
Direct synthesis of C-glycosides from unprotected 2-N-acyl-aldohexoses via aldol condensation–oxa-Michael reactions with unactivated ketones†
Sherida
Johnson
and
Fujie
Tanaka
*
Chemistry and Chemical Bioengineering Unit, Okinawa Institute of Science and Technology Graduate University, 1919-1 Tancha, Onna, Okinawa 904-0495, Japan. E-mail: ftanaka@oist.jp
First published on 2nd November 2015
Abstract
C-glycosides are important compounds as they are used as bioactive molecules and building blocks. We have developed methods to concisely synthesize C-glycosides from unprotected 2-N-acyl-aldohexoses and unactivated ketones; we designed aldol-condensation–oxa-Michael addition reactions catalyzed by amine-based catalysts using additives. Depending on the conditions used, C-glycosides were stereoselectively obtained. Our methods allowed the C–C bond formations at the anomeric centers of unprotected carbohydrates under mild conditions to lead the C-glycosides in atom- and step-economical ways.
Introduction
Methods for the C–C bond formation at the anomeric centers of unprotected carbohydrates are necessary to provide concise access to C-glycosides, glycoconjugates, carbon chain-elongated carbohydrates, and related compounds.1–3 These molecules are used as therapeutics, bioactives, bioactive candidates, probes, synthons, and building blocks.1–3 Some pioneering chemical (i.e., non-enzymatic) methods for the C–C bond formation at the anomeric carbons or hemiacetal carbons of unprotected carbohydrates have been reported; these include reactions of unprotected carbohydrates, directly or via in situ-formed imines/iminium ions, with nucleophiles.4,5 However, the reactions are relatively limited.4–6 The reactions of unprotected aldohexoses that form 6-membered hemiacetals have been less demonstrated.2,4–6 In particular, reactions of 2-N-acyl-substituted C6-carbohydrates (such as N-acetyl-D-mannosamine and N-acetyl-D-glucosamine) have rarely been included in the reported examples in spite of the importance of the products. Here we report direct C–C bond formation at the anomeric carbon of unprotected 2-N-acyl-C6-aldopyranoses with unactivated ketones to give C-glycosides (Scheme 1).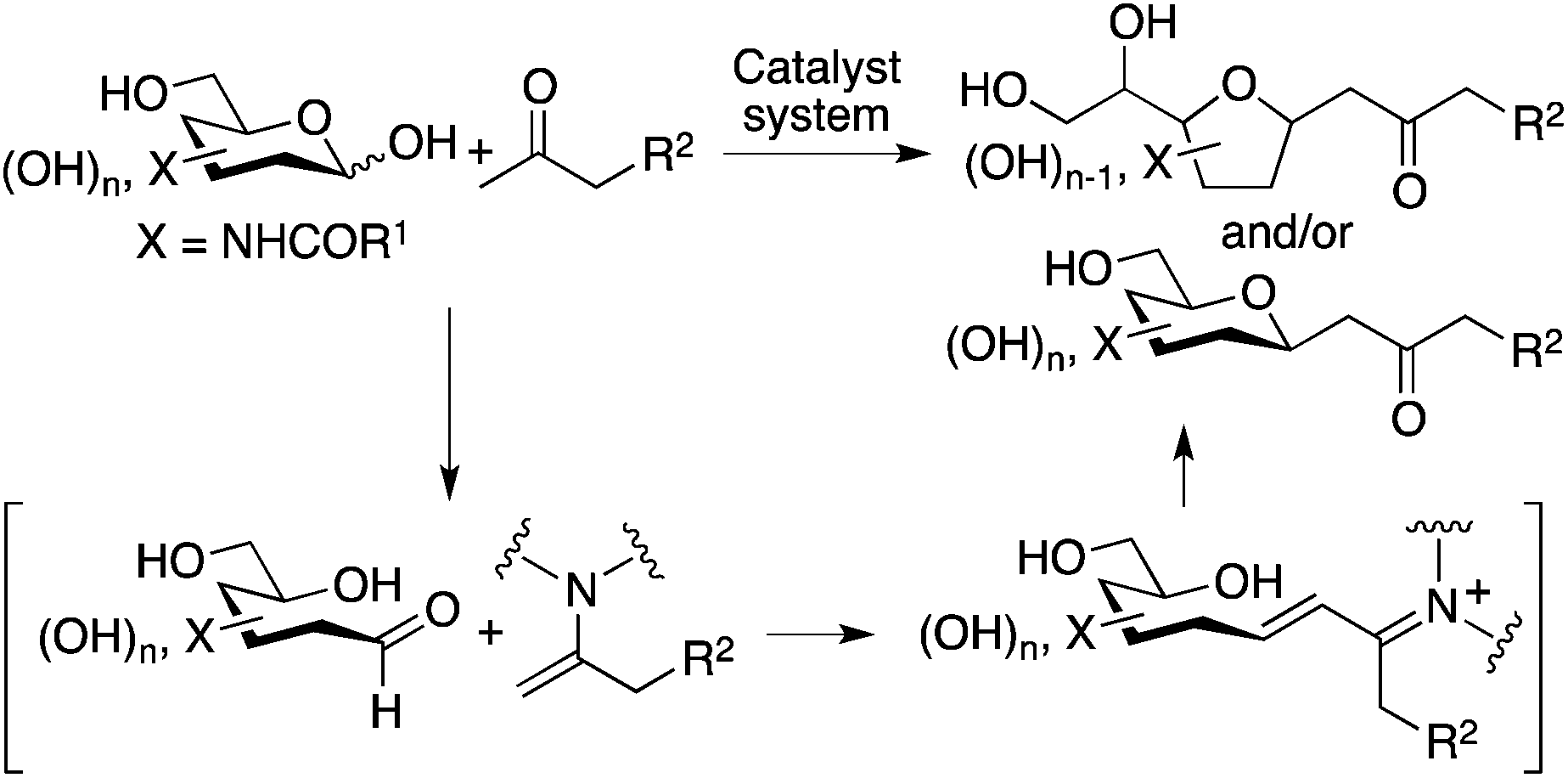 | ||
| Scheme 1 C–C bond forming reactions at the anomeric carbon of unmodified aldopyranoses to lead to C-glycosides. | ||
Our design to generate the C-glycosides involves aldol condensations of unprotected carbohydrates with simple ketones such as acetone, followed by oxa-Michael cyclization. To prepare this type of C-glycoside, reactions were previously performed with 1,3-diketones via Knoevenagel condensation followed by the elimination of the acyl group.5d–i There have been no reports of direct, non-enzymatic reactions of unprotected aldohexoses that form 6-membered cyclic hemiacetals, with unactivated ketones.5j Note that 6-membered hemiacetals are usually stable; usually no reactions on the anomeric centers proceed.7 To perform the reactions of unmodified 2-N-acyl-aldopyranoses with unactivated ketones, we designed strategies that use amine-based catalysts to generate enamines of the ketones and simultaneously that provide interactions for the carbohydrates to open the cyclic hemiacetals to generate the aldehyde groups, by mimicking enzyme strategies.
