
An unprecedented Pd-catalyzed decarboxylative coupling reaction of aromatic carboxylic acids in aqueous medium under air: synthesis of 3-aryl-imidazo[1,2-a]pyridines from aryl chlorides†
Bing
Mu
ab,
Yusheng
Wu
*acd,
Jingya
Li
ce,
Dapeng
Zou
a,
Junbiao
Chang
a and
Yangjie
Wu
a
aThe College of Chemistry and Molecular Engineering, Zhengzhou University, Zhengzhou, PR China. E-mail: yusheng.wu@tetranovglobal.com; Fax: +1 732 253 7327; Tel: +1 732 253 7326
bThe College of Chemistry and Chemical Engineering, Zhengzhou Normal University, Zhengzhou 450044, PR China
cCollaborative Innovation Center of New Drug Research and Safety Evaluation, Henan Province, China
dTetranov International, Inc, 100 Jersey Avenue, Suite A340, New Brunswick, NJ 08901, USA
eTetranov Biopharm, LLC, 75 Daxue Road, Zhengzhou, PR China
First published on 4th November 2015
Abstract
An efficient and practical protocol for palladium-catalyzed decarboxylative arylation of imidazo[1,2-a]pyridine-3-carboxylic acids with aryl chlorides has been developed. Note that the reaction could proceed smoothly without an additive in aqueous medium under an ambient atmosphere, and the addition of H2O could effectively promote the decarboxylative arylation. Particularly noteworthy is that these results represent the first examples of Pd-catalyzed decarboxylative coupling reactions of (hetero) aromatic carboxylic acids in aqueous medium under air, and the first successful examples of the synthesis of 3-aryl-imidazo[1,2-a]pyridines using cheap, diverse aryl chlorides and heteroaryl chlorides as the starting materials.
Introduction
Imidazo[1,2-a]pyridines are among the most important classes of organic compounds due to the abundance of the imidazo[1,2-a]pyridine structural motif in agrochemicals,1 functional materials,2 and pharmaceutical products.3 In particular, 3-arylimidazo[1,2-a]pyridines show excellent antiinflammatory,4 anticancer,5 antiprotozoal,6 antibacterial,7 antiulcer,8 antiviral,9 and antifungal activities.10 Owing to the attractive biological properties of 3-arylimidazo[1,2-a]pyridine derivatives, a variety of synthetic strategies have been developed for the construction of 3-arylimidazo[1,2-a]pyridine scaffolds. The traditional route to 3-arylimidazo[1,2-a]pyridines, such as the condensation between 2-aminopyridines and 2-bromo-1,2-diarylethanone,11–14 and the oxidative coupling of 2-aminopyridine with nitroalkenes15/alkynes16/chalcones,17 usually suffers from the need for prior preparation of starting materials and the limited substrate scope, thus restraining the applications of these methodologies. Additionally Suzuki-type cross-coupling between 3-halo-2-arylimidazo[1,2-a]pyridines and arylboronic acid is also an important strategy for the synthesis of these derivatives.18–22 However, the reaction usually needs the preactivation of organoboron nucleophiles and aryl iodides/aryl bromides that are often not commercially available and cause severe environmental problems. Very recently, a more streamlined and attractive strategy was established in which the reaction can proceed via C–H arylations of imidazo[1,2-a]pyridines at C-3,23 but these reactions are usually performed efficiently in dry organic solvents under a nitrogen atmosphere, and the substrate scope is mainly limited to aryl iodides/bromides or C2-substituted imidazo[1,2-a]pyridines.24–29In the past few years, a rapidly growing number of decarboxylative coupling reactions as a new synthetic strategy have been reported that give access to various valuable product classes,30–33 since carboxylic acids have distinguishing features of being a solid without pungent smell, non-toxic, low cost, readily available, and easy to store, transport and handle.34–37 However, Pd-catalyzed decarboxylative coupling reactions of (hetero) aromatic carboxylic acids in aqueous medium under air have not been reported, as far as we know. Therefore, Pd-catalyzed decarboxylative coupling reactions in aqueous medium would be challenging. To the best of our knowledge, the decarboxylative coupling of imidazo[1,2-a]pyridine-3-carboxylic acids with aryl halides was only reported by Lee for the syntheses of 3-arylimidazo[1,2-a]pyridine derivatives.38 The method suffers from significant moisture and air sensitivity, and the substrate scope is mainly limited to arylbromides.
Therefore, a catalyst system which allows the construction of C2-unsubstituted 3-arylimidazo[1,2-a]pyridine derivatives in aqueous medium under air, and makes the coupling partner more economical and practical is still in demand since such a catalyst system has the potential to be used in industrial processes. In continuation of our former studies on palladium-catalyzed decarboxylative cross-coupling reactions,39 herein, we attempt to develop a convenient and efficient protocol for the synthesis of 3-aryl-imidazo[1,2-a]pyridines in aqueous medium under air via palladium-catalyzed decarboxylative coupling of imidazo[1,2-a]pyridine-3-carboxylic acids with cheap aryl/heteroaryl chlorides.
