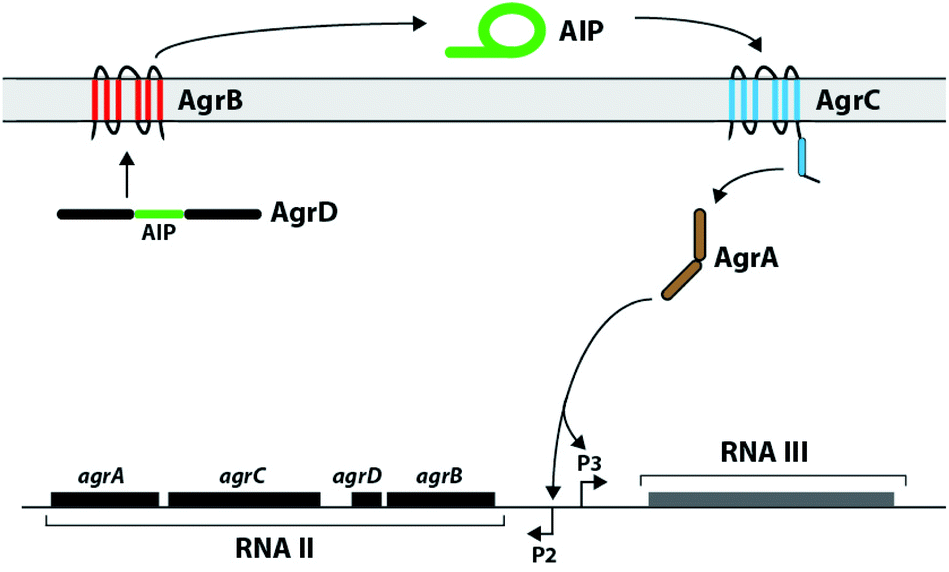 | ||
| Fig. 1 Schematic of the agr QS circuit. The agr locus is comprised of two transcripts termed RNA II and RNA III. The RNA II transcript encodes the four agr components, and the RNA III transcript is the main effector of virulence. AgrD is the precursor of the AIP signal and is processed and exported by AgrB. At high concentrations, the AIP signal binds and activates the transmembrane histidine kinase, AgrC. Activation of AgrC leads to phosphorylation of the response regulator, AgrA, which then binds and activates the P2 and P3 promoters. | ||
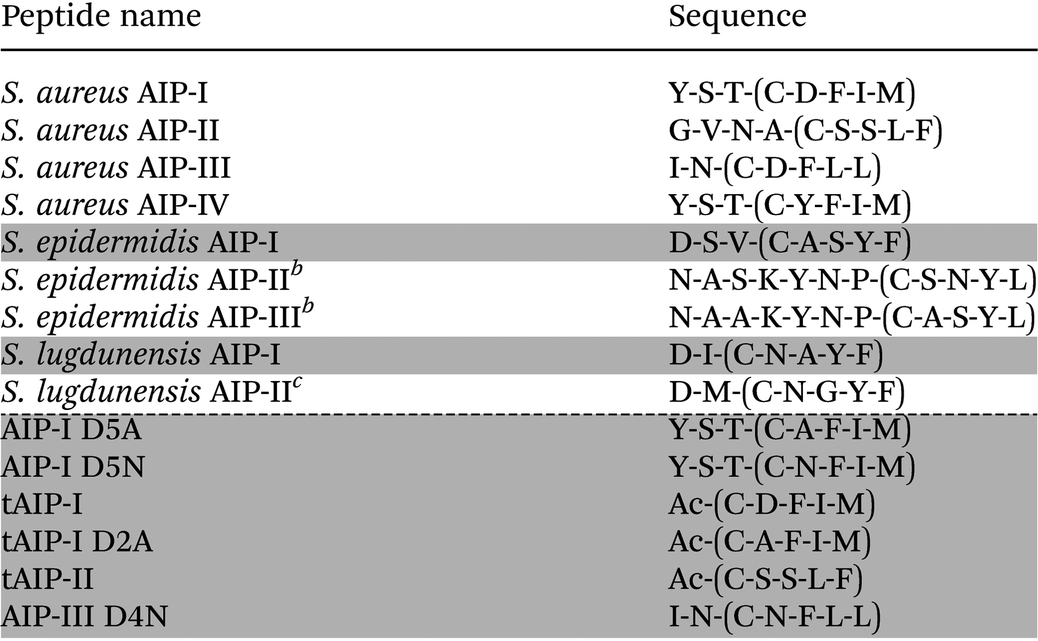
To date, four specificity groups of S. aureus strains have been identified (groups I–IV), each having distinct AIP signals (I–IV) and corresponding cognate AgrC receptors (I–IV).3,4 In addition, the agr QS circuit is conserved in many staphylococcal species (numbering over 20 so far), with each producing its own unique AIP.4 The structures of the AIP signals from S. aureus, S. epidermidis, and S. lugdunensis are shown in Table 1. Interestingly, each of the native AIPs in S. aureus is capable of inhibiting the non-cognate AgrC receptors in the other three S. aureus groups.4–6,19 Furthermore, the AIPs used by S. epidermidis (group I) and S. lugdunensis (group I) have been reported to cross-inhibit the AgrC receptors of selected S. aureus groups (groups I–III).19–21 These observations have prompted the hypothesis that staphylococcal species use their QS systems to not only assess their own local population density, but also to interfere with the QS systems of other bacteria residing nearby. Such interference could, for example, allow one group or species to preferentially colonize an environmental niche on a host. We are particularly interested in delineating possible intergroup and interspecies QS interference in S. aureus and in other related bacteria. Specifically, we seek to identify non-native molecules capable of selectively modulating individual AgrC receptors for use as mechanistic probes to attenuate QS signalling in mixed microbial populations. The design of such molecules requires a detailed understanding of the structure–activity relationships (SARs) between the AIPs and the different AgrC receptors.
So far, the majority of the SAR studies on AIPs have involved systematic replacement of amino acids with alanine (i.e., alanine scans),9,13,14D-amino acids (D-AA scans),9,14N-methylated amino acids (N-methyl scans)11,16 or N-alkylated glycine derivatives (peptoid scans).11,12 These systematic analyses have provided valuable information regarding the importance of different chemical elements (i.e., side chains, stereochemistry, and hydrogen bonds) to the overall activity of the AIP signals; however, the lack of 3-D structural information for the different AIP analogues has hindered delineation of the structural motifs required for both activation and inhibition of the AgrC receptors. Towards this goal, we recently reported the 3-D solution-phase structures of the four native AIP signals (I–IV) in S. aureus and several synthetic AIP-III analogues as determined using NMR spectroscopy.10 This past study allowed us to identify two critical structural motifs within AIP-type ligands that confer inhibition and activation of the AgrC-III receptor – (i) a hydrophobic patch (or “knob”) on the macrocycle essential for receptor binding and (ii) an additional hydrophobic contact or “anchor” on the N-terminal tail critical for receptor activation. In the absence of the anchor, peptides containing a hydrophobic knob were found to inhibit the AgrC-III receptor, presumably by outcompeting the native ligand.
