
The structure of tagetitoxin†
Abil E.
Aliev
*a,
Kersti
Karu
a,
Robin E.
Mitchell
b and
Michael J.
Porter
a
aDepartment of Chemistry, University College London, 20 Gordon Street, London WC1H 0AJ, UK. E-mail: A.E.Aliev@ucl.ac.uk
bPlant and Food Research, Private Bag 92619, Auckland 1142, New Zealand
First published on 27th October 2015
Abstract
Based on detailed analysis of newly acquired NMR data, we show that the previously revised structure of tagetitoxin is incorrect. A new structure of tagetitoxin is proposed which is consistent with the NMR and MS data.
Introduction
Tagetitoxin is a toxin isolated from the plant pathogenic bacterium Pseudomonas syringae pv. tagetis.1 It is known to cause chlorosis in young plant leaves, which has been attributed to inhibition of RNA polymerase in chloroplasts.2 Tagetitoxin also inhibits bacterial RNA polymerase,2 and is the only natural product known to inhibit eukaryotic RNA polymerase III in a specific manner.3 Recently, Yuzenkova et al. have shown that tagetitoxin neither affects the chemistry of RNA synthesis nor competes with the nucleoside triphosphate in the active centre.4 Instead, tagetitoxin increases the stability of the pre-translocated state of the elongation complex, thus slowing down addition of the following nucleotide.4The first published structure of tagetitoxin by Mitchell and Hart in 1983 consisted of an eight-membered heterocycle with a sulfur atom (structure 1 in Fig. 1, molecular weight 435, C11H18NO13PS).5
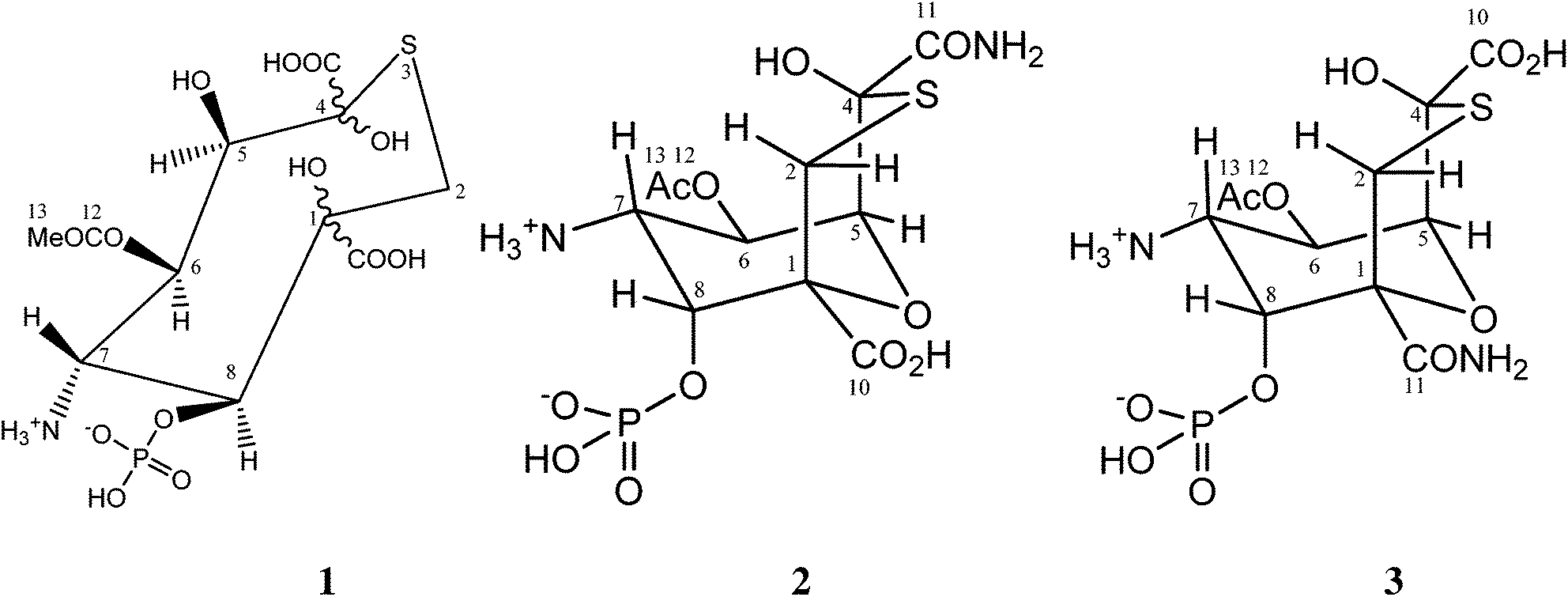 | ||
| Fig. 1 Previously proposed structures of tagetitoxin. | ||
It was found that heteroatomic components comprised of oxygen, nitrogen in an amine, phosphorus in a phosphate ester and sulfur. Investigations of the structure of tagetitoxin continued based on new MS and NMR data, after attempts to obtain crystals for X-ray analysis failed.5 In 1989, a revised bicyclic structure of tagetitoxin based on the 9-oxa-3-thiabicyclo[3.3.1]nonane ring system was proposed by Mitchell et al. (Fig. 1).6 FAB mass spectrometry showed (M + H)+ = 417.0361 (C11H18N2O11PS requires 417.0369) indicating that tagetitoxin has a molecular formula C11H17N2O11PS. Structure 2 was favoured, although the spectroscopic data did not rule out the closely related structure 3 (Fig. 1).6
In 2005, a crystal structure of the RNA polymerase from Thermus thermophilus with tagetitoxin bound to the active site was published by Vassylyev et al.7 Although the difference electron density map revealed an electron density attributed to tagetitoxin in this crystal structure, the structure of tagetitoxin was not investigated and a stereoisomer of structure 2 was used by Vassylyev et al. without further verification.7 In the same year, Gronwald et al. published their purification protocol and partial characterization of tagetitoxin.8 According to their analysis, the revised structure of Mitchell et al.6 is incorrect. Based on electrospray ionization mass spectrometry in 50% methanol![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :H2O, Gronwald et al. reported that the molecular weight of tagetitoxin is 678, although the NMR spectrum of tagetitoxin published by them indicated that tagetitoxin contained additional peaks at 2.53 ppm and 1.75 ppm not observed previously by Mitchell et al.5,6 Despite the ambiguity of the structure of tagetitoxin, several reports have been published to date, detailing synthetic approaches to tagetitoxin and its analogues with the basic bicyclic ring structure 2,9 though none of these has successfully delivered the full structure 2.