Results and discussion
We first investigated reactions of N-acetyl-D-mannosamine (1) with acetone. Before testing enzyme-mimicking strategies, we started to perform the reaction under commonly used enamine-based catalysis conditions. Selected reaction conditions tested and the results are shown in Table 1. When L-proline was used as the catalyst,8 product 2a was obtained although the yield was very low (entry 1). On the other hand, in the presence of D-proline as the catalyst, 2 was not obtained (entry 2). For these reactions, carbohydrate 1 mostly remained unreacted. The proline catalysis conditions,8 widely used for aldol reactions of aldehydes and acetone, did not efficiently catalyze the reactions of 1. When amines were used as additives to the L-proline-catalyzed reaction, depending on amine additive, reaction rates and product stereoselectivities varied (entries 3–6). We expected the amine additives to act as bases in the formation of the enamines with the proline. In addition, the amine additives may be protonated and act as acids. We also expected that addition of amines would make the reaction conditions more basic to promote the formation of the aldehyde from the hemiacetal.9 With N,N-diisopropylethylamine (DIPEA) additive, 5-membered C-glycoside 2a was obtained in reasonable yield (62%) with an excellent stereoselectivity (>10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) after 4 days (entry 6). The structure of C-glycoside 2a was confirmed by X-ray crystal structure analysis.10
:1) after 4 days (entry 6). The structure of C-glycoside 2a was confirmed by X-ray crystal structure analysis.10
|
|
|||||
|---|---|---|---|---|---|
| Entry | Catalyst | Additiveb | Time (h) | Yieldc (%) | Ratio 2a:2b |
| a Reaction conditions: N-acetyl-D-mannosamine (1) (0.4 mmol, 1.0 equiv.), acetone (0.6 mL, 8 mmol, 20 equiv.), catalyst (0.2 mmol, 0.5 equiv.), and amine additive (if added, 0.2 mmol, 0.5 equiv.) in DMSO (1.0 mL), at 25 °C, except noted. b Additives: EDA = ethylenediamine, TMEDA = tetramethylethylenediamine, DIPEA = N,N-diisopropylethylamine, compound 3 = cis-4-hydroxycyclohexanecarboxylic acid, compound 4 = trans-4-hydroxycyclohexanecarboxylic acid, DBU = 1,8-diazabicyclo[5.4.0]undec-7-ene. c Isolated yield; nd = formation of 2 was not detected. d Reaction at 40 °C. e DIPEA (0.6 mmol, 1.5 equiv.) and acid additive (0.4 mmol, 1.0 equiv.). f H3BO3 (0.4 mmol). | |||||
| 1 | L-Proline | None | 24 | 6 | >10:1 |
| 2 | D-Proline | None | 24 | nd | — |
| 3 | L-Proline | Et3N | 96 | 31 | 3:1 |
| 4 | L-Proline | EDA | 96 | 54 | 3:1 |
| 5 | L-Proline | TMEDA | 120 | 28 | 9:1 |
| 6 | L-Proline | DIPEA | 96 | 62 |
>10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/b_char_2009.gif) :1 :1
|
| 7 | D-Proline | DIPEA | 96 | nd | — |
| 8 | Pyrrolidine + CH3COOH | DIPEA | 96 | nd | — |
| 9 | None | DIPEA | 96 | nd | — |
| 10 | L-Proline | DIPEA | 48 | 27 | >10:1 |
| 11d | L-Proline | DIPEA | 48 | 82 | 1:1 |
| 12 | L-Proline | DIPEA + 3 | 48 | 44 |
>10:1
|
| 13e | D-Proline | DIPEA + 3 | 48 | nd | — |
| 14e | L-Proline | DIPEA + 4 | 48 | 12 | >10:1 |
| 15e | L-Proline | DIPEA + CH3COOH | 96 | 29 | 2:1 |
| 16e | L-Proline | DIPEA + H3BO3 | 48 | 29 | 1:>10 |
| 17 | L-Proline | DBU | 96 | 59 | 1:>10 |
| 18 | Pyrrolidine | H 3 BO 3 | 24 | 57 |
1:>10 |
| 19 | Pyrrolidine | DBU | 96 | 13 | — |
| 20 | Pyrrolidine | None | 24 | nd | — |
| 21f | None | H3BO3 | 24 | nd | — |
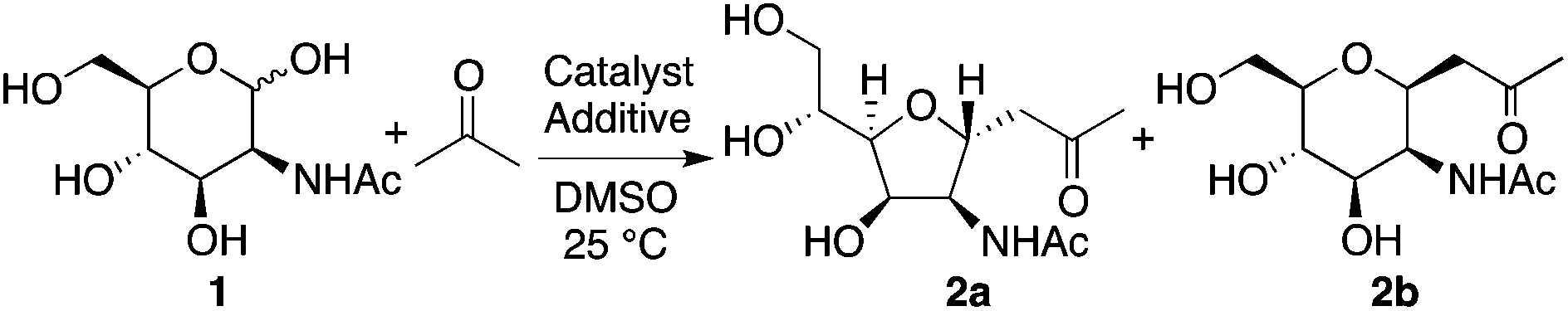
Both the L-proline and DIPEA are essential for the catalysis: use of D-proline or pyrrolidine-acetic acid instead of L-proline or no pyrrolidine-based catalyst in the presence of DIPEA did not give the aldol product (entries 7–9). Whereas the L-proline catalyzed reaction with DIPEA afforded 2a, the reaction needed 4 days to give the product in a reasonable yield; the reaction for 48 h only gave the product in a low yield (entry 10). Heating at 40 °C resulted in faster reaction than the reaction at 25 °C, but the stereoselectivity was eroded (entry 11).
It has been suggested that an enzyme that catalyzes the aldol reaction of 1 (and the retro-aldol reaction of N-acetylneuraminic acid) binds the aldehyde form of 1 using the carboxylate of a glutamic acid residue and a threonine hydroxy group during the catalysis (Scheme 2a).11 Although the complex structure has been discussed as a result of the retro-aldol reaction and has not been explained for the corresponding aldol reaction, we interpret that the aldehyde-bound crystal structure suggests that formation of the aldehyde form is one of the factors for accelerating the aldol reaction by the enzyme. Based on this consideration, we tested additives to provide similar interactions to those observed in the enzyme complex for increasing the formation of the aldehyde to accelerate the reaction (Scheme 2b). The reaction using L-proline as the catalyst in the presence of DIPEA and cis-4-hydroxycyclohexanecarboxylic acid (3) gave product 2a in reasonable yield in shorter reaction times than that in the absence of additive 3 (entry 12 versus entry 10). The reaction in the presence of L-proline, DIPEA, and acid 3 was faster at the initial stage of the reaction than the reaction in the presence of only L-proline and DIPEA (entry 12 versus entry 10). The high stereoselectivity to give 2a was retained in the reaction. In contrast, the use of trans-4-hydroxycyclohexanecarboxylic acid (4) or acetic acid instead of 3 was not effective to give 2 (entry 12 versus entries 14 and 15). These results indicate that compound 3 provides specific interactions with 1 to enhance the reaction, although the exact role of 3 needs to be determined.