The current study had three parallel aims focused on further expanding our understanding of AIP:AgrC interactions. First, we sought to identify structural motifs that dictate inhibition and activation of the other AgrC receptors (beyond AgrC-III) used by different S. aureus groups. To this end, we chose several AIP-I and AIP-II analogues that were previously reported to exhibit different activity trends in certain AgrC receptors and determined their 3-D solution-phase structures using NMR. Second, we sought to characterize structural motifs involved in potential inter-staphylococcal AIP:AgrC receptor interactions; thus, we also determined the structures of native AIP signals from S. epidermidis and S. lugdunensis and compared them to a native AIP signal in S. aureus. Third, we evaluated the structure of a new AIP-III analogue, AIP-III D4N, which we previously hypothesized should act as an AgrC-III agonist,10 and found that it indeed acts as an AgrC-III agonist and adopts the expected 3-D conformation, strengthening our hypothesis regarding the structural requirements needed for AgrC-III receptor activation. Overall, we were able to identify and refine a series of different structural motifs that are required for inhibition or activation of different S. aureus AgrC receptors (I–III) by AIP-type ligands. These results are significant, as structural data for AIPs and analogues thereof still remains scarce. In addition, these results can be used to guide the design of new peptide, and potentially non-peptide, QS modulators in S. aureus with enhanced receptor selectivities and potencies.
Experimental
Chemical reagents and instrumentation
All chemical reagents were purchased from commercial sources (Alfa-Aesar, Sigma-Aldrich, and Acros) and used without further purification. Solvents were purchased from commercial sources (Sigma-Aldrich and J.T. Baker) and used as obtained. Water (18 MΩ) was purified using a Millipore Analyzer Feed System. Solid-phase resin was purchased from NovaBiochem.Reversed-phase high performance liquid chromatography (RP-HPLC) was performed using a Shimadzu system equipped with an SCL-10Avp controller, an LC-10AT pump, an FCV-10ALvp solvent mixer, and an SPD-10MAvp UV/vis diode array detector. MALDI-TOF mass spectrometry (MS) data were obtained on a Bruker RELEX II spectrometer equipped with a 337 nm laser and a reflectron. In positive ion mode, the acceleration voltage was 25 kV. Exact mass (EM) data were obtained on a Waters (Micromass) LCT ESI-TOF spectrometer. The samples were sprayed with a sample cone voltage of 20 V.
Peptide synthesis
Table 1 lists the peptides analysed in this study. Linear peptides were synthesized on 3-(Fmoc-amino)-4-aminobenzoyl aminomethyl polystyrene resin (Dawson Dbz AM resin; 0.42 mmol g−1) using standard Fmoc-based solid-phase synthesis protocols. Cleavage from the resin was conducted using the Dawson protocol to afford C-terminal peptide-N-acyl-benzimidazolinone (Nbz) derivatives.23 Following purification of the peptide-Nbz by RP-HPLC, peptide macrocyclization was conducted using our previously reported solution-phase protocol.9 All cyclic peptides were purified to homogeneity (>98%) by RP-HPLC. Peptide characterization data (MS and HPLC) are provided in the ESI.†Biological reagents and strain information
All biological reagents were purchased from Sigma-Aldrich and used according to enclosed instructions. The bacterial strains used in this study are listed in Table 2. Bacterial cultures were grown in a standard laboratory incubator at 37 °C with shaking (at 200 rpm). Absorbance and fluorescence measurements were obtained using a Biotek Synergy 2 microplate reader running Gen5 data analysis software. All biological assays were performed in triplicate. IC50 and EC50 values were calculated using GraphPad Prism software (v. 4.0) using a sigmoidal curve fit.Compound handling protocol
Stock solutions of peptides (1 mM) were prepared in DMSO and stored at 4 °C in sealed vials. The amount of DMSO used in biological assays did not exceed 4% (v/v). Black or clear polystyrene 96-well microtiter plates (Costar) were used for bacteriological assays.Fluorescence and β-lactamase assays
The fluorescence and β-lactamase/nitrocefin assays measuring AgrC receptor activity were conducted as previously described using S. aureus GFP and β-lactamase reporter strains, respectively (Table 2).11NMR methods and structural analyses
All NMR spectra were recorded on a Varian Inova 600 MHz spectrometer at 298 K, using 1.0–1.5 mM solutions of the peptides in 70% H2O/30% CD3CN, pH 3.65 (note, the peptides are insoluble in aqueous solutions with lower percentages of CD3CN at the concentrations needed for the NMR experiments). Spectra were processed using the Vnmr software package (v. 6.1C; Varian) and NMRPipe software.25 Chemical shifts were referenced to CD3CN at 1.94 ppm.Two-dimensional (2-D) homonuclear experiments gcosy, wgtocsy (tocsy26,27 using DIPSI spinlock and the 3-9-19 water suppression sequence) and wgroesy (rotating frame NOE experiment with pulsed T-Roesy28 spin lock and the 3-9-19 water suppression sequence) were acquired. The gcosy experiments were collected with 1754 and 512 real data points in the direct and indirect dimensions, respectively, with 16 scans per data point. For the wgtocsy experiments, 1536 and 256 real data points were collected in the direct and indirect dimensions, respectively, with 16 scans per data point. A relaxation delay of 2 s was used for both the gcosy and wgtocsy experiments, with a mixing time of 80 ms for the wgtocsy. For the wgroesy experiments, 3080 real data points were used in the direct dimension, with 300 data points in the indirect dimension, and 64 scans per data point. A relaxation delay of 3 s and a mixing time of 300 ms were used. Presaturation water suppression was used in the 32 scan 1H 1-D experiments with 30272 real data points.