:H2O, Gronwald et al. reported that the molecular weight of tagetitoxin is 678, although the NMR spectrum of tagetitoxin published by them indicated that tagetitoxin contained additional peaks at 2.53 ppm and 1.75 ppm not observed previously by Mitchell et al.5,6 Despite the ambiguity of the structure of tagetitoxin, several reports have been published to date, detailing synthetic approaches to tagetitoxin and its analogues with the basic bicyclic ring structure 2,9 though none of these has successfully delivered the full structure 2.
Here, we report the results of our analysis of NMR and MS data for tagetitoxin and show that neither of the published structures of tagetitoxin is correct. A new structure of tagetitoxin is reported which is in agreement with NMR and MS data.
Results and discussion
The sample studied was that originally isolated and purified by Mitchell.1,5 In order to illustrate the purity of the compound studied, the proton NMR spectrum of tagetitoxin in D2O is shown in Fig. 2. Note that additional peaks of smaller intensity appeared in 1H NMR spectrum of tagetitoxin kept in D2O solution over 4–6 weeks (see Fig. S2 in ESI†), suggesting that tagetitoxin gradually decomposes in aqueous solutions. Although most of the spectral features in Fig. 1 resemble those observed in the 1H NMR spectrum of tagetitoxin by Gronwald et al.,8 no peaks are observed at 1.75 and 2.53 ppm. Similarly, no 13C peak was observed at 181.45 ppm. These observations suggest the material studied by Gronwald et al.8 was less pure compared to that extracted by Mitchell et al.5,6 From the analysis of the MS data obtained in this work (see ESI† for full details), no species with the molecular weight of 678 were found, which is reported by Gronwald et al.8 as a revised molecular weight of tagetitoxin.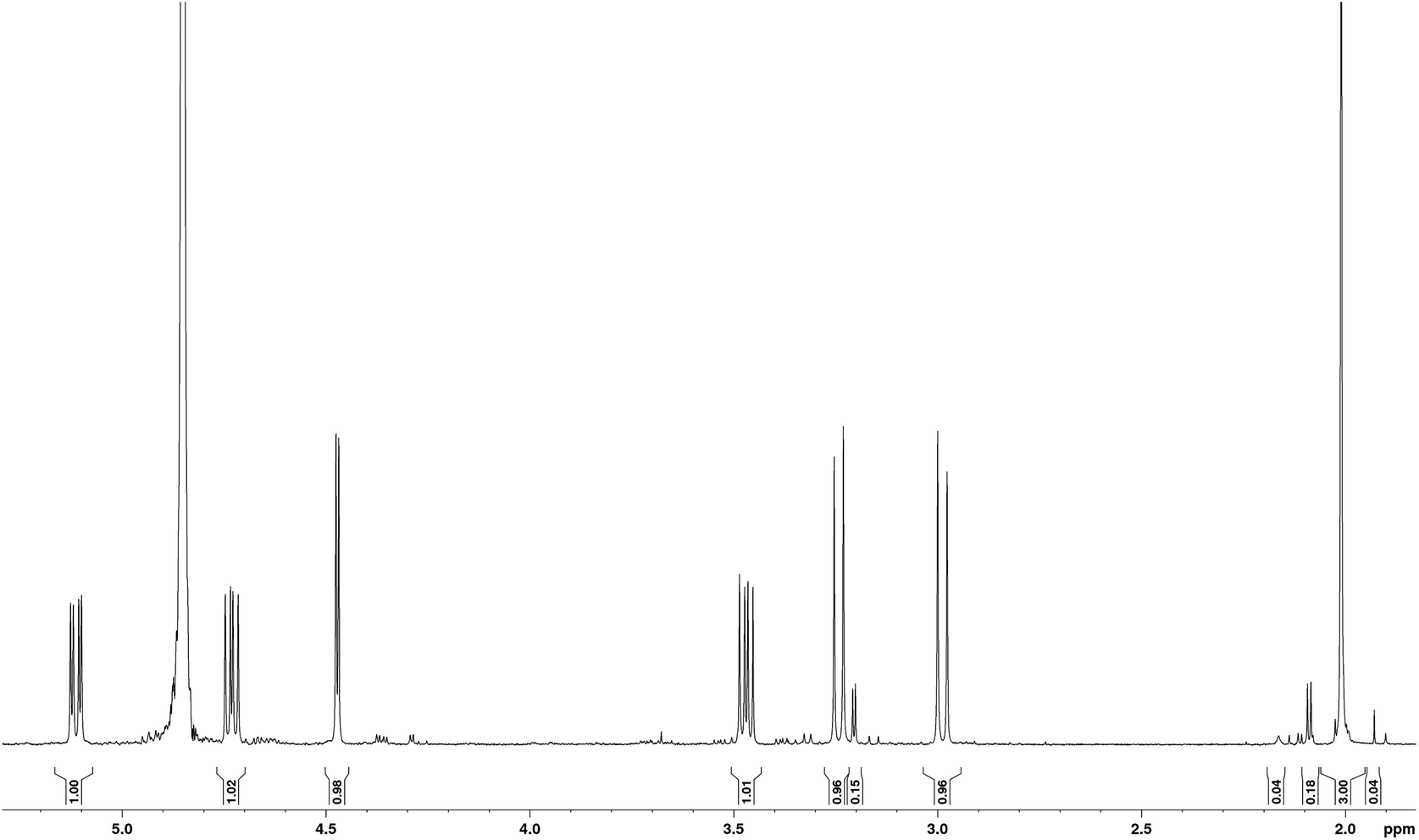 | ||
| Fig. 2 1H NMR spectrum of tagetitoxin in D2O at 293 K (600 MHz). Integral intensities are shown between the chemical shift axis and the spectrum. The most significant impurity peaks are observed as doublets at 3.21 ppm with 4.0 Hz splitting (integral intensity 0.15) and at 2.09 ppm with 5.0 Hz splitting (integral intensity 0.18). | ||
In 1989, the revised structure 2 (Fig. 1) was deduced based on the analysis of 1H and 13C NMR spectra, 1H NOEs and the COLOC spectrum for 1H–13C long-range correlations.6 The latter is expected to provide information similar to that from the HMBC spectrum, although it is significantly less sensitive than HMBC, which is usually used for identification of 2 or 3 bond correlations between 1H and 13C nuclei. The HMBC spectrum shown in Fig. 3, as well as the values of long-range nJCH couplings (Table 1), revealed several correlations which allowed us to rule out structures 1–3 shown in Fig. 1. In particular, some of the disagreements are as follows:
(1) A cross-peak is observed for the C11–H8 pair which is in disagreement with structure 2 with six bonds between C11 and H8.