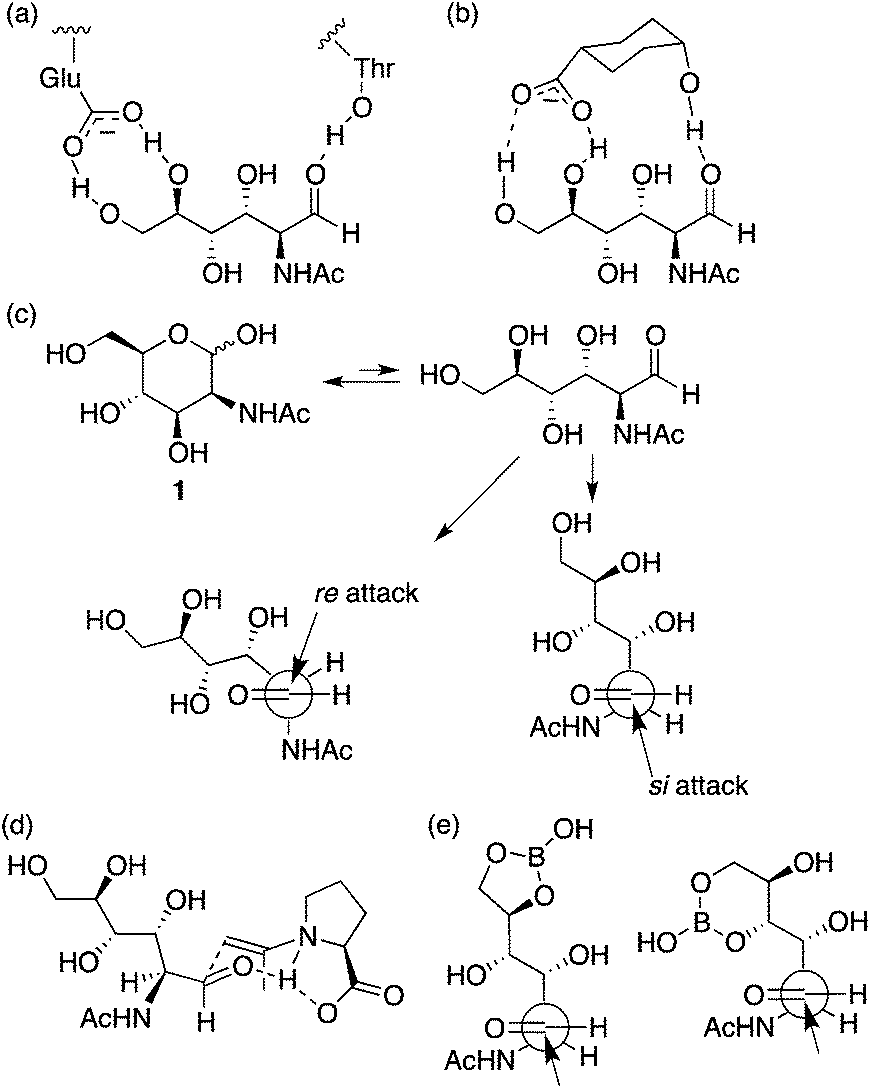 | ||
| Scheme 2 (a) A schematic representation of interactions, observed in an X-ray crystal structure, between the aldehyde form of N-acetyl-D-mannosamine (1) and enzyme N-acetylneuraminic acid lyase (a mutant with low catalytic activity to obtain the ligand complex) that catalyzes reversible condensation between pyruvates and 1.11 (b) A proposed interactions between the aldehyde form of 1 and additive acid 3. (c) Re- and si-attacks on the aldehyde generated from carbohydrate 1. (d) A plausible transition state catalyzed by L-proline for the C–C bond forming step, in which the enamine attacks re-face of the aldehyde. (e) Proposed role of boric acid in the reaction. | ||
The stereochemistry of product 2 may be determined at the oxa-Michael step, but the reaction rate of the formation of product 2 may be affected by the rate of the initial nucleophilic attack of the enamine on the aldehyde group. Proline stereochemistry was also important to accelerate the reaction under the conditions with 3 (entry 12 versus entry 13); the most favoured transition state for the C–C bond formation at the aldol step may be involved in the re-attack to the aldehyde by the acetone enamine with L-proline8 (Scheme 2c and d). Addition of 3 may increase the aldehyde form of carbohydrate 1 without altering the transition state of the C–C bond-forming reaction on the aldehyde group. The aldol condensation product, an intermediate that affords 2, may also be generated by a Mannich route; in this case, the aldehyde generated from the hemiacetal may form an iminium ion with proline before the C–C bond formation.12
We also tested boric acid as an additive in L-proline catalysis with DIPEA. In this case, the major product was altered; 6-membered β-C-glycoside 2b was obtained as the major product (entry 16). Boron derivatives have been used to form B–O covalent bonds with unprotected carbohydrates.13 We propose that the B–O bond formation also mimics the enzyme's role to generate the aldehyde group from the hemiacetal (Scheme 2e). The reaction catalyzed by pyrrolidine-boric acid also afforded 2b as the major product isomer (entry 18). Reactions using pyrrolidine alone or boric acid alone as a catalyst did not give 2 nor the corresponding aldol product (entries 20 and 21). When the amine additive was DBU in the L-proline catalysis, the major product was also 2b (entry 17).
For the reaction of 1 to generate 2a, the best conditions tested include the catalysis by L-proline with DIPEA at 25 °C when 4 days reaction time is acceptable (entry 6). To obtain 2a in reasonable yield in 2 days, the best conditions include the catalysis by L-proline with DIPEA and 3 at 25 °C (entry 12). To obtain 2b, the best conditions include the catalysis by pyrrolidine-boric acid at 25 °C (entry 18).
Plausible transition states to afford 2a and 2b from the aldol condensation product via oxa-Michael reactions14 are shown in Scheme 3. In transition states A, B, C, and D, the C–N bond of the CNHAc overlaps with the C–C double bond π-system; this may favour the oxygen nucleophilic attack stereoelectronically. In transition states E and F, there is no such overlap. In proline catalysis with DIPEA (with or without 3), transition state A that leads to 2a may be stereoelectronically and sterically more favoured than transition state B leading to 2a-2 and other transition states leading to 6-membered products 2b and 2b-2. Transition state B may be less favoured than A because of the pseudo axial positions of the C–C double bond and of a hydroxy group. Under the pyrrolidine-boric acid conditions, 2b may be formed by thermodynamic control by the isomerization of 2a through a reversible ring opening and closing. In fact, when 2a was treated with the pyrrolidine-boric acid catalysis conditions, 2a was completely converted to 2b.