Results and discussion
The decarboxylative cross-coupling reaction of imidazo[1,2-a]pyridine-3-carboxylic acid with chlorobenzene was chosen as model reaction to optimize the reaction conditions as shown in Table 1. To make our present methodology more convenient and practical, we initiated our studies by examining the influence of moisture and air on the conversion. First, we performed the reaction in dry DMA under nitrogen in the presence of 0.5 equiv. of Cu2O at 160 °C, and the desired product was obtained in 91% isolated yield (entry 1). With the same reaction conditions as above, only 52% GC yield was obtained in the absence of Cu2O (entry 2). Then, we carried out the reaction in DMA under air at 160 °C with Cu2O, and the desired coupling product was obtained in 61% yield (entry 3). However, the yield increased slightly without Cu2O in 70% yield, indicating that the addition of Cu2O is not necessary under air (entry 4). Lowering the reaction temperature to 150 °C led to poor substrate conversion (entry 4 vs. entry 5). Subsequently, the effects of different solvents were tested at 150 °C, and no improvement of the yield was observed (entries 6–8). To our delight, the yields significantly changed with the addition of H2O to these catalytic systems (entry 9). However, when the reaction was conducted in DMA/H2O (3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1), only trace amounts of product 3a were detected (entry 10). Obviously, DMA/H2O (40:1) as the solvent is the best choice for this reaction (entries 5–11). Then, a variety of bases were investigated, such as K2CO3, Na2CO3, KOAc, K3PO4·7H2O, Cs2CO3 and KOtBu (see Table S1 in the ESI†). K2CO3 was identified as the best base. Other ligands such as S-Phos, X-Phos, and Ru-Phos were also screened (Fig. 1), and obviously S-Phos was more effective than the other ligands in this reaction affording 96% isolated yield (entries 12–15). Finally, the replacement of Pd(OAc)2 with PdCl2, Pd2(dba)3, Pd(acac)2 or Pd(CF3CO2)2 leads to a drop in yield (see Table S1 in the ESI†).
:1), only trace amounts of product 3a were detected (entry 10). Obviously, DMA/H2O (40:1) as the solvent is the best choice for this reaction (entries 5–11). Then, a variety of bases were investigated, such as K2CO3, Na2CO3, KOAc, K3PO4·7H2O, Cs2CO3 and KOtBu (see Table S1 in the ESI†). K2CO3 was identified as the best base. Other ligands such as S-Phos, X-Phos, and Ru-Phos were also screened (Fig. 1), and obviously S-Phos was more effective than the other ligands in this reaction affording 96% isolated yield (entries 12–15). Finally, the replacement of Pd(OAc)2 with PdCl2, Pd2(dba)3, Pd(acac)2 or Pd(CF3CO2)2 leads to a drop in yield (see Table S1 in the ESI†).
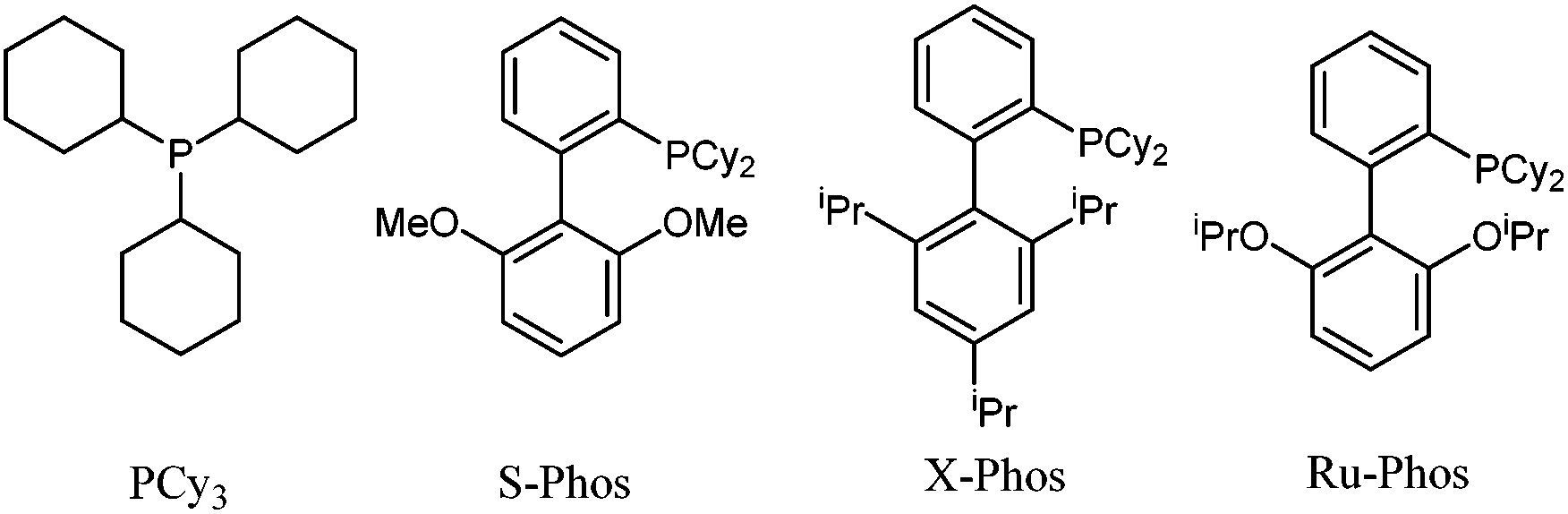 | ||
| Fig. 1 Various ligands used in Pd-catalyzed decarboxylative arylation. | ||
|
|
||||
|---|---|---|---|---|
| Entry | Ligand | Solvent | Temp (°C) | Yield b(%) |