All spectra were analysed with SPARKY.29 Assignment of resonances for each peptide (listed in Tables S-3–S-10†) was achieved using standard sequential assignment methodology.30 The numbers of ROEs observed for each peptide are listed in Table S-11.† The volumes of the ROE peaks were calculated by SPARKY and converted into a continuous distribution of interproton distance restraints, with a uniform 20% distance error applied to take into account spin diffusion. Three-dimensional (3-D) structure calculations and refinements made use of the torsion angle molecular dynamics and the internal variable dynamics modules31 of Xplor-NIH (v. 2.31),32 with patches for the thioester bridge and ring closure. The target function minimized was comprised of the experimental NMR restraints (ROE-derived interproton distances and torsion angles), a repulsive van der Waals potential for the non-bonded contacts,33 a torsion angle database potential of mean force,34 and a gyration volume potential.35 PyMOL36 and Chimera37 were used for visual analysis and presentation of the peptide structures. The pdb files for each peptide structure are included in the ESI.†
Results and discussion
Evaluation of AIP-I analogues
Our first objective was to identify structural features on AIP ligands that are required for modulation of the AgrC-I receptor from group-I S. aureus. We chose to study four synthetic AIP-I analogues that were previously reported to modulate AgrC-I and displayed a range of activities (structures shown in Table 1): one agonist, AIP-I D5N;15 two potent antagonists, AIP-I D5A and tAIP-I D2A;15 and one relatively inactive analogue, tAIP-I (t = truncated, or lacking an N-terminal exocyclic tail).15 To better gauge their relative activities and receptor selectivities (if any), we first examined these four AIP-I analogues for agonistic and antagonistic activity in the four AgrC receptors (I–IV) using cell-based reporter assays and compared our findings to prior reports. For AgrC inhibition, we used S. aureus strains (groups I–IV) possessing P3-gfp reporter plasmids (see Experimental section).24 In these strains, activation of AgrC by the AIP signal results in AgrA phosphorylation, which in turn binds the P3 promoter and induces gfp transcription (Fig. 1). These strains are capable of native AIP production, and thus GFP is produced at quorate cell densities. QS inhibition by exogenous peptides was quantified by measuring reduction in GFP fluorescence. For AgrC activation, we used a set of S. aureus agr-null strains each harbouring two plasmids, a P3-blacZ reporter and agrCA from groups-I, -II, -III, or -IV.15 QS activation was quantified by measuring β-lactamase activity. The biological assay results for the four peptides are summarized in Table 3.| Peptide name | Activation (EC50 nM)b | Inhibition (IC50 nM)b | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| AgrC-I | AgrC-II | AgrC-III | AgrC-IV | AgrC-I | AgrC-II | AgrC-III | AgrC-IV | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| a See Experimental section for details of reporter strains and methods. All assays performed in triplicate. b EC50 and IC50 values determined by testing AIPs over a range of concentrations (200 fM–40 μM). See ESI for plots of agonism and antagonism dose response curves and 95% confidence ranges for EC50 and IC50 values (Table S-2). c Agonism dose response exceeded 100% activation compared to the native ligand. d Antagonism dose response did not reach 100% inhibition. e Agonism dose response did not reach 100% activation compared to the native ligand. f Data reproduced from ref. 11. IC50 values for native AIPs I–III are reported in ref. 9 and omitted here for brevity. | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| AIP-I D5N | 40.4 | — | 1120c | — | 72.4d | 4.53 | — | 2.80 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| AIP-I D5A | 386e | — | — | — | 0.214 | 0.613 | 1.30 | 0.0827 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| tAIP-I D2A | — | — | — | — | 3.06 | 10.1 | 0.260 | 0.353 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| tAIP-I | — | — | — | — | >1000 | 9.42 | 516 | >1000 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| tAIP-II | — | — | — | — | 95.9 | 77.5 | 0.616 | 11.7 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| S. epidermidis AIP-I | — | — | — | — | 166 | >1000 | 13.0 | >1000 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| S. lugdunensis AIP-I | — | — | — | — | 384 | 419 | 36.6 | >1000 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| AIP-III D4N | — | — | 967e | — | 1.17 | 0.319 | — | 0.294 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| AIP-I | 3.21f | — | — | — | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| AIP-II | — | 40.9f | — | — | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| AIP-III | — | — | 406f | — | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Our reporter assay results for the AIP-I analogues corroborated previously reported data,14,15 with two minor exceptions. First, we observed AIP-I D5A was not only a pan-group AgrC inhibitor at low concentrations (as was reported),15 but also was a cognate receptor (AgrC-I) activator at higher concentrations (∼60% activation of AgrC-I compared to AIP-I). We note that Muir, Novick, and co-workers have recently reported similar observations using an in vitro AgrC-I binding and phosphorylation assay employing nano-discs.7,17 They hypothesized that the AgrC-I receptor has multiple binding pockets for AIP-I D5A: a “high affinity” binding pocket that leads to competitive inhibition, and a “low affinity” pocket that leads to receptor activation at high concentrations. Our assay data for AIP-I D5A are congruent with this in vitro activity profile. Second, we found that AIP-I D5N was not only an AgrC-I agonist (100% activation compared to AIP-I; ∼10-fold higher EC50 value), but also was capable of partial AgrC-I inhibition (∼60% inhibition) in the GFP reporter; previous reports indicated this analogue was only an AgrC-I agonist.15 This activity profile may be due to our use of different S. aureus strains to assess AgrC-I activity: the β-lactamase reporter is in a relatively standard lab strain of group-I S. aureus,15,38,39 while the GFP reporter is in a high-toxin producing group-I strain known for elevated expression of the agr system (USA300 LAC).24,40 Accordingly, we suspect that, while AIP-I D5N is capable of fully activating AgrC-I in the former strain, it is likely only capable of partial activation of AgrC-I in the latter strain, and this results in reduced levels of GFP production relative to the background level at high AIP-I D5N concentrations (read-out as partial AgrC-I inhibition). In terms of receptor selectivity, tAIP-I displayed the most notable trend of the peptides in Table 3, being a moderate inhibitor of AgrC-II only and having little to no effect on the other receptors.
With this set of biological assay data in hand, we next characterized the four AIP-I analogues by NMR spectroscopy and determined their 3-D solution-phase structures (see Experimental section), with a primary intent of identifying structural motifs required for AgrC-I receptor inhibition and activation.
We first analysed the structures of the two full-length AIP-I analogues, AIP-I D5A and AIP-I D5N, and compared them to the structure of the native AIP-I that we recently reported.10 AIP-I D5A was found to be less structured relative to AIP-I (RMSD values over 3-fold higher, Table S-11; see Fig. S-1A† for 10-structure ensemble of AIP-I D5A), but within the macrocycle a similar triangular hydrophobic knob motif was apparent, composed of the Phe6, Ile7, and Met8 side chains projecting from one face (Fig. 2A–C). The exocyclic tail projects back from the macrocycle in both AIP-I D5A and AIP-I; however, the orientation of the tail differs between the two structures. In AIP-I, the terminal Tyr1 side chain is positioned on the opposite face of the macrocycle, while in AIP-I D5A the same side chain is facing the macrocycle (see overlay in Fig. 2D).