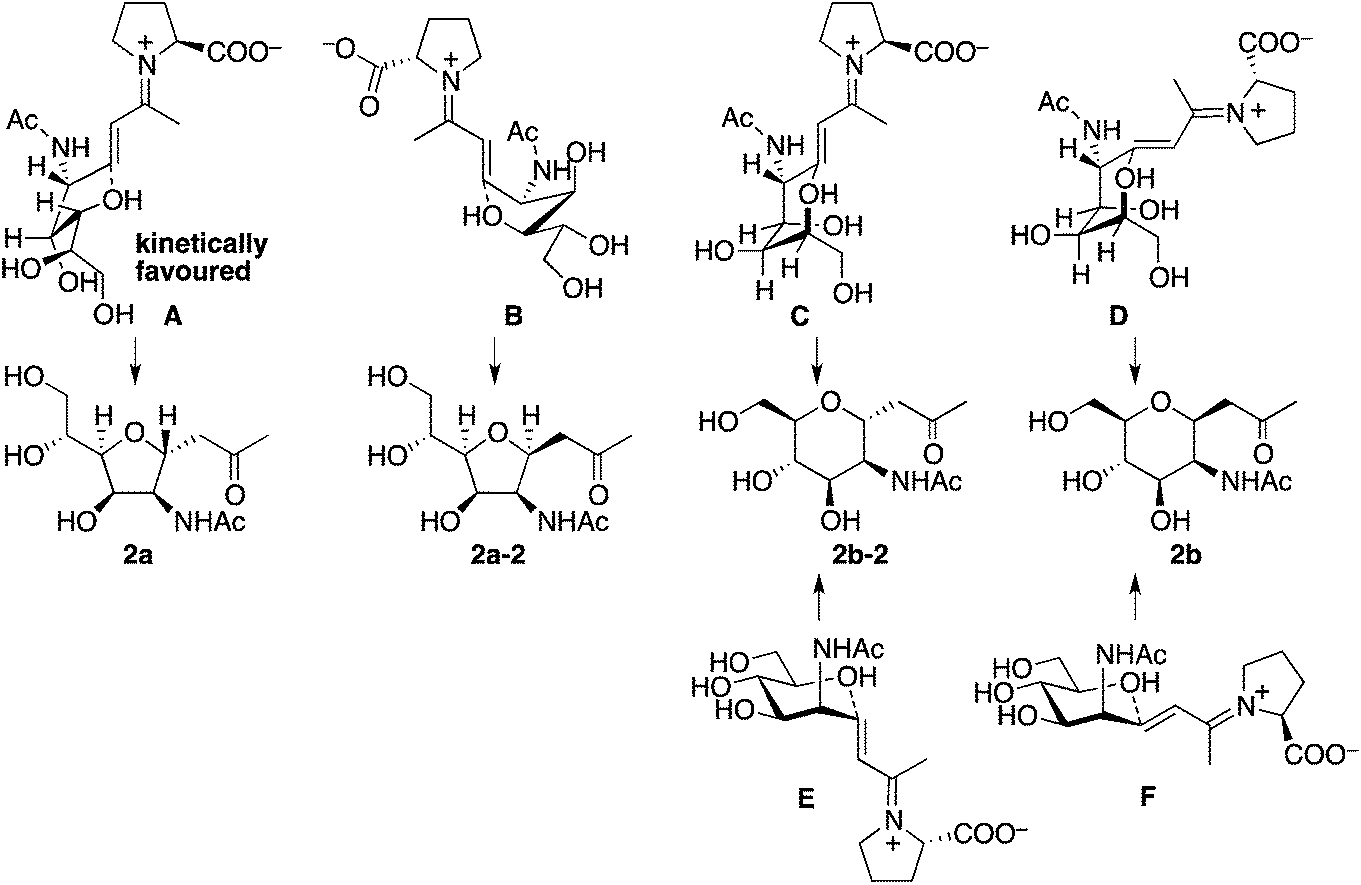 | ||
| Scheme 3 Plausible transition states to afford 2via oxa-Michael reactions. Under the catalysis by the proline-DIPEA (with or without 3), transition state A may be most favoured. | ||
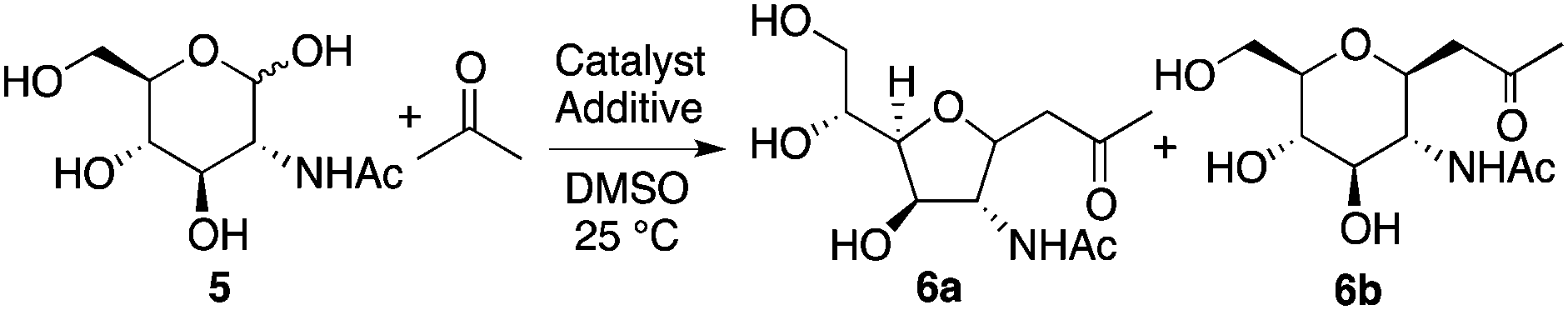
Next, the reaction of N-acetyl-D-glucosamine (5) with acetone was investigated. Selected results are shown in Table 2. Whereas the reaction of 1 with acetone worked with L-proline catalysis to afford product 2 (Table 1, entry 1 versus entry 2), reaction of 5 to give product 6 worked under D-proline catalysis (Table 2, entry 2 versus entry 1). The reaction of glucosamine derivative 5 with acetone under the D-proline-DIPEA catalysis gave products 6a and 6b, but was difficult compared to the reaction of mannosamine derivative 1 using L-proline-DIPEA catalysis (Table 2, entry 4 versusTable 1, entry 6). Addition of additive acid 3 was not effective for the reaction of 5 (Table 2, entry 5). Depending on the stereochemistry of the reactant carbohydrate, suitable additive molecules that generate the aldehyde group may be different. Gluco-type carbohydrates may be more difficult to open the cyclic hemiacetal to generate the aldehyde group than manno- and other types of carbohydrates.15 On the other hand, pyrrolidine-boric acid catalysis worked well to afford product 6b (Table 2, entries 6 and 7). The structure of 6b was confirmed by X-ray crystal structure analysis.10
|
|
|||||
|---|---|---|---|---|---|
| Entry | Catalyst | Additive | Time (h) | Yieldb (%) | Ratio 6a:6b |
| a Reaction conditions: N-Acetyl-D-glucosamine (5) (0.45 mmol, 1.0 equiv.), acetone (0.66 mL, 9.0 mmol, 20 equiv.), catalyst (0.23 mmol, 0.5 equiv.), and amine additive (if added, 0.23 mmol) in DMSO (1.0 mL), at 25 °C, except noted. b Isolated yield; nd = formation of 6 was not detected. c Three times-scale reaction. d DIPEA (1.5 equiv. to 5) and 3 (1 equiv. to 5). e H3BO3 (1 equiv. to 5). f Six times-scale reaction. g H3BO3 (2 equiv. to 5). | |||||
| 1 | L-Proline | None | 24 | nd | — |
| 2 | D-Proline | None | 24 | 4 | — |
| 3 | L-Proline | DIPEA | 96 | nd | — |
| 4c | D-Proline | DIPEA | 96 | 2 | — |
| 5d | D-Proline | DIPEA + 3 | 96 | nd | — |
| 6e | Pyrrolidine | H3BO3 | 24 | 22f | >10:1 |
| 7g | Pyrrolidine | H3BO3 | 24 | 68 | 1:>10 |
| 8g | None | H3BO3 | 24 | nd | — |
When N-valeryl-D-glucosamine (7) was used for the D-proline-catalyzed reaction in the presence of DIPEA, product 8a was obtained as a single diastereomer, and the reaction of 7 under pyrrolidine-boric acid conditions selectively gave 8b (Scheme 4).