| a Reaction conditions: 0.3 mmol of imidazo[1,2-a]pyridine-3-carboxylic acid, 0.9 mmol of chlorobenzene, 0.9 mmol of K2CO3, 5 mol% of Pd(OAc)2, 6 mol% of ligand, and 4 mL of solvent under air for 24 h. b GC yields. c Isolated yields. d 0.15 mmol of Cu2O. e 4 mL of dry DMA, 200 mg of 4 Å molecular sieves, under nitrogen. f 7.5 mol% of ligand. | ||||
| 1d,e | PCy3 | DMA | 160 | 91c |
| 2e | PCy3 | DMA | 160 | 52 |
| 3d | PCy3 | DMA | 160 | 61 |
| 4 | PCy3 | DMA | 160 | 70 |
| 5 | PCy3 | DMA | 150 | 26 |
| 6 | PCy3 | DMF | 150 | 20 |
| 7 | PCy3 | DMSO | 150 | Trace |
| 8 | PCy3 | Dioxane | 110 | Trace |
| 9 | PCy3 | DMA/H2O (40/1) | 150 | 67 |
| 10 | PCy3 | DMA/H2O (3/1) | 150 | 5 |
| 11 | PCy3 | Xylene/H2O (1/1) | 120 | NR |
| 12 | S-Phos | DMA/H2O (40/1) | 150 | 81 |
| 13f | S-Phos | DMA/H2O (40/1) | 150 | 100 (96c) |
| 14f | X-Phos | DMA/H2O (40/1) | 150 | 59 |
| 15f | Ru-Phos | DMA/H2O (40/1) | 150 | 30 |
| 16f | S-Phos | DMA/H2O (40/1) | 140 | 11 |
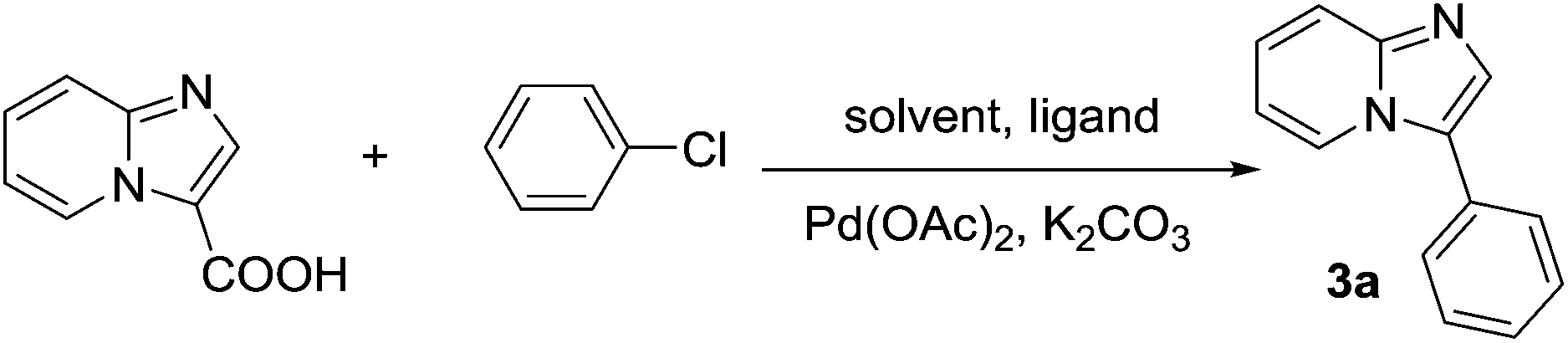
With the optimized conditions in hand, the scope of the coupling of aryl chlorides with imidazo[1,2-a]pyridine-3-carboxylic acid is summarized in Scheme 1. The para-substituted aryl chlorides such as 4-chlorotoluene, 4-chloroanisole or 4-chloroacetanilide gave the corresponding products in 94–96% yields (products 3b, 3e and 3g). Electron-deficient aryl chlorides such as 4-chloroacetophenone or 4-chloronitrobenzene gave the coupling product in 75 and 70% yields, respectively (products 3m and 3n). 4-Chloroaniline and 1-chloronaphthalene were also found to be suitable coupling partners, and gave the product in 59% and 66% yields, respectively (products 3k and 3l). For meta or ortho-substituted aryl chlorides such as 2-chloroanisole, 3-chloroacetanilide, 2-chloroacetanilide or 3-chloroaniline, products 3f, 3h–j were obtained in slightly lower yields of 53–73%. Notably, the sterically hindered 2-chlorotoluene and 3-chlorotoluene also proceeded smoothly, giving the desired arylation products 3d and 3c in 88% and 99% yields, respectively. Moreover, heteroaromatic chlorides such as 4-chloropyridine and 3-chloropyridine were also reactive partners in this coupling to give the product in 94 and 77% yields, respectively (products 3o and 3p). Subsequently, the scope of imidazo[1,2-a]pyridine-3-carboxylic acids was also explored, and the results are summarized in Scheme 1. For example, 2-methyl, 2-phenyl and 2,8-dimethyl imidazo[1,2-a]pyridine-3-carboxylic acids could be efficiently converted into the corresponding products in good yields (products 3r–3t). However, 6-fluoroimidazo[1,2-a]pyridine-3-carboxylic acid had a relatively low reactivity, only affording the product in moderate yield (product 3u). To demonstrate the scalability of the reaction, we chose the decarboxylative reaction of imidazo[1,2-a]pyridine-3-carboxylic acid with chlorobenzene on a 1.0 g scale. The desired product was isolated in 85% yield (product 3a).
