
Total synthesis of a piperidine alkaloid, microcosamine A†
Chada
Raji Reddy
*a,
Bellamkonda
Latha
a,
Kamalkishor
Warudikar
a and
Kiran Kumar
Singarapu
b
aDivision of Natural Products Chemistry, CSIR-Indian Institute of Chemical Technology, Hyderabad, India 500 007. E-mail: rajireddy@iict.res.in
bCentre for NMR & Structural Chemistry, CSIR-Indian Institute of Chemical Technology, Hyderabad, 500 007, India
First published on 3rd November 2015
Abstract
The first asymmetric total synthesis of a new natural piperidine alkaloid, microcosamine A, has been accomplished from D-serine and D-methyl lactate as chiral pool starting materials. Key features of the strategy include the utility of Horner–Wadsworth–Emmons reaction, Luche reduction, intramolecular carbamate N-alkylation to form the piperidine framework and Julia–Kocienski olefination to install the triene side-chain.
Introduction
2-Methyl-3-hydroxy-6-alkylated piperidines constitute an important class of natural alkaloids due to their interesting biological and pharmacological properties (anaesthetic, analgesic, antitumor, antibiotic, CNS stimulating biological properties, antihypertensive and antifungal activities etc.).1–6 These molecules possess different stereochemical patterns and C6 side chains with either saturated or unsaturated alkyl groups. To date three types of 2-methyl-3-hydroxy natural piperidines having different unsaturated side chains at C6 carbon, such as corydendramines A & B (1a, 1b),4 microgrewiapine A (1c)5 and microcosamines A & B (2a, 2b)6 have been isolated (Fig. 1). However, there is no synthesis reported for any of these molecules. The interesting structural feature of these molecules is the presence of a chiral hydroxy group at the C3-position of the piperidine ring with trans stereochemistry with respect to the C2 and C6 carbons, which is a rare substructure in piperidine alkaloids. Structurally, these compounds possess a polar head group with hydroxyl and amine functionalities and a hydrophobic aliphatic tail, which can be considered as cyclic analogues of the lipid sphingosine membrane.7 The above observations combined with our interest in the synthesis of alkaloids8 have driven us for the synthesis of microcosamine A (2a).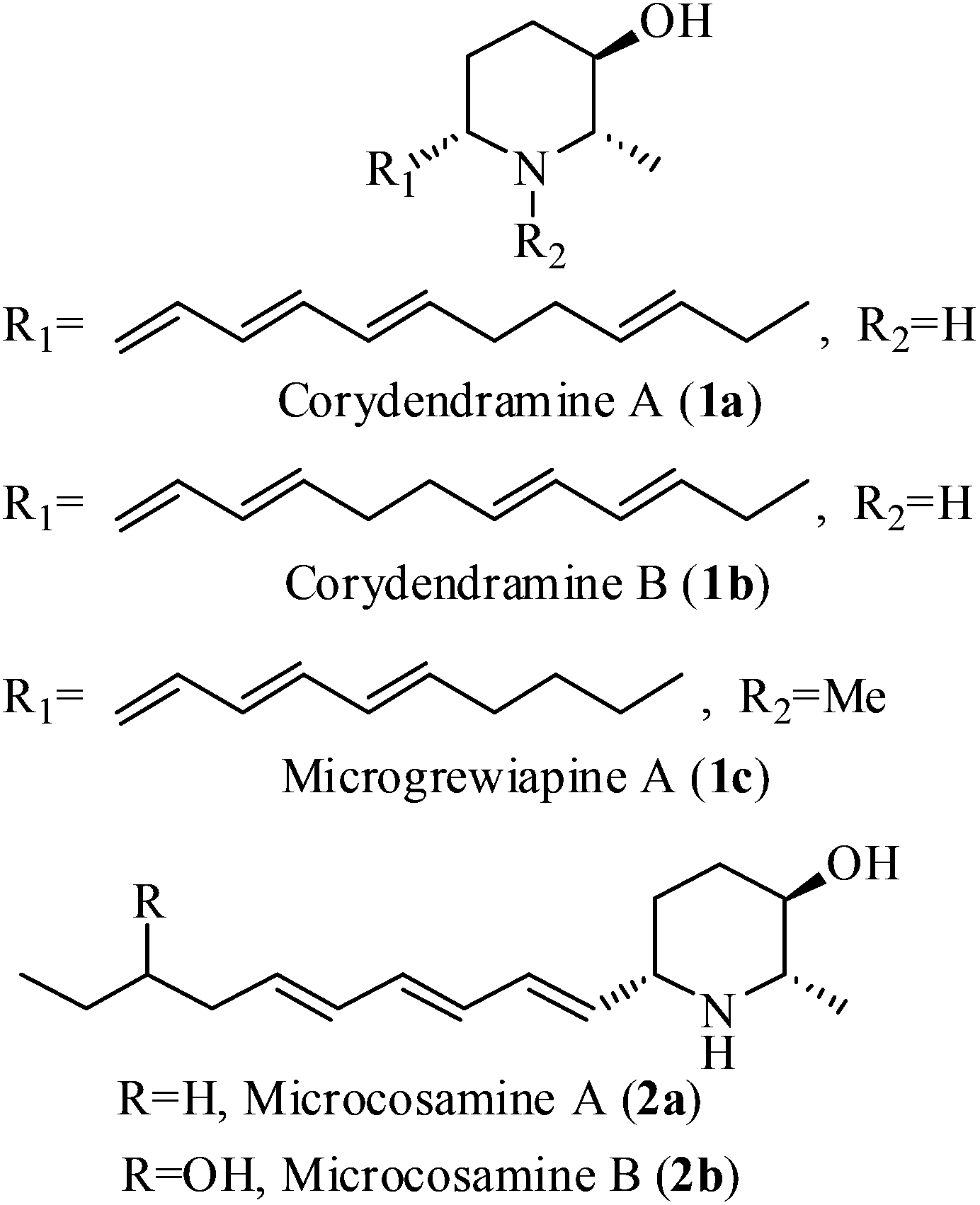 | ||