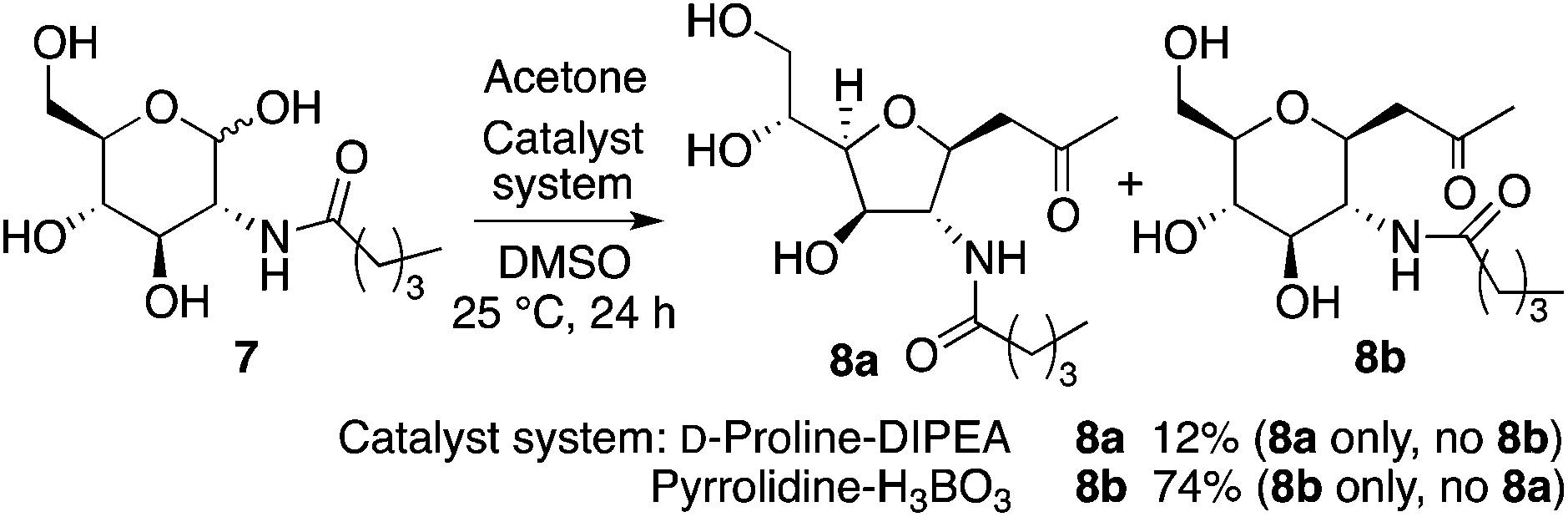 | ||
| Scheme 4 Reactions of N-valeryl-D-glucosamine (7) to afford 8. | ||
In the reaction of 5 to yield 6 (Table 2, entry 4), transition states G, H, and I may be used (Scheme 5). In transition states G and H, the CH(OH)CH2OH group is at the pseudo-axial position; because of this, transition states G and H leading to 5-membered product 6a may not be as kinetically favoured as transition state I leading to 6-membered product 6b. In the reaction of 7, which has a bulkier acyl group than 5, hemiacetal ring opening may be faster than for 5, resulting in the formation of product 8a from 7 in better yield than the formation of 6a from 5. Because of the bulky acyl group of 7, for the reaction of 7, transition state H may be more favoured than transition state G. Under the pyrrolidine-boric acid catalysis in which boric acid was sufficiently used, the reaction of 5 or of 7 gave thermodynamically stable 6-membered product 6b or 8b, respectively (Schemes 4 and 5).
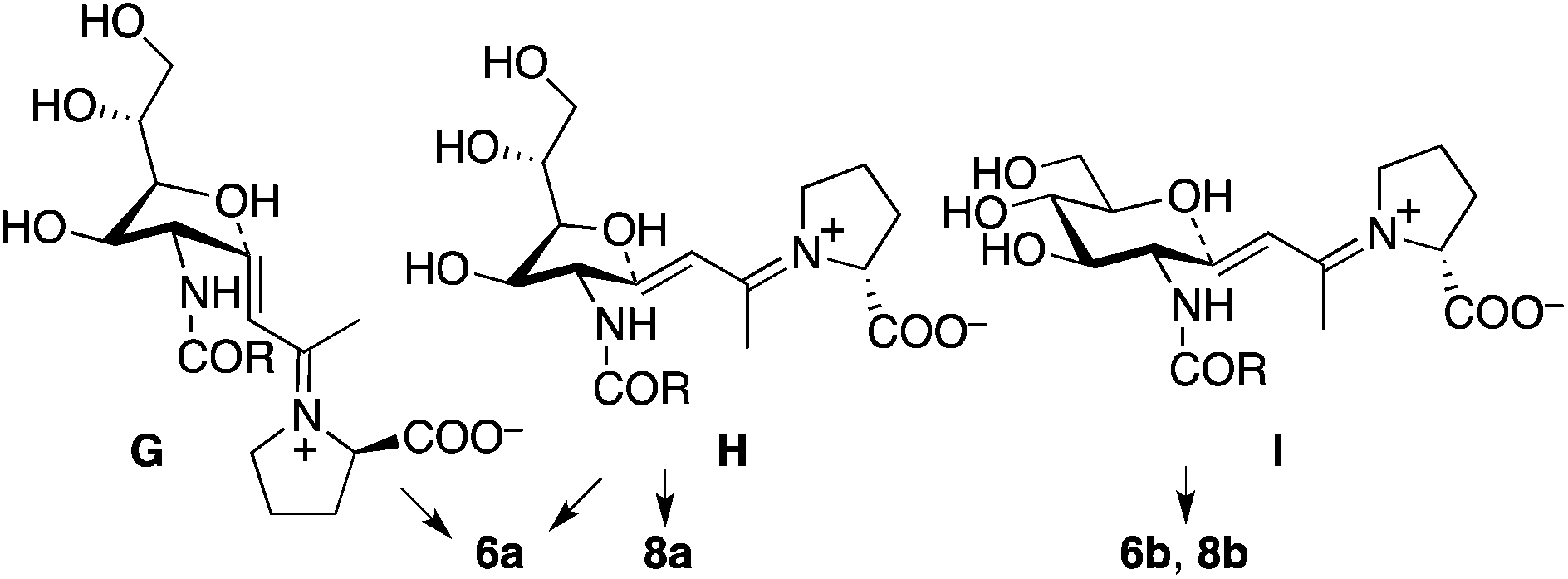 | ||
| Scheme 5 Plausible transition states for the formation of 6 and 8. | ||
The reaction of N-acetyl-D-galactosamine (9) with acetone also provided product 10a under the D-proline-DIPEA catalysis at 25 °C, and product 10b was formed when the reaction was performed at 60 °C (Scheme 6).