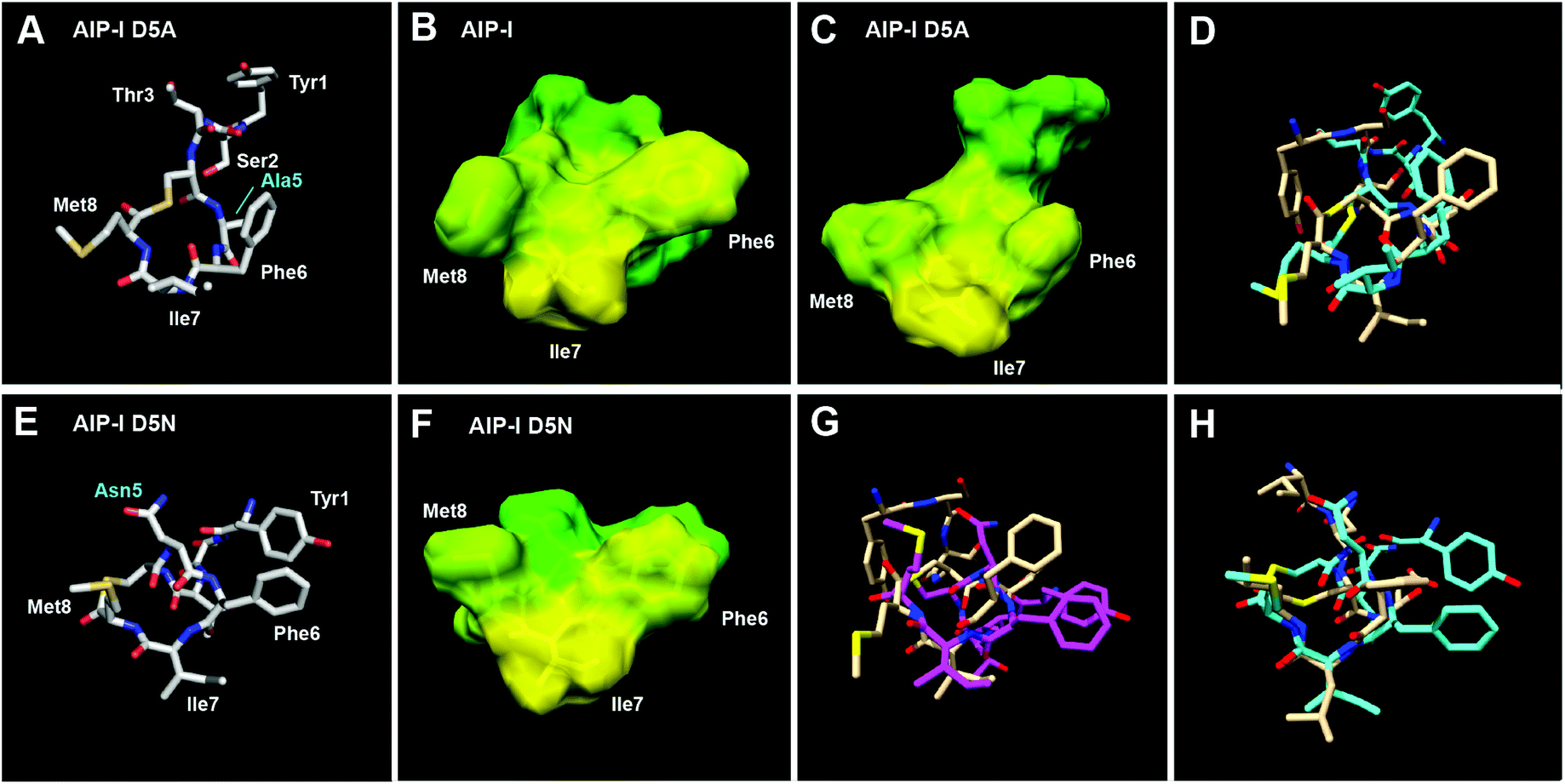 | ||
| Fig. 2 (A) Heavy atom lowest energy structure of AIP-I D5A. Altered residue labelled in cyan. Space filling models of (B) AIP-I and (C) AIP-I D5A displaying hydrophobic (yellow) and hydrophilic (green) surfaces. (D) Overlay of AIP-I D5A (cyan) and AIP-I (tan) structures. (E) Heavy atom lowest energy structure of AIP-I D5N. Altered residue labelled in cyan. (F) Space filling model of AIP-I D5N displaying hydrophobic (yellow) and hydrophilic (green) surfaces. (G) Overlay of AIP-I D5N (magenta) and AIP-I (tan). (H) Overlay of AIP-I D5N (cyan) and AIP-III (tan) structures. AIP-I and AIP-III structures reproduced from ref. 10. | ||
AIP-I D5N was found to be more structured than AIP-I D5A (see RMSD values on Table S-11, and Fig. S-1B† for 10-structure ensemble of AIP-I D5N). This analogue also maintained the triangular knob motif; however, analysis of an overlay with AIP-I reveals the side chains comprising the knob are twisted clockwise (Fig. 2E–G). Interestingly, unlike AIP-I and AIP-I D5A, the exocyclic tail of AIP-I D5N projects towards the macrocycle (see Fig. S-1C† for an overlay of AIP-I, AIP-I D5A and AIP-I D5N), similar to the structure of the group-III native signal, AIP-III. The AIP-I D5N tail projects more toward the upper right side of the macrocycle, while the tail of AIP-III projects toward the upper left side of the macrocycle (Fig. 2H).10 The closer similarity of the structure of AIP-I D5N to that of AIP-III, compared to AIP-I and AIP-I D5A, correlates with the ability of AIP-I D5N to activate the AgrC-III receptor, while both AIP-I and AIP-I D5A are potent AgrC-III inhibitors (Table 3). Indeed, the presence of the knob motif in all three AIP-I analogues supports our previous hypothesis regarding the requirement for the triangular knob motif for AgrC-III modulation (both inhibition and activation).10 With regards to AgrC-I modulation, in our hands both AIP-I D5A and AIP-I D5N were found to activate AgrC-I to some extent, although these two full-length analogues were also capable of AgrC-I inhibition in the GFP reporter (Table 3).
Collectively, this structural comparison of AIP-I and the two analogues, AIP-I D5A and AIP-I D5N, reveals that the conformation of the tail region varied significantly between the peptides, while the macrocycle segment was relatively conserved. Thus, we speculate that, similar to AgrC-III, the macrocyclic hydrophobic triangular knob motif present in all three peptides is responsible for initial AgrC-I binding, and a correctly positioned exocyclic tail contact is responsible for AgrC-I activation. This mechanism is well aligned with that proposed by Muir, Novick and co-workers regarding the roles of the AIP macrocycle and the exocyclic tail in AgrC-I activation.17 Therefore, it is reasonable to assume that all three peptides bind the AgrC-I receptor, but it is the orientation of the tail (when bound; to the same or a different pocket) that dictates the resulting different degrees of activation.