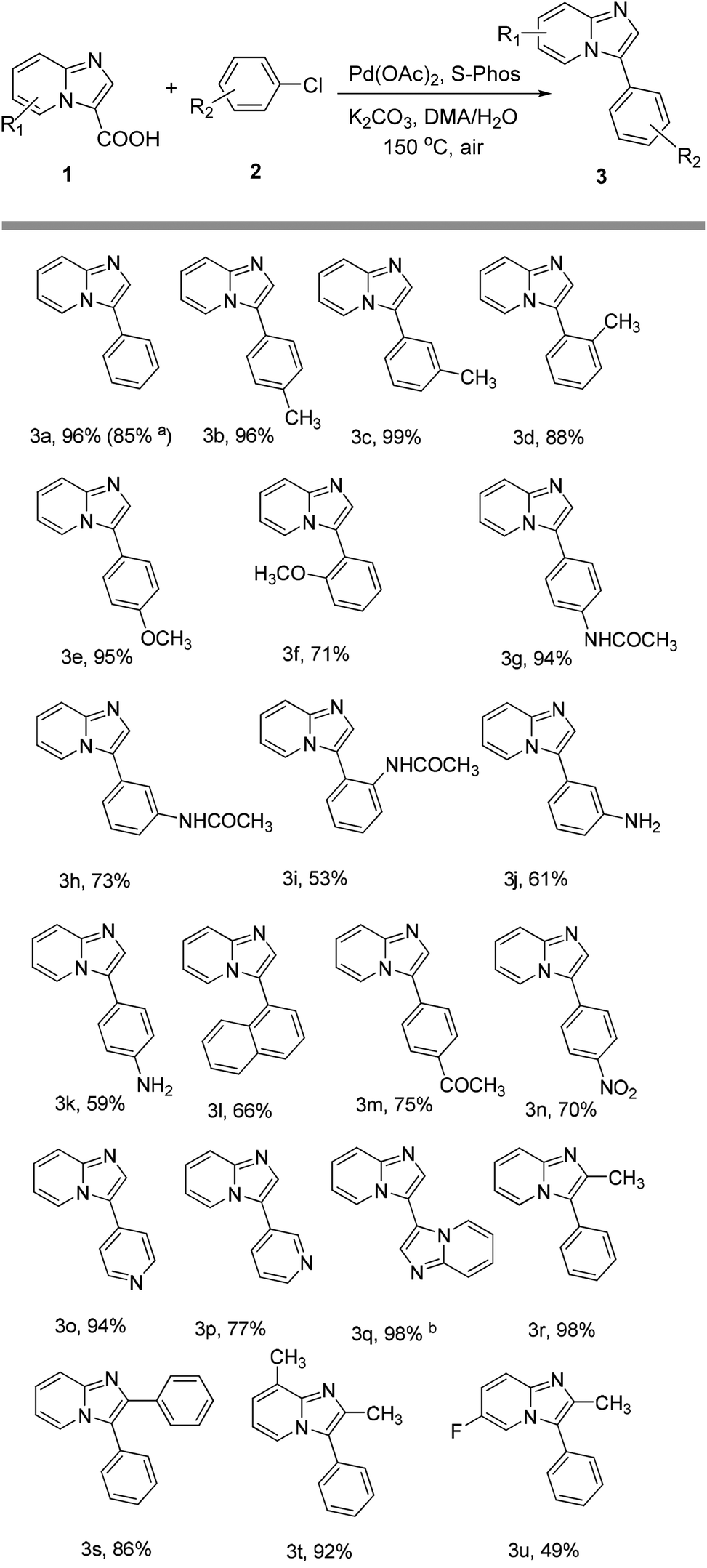 | ||
| Scheme 1 Decarboxylative arylation of imidazo[1,2-a]pyridine-3-carboxylic acids with aryl chlorides. Reaction conditions: 0.3 mmol of imidazo[1,2-a]pyridine-3-carboxylic acids, 0.9 mmol of aryl chlorides, 0.9 mmol of K2CO3, 5 mol% of Pd(OAc)2, 7.5 mol% of S-Phos, 4 mL of DMA, and 0.1 mL of H2O at 150 °C under air for 24 h. Isolated yields. a6.2 mmol (1.0 g) of imidazo[1,2-a]pyridine-3-carboxylic acid. bWithout aryl chlorides. | ||
Finally, the investigation was aimed at revealing the decarboxylative arylation of imidazo[1,2-a]pyridine-3-carboxylic acid as shown in Scheme 2. With the optimized conditions, the decarboxylative arylation of imidazo[1,2-a]pyridine-3-carboxylic acid with chlorobenzene gave the product in 96% yield, but the direct arylation of imidazo[1,2-a]pyridine with chlorobenzene was still very sluggish, affording only a 34% GC yield of the arylation product. Under the same conditions without added aryl chlorides, imidazo[1,2-a]pyridine from hydrodecarboxylation of imidazo[1,2-a]pyridine-3-carboxylic acid was also not detected for 2 h, and decarboxylative homocoupling of imidazo[1,2-a]pyridine-3-carboxylic acid was performed in 98% yield for 24 h. On the basis of the previous reports,38 the decarboxylative arylation of imidazo[1,2-a]pyridine-3-carboxylic acid with bromobenzene gave the product in 73% yield for 24 h, and without added aryl chlorides imidazo[1,2-a]pyridine from hydrodecarboxylation of imidazo[1,2-a]pyridine-3-carboxylic acid was afforded in a high yield of 99% for 2 h. The results indicated that the arylation products of imidazo[1,2-a]pyridine-3-carboxylic acid were not generated by the hydrodecarboxylation in our catalytic system.