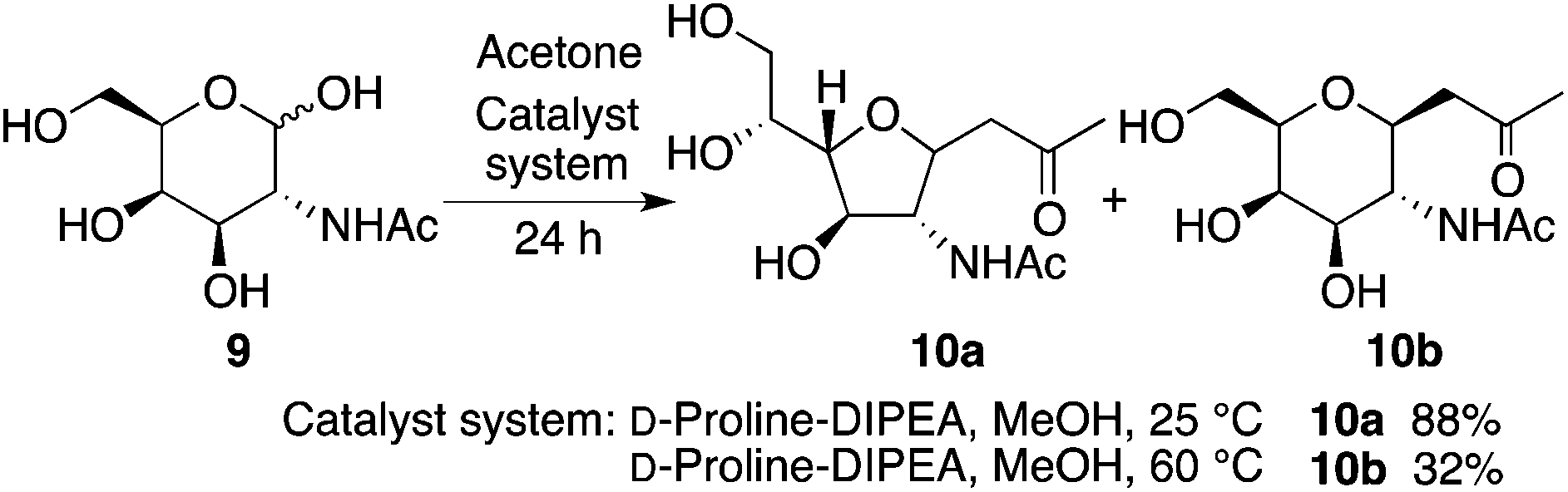 | ||
| Scheme 6 Reaction of N-acetyl-D-galactosamine (9) to afford 10. | ||
Reactions with ketones other than acetone also gave the corresponding C-glycosides. For example, C-glycoside 11 was synthesized from 5 and methoxyacetone (Scheme 7).
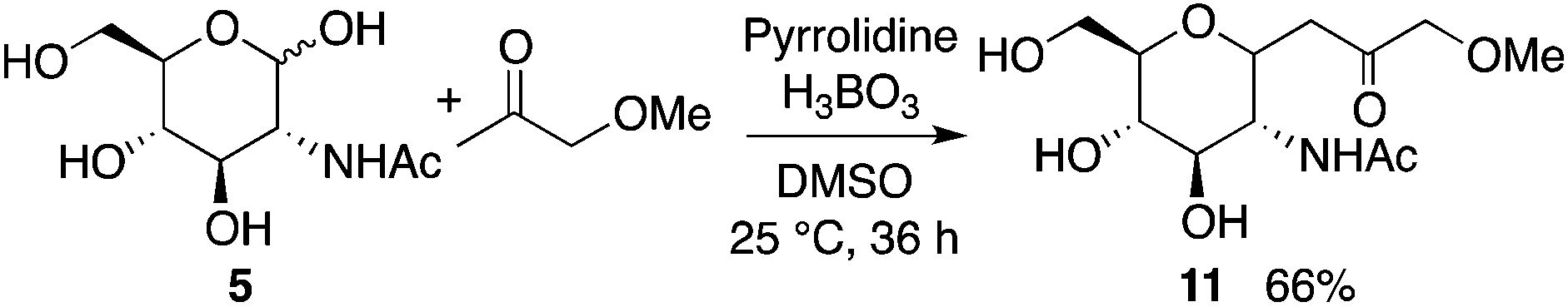 | ||
| Scheme 7 Reaction of 5 with methoxyacetone to afford 11. | ||
Further, C-glycosides were transformed by retaining the C-glycoside structures. For example, compound 12 was synthesized by allylation of 2a (Scheme 8) (see the ESI† for additional transformations). As ketone-moiety-bearing C-glycosides have been used for syntheses of many C-glycoside derivatives and related molecules,16 our methods to synthesize C-glycosides reported here can aid to provide easy access to those molecules and related molecules.