(2) A strong cross-peak C10–H2′ is in disagreement with structure 3 with four bonds between C10 and H2′. In principle, 4JCH correlations can be observed in HMBC spectra, however, the value of JC10H2′ coupling derived from the HMBC-JC spectrum is 5.0 Hz, which cannot be attributed to a 4JCH coupling.
(3) Cross-peaks are observed for C7–H2 (JCH = 5 Hz) and C7–H2′ (JCH = 3 Hz), which are in disagreement with all three structures shown in Fig. 1, with four bond separation between C7 and H2.
(4) Dihedral angle between C4 and H6 is ∼180° in structure 2, while only a weak HMBC cross-peak is observed in the HMBC spectrum. From HMBC-JC, the value of JC4H6 is 1.4 Hz.
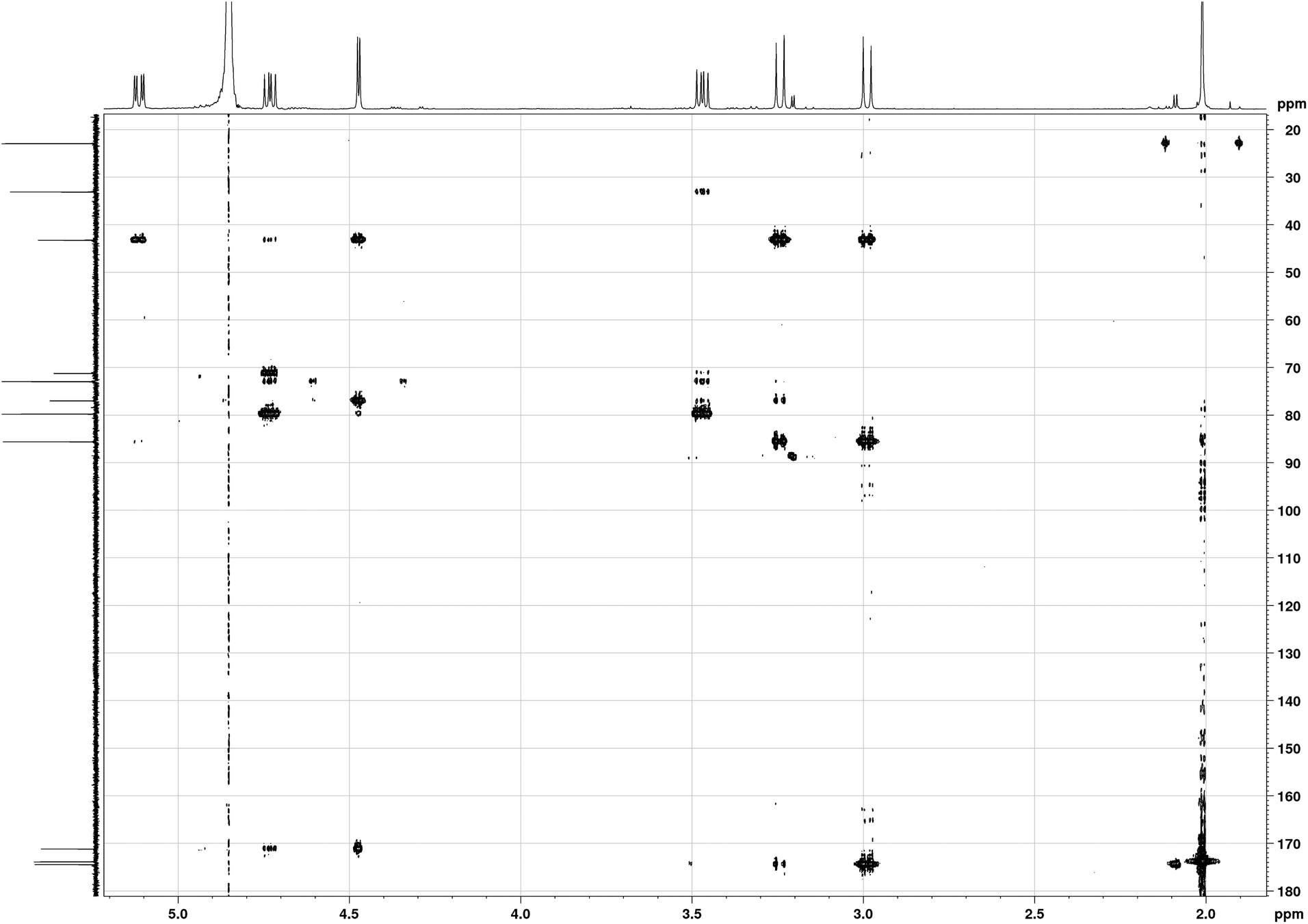 | ||
| Fig. 3 1H–13C HMBC NMR spectrum of tagetitoxin in D2O at 293 K (600 MHz). | ||
| Exper. nJCH/Hz | 4-Chair Calc. nJCH/Hz | 4-Tw.-chair Calc. nJCH/Hz | 4 Calc. 〈nJCH〉/Hz | |
|---|---|---|---|---|
| C1–H7 | 1.3 | 1.20 | 2.05 | 1.41 |
| C1–H8 | 2.4 | −2.07 | −2.46 | −2.17 |
| C1–H5 | ∼0 | −0.62 | −0.88 | −0.69 |
| C1–H6 | ∼0 | 0.16 | −0.01 | 0.12 |
| C2–H7 | 1.5 | 2.82 | 0.02 | 2.10 |
| C4–H2 | 2.3 | −2.32 | −1.63 | −2.15 |
| C4–H2′ | 4.1 | −4.02 | −2.20 | −3.56 |
| C4–H13 | 0.8 | 0.53 | 0.48 | 0.51 |
| C4–H6 | 1.4 | 0.79 | −0.10 | 0.56 |
| C5–H7 | (+)1.1 | 1.28 | 1.02 | 1.22 |
| C5–H8 | (+)0.9 | 0.72 | 0.41 | 0.64 |
| C5–H6 | (+)0.2 | 0.17 | 0.95 | 0.37 |
| C6–H5 | (−)0.7 | −0.14 | −0.01 | −0.11 |
| C6–H7 | (−)5.6 | −5.44 | −5.65 | −5.49 |
| C6–H8 | (+)8.0 | 7.26 | 6.32 | 7.02 |
| C7–H2 | 5.0 | 6.98 | −0.10 | 5.17 |
| C7–H2′ | 3.0 | 2.44 | 5.99 | 3.35 |
| C7–H5 | (+)6.1 | 5.53 | 5.31 | 5.47 |
| C7–H6 | (−)2.7 | −2.36 | −1.60 | −2.17 |
| C7–H8 | (−)1.1 | −2.12 | −1.96 | −2.08 |
| C8–H2 | 1.4 | 1.39 | −0.17 | 0.99 |
| C8–H5 | (+)6.2 | 6.35 | 6.40 | 6.36 |
| C8–H6 | (+)0.3 | 0.50 | 0.22 | 0.43 |
| C8–H7 | (−)0.4 | −1.12 | −0.44 | −0.95 |
| C10–H2 | 1.2 | 1.35 | 1.64 | 1.42 |
| C10–H2′ | 5.0 | 7.03 | 1.54 | 5.63 |
| C11–H8 | 1.5 | 1.86 | 2.89 | 2.12 |
| C11–H5 | 2.7 | 2.12 | 1.95 | 2.07 |
| C12–H13 | 6.0 | −5.51 | −5.52 | −5.51 |
| C12–NH | 3.7 | 4.57 | 5.14 | 4.72 |
| rmsJ/Hz | — | 0.72 | 1.50 | 0.52 |