We next characterized the structures of the two truncated AIP-I analogues, tAIP-I and tAIP-I D2A by NMR. Both of these peptides lack exocyclic tails and failed to activate AgrC-I in our reporter assays (Table 3), strengthening our structural hypothesis for AgrC-I receptor activation. With regards to the inhibitory activity of these truncated analogues, tAIP-I is a very weak AgrC-I inhibitor, while tAIP-I D2A is a potent inhibitor. We therefore reasoned that tAIP-I D2A would assume a conformation having a triangular knob, while tAIP-I would not. Surprisingly, both peptides assume a conformation in which the three hydrophobic residues form a more planar hydrophobic face (Fig. 3 and S-2†). Nevertheless, the lack of triangular knob motif may explain the 10-fold reduced activity of tAIP-I D2A compared to its full-length analogue, AIP-I D5A, against the AgrC-I receptor (Table 3). As the structures of tAIP-I and tAIP-I D2A are relatively similar while their activity trends vary significantly (see Fig. 3C for overlay), we reason that the disparity in activity is caused by the replacement of the Asp2 side chain with that of Ala and not due to a significant conformational change. As previous studies have shown that this position is critical for activation by AIP-I,17 it is likely that the Asp side chain in AIP-I can form key contacts with the AgrC-I receptor. Further experiments are needed to test these hypotheses and the role of this Asp residue in AgrC-I activation (and inhibition) by AIPs in general.
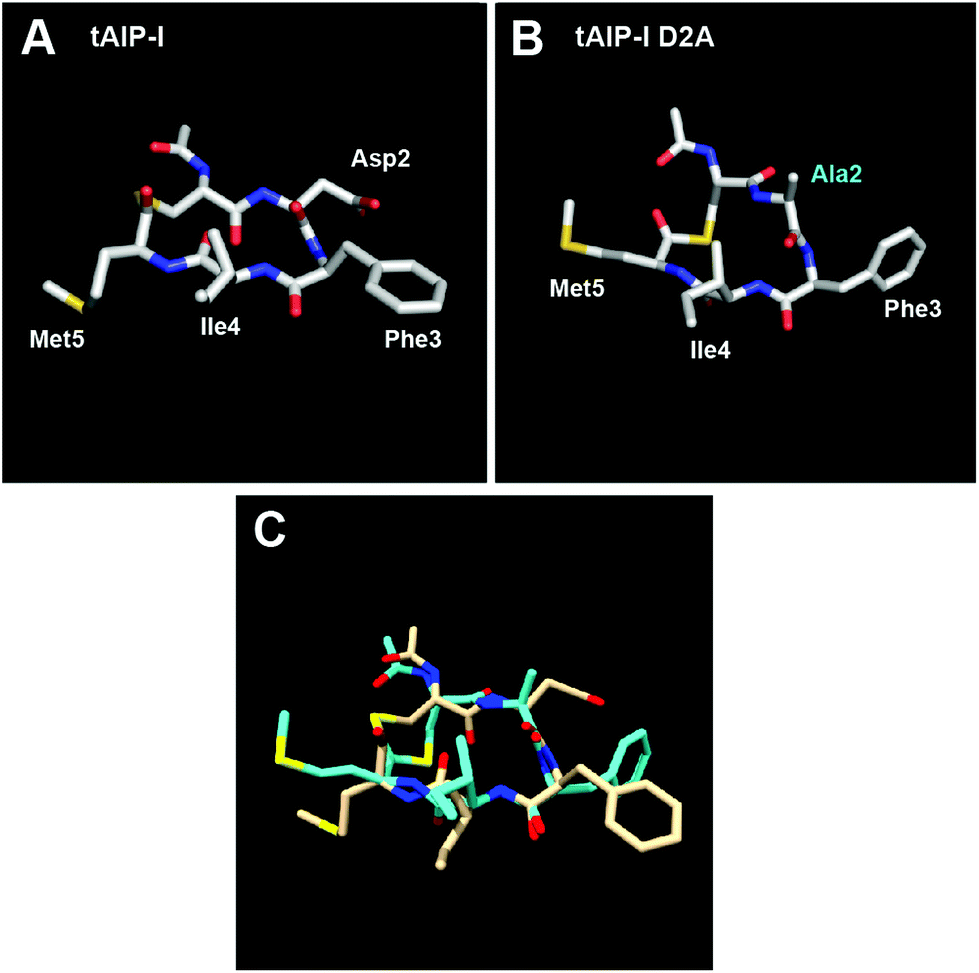 | ||
| Fig. 3 Heavy atom lowest energy structure for (A) tAIP-I; and (B) tAIP-I D2A. Altered residue labelled in cyan. (C) Overlay of tAIP-I (tan) and tAIP-I D2A (cyan) structures. | ||
Evaluation of AIP-II analogues
Our next objective was to assess the structural requirements for modulating the AgrC-II receptor by AIP-type ligands. In our previous study, we determined the solution-structure of AIP-II by NMR and found that this peptide was relatively unstructured (at least under our experimental conditions).10 However, a closer evaluation of the two hydrophobic residues within the macrocycle (Leu8 and Phe9) revealed that their side chains projected in the same direction from the ring in all of the calculated structures (total 20 structures).10 In view of this structural trend, we reasoned that the macrocyclic portion of AIP-II may be capable of adopting a defined structure in the absence of the exocyclic tail. We thus determined the solution structure of the truncated version of AIP-II, tAIP-II, a known AgrC-II inhibitor15 (our assays also supported this activity profile; Table 3). In congruence with our hypothesis, this peptide adopted a very well defined structure relative to the native AIP-II (Fig. 4A and S-3†), with the lowest RMSD values of the peptides evaluated in this study (Table S-11†). When overlaid on the lowest energy structure of AIP-II, it appears that the hydrophobic residue side chains of tAIP-II (i.e., Leu4 and Phe5) point in the same direction and are in close proximity, similar to the same residues in AIP-II (Leu8 and Phe9), although with a slightly different angle compared to the macrocycle backbone (overlay shown in Fig. 4B). The discrepancy in flexibility between the two peptides suggests that one of the roles of the exocyclic tail in the native AIP-II may be to disrupt the well-defined structure of the macrocycle segment. Further, it appears that the minimal requirement for effective AgrC-II binding, whether leading to receptor activation or inhibition, is two endocyclic hydrophobic residues pointing their side chains in a similar direction and in close proximity.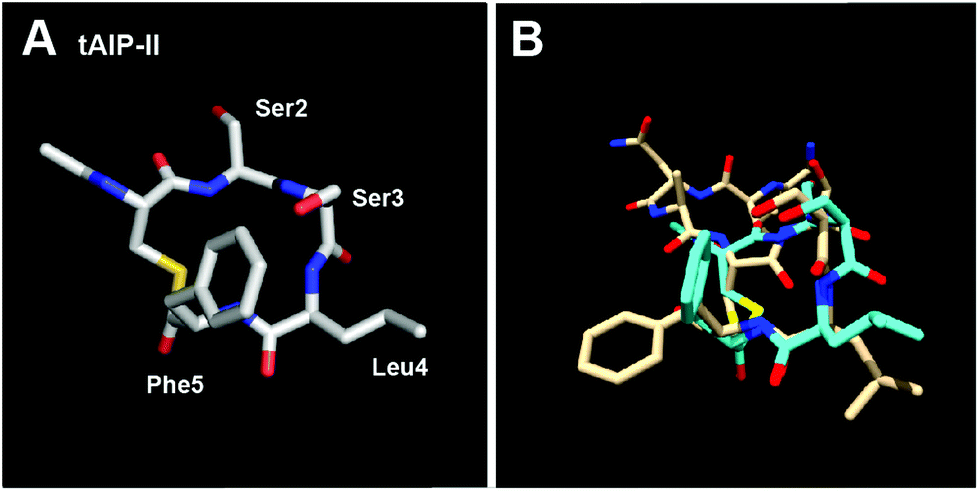 | ||
| Fig. 4 (A) Heavy atom lowest energy structure of tAIP-II. (B) Overlay of AIP-II (tan) and tAIP-II (cyan) structures. AIP-II structure reproduced from ref. 10. | ||