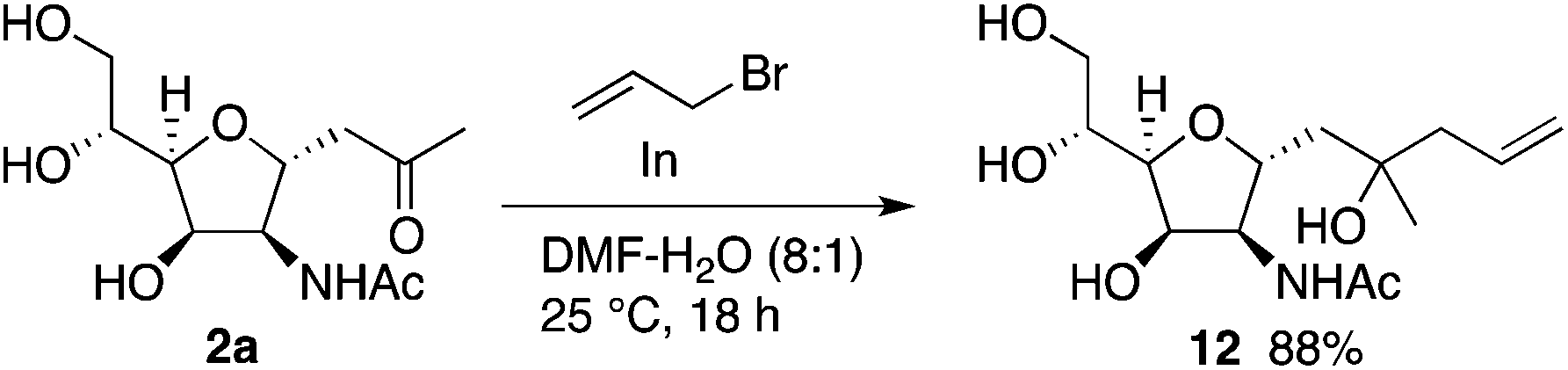 | ||
| Scheme 8 Transformation of 2a. | ||
Conclusions
We have developed methods to synthesize C-glycosides from unprotected carbohydrates and unactivated ketones under mild conditions via the C–C bond formation at the anomeric carbon of the carbohydrates. These reactions are atom- and step-economical. Depending on the carbohydrate and the conditions used, 5- and 6-membered C-glycosides were synthesized in highly stereoselective manners. Our strategies allowed access to C-glycosides that were unable to be synthesized by the previously reported methods. We are investigating the roles of the components of the catalyst systems used for the reactions. We are also expanding our strategies to synthesize a series of C-glycosides from unprotected carbohydrates. These results will be reported in due course.Acknowledgements
We thank Dr Michael Chandro Roy, Research Support Division, Okinawa Institute of Science and Technology Graduate University for the mass analyses. This study was supported by the Okinawa Institute of Science and Technology Graduate University and a Grant-in-Aid for Scientific Research on Innovative Areas “Advanced Molecular Transformations by Organocatalysts” from the MEXT (Japan).Notes and references
- I. S. Young and P. S. Baran, Nat. Chem., 2009, 1, 193 CrossRef CAS PubMed.
- R. Mahreald, Chem. Commun., 2015, 51, 13868 RSC.
- Enzyme-catalyzed C–C bond formation at the anomeric carbon of unprotected carbohydrates: (a) M. J. Kim, W. J. Hennen, M. Sweers and C.-H. Wong, J. Am. Chem. Soc., 1988, 110, 6481 CrossRef CAS; (b) E. Simon, M. D. Bednarski and G. Whitesides, J. Am. Chem. Soc., 1988, 110, 7159 CrossRef CAS; (c) H. Yu, S. Huang, H. Chokhawala, M. Sun, H. Zhang and X. Chen, Angew. Chem., Int. Ed., 2006, 45, 3938 CrossRef CAS PubMed; (d) H. C. Chokhawala, H. Cao, H. Yu and X. Chen, J. Am. Chem. Soc., 2007, 129, 10630 CrossRef CAS PubMed; (e) M. D. Bednarski, H. K. Chenault, E. S. Simon and G. M. Whitesides, J. Am. Chem. Soc., 1987, 109, 1283 CrossRef CAS; (f) U. Kragl, D. Gygax, O. Ghisalba and C. Wandrey, Angew. Chem., Int. Ed. Engl., 1991, 30, 827 CrossRef; (g) C.-S. Tsai, H.-Y. Yen, M. I. Lin, T.-I. Tsai, S.-Y. Wang, W.-I. Huang, T.-L. Hsu, Y.-S. E. Cheng, J.-M. Fang and C.-H. Wong, Proc. Natl. Acad. Sci. U S. A., 2013, 110, 2466 CrossRef CAS PubMed.
- (a) H. B. Wood and H. G. Fletcher, J. Org. Chem., 1961, 26, 1969 CrossRef CAS; (b) J. Sowden and H. O. L. Fischer, J. Am. Chem. Soc., 1947, 69, 1963 CrossRef CAS PubMed; (c) H. A. van Kalkeren, S. van Rootselaar, F. S. Haasjes, F. P. J. T. Rutjes and F. L. van Delft, Carbohydr. Res., 2012, 362, 30 CrossRef CAS PubMed; (d) A. Ranoux, L. Lemiegre, M. Benoit, J.-P. Guegan and T. Benvegnu, Eur. J. Org. Chem., 2010, 1314 CrossRef CAS; (e) Z. Hong, L. Liu, C.-C. Hsu and C.-H. Wong, Angew. Chem., Int. Ed., 2006, 45, 7417 CrossRef CAS PubMed; (f) J.-B. Behr, A. Hottin and A. Ndoye, Org. Lett., 2012, 14, 1536 CrossRef CAS PubMed; (g) W. Schmid and G. M. Whitesides, J. Am. Chem. Soc., 1991, 113, 6674 CrossRef CAS; (h) J.-B. Behr, A. Hottin and A. Ndoye, Org. Lett., 2012, 14, 1536 CrossRef CAS PubMed; (i) A. Palmelund and R. Madsen, J. Org. Chem., 2005, 70, 8248 CrossRef CAS PubMed; (j) Y. Kimura, S. Ito, Y. Shimizu and M. Kanai, Org. Lett., 2013, 15, 4130 CrossRef CAS PubMed; (k) K. Toshima, G. Matsuo, T. Ishizuka, Y. Ushiki, M. Nakata and S. Matsumura, J. Org. Chem., 1998, 63, 2307 CrossRef CAS.
- (a) B. Voigt, U. Scheffler and R. Mahrwald, Chem. Commun., 2012, 48, 5304 RSC; (b) B. Voigt, A. Matviitsuk and R. Mahrwald, Tetrahedron, 2013, 69, 4302 CrossRef CAS; (c) B. Voigy and R. Mahrwald, Chem. Commun., 2014, 50, 817 RSC; (d) N. Bragnier and M.-C. Scherrmann, Synthesis, 2005, 814 CAS; (e) R. Winzar, J. Philips and M. J. Kiefel, Synlett, 2010, 583 CAS; (f) J. Wang, Q. Li, Z. Ge and R. Li, Tetrahedron, 2012, 68, 1315 CrossRef CAS; (g) M.-C. Scherrmann, Top. Curr. Chem., 2010, 295, 1 CrossRef CAS PubMed; (h) I. Riemann, M. A. Papadopoulos, M. Knorst and W.-D. Fessner, Aust. J. Chem., 2002, 55, 147 CrossRef CAS; (i) F. Rodrigues, Y. Canac and L. Andre, Chem. Commun., 2000, 2049 RSC; (j) Recently, reactions of aldopentoses with unactivated ketones to give C-glycosides have been demonstrated: S. N. Witte, B. Voigt and R. Mahrwald, Synthesis, 2015, 47, 2249 CrossRef CAS.