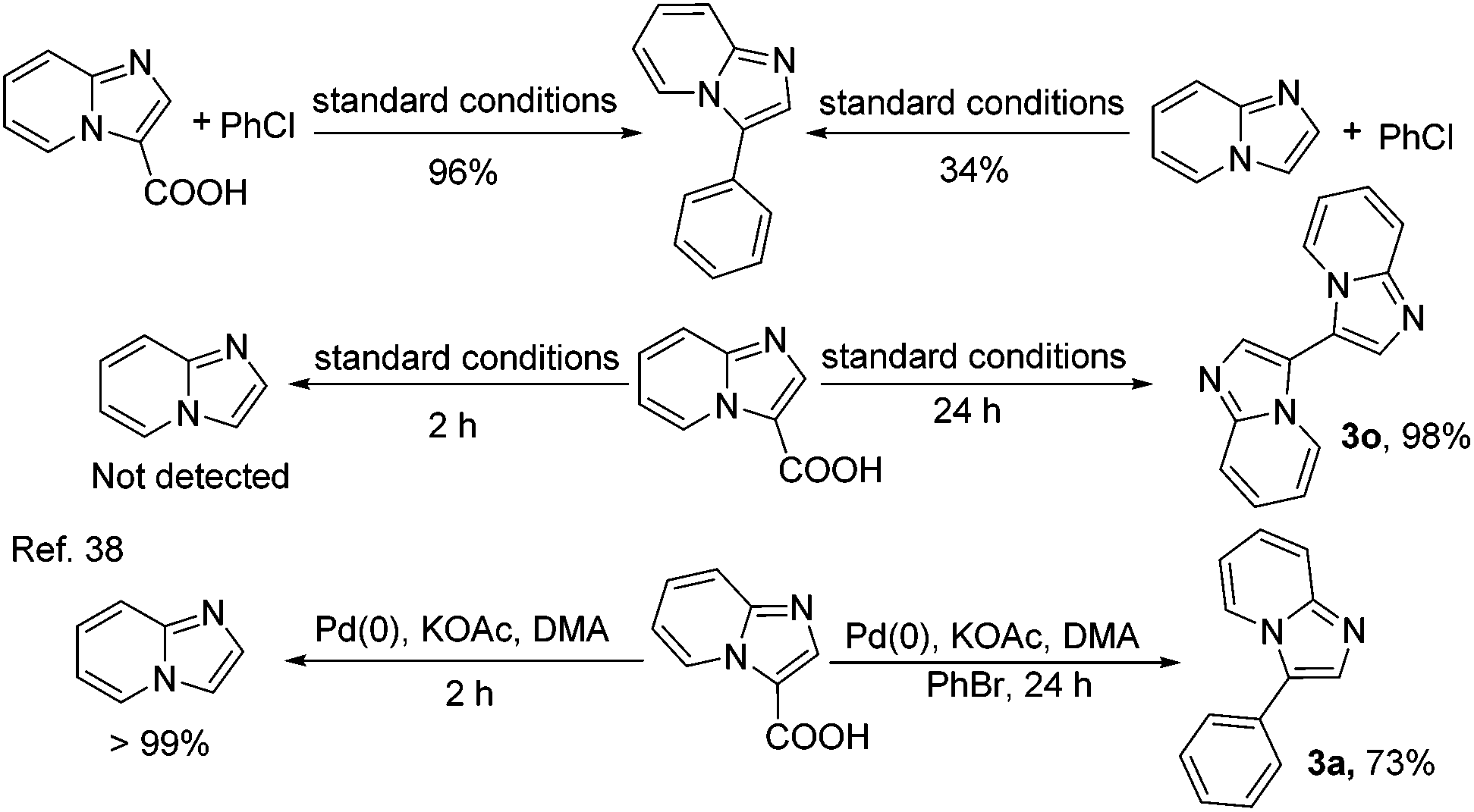 | ||
| Scheme 2 Comparative experiments under different reaction conditions. | ||
On the basis of the above-mentioned results, a possible mechanism of palladium-catalyzed decarboxylative arylation is outlined in Scheme 3. The first step would be the release of Pd(0) species from Pd(OAc)2, which is possibly through the ligand-exchange of imidazo[1,2-a]pyridine-3-carboxylic acid with the acetic acid anion followed by the decarboxylative reaction and reductive elimination to afford the homocoupling product and the catalytically active Pd(0)L2 (I) with the assistance of the phosphine ligand. Then, the oxidative addition of aryl chloride (2) to Pd(0)L2 would take place to form the intermediate II. Subsequently, the ligand-exchange reaction between intermediate II and the carboxylic acid anion which was generated by the reaction of carboxylic acid and K2CO3 could occur to afford intermediate III. After the intermediate III underwent the decarboxylative reaction to release one molecular CO2 to form the intermediate IV, the reductive elimination of the intermediate IV would afford the corresponding products (3) and regenerate the catalytically active catalyst Pd(0)L2 to fulfill the catalytic cycle.
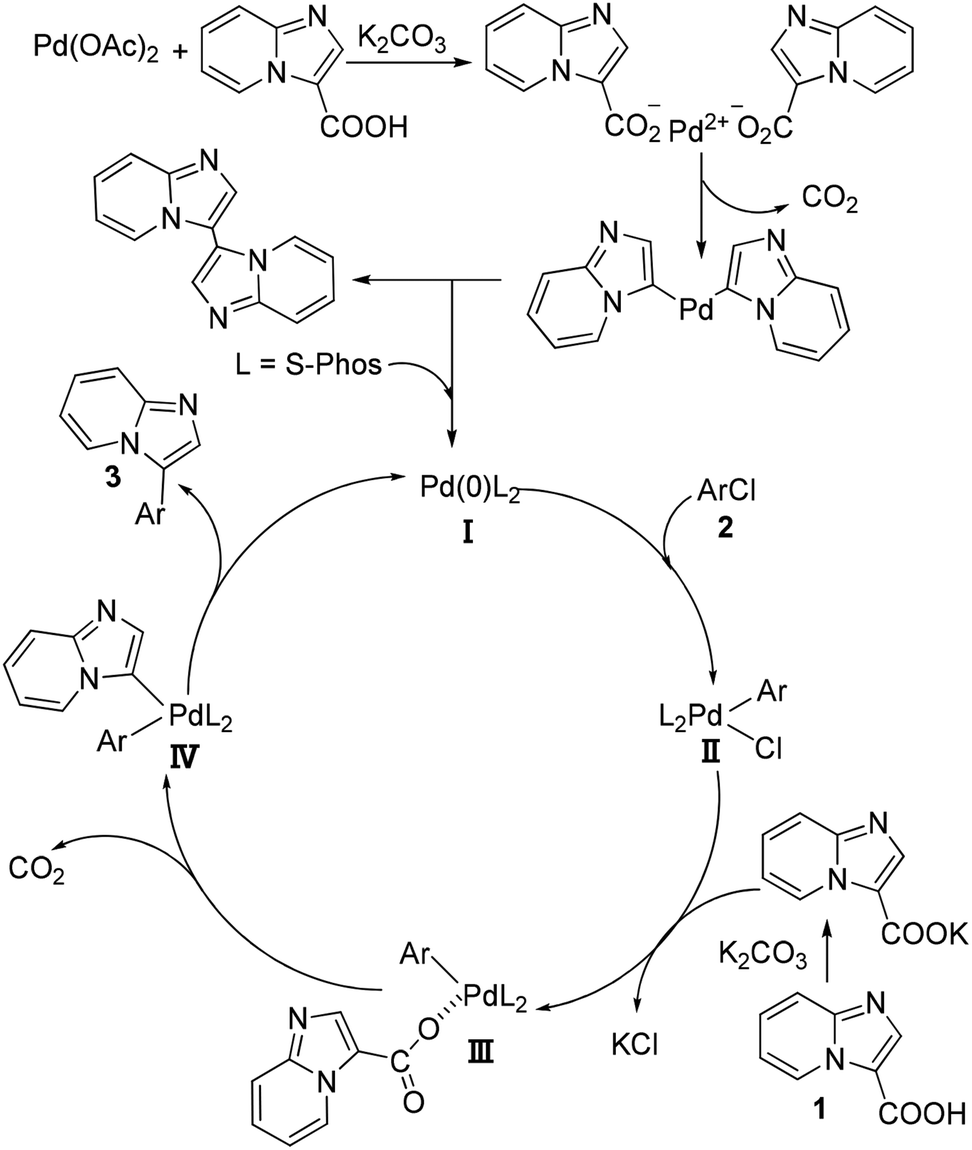 | ||
| Scheme 3 Proposed mechanism of palladium-catalyzed decarboxylative arylation. | ||
Experimental
General procedure for palladium-catalyzed decarboxylative arylation of imidazo[1,2-a]pyridine-3-carboxylic acid with aryl chlorides