To further strengthen our claim regarding the minimal requirement for AgrC-II modulation by AIP-type ligands, we set out to structurally evaluate additional AIPs bearing only two hydrophobic, endocyclic residues. To this end, we chose to evaluate the 3-D structures of native AIPs of other staphylococcal species that contain such residues: AIP-I of S. epidermidis and AIP-I of S. lugdunensis (Table 1). Interestingly, both of these native AIPs have previously been reported to inhibit QS in S. aureus group II.19,21 In our hands, we observed that they were only weak to moderate AgrC-II inhibitors compared to tAIP-II (Table 3). Analysis of the solution-phase structures of these two AIPs revealed that the two peptides assume similar backbone conformations, although the side chain residues are tilted compared to each other (Fig. 5A–C and S-4†). This observation may explain their similar activity trends. More interestingly, in both peptides the two hydrophobic residues (Tyr7 and Phe8 in S. epidermidis; Tyr6 and Phe7 in S. lugdunensis) project their side chains in different directions. Further, when overlaid on tAIP-II, it appears that the hydrophobic side chains are significantly separated compared to tAIP-II (Fig. 5D). The lack of two hydrophobic residues in close proximity in both of the S. epidermidis and S. lugdunensis AIPs corroborates the weak to moderate inhibitory activity of these peptides against AgrC-II and serves to foster our hypothesis regarding the minimal structural requirement for effective AgrC-II modulation by AIP ligands.
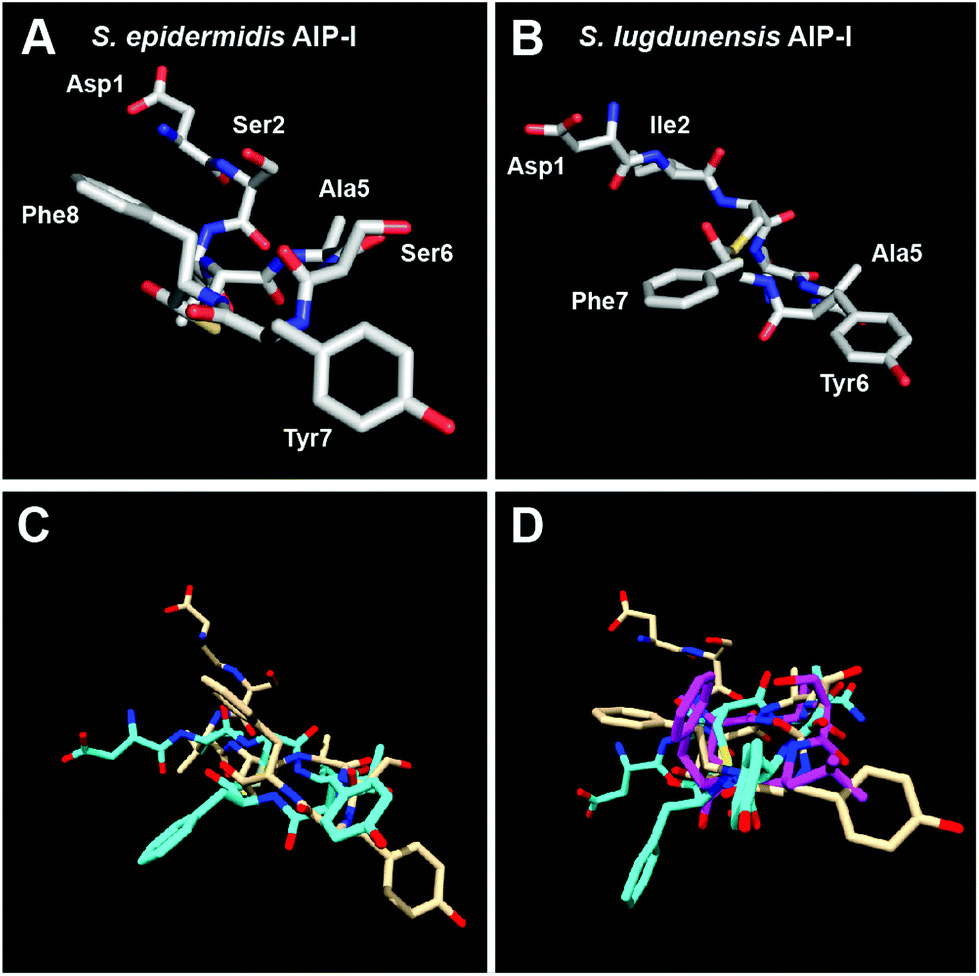 | ||
| Fig. 5 Heavy atom lowest energy structure of: (A) S. epidermidis AIP-I; and (B) S. lugdunensis AIP-I. Overlay of: (C) S. epidermidis AIP-I (tan) and S. lugdunensis AIP-I (cyan) structures; and (D) S. epidermidis AIP-I (tan), S. lugdunensis AIP-I (cyan) and tAIP-II (magenta) structures. | ||
Evaluation of AIP-III D4N
As the last aim of this study, we examined the structure and activity of a new AIP-III analogue. In our previous analysis of AIP-III SARs,10 we hypothesized that the role of the polar Asp4 residue (in the same position as Asp5 in AIP-I) is mainly to correctly orient the macrocycle and exocyclic tail for AgrC-III activation, and that replacement of Asp4 with the non-polar Ala causes a repositioning of the tail to be in closer proximity to the macrocycle, yielding a conformation capable of strong AgrC-III inhibition. We therefore reasoned that replacing Asp4 with Asn – a residue of more similar polarity and size relative to Ala – should result in an AIP-III analogue still capable of AgrC-III activation. In support of this hypothesis, Muir and co-workers previously showed that an AIP-I analogue with the same modification (AIP-I D5N) could activate both AgrC-I and -III (which we also observe; Table 3).15 Accordingly, we sought to explore whether a similar replacement in AIP-III would result in an analogue capable of activating both receptors. As expected, we observed AIP-III D4N to activate the AgrC-III receptor in our reporter assay (with an EC50 only ∼2-fold higher than native AIP-III;10Table 3); however, this analogue failed to activate the non-cognate receptor, AgrC-I. This result may imply that while the structural requirements for AgrC-III activation by AIPs are mainly the presence of two conserved structural motifs (the hydrophobic knob and an exocyclic hydrophobic anchor point) and not the identity of the residues forming or positioning (i.e., Asp4) these two motifs, the requirements for AgrC-I activation by AIPs are likely a combination of structural motifs and specific side chain contacts (as we highlighted above).Based on these results, we anticipated that the structure of AIP-III D4N would share the two structural motifs of AIP-III. Indeed, the NMR solution structure of AIP-III D4N was quite similar to that of AIP-III (Fig. 6 and S-5†).10 The main difference was in the orientation of the exocyclic tail, which projected a bit back and away from the AIP-III D4N macrocycle in comparison to AIP-III (overlay in Fig. 6B). This altered position may explain its reduced agonistic activity relative to the native AIP-III. Combined, these NMR and biological assay data for AIP-III D4N serve to further reinforce the two structural features required for AgrC-III receptor activation by AIP-III-type ligands.