Furthermore, the 1D NOESY spectrum with selective excitation of methyl protons at 2.01 ppm showed a negative exchange enhancement at 2.16 ppm with the integral intensity ratio 68:1 for singlets at 2.01 and 2.16 ppm in the 1H NMR spectrum (Fig. S3 in ESI†). Such a slow exchange at room temperature between two sites with unequal populations is characteristic for an amide group NHCOMe, but not for OCOMe shown in structures 1–3 (Fig. 1). 1H NMR spectrum recorded in H2O:D2O (9:1) showed a singlet at 8.47 ppm (Fig. S13 in ESI†), which is in agreement with the presence of the NHCOMe group. In addition, the 1H–15N HMBC spectrum in D2O (Fig. S20 in ESI†) showed a correlation for the methyl protons with the 15N signal at 140.5 ppm, in agreement with the expected 15N chemical shift for a secondary amide in the range 110–160 ppm (relative to liquid NH3).
Based on mainly 1H–13C HMBC correlations and the values of long-range JCH couplings a new structure was derived shown in Fig. 4. The above noticed disagreements (1)–(4) for structures 1–3 were verified for structure 4:
(1) The cross-peak observed for C11–H8 pair is due to 3JCH coupling in 4;
(2) The cross-peak C10–H2′ is due to 3JCH coupling in 4.
(3) Cross-peaks observed for C7–H2 (JCH = 5 Hz) and C7–H2′ (JCH = 3 Hz) are due to 3JCH coupling in 4.
(4) Assuming a chair conformation of the six membered ring, the dihedral angle between C4 and H6 is ∼60° in structure 4, in agreement with the measured value of 3JC4H6 = 1.4 Hz based on the Karplus-type relationship for 3JCH couplings.
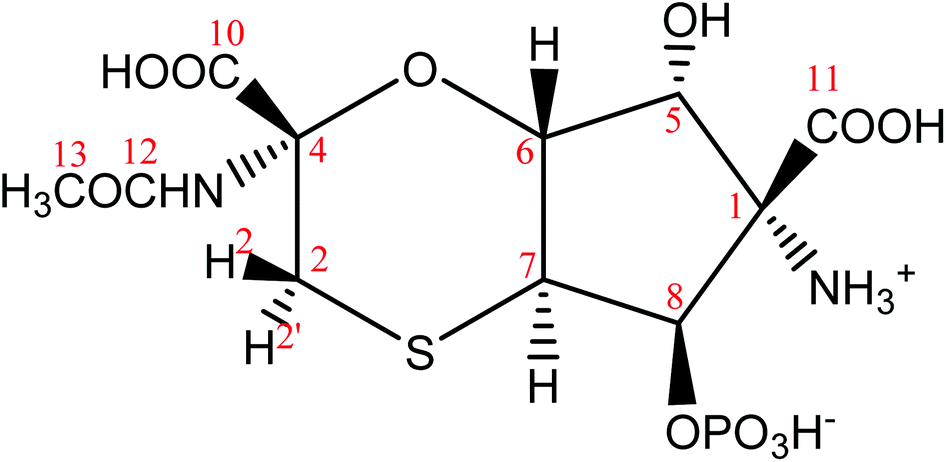 | ||
| Fig. 4 The proposed revised structure of tagetitoxin based mainly on the analysis of 1H–13C HMBC correlations and the values of long-range JCH couplings. The atom numbering used corresponds to that in 2 (Fig. 1). | ||
In a similar fashion, we have analysed the measured values of all the vicinal 3JCH couplings, which show good agreement with structure 4. There are relatively few JHH couplings in tagetitoxin. Nevertheless, the large value of the 3JH6H7 = 12.2 Hz is in favour of the trans fusion of two cycles with both protons occupying axial orientations. Furthermore, from the measured signal enhancements in 1D NOESY spectra (Table 2), the NOE is relatively small for the H6–H7 pair (0.5%) compared to, for example, H5–H6 (1.4–1.5%) or H7–H8 (1.2–1.3%). Combined with the values of vicinal couplings (3JH5H6 = 4.1 Hz and 3JH7H8 = 7.8 Hz), these NOEs are in favour of the trans configuration of protons H6 and H7, the cis configuration of protons H5 and H6 and the cis configuration of protons H7 and H8. A very small enhancement (0.1%) observed for the H6–H8 pair is in agreement with their trans configuration in the five-membered ring.