Imidazo[1,2-a]pyridine-3-carboxylic acid (0.3 mmol), aryl chloride (0.9 mmol, 3 equiv.), K2CO3 (0.9 mmol, 3 equiv.), Pd(OAc)2 (5 mol%) and S-Phos (7.5 mol%) were added to a 10 mL round-bottomed flask, and then a mixed solvent of 4.0 mL of DMA and 0.1 mL of H2O was added. The mixture was stirred at 150 °C for 24 h under air. After the reaction was complete, the mixture was washed with water and extracted with ethyl acetate three times. The combined organic layer was dried with anhydrous MgSO4 and filtered. The filtrate was concentrated in vacuo. The crude product was purified by flash chromatography on silica gel using ethyl acetate or hexane/ethyl acetate as the eluent to give the pure product. The products were characterized by 1H NMR, 13C NMR, m.p., MS and HRMS.Conclusions
In conclusion, we have developed an efficient and facile protocol for the synthesis of 3-aryl-imidazo[1,2-a]pyridines via palladium-catalyzed decarboxylative arylation of imidazo[1,2-a]pyridine-3-carboxylic acids with aryl chlorides. It is worth noting that the reaction proceeded smoothly without an additive in aqueous medium under an ambient atmosphere, and the scope of the substrate could be extended to electron-poor, electron-neutral, electron-rich, even sterically hindered aryl chlorides and aromatic heterocyclic chlorides. Remarkably, the decarboxylative arylation was quite effectively promoted by the addition of H2O. This economical and practical synthetic protocol for 3-arylimidazo[1,2-a]pyridines may have wide applications to industrial processes in the future.Acknowledgements
We are grateful to the Research Program of Fundamental and Advanced Technology of Henan Province (122300413203), Technology Research and Development Funds of Zhengzhou (141PRCYY516), the Postdoctoral Science Foundation of Henan Province (2014003), and the Science and Technology Foundation of Zhengzhou Science and Technology Bureau (131PPTGC419-3) for financial support.References