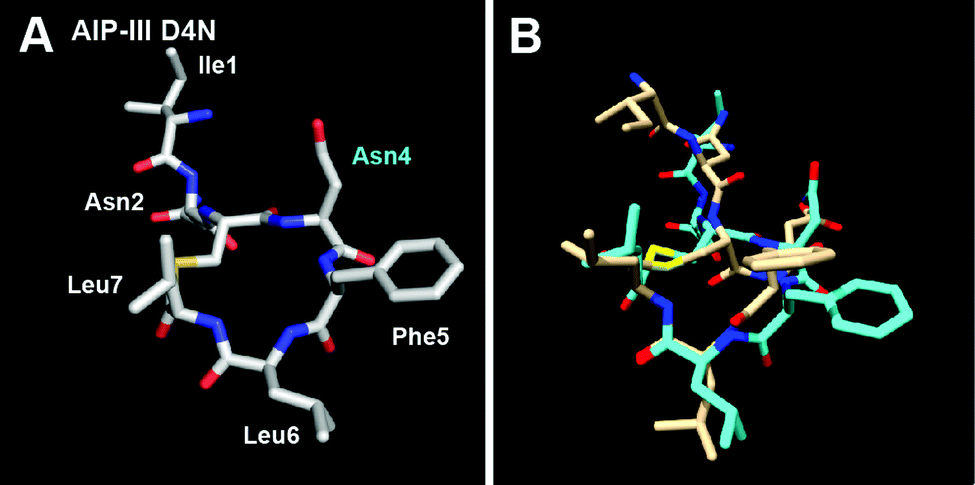 | ||
| Fig. 6 (A) Heavy atom lowest energy structure of AIP-III D4N. Altered residue labelled in cyan. (B) Overlay of AIP-III (tan) and AIP-III D4N (cyan) structures. AIP-III structure reproduced from ref. 10. | ||
Summary and conclusions
In summary, we have determined the solution-phase structures of several native AIPs and synthetic AIP analogues using NMR spectroscopy. Analysis of these structures, in combination with their biological activity data in cell-based reporter assays for AgrC receptor agonism and antagonism, enabled us to identify minimal structural elements necessary for effective modulation of different AgrC receptors in S. aureus. Notably, we determined different structural motifs that are required for the modulation of different AgrC receptors by AIP-type ligands: namely, (i) a triangular, hydrophobic knob motif for both AgrC-I inhibition and activation, and (ii) the presence of two hydrophobic residues projecting in the same direction and in close proximity for AgrC-II inhibition. We also obtained further data in support of the triangular knob and exocyclic tail combinations required for AgrC-I and AgrC-III activation. It is of course important to note that the structural data reported herein are for peptides in solution, and that these conformations may be altered upon binding to the different AgrC receptors. Further, the conditions used for the NMR experiments are different than those used for the bioassays. However, as the macrocycles in the peptides examined in this study should be relatively rigid (no matter the solvent) and the conformational data correlate well with the biological activity trends for these peptides, we believe these structures are likely biologically relevant and represent conformations similar to the AgrC-bound forms.Looking forward, the structural motifs delineated by this study should prove useful for the design and construction of new AIP analogues that possess only the minimal structural requirements for interactions with specific AgrC receptors. Such receptor selective analogues could be useful to interfere with QS in one S. aureus group without affecting agr signalling in other groups or staphylococcal species, and may reveal new pathways for infection control. These AIP mimetics may be constructed by incorporating only the essential hydrophobic residues (e.g., two to four residues, depending on the targeted receptor) and tuning the conformation by altering the macrocycle ring size and ring chemistry. Further studies are still needed to better understand the role of specific residues (e.g., Asp5 in AIP-I, and others) in selective receptor activation and inhibition; this information will allow us to best fine-tune the structures of future mimetics, and may be most easily obtained using in vitro experiments.7,17 These and related experiments are on-going in our laboratory, and will be reported in due course.
Acknowledgements
This work was supported by the Office of Naval Research (N00014-14-1-0791), Kimberly-Clark Corporation, and Burroughs Welcome Fund. The National Magnetic Resonance Facility at Madison is supported by the NIH (P41 GM103399). NMR facilities in the Department of Chemistry are supported in part by the NIH (1 S10 RR13866-01). We thank Prof. Richard Novick and Prof. Alexander Horswill for generously providing S. aureus strains.References
- S. T. Rutherford and B. L. Bassler, Cold Spring Harbor Perspect. Med., 2012, 2, a012427 CrossRef PubMed.
- H. F. Chambers and F. R. DeLeo, Nat. Rev. Microbiol., 2009, 7, 629 CrossRef CAS PubMed.
- R. P. Novick and E. Geisinger, Annu. Rev. Genet., 2008, 42, 541 CrossRef CAS PubMed.
- M. Thoendel, J. S. Kavanaugh, C. E. Flack and A. R. Horswill, Chem. Rev., 2011, 111, 117 CrossRef CAS PubMed.