| “Touched” protons | ||||||
|---|---|---|---|---|---|---|
| H2′ | H2 | H7 | H5 | H8 | H6 | |
| H2′ | — | 5.4 (1.77/1.77) | 0.8 (2.37/3.92) | 0.1 (3.95/3.73) | ||
| H2 | 5.6 (1.77/1.77) | — | 0.6 (4.12/2.30) | |||
| H7 | 1.1 (2.37/3.92) | — | 0.1(3.72/3.74) | 1.2 (2.26/2.21) | 0.5 (3.04/3.03) | |
| H5 | 0.1 (3.72/3.74) | — | 1.5 (2.44/2.45) | |||
| H8 | 1.3 (2.26/2.21) | — | 0.1 (3.76/3.80) | |||
| H6 | 0.7 (4.12/2.30) | 0.5 (3.04/3.03) | 1.4 (2.44/2.45) | 0.1 (3.76/3.80) | — | |
Protons of the methylene group in tagetitoxin are labelled as 2 and 2′ (Fig. 4). The methylene proton with the cis configuration relative to proton H6 is denoted as H2 (the high-frequency CH2 signal in the 1H NMR spectrum), while the other methylene proton with the trans configuration relative to H6 is denoted as H2′ (the low-frequency CH2 signal in the 1H NMR spectrum). Thus, in a chair conformation of the six-membered ring with the axial orientation of H6, such a definition of H2 and H2′ corresponds to the equatorial orientation of H2 and the axial orientation of H2′. Relatively strong NOE (0.6%) was observed for proton pair H2–H6 (Table 2), which led to a consideration of a twisted chair conformation for the six-membered ring. Both chair and twisted-chair conformations were included into our computational analysis and the final lowest energy conformations derived from DFT M06-2X/def2-TZVP geometry optimisations are shown in Fig. 5. The free energy of the twisted-chair conformation relative to that of the chair conformation is +1.22 kcal mol−1. On the assumption of a two-site fast exchange (in the NMR timescale) between chair and twisted-chair conformations, the predicted populations by DFT M06-2X/def2-TZVP calculations are 89% and 11% for chair and twisted-chair conformations, respectively. A more reliable estimate of the conformational populations was achieved using experimental values of 30 long-range JCH couplings and predicted values of corresponding coupling constants in chair and twisted-chair conformations at the DFT B3LYP/6-311+G(2d,p) level of theory (Table 1; regarding the performance of B3LYP calculations for predictions of J couplings, see ref. 10). The populations of conformers derived from this analysis were 75% and 25% for chair and twisted-chair conformations, respectively.
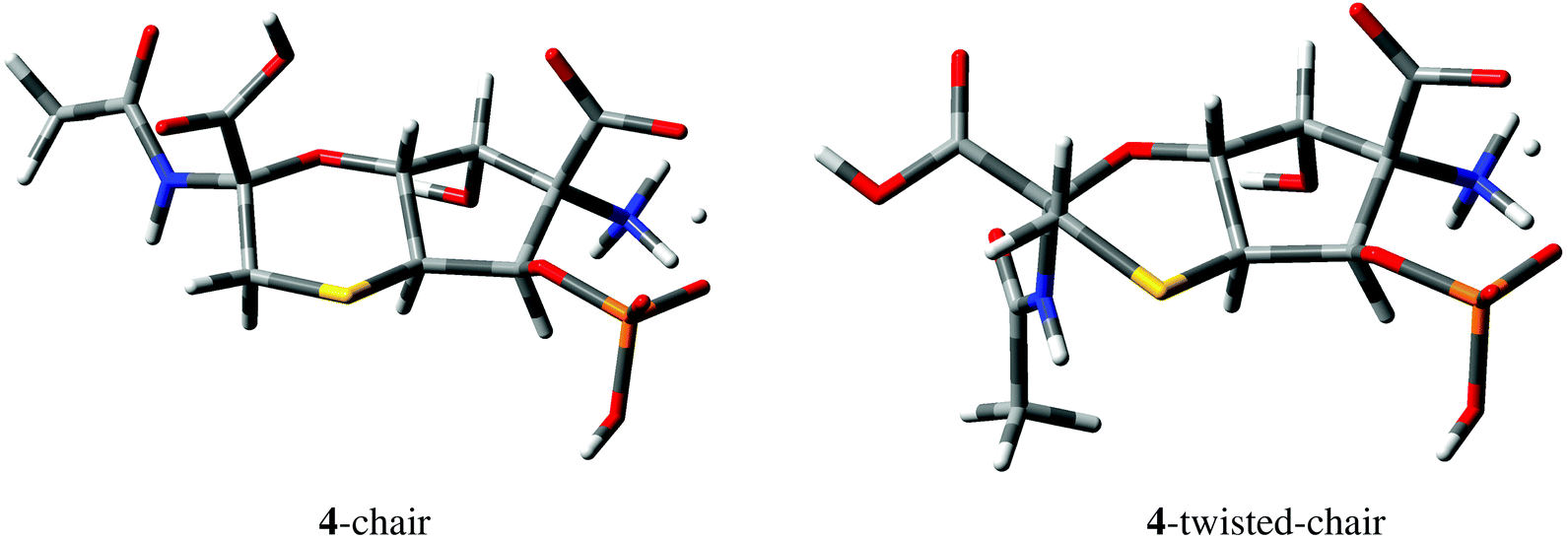 | ||
| Fig. 5 Geometries of 4-chair and 4-twisted-chair conformations derived from DFT M06-2X/def2-TZVP calculations. One of the carboxylic protons of structure 4 (Fig. 4) is delocalized between COO− and OPO3H− groups in both conformations. | ||
No HMBC correlations were observed for C1–H5 and C1–H6 pairs separated by two and three bonds, respectively, in structure 4. DFT calculations confirmed that the expected values of the corresponding 2,3JCH couplings are indeed small, e.g., −0.62 Hz and 0.16 Hz in the 4-chair conformation shown in Fig. 5 (−0.88 Hz and −0.01 Hz in the 4-twisted-chair conformation).
In order to determine orientations of substituents in position 4, we have used weak NOEs observed for the amide NH proton with H7 in H2O + D2O (9:1) solution, as well as the fact that the NOE for the NH–H2′ pair is significantly stronger than that for the NH–H2 pair (∼4.4 times based on the volume integration of the corresponding cross-peaks; the volume integration ratio for the cross-peaks of the amide proton NH with H7, H2, H2′, Me was 1.0:2.3:10.1:9.2; Fig. S13 in ESI†). Furthermore, the cis orientation of the NHCOMe group relative to proton H7 was confirmed by the analysis of vicinal JCH couplings of the adjacent carboxylic carbon based on the Karplus-type relationship: 3JC10H2 = 1.2 Hz and 3JC10H2′ = 5.0 Hz. These agree well with the DFT predicted values of 1.4 and 5.6 Hz on the assumption of the equilibrium between 4-chair (75%) and 4-twisted-chair (25%) conformations (Fig. 5 and Table 1).