- U. Egner, K. P. Gerbling, G. A. Hoyer, G. Krüger and P. Wegner, Pestic. Sci., 1996, 47, 145 CrossRef CAS.
- J. S. Bae, D. W. Lee, D. H. Lee and D. S. Jeong, PCT Int. Appl, WO2007011163A1, 2007 Search PubMed.
- G. C. Moraski, L. D. Markley, P. A. Hipskind, H. Boshoff, S. Cho, S. G. Franzblau and M. J. Miller, ACS Med. Chem. Lett., 2011, 2, 466 CrossRef CAS PubMed.
- R. B. Lacerda, C. K. F. de Lima, L. L. da Silva, N. C. Romeiro, A. L. P. Miranda, E. J. Barreiro and C. A. M. Fraga, Bioorg. Med. Chem., 2009, 17, 74 CrossRef CAS PubMed.
- W. M. El-Sayed, W. A. Hussin, Y. S. Al-Faiyz and M. A. Ismail, Eur. J. Pharmacol., 2013, 715, 212 CrossRef CAS PubMed.
- M. A. Ismail, R. K. Arafa, T. Wenzler, R. Brun, F. A. Tanious, W. D. Wilson and D. W. Boykin, Bioorg. Med. Chem., 2008, 16, 683 CrossRef CAS PubMed.
- N. M. Shukla, D. B. Salunke, E. Yoo, C. A. Mutz, R. Balakrishna and S. A. David, Bioorg. Med. Chem., 2012, 20, 5850 CrossRef CAS.
- J. J. Kaminski and A. M. Doweyko, J. Med. Chem., 1997, 40, 427 CrossRef CAS PubMed.
- J. B. Véron, H. Allouchi, C. Enguehard-Gueiffier, R. Snoeck, G. Andrei, E. De Clercq and A. Gueiffier, Bioorg. Med. Chem., 2008, 16, 9536 CrossRef PubMed.
- Y. Rival, G. Grassy, A. Taudon and R. Ecalle, Eur. J. Med. Chem., 1991, 26, 13 CrossRef CAS.
- C. Enguehard-Gueiffier, S. Musiu, N. Henry, J. B. Véron, S. Mavel, J. Neyts, P. Leyssen, J. Paeshuyse and A. Gueiffier, Eur. J. Med. Chem., 2013, 64, 448 CrossRef CAS PubMed.
- J. G. Kettle, S. Brown, C. Crafter, B. R. Davies, P. Dudley, G. Fairley, P. Faulder, S. Fillery, H. Greenwood, J. Hawkins, M. James, K. Johnson, C. D. Lane, M. Pass, J. H. Pink, H. Plant and S. C. Cosulich, J. Med. Chem., 2012, 55, 1261 CrossRef CAS PubMed.