- Synthesis of C-glycosides and long carbon-chain carbohydrates via the C–C bond forming reactions on protected carbohydrates and carbohydrate derivatives: (a) C. Zhao, X. Jia, X. Wang and H. Gong, J. Am. Chem. Soc., 2014, 136, 17645 CrossRef CAS PubMed; (b) J. R. Kramer and T. J. Deming, J. Am. Chem. Soc., 2010, 132, 15068 CrossRef CAS PubMed; (c) G. McGarvery, C. A. LeClair and B. A. Schmidtmann, Org. Lett., 2008, 10, 4727 CrossRef PubMed; (d) H. Gong and M. R. Gagne, J. Am. Chem. Soc., 2008, 130, 12177 CrossRef CAS PubMed; (e) D. Enders and T. Gasperi, Chem. Commun., 2007, 88 RSC; (f) D. G. Gillingham, P. Stallforth, A. Adibekian, P. H. Seeberger and D. Hilvert, Nat. Chem., 2010, 2, 102 CrossRef CAS PubMed; (g) C.-H. Hsu, M. Schelwies, S. Enck, L.-Y. Huang, S.-H. Huang, Y.-F. Chang, T.-J. R. Cheng, W.-C. Cheng and C.-H. Wong, J. Org. Chem., 2014, 79, 8629 CrossRef CAS PubMed; (h) V. N. Thota, M. Brahmaiah and S. S. Kulkarmi, J. Org. Chem., 2013, 78, 12082 CrossRef CAS PubMed; (i) R. S. Andrew, J. J. Becker and M. R. Gagne, Angew. Chem., Int. Ed., 2012, 51, 4140 CrossRef PubMed; (j) R. S. Andrew, J. J. Becker and M. R. Gagne, Angew. Chem., Int. Ed., 2010, 49, 7274 CrossRef PubMed; (k) G. Yang, J. Schmieg, M. Tsuji and R. W. Frank, Angew. Chem., Int. Ed., 2004, 43, 3818 CrossRef CAS PubMed; (l) G. D. McAllister, D. E. Paterson and R. J. K. Taylor, Angew. Chem., Int. Ed., 2003, 42, 1387 CrossRef CAS PubMed; (m) S. Tamura, H. Abe, A. Matsuda and S. Shuto, Angew. Chem., Int. Ed., 2003, 42, 1021 CrossRef PubMed; (n) J. L. Chiara and E. Sesmilo, Angew. Chem., Int. Ed., 2002, 41, 3242 CrossRef CAS; (o) G. Cheng, R. Fan, J. M. Hernandez-Torres, F. P. Boulineau and A. Wei, Org. Lett., 2007, 9, 4849 CrossRef CAS PubMed; (p) G. Yang, R. W. Frank, R. Bittmann, P. Samadder and G. Arthur, Org. Lett., 2001, 3, 197 CrossRef CAS PubMed; (q) R. W. Frank and M. Tsuji, Acc. Chem. Res., 2006, 39, 692 CrossRef PubMed and references cited therein.
- (a) T. D. Machajewski and C.-H. Wong, Angew. Chem., Int. Ed., 2000, 39, 1352 CrossRef CAS; (b) X. Garrabou, J. A. Castillo, C. Guerard-Helaine, T. Parella, J. Joglar, M. Lemaiire and P. Clapes, Angew. Chem., Int. Ed., 2009, 48, 5521 CrossRef CAS PubMed; (c) A. B. Northrup and D. W. C. MacMillan, Science, 2004, 305, 1752 CrossRef CAS PubMed; (d) N. S. Chowdari, D. B. Ramachary, A. Cordova and C. F. Barbas III, Tetrahedron Lett., 2002, 43, 9591 CrossRef CAS; (e) J. Cases, M. Engqvist, I. Ibraham, B. Kaynak and A. Cordova, Angew. Chem., Int. Ed., 2005, 44, 1343 CrossRef PubMed.
- (a) B. List, R. A. Lerner and C. F. Barbas III, J. Am. Chem. Soc., 2000, 122, 2395 CrossRef CAS; (b) S. Bahmanyar and K. N. Houk, Org. Lett., 2003, 5, 1249 CrossRef CAS PubMed; (c) P. H.-Y. Cheong, C. Y. Legault, J. M. Um, N. Celebi-Olcum and K. N. Houk, Chem. Rev., 2011, 111, 5043 CrossRef PubMed.
- P. Delahay and J. E. Strassner, J. Am. Chem. Soc., 1952, 74, 893 CrossRef CAS.
- CCDC 1422504 (compound 2a) and CCDC 1422505 (compound 6b) contain the supplementary crystallographic data for this paper.
- A. D. Daniel, I. Campeotto, M. W. van der Kamp, A. H. Bolt, C. H. Trinh, S. E. V. Phillips, A. R. Pearson, A. Nelson, A. J. Mulholland and A. Berry, ACS Chem. Biol., 2014, 9, 1025 CrossRef PubMed.
- S. M. Opalka, J. L. Steinbacher, B. A. Lambiris and D. T. McQuade, J. Org. Chem., 2011, 76, 6503 CrossRef CAS PubMed.
- (a) Ref. 4j; ; (b) J. F. Teichert, D. Mazunin and J. W. Bode, J. Am. Chem. Soc., 2013, 135, 11314 CrossRef CAS PubMed.
- Intramolecular oxa-Michael cyclization to give tetrahydropyrans, tetrahydrofurans, and other oxygen-containing heterocycles via the formation of iminium ions of enones: (a) S. R. Byeon, H. Park, H. Kim and J. Hong, Org. Lett., 2011, 13, 5816 CrossRef CAS PubMed; (b) Y. Lu, G. Zou and G. Zhao, ACS Catal., 2013, 3, 1356 CrossRef CAS; (c) W. Wu, X. Li, H. Huang, X. Yuan, J. Lu, K. Zhu and J. Ye, Angew. Chem., Int. Ed., 2013, 52, 1743 CrossRef CAS PubMed. Intramolecular oxa-Michael cyclizations of enals via iminium activations: (d) S. R. Byeon, H. Park, H. Kim and J. Hong, Org. Lett., 2011, 13, 5816 CrossRef CAS PubMed; (e) P. G. McGarraugh and S. E. Brenner-Moyer, Org. Lett., 2011, 13, 6460 CrossRef CAS PubMed; (f) P. G. McGarraugh, R. C. Johnston, A. Martines-Munoz, P. H.-Y. Cheong and S. E. Brenner-Moyer, Chem. – Eur. J., 2012, 18, 10742 CrossRef CAS PubMed; (g) K. Lee, H. Kim and J. Hong, Eur. J. Org. Chem., 2012, 1025 CrossRef CAS. Other organocatalytic intramolecular oxa-Michael cyclizations of enones: (h) K. Asano and S. Matsubara, J. Am. Chem. Soc., 2011, 133, 16711 CrossRef CAS PubMed; (i) Y. Fukata, R. Miyaji, T. Okamura, K. Asano and S. Matsubara, Synthesis, 2013, 45, 1627 CrossRef CAS.
- (a) H. F. Bunn and P. J. Higgins, Science, 1981, 213, 222 CAS; (b) J. P. Dworkin and S. L. Miller, Carbohydr. Res., 2000, 329, 359 CrossRef CAS PubMed.
- (a) K. Lalitha, K. Muthusamy, Y. S. Prasad, P. K. Vemula and S. Nagarajan, Carbohydr. Res., 2015, 402, 158 CrossRef CAS PubMed and references cited therein; (b) L. Liu, B. A. Motaal, M. Schmidt-Supprian and N. L. B. Pohl, J. Org. Chem., 2012, 77, 1539 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Synthesis and characterization of compounds, and X-ray crystal structures of compounds 2a and 6b. CCDC 1422504 and 1422505. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5ob02094h |
| This journal is © The Royal Society of Chemistry 2016 |