The determination of orientations of substituents in position 1 required consideration of both alternatives in DFT calculations and the analysis of 3JCH couplings of the carboxylic carbon C11 with protons H5 and H8. Structures of 5-chair and 5-twisted-chair, in which the orientations of N+H3 and COOH are interchanged at C1 compared to 4, are shown in Fig. S1 (ESI†). The free energy of the 5-twisted-chair conformation relative to that of the 5-chair conformation is +0.81 kcal mol−1. On the assumption of a two-site exchange between chair and twisted-chair forms, the predicted populations by DFT M06-2X/def2-TZVP calculations are 81% and 19% for 5-chair and 5-twisted-chair conformations, respectively. From the analysis of experimental values of 30 long-range JCH couplings and predicted values of corresponding coupling constants in 5-chair and 5-twisted-chair conformations at the DFT B3LYP/6-311+G(2d,p) level of theory (Table S3†), the populations of conformers were 78% and 22% for 5-chair and 5-twisted-chair conformations, respectively. However, the rms deviation between experimental and calculated couplings is 1.24 Hz for the conformational equilibrium 5-chair/5-twisted-chair, compared to 0.52 Hz for the conformational equilibrium 4-chair/4-twisted-chair. The predicted values for 3JC11H8 and 3JC11H5 were 3.7 and 0.3 Hz for the 5-chair/5-twisted-chair equilibrium, which are in disagreement with the experimental values of 1.5 and 2.7 Hz. In the case of the 4-chair/4-twisted-chair equilibrium, the predicted values for 3JC11H8 and 3JC11H5 were 2.1 and 2.1 Hz. Thus, the cis configuration of the phosphate and carboxylic groups at C8 and C1, respectively, can be deduced based on the analysis of experimental and calculated JCH couplings.
In addition to the measured values, we have also determined the sign of some of the JCH couplings (Table 1). It is well known that 13C–1H couplings over one and three bonds are positive, while those over two or four bonds (2JCH or 4JCH) are either positive or negative. Thus, if we know that the sign of nJCH is negative, then the number of bonds between C and H cannot be three and using the absolute value of the coupling constant we could deduce whether it corresponds to 2JCH or 4JCH. We have used the HSQC-HECADE spectrum for sign determinations.11 Note that the sign of only some of the nJCH couplings are available from this spectrum (e.g., nJCH correlations of quaternary carbons are not detectable).11 Nevertheless, all the measured negative values (for spin pairs C6–H5, C6–H7, C7–H6 and C8–H7) can be attributed to geminal 2JCH couplings, while 3JCH couplings have a positive sign (for spin pairs C5–H7, C5–H8, C6–H8, C7–H5, C8–H5 and C8–H6). Thus, these results additionally support the sequence in which the corresponding C and H atoms are arranged. The signs of these couplings predicted by the DFT calculations were in agreement with the HSQC-HECADE measurements (Table 1).
The EASY-ROESY method was also used, which is known to provide accurate integration of cross-peaks for quantitative estimates.12 We have analysed the observed rotational Overhauser effects (ROEs) using a simplified version of the growth rates method in order to estimate internuclear 1H–1H distances.13 The satisfactory performance of the simplified growth rates method has been demonstrated previously for cyclic organic compounds.14 The standard deviations for distance measurements were typically 10% of the corresponding mean values.14 Volume integrals of ROE cross-peaks for 6 proton pairs were measured for tagetitoxin (Table 3 and Fig. S14 in ESI†). Using r = 1.77 Å as the reference value for the geminal H2–H2′ pair, internuclear distances for other proton pairs were calculated using the r−6 dependence of ROEs.13 In Table 3, we compare experimental values with those from interatomic distances (ricalc, Å) derived from M062X/def2-TZVP-optimised geometries. The individual chair and twisted-chair conformers showed the rms deviations (rmsd, Å) of 0.62 Å and 0.71 Å, respectively, for five pairs of protons (excluding the reference geminal pair). Significantly improved agreement is observed with rmsd = 0.11 Å on considering a two-site fast exchange between 4-chair (75%) and 4-twisted-chair (25%), with the populations determined from the analysis of nJCH couplings above.
| Proton pair | NMR 〈rexp〉a (Å) | 4-Chair rcalc (Å) | 4-Twisted-chair rcalc (Å) | 4 〈rcalc〉b (Å) |
|---|---|---|---|---|
| a Uncertainties in experimental values were estimated using volume integrations of cross-peaks above and below the diagonal. b In calculations of averaged values of 〈rcalc〉, the calculated ROEs were weighted using populations of conformers 4-chair (75%) and 4-twisted-chair (25%). | ||||
| 2–2′ | 1.77 | 1.77 | 1.77 | 1.77 |
| 7–8 | 2.21 ± 0.02 | 2.26 | 2.21 | 2.24 |
| 5–6 | 2.31 ± 0.01 | 2.44 | 2.45 | 2.44 |
| 6–7 | 2.85 ± 0.02 | 3.04 | 3.03 | 3.03 |
| 2–6 | 2.76 ± 0.03 | 4.12 | 2.30 | 2.85 |
| 2′–7 | 2.43 ± 0.01 | 2.37 | 3.92 | 2.47 |
| rmsd | — | 0.62 | 0.71 | 0.11 |
The populations of chair (75%) and twisted chair (25%) conformers obtained from the analysis of 3JCH couplings also agree well with the combined analysis of experimental and calculated 1H and 13C chemical shifts. The methodology used here has been verified previously for cyclic organic compounds with known structures.14,15 In particular, optimised geometries of 4-chair and 4-twisted-chair were used in GIAO B3LYP/6-311+G(2d,p) chemical shielding calculations. The conformationally averaged values of the isotropic shieldings 〈σcalc(i)〉 were calculated. The averaged values of the isotropic shieldings were then converted into conformationally averaged values of chemical shifts, 〈δcalc(i)〉, using both the slope and the intercept of the σcalcvs. δexp plot, as described previously.14 From the results obtained (Table 4), the rmsδ values for 4 were 0.08 ppm (1H) and 2.2 ppm (13C). For comparison, the rmsδ values for a closely related structure 5 (78% chair and 22% twisted chair) were 0.21 ppm (1H) and 2.8 ppm (13C), showing high sensitivity of both 1H and 13C chemical shifts to the change in the orientation of substituents. Overall, the relatively small values of rms deviations for chemical shifts (rmsδ1H 0.08 ppm and 13C 2.2 ppm), together with the ROE analysis of interproton distances (rmsd 0.11 Å), further support the validity of structure 4 for tagetitoxin. The NMR-derived structure 4 of tagetitoxin was also consistent with the accurate mass measurements and gas-phase fragmentation patterns, full details of which are included in ESI.†