- L. Yin, J. Liebscher and F. Erdmann, J. Heterocycl. Chem., 2005, 42, 1369 CrossRef CAS.
- H. S. Patel, J. A. Linn, D. H. Drewry, D. A. Hillesheim, W. J. Zuercher and W. J. Hoekstra, Tetrahedron Lett., 2003, 44, 4077 CrossRef CAS.
- K. Monir, A. K. Bagdi, M. Ghosh and A. Hajra, Org. Lett., 2014, 16, 4630 CrossRef CAS.
- J. Zeng, Y. J. Tan, M. L. Leow and X. W. Liu, Org. Lett., 2012, 14, 4386 CrossRef CAS PubMed.
- K. Monir, A. K. Bagdi, S. Mishra, A. Majee and A. Hajra, Adv. Synth. Catal., 2014, 356, 1105 CrossRef CAS.
- S. C. Goodacre, L. J. Street, D. J. Hallett, J. M. Crawforth, S. Kelly, A. P. Owens, W. P. Blackaby, R. T. Lewis, J. Stanley, A. J. Smith, P. Ferris, B. Sohal, S. M. Cook, A. Pike, N. Brown, K. A. Wafford, G. Marshall, J. L. Castro and J. R. Atack, J. Med. Chem., 2006, 49, 35 CrossRef CAS.
- C. Enguehard, J. L. Renou, V. Collot, M. Hervet, S. Rault and A. Gueiffier, J. Org. Chem., 2000, 65, 6572 CrossRef CAS PubMed.
- J. M. Crawforth, S. C. Goodacre, D. J. Hallett, T. Harrison, A. P. Owens, M. Rowley and M. R. Teall, PCT Int. Appl, WO2001038326A2, 2001 Search PubMed.
- M. T. Bilodeau, M. E. Fraley and Z. Wu. PCT Int. Appl, WO2003092595A2, 2003 Search PubMed.
- N. Masuda, S. Miyamoto, S. Kikuchi, K. Samizu, F. Sato, Y. Shiina, W. Hamaguchi, R. Seo and T. Mihara, PCT Int. Appl, WO2012133607A1, 2012 Search PubMed.
- J. Yamaguchi, A. D. Yamaguchi and K. Itami, Angew. Chem., Int. Ed., 2012, 51, 8960 CrossRef CAS PubMed.
- J. Lee, J. Chung, S. M. Byun, B. M. Kim and C. Lee, Tetrahedron, 2013, 69, 5660 CrossRef CAS.
- H. Y. Fu, L. Chen and H. Doucet, J. Org. Chem., 2012, 77, 4473 CrossRef CAS PubMed.
- H. Cao, H. Y. Zhan, Y. G. Lin, X. L. Lin, Z. D. Du and H. F. Jiang, Org. Lett., 2012, 14, 1688 CrossRef CAS PubMed.
- S. Marhadour, M. A. Bazin and P. Marchand, Tetrahedron Lett., 2012, 53, 297 CrossRef CAS.
- H. Cao, Y. G. Lin, H. Y. Zhan, Z. D. Du, X. L. Lin, Q. M. Liang and H. Zhang, RSC Adv., 2012, 2, 5972 RSC.
- P. Y. Choy, K. C. Luk, Y. N. Wu, C. M. So, L. L. Wang and F. Y. Kwong, J. Org. Chem., 2015, 80, 1457 CrossRef CAS PubMed.
- L. J. Goossen, F. Collet and K. Goossen, Isr. J. Chem., 2010, 50, 617 CrossRef CAS.
- K. Park and S. Lee, RSC Adv., 2013, 3, 14165 RSC.
- N. Rodríguez and L. J. Goossen, Chem. Soc. Rev., 2011, 40, 5030 RSC.
- F. Bilodeau, M. C. Brochu, N. Guimond, K. H. Thesen and P. Forgione, J. Org. Chem., 2010, 75, 1550 CrossRef CAS PubMed.
- D. Yu and Y. Zhang, Green Chem., 2011, 13, 1275 RSC.
- X. Zhang, W. Z. Zhang, X. Ren, L. L. Zhang and X. B. Lu, Org. Lett., 2011, 13, 2402 CrossRef CAS PubMed.
- L. J. Gooßen, N. Rodríguez, F. Manjolinho and P. P. Lange, Adv. Synth. Catal., 2010, 352, 2913 CrossRef.
- D. Yu, M. X. Tan and Y. Zhang, Adv. Synth. Catal., 2012, 354, 969 CrossRef CAS.
- D. Nandi, Y. M. Jhou, J. Y. Lee, B. C. Kuo, C. Y. Liu, P. W. Huang and H. M. Lee, J. Org. Chem., 2012, 77, 9384 CrossRef CAS PubMed.
- X. J. Li, D. P. Zou, F. Q. Leng, C. X. Sun, J. Y. Li, Y. J. Wu and Y. S. Wu, Chem. Commun., 2013, 49, 312 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5ob02112j |
| This journal is © The Royal Society of Chemistry 2016 |