- N. Amara, B. P. Krom, G. F. Kaufmann and M. M. Meijler, Chem. Rev., 2011, 111, 195 CrossRef CAS PubMed.
- C. P. Gordon, P. Williams and W. C. Chan, J. Med. Chem., 2013, 56, 1389 CrossRef CAS PubMed.
- B. Wang, A. Zhao, R. P. Novick and T. W. Muir, Mol. Cell, 2014, 53, 929 CrossRef CAS PubMed.
- B. A. Khan, A. J. Yeh, G. Y. C. Cheung and M. Otto, Expert Opin. Invest. Drugs, 2015, 24, 689 CrossRef CAS PubMed.
- Y. Tal-Gan, D. M. Stacy, M. K. Foegen, D. W. Koenig and H. E. Blackwell, J. Am. Chem. Soc., 2013, 135, 7869 CrossRef CAS PubMed.
- Y. Tal-Gan, M. Ivancic, G. Cornilescu, C. C. Cornilescu and H. E. Blackwell, J. Am. Chem. Soc., 2013, 135, 18436 CrossRef CAS PubMed.
- Y. Tal-Gan, D. M. Stacy and H. E. Blackwell, Chem. Commun., 2014, 50, 3000 RSC.
- S. A. Fowler, D. M. Stacy and H. E. Blackwell, Org. Lett., 2008, 10, 2329 CrossRef CAS PubMed.
- P. Mayville, G. Ji, R. Beavis, H. Yang, M. Goger, R. P. Novick and T. W. Muir, Proc. Natl. Acad. Sci. U. S. A., 1999, 96, 1218 CrossRef CAS.
- P. McDowell, Z. Affas, C. Reynolds, M. T. Holden, S. J. Wood, S. Saint, A. Cockayne, P. J. Hill, C. E. Dodd, B. W. Bycroft, W. C. Chan and P. Williams, Mol. Microbiol., 2001, 41, 503 CrossRef CAS.
- G. J. Lyon, J. S. Wright, T. W. Muir and R. P. Novick, Biochemistry, 2002, 41, 10095 CrossRef CAS PubMed.
- E. A. George, R. P. Novick and T. W. Muir, J. Am. Chem. Soc., 2008, 130, 4914 CrossRef CAS PubMed.
- J. G. Johnson, B. Wang, G. T. Debelouchina, R. P. Novick and T. W. Muir, ChemBioChem, 2015, 16, 1093 CrossRef CAS PubMed.
- S. M. Yerushalmi, M. E. Buck, D. M. Lynn, N. G. Lemcoff and M. M. Meijler, Chem. Commun., 2013, 49, 5177 RSC.
- G. Ji, R. Beavis and R. P. Novick, Science, 1997, 276, 2027 CrossRef CAS PubMed.
- M. Otto, R. Sussmuth, G. Jung and F. Gotz, FEBS Lett., 1998, 424, 89 CrossRef CAS PubMed.
- M. Otto, H. Echner, W. Voelter and F. Gotz, Infect. Immun., 2001, 69, 1957 CrossRef CAS PubMed.
- M. E. Olson, D. A. Todd, C. R. Schaeffer, A. E. Paharik, M. J. Van Dyke, H. Büttner, P. M. Dunman, H. Rohde, N. B. Cech, P. D. Fey and A. R. Horswill, J. Bacteriol., 2014, 196, 3482 CrossRef CAS PubMed.
- J. B. Blanco-Canosa and P. E. Dawson, Angew. Chem., Int. Ed., 2008, 47, 6851 CrossRef CAS PubMed.
- R. N. Kirchdoerfer, A. L. Garner, C. E. Flack, J. M. Mee, A. R. Horswill, K. D. Janda, G. F. Kaufmann and I. A. Wilson, J. Biol. Chem., 2011, 286, 17351 CrossRef CAS PubMed.
- F. Delaglio, S. Grzesiek, G. W. Vuister, G. Zhu, J. Pfeifer and A. Bax, J. Biomol. NMR, 1995, 6, 277 CrossRef CAS PubMed.
- A. Bax and D. G. Davis, J. Magn. Reson., Ser. A, 1985, 65, 355 CAS.
- M. H. Levitt, R. Freeman and T. Frenkiel, J. Magn. Reson., 1982, 47, 328 CAS.
- T. L. Hwang and A. J. Shaka, J. Am. Chem. Soc., 1992, 114, 3157 CrossRef CAS.
- T. D. Goddard and D. G. Kneller, University of California, San Francisco, 3.114 edn., 2007.
- K. Wüthrich, NMR of Proteins and Nucleic Acids, John Wiley & Sons, New York, 1986 Search PubMed.
- C. D. Schwieters and G. M. Clore, J. Magn. Reson., 2001, 152, 288 CrossRef CAS PubMed.
- C. D. Schwieters, J. J. Kuszewski, N. Tjandra and G. M. Clore, J. Magn. Reson., 2003, 160, 65 CrossRef CAS PubMed.
- M. Nilges, G. M. Clore and A. M. Gronenborn, FEBS Lett., 1988, 229, 317 CrossRef CAS PubMed.
- G. M. Clore and J. Kuszewski, J. Am. Chem. Soc., 2002, 124, 2866 CrossRef CAS PubMed.
- C. D. Schwieters and G. M. Clore, J. Phys. Chem. B, 2008, 112, 6070 CrossRef CAS PubMed.
- The PyMOL Molecular Graphics System, Version 1.3, Schrödinger, LLC.
- E. F. Pettersen, T. D. Goddard, C. C. Huang, G. S. Couch, D. M. Greenblatt, E. C. Meng and T. E. Ferrin, J. Comput. Chem., 2004, 25, 1605 CrossRef CAS PubMed.
- G. J. Lyon, P. Mayville, T. W. Muir and R. P. Novick, Proc. Natl. Acad. Sci. U. S. A., 2000, 97, 13330 CrossRef CAS PubMed.
- R. P. Novick, H. F. Ross, S. J. Projan, J. Kornblum, B. Kreiswirth and S. Moghazeh, EMBO J., 1993, 12, 3967 CAS.
- C. P. Montgomery, S. Boyle-Vavra, P. V. Adem, J. C. Lee, A. N. Husain, J. Clasen and R. S. Daum, J. Infect. Dis., 2008, 198, 561 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Peptide characterization data, dose response curves, full biological assay data, tables of resonance assignments and RMSD values, additional structural figures, and pdb files. See DOI: 10.1039/c5ob01735a |
| ‡ Current address: Department of Chemistry, University of Nevada, Reno, 1664 N. Virginia Street, Reno, NV 89557, USA. |
| § Current address: Department of Chemistry, University of Vermont, 82 University Place, Burlington, VT 05405, USA. |
| This journal is © The Royal Society of Chemistry 2016 |