| Proton | δ expH (ppm) | 〈δcalcH〉a (ppm) | Carbon | δ expC (ppm) | 〈δcalcC〉a (ppm) |
|---|---|---|---|---|---|
| a Calculated chemical shifts [δcalc(i) = (σcalc(i) − b)/a] were determined using the slope [a(1H) = −1.14 and a(13C) = −0.98] and the intercept [b(1H) = 32.31 ppm and b(13C) = 177.66 ppm] derived from the least squares fittings [σcalc(i) = aδexp(i) + b]. | |||||
| 2 | 3.25 | 3.23 | 1 | 71.2 | 73.1 |
| 2′ | 2.98 | 2.87 | 2 | 33.1 | 36.2 |
| 5 | 4.46 | 4.39 | 4 | 85.6 | 82.8 |
| 6 | 5.13 | 5.15 | 5 | 72.9 | 72.8 |
| 7 | 3.48 | 3.65 | 6 | 79.8 | 80.4 |
| 8 | 4.73 | 4.72 | 7 | 43.2 | 44.8 |
| 13 | 2.01 | 2.02 | 8 | 77.0 | 78.4 |
| 10 | 174.4 | 174.4 | |||
| 11 | 171.2 | 170.1 | |||
| 12 | 173.8 | 174.3 | |||
| 13 | 22.9 | 17.7 | |||
| rmsδ | — | 0.08 | rmsδ | — | 2.2 |
| Δ max | — | 0.17 | Δ max | — | −5.2 |
Experimental
NMR spectroscopy
Purified tagetitoxin was received in non-crystallised solid form from Robin Mitchell.1,5,61H and 13C NMR spectra were recorded on a Bruker Avance III 600 MHz NMR spectrometer equipped with a 5 mm cryoprobe (1H 600.13 MHz and 13C 150.90 MHz). These spectra showed no change from the data of Mitchell et al.615N and 31P NMR spectra were recorded on a Bruker Avance III 400 MHz NMR spectrometer equipped with a 5 mm 1H–13C–15N–31P probe (1H 400.13 MHz, 15N 40.55 MHz and 31P 161.98 MHz). Data acquisition and processing were performed using standard TopSpin software (versions 2.1 and 3.2). 1H and 13C chemical shifts were calibrated indirectly, using dioxane shifts in D2O (1H 3.75 ppm, 13C 67.19 ppm). 15N and 31P NMR chemical shifts were calibrated using 15N2-urea dissolved in DMSO-d6 (77.6 ppm relative to liquid NH3) and 85% H3PO4 (0 ppm). Unless otherwise specified, NMR measurements were carried out at 293 K. Temperature calibration was carried out using a sample of 99.8% deuterated MeOD in a 5 mm NMR tube. In addition to standard 1D and 2D spectra, additional techniques were employed for measuring long-range JCH couplings, including HMBC-JC16 and HSQC-HECADE.11One- and two-dimensional NOE measurements were undertaken for establishing spatial proximities of protons.13 Standard pulse sequences and those with the elimination of strong interference caused by zero-quantum coherence were employed.17 2D EASY-ROESY spectra were also acquired. The main advantage of this experiment is that artifacts due to J-couplings are minimised. It has also been shown to yield reliable intramolecular distances without a sample-specific setup.12
Calculations
Initial structures for quantum-mechanical calculations were built and optimized using PCMODEL (version 8.5).18 The MMX force field was used for energy evaluations.18,19 Relaxed grid search (RGS) analysis18 was carried out for each conformer considered using PCMODEL. RGS is a systematic method, which involves creation of a large number of starting configurations and mapping out the shape of the potential energy surface. In this method the rotatable bonds of interest are first identified. The calculation starts by evaluating the energy when all the rotatable bonds are set to 180°. The bonds are then rotated sequentially and all the structures are minimized and sorted based on their total energy, with any duplicate configurations removed. Since the total number of energy evaluations can be very large (usually several hundreds or thousands depending on the number of rotatable bonds), the energies of conformers were calculated using molecular mechanics method and the MMX force field.In some cases, the RGS derived structures were further optimized via semi-empirical PM620 calculations using Gaussian 09. The reaction field method IEFPCM21 was used to account for water solvent effects in PM6 calculations.
All quantum mechanical calculations were carried out using Gaussian 09.22 For geometry optimizations using density functional theory (DFT), the M06-2X23 functional with def2-TZVP basis set was used.24 The performance of M06-2X functional has been compared extensively to other DFT methods and MP2.23,25 Its superior performance has been illustrated in a comprehensive review article by Zhao and Truhlar,25 in which they have included comparisons of M06-2X with SCS-MP2 and B2PLYP-D. The choice of the def2-TZVP basis set is dictated primarily by the presence of sulfur and phosphorus atoms in tagetitoxin. At the DFT level the def2-TZVP basis set has been shown to produce results that are not too far from the DFT basis set limit.24 For optimization of structure parameters, the def2-TZVP errors in bond lengths are typically smaller than 1 pm and that in bond angles are smaller than 1°.24 The ultrafine numerical integration grid (with 99 radial shells and 590 angular points per shell) was used in our M06-2X/def2-TZVP geometry optimisations, combined with the “verytight” convergence condition (requesting the root-mean-square forces to be smaller than 1 × 10−6 Hartree Bohr−1). Additional frequency calculations were also undertaken in order to verify that the optimized geometries correspond to true minima. The reaction field method IEFPCM21 was used to account for water solvent effects. NMR chemical shieldings and J couplings were computed at the B3LYP/6-311+G(2d,p) level using the GIAO method.26 Water solvent effects were used in all the quantum mechanical calculations using the reaction field method IEFPCM.21
Conformationally averaged interatomic distances from the QM calculations were determined in a way similar to that used in NMR measurements: (i) internuclear distances (ri) for pairs of hydrogen atoms were measured in each conformer i; (ii) a quantity equal to ri−6 was calculated as a measure of the expected NOE in each conformer, ηi; (iii) the sum of piri−6 was calculated, where values of populations pi were derived from the analysis of experimental long range JCH couplings using their QM-predicted boundary values in each conformer i; (iv) using r = 1.77 Å as the reference H2–H2′ distance for geminal protons, internuclear distances for other proton pairs were calculated using the η ∼ r−6 relationship.13
Acknowledgements
We thank University College London (UCL) for the provision of computational facilities. The authors acknowledge the use of the UCL Legion High Performance Computing Facility (Legion@UCL), and associated support services, in the completion of this work.References
- R. E. Mitchell and R. D. Durbin, Physiol. Plant Pathol., 1981, 18, 157–168 CrossRef CAS.
- D. E. Mathews and R. D. Durbin, J. Biol. Chem., 1990, 265, 493–498 CAS.
- (a) T. H. Steinberg, D. E. Mathews, R. D. Durbin and R. R. Burgess, J. Biol. Chem., 1990, 265, 499–505 CAS; (b) L. P. Wu, J. Pan, V. Thoroddsen, D. R. Wysong, R. K. Blackman, C. E. Bulawa, A. E. Gould, T. D. Ocain, L. R. Dick, P. Errada, P. K. Dorr, T. Parkinson, T. Wood, D. Kornitzer, Z. Weissman, I. M. Willis and K. McGovern, Eukaryotic Cell, 2003, 2, 256–264 CrossRef CAS PubMed.
- Y. Yuzenkova, M. Roghanian, A. Bochkareva and N. Zenkin, Nucleic Acids Res., 2013, 41, 9257–9265 CrossRef CAS PubMed.
- R. E. Mitchell and P. A. Hart, Phytochemistry, 1983, 22, 1425–1428 CrossRef CAS.
- R. E. Mitchell, J. M. Coddington and H. Young, Tetrahedron Lett., 1989, 30, 501–504 CrossRef CAS.
- D. G. Vassylyev, V. Svetlov, M. N. Vassylyeva, A. Perederina, N. Igarashi, N. Matsugaki, S. Wakatsuki and I. Artsimovitch, Nat. Struct. Mol. Biol., 2005, 12, 1086–1093 CAS.
- J. W. Gronwald, K. L. Plaisance, S. Marimanikkuppam and B. G. Ostrowski, Physiol. Mol. Plant Pathol., 2005, 67, 23–32 CrossRef CAS.
- (a) T. Sammakia, T. B. Hurley, D. M. Sammond, R. S. Smith, S. B. Sobolov and T. R. Oeschger, Tetrahedron Lett., 1996, 37, 4427–4430 CrossRef CAS; (b) B. R. Dent, R. H. Furneaux, G. J. Gainsford and G. P. Lynch, Tetrahedron, 1999, 55, 6977–6996 CrossRef CAS; (c) M. Ioannou, M. J. Porter and F. Saez, Chem. Commun., 2002, 346–347 RSC; (d) M. Ioannou, M. J. Porter and F. Saez, Tetrahedron, 2005, 61, 43–50 CrossRef CAS; (e) J. R. H. Plet and M. J. Porter, Chem. Commun., 2006, 1197–1199 RSC; (f) A. J. P. Mortimer, A. E. Aliev, D. A. Tocher and M. J. Porter, Org. Lett., 2008, 10, 5477–5480 CrossRef CAS PubMed; (g) J. R. H. Plet, A. K. Sandhu, M. Sehailia and M. J. Porter, Synlett, 2009, 3258–3262 CAS; (h) A. J. P. Mortimer, J. R. H. Plet, O. A. Obasanjo, N. Kaltsoyannis and M. J. Porter, Org. Biomol. Chem., 2012, 10, 8616–8627 RSC; (i) H. Yamada, M. Adachi and T. Nishikawa, Chem. Commun., 2013, 49, 11221–11223 RSC.
- (a) A. E. Aliev and D. Courtier-Murias, J. Phys. Chem. B, 2007, 111, 14034–14042 CrossRef CAS PubMed; (b) A. E. Aliev, Z. A. Mia, M. J. Busson, R. J. Fitzmaurice and S. Caddick, J. Org. Chem., 2012, 77, 6290–6295 CrossRef CAS PubMed; (c) A. E. Aliev and D. Courtier-Murias, J. Phys. Chem. B, 2010, 114, 12358–12375 CrossRef CAS PubMed.
- W. Koźmiński and D. Nanz, J. Magn. Reson., 1997, 124, 383–392 CrossRef.
- C. M. Thiele, K. Petzold and J. Schleucher, Chem. – Eur. J., 2009, 15, 585–588 CrossRef CAS PubMed.
- (a) D. Neuhaus and M. P. Williamson, The Nuclear Overhauser Effect in Structural and Conformational Analysis, Wiley-VCH, New York, 2nd edn, 2000 Search PubMed; (b) T. Claridge, High-Resolution NMR Techniques in Organic Chemistry, in Tetrahedron Organic Chemistry Series, Pergamon Press, Oxford, 1999, vol. 19 Search PubMed; (c) M. Reggelin, H. Hoffman, M. Köck and D. F. Mierke, J. Am. Chem. Soc., 1992, 114, 3272–3277 CrossRef CAS.
- A. E. Aliev, Z. A. Mia, H. S. Khaneja and F. D. King, J. Phys. Chem. A, 2012, 116, 1093–1109 CrossRef CAS PubMed.
- (a) R. Jain, T. Bally and P. R. Rablen, J. Org. Chem., 2009, 74, 4017–4023 CrossRef CAS PubMed; (b) C. R. Mooney, M. E. Sanz, A. R. McKay, R. J. Fitzmaurice, A. E. Aliev, S. Caddick and H. H. Fielding, J. Phys. Chem. A, 2012, 116, 7943–7949 CrossRef CAS PubMed.
- A. Meissner and O. W. Sørensen, Magn. Reson. Chem., 2001, 39, 49–52 CrossRef CAS.
- M. J. Thrippleton and J. Keeler, Angew. Chem., Int. Ed., 2003, 42, 3938–3941 CrossRef CAS PubMed.
- K. E. Gilbert, PCMODEL, version 8.5, Serena Software, Bloomington Search PubMed.
- M. F. Schlecht, Molecular Modeling on the PC, Wiley-VCH, New York, 1998 Search PubMed.
- J. J. P. Stewart, J. Mol. Model, 2007, 13, 1173–1213 CrossRef CAS PubMed.
- (a) E. Cances and B. Mennucci, J. Math. Chem., 1998, 23, 309–326 CrossRef CAS; (b) M. Cossi, N. Rega, G. Scalmani and V. Barone, J. Comput. Chem., 2003, 24, 669–681 CrossRef CAS PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr., J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Ö. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Revision D.01, Gaussian, Inc., Wallingford CT, 2009 Search PubMed.
- (a) Y. Zhao and D. G. Truhlar, Theor. Chem. Acc., 2008, 120, 215–241 CrossRef CAS; (b) Y. Zhao and D. G. Truhlar, J. Chem. Theory Comput., 2008, 4, 1849–1868 CrossRef CAS.
- F. Weigend and R. Ahlrichs, Phys. Chem. Chem. Phys., 2005, 7, 3297–3305 RSC.
- Y. Zhao and D. G. Truhlar, Chem. Phys. Lett., 2011, 502, 1–13 CrossRef CAS.
- J. R. Cheeseman, G. W. Trucks, T. A. Keith and M. J. Frisch, J. Chem. Phys., 1996, 104, 5497–5509 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5ob02076j |
| This journal is © The Royal Society of Chemistry 2016 |