| Fig. 1 Structures of corydendramines A & B, microgrewiapine A and microcosamines A & B. | ||
Microcosamine A (2a), was first isolated by Lin and co-workers in 2008, from the chloroform extraction of the leaves of Microcos paniculata along with microcosamine B (2b) and found their insecticidal activity against the larvae of Culex quinquefasciatus with LC50 values of 5.2 and 17.0 μg mL−1, respectively.6Microcos paniculata, a large shrub or a small tree that grows in South and Southeast Asian countries, is found to be a rich source of bio-active compounds and several parts such as roots, stem bark, leaves and fruits are being used traditionally to treat diarrhea and fever, as herbal tea to treat cold, enteritis, and skin rashes and as insecticides.9 There is a good number of 2,3,6-trisubstituted piperidine alkaloids which have been isolated from this species.10 Later, in 2013, 2a was again isolated from the same plant along with some other piperidine alkaloids by Kinghorn et al. and examined for their effects on neuronal nicotinic acetylcholine receptors (nAChRs).5 Microcosamine A (2a) exhibited approximately 53.7% and 59% of hα3β4 and hα3β2 nAChR activity, respectively. Herein, we present the first total synthesis of microcosamine A (2a).
Results and discussion
Scheme 1 outlines the retrosynthetic strategy of 2a. We planned the installation of the triene-side chain at the C6 position of hydroxy piperidine ring 4 by using 3via oxidation followed by Julia–Kocienski olefination.11 Synthesis of the sulfone fragment 3 was achieved from 1-octyne (8) by using a known conjugated alcohol 5.12 The construction of piperidine 4 was designed through the intramolecular carbamate N-alkylation of its precursor which could be obtained from N,O-protected D-serine ester 6 and β-keto phosphonate 7 using Horner–Wadsworth–Emmons (HWE) olefination13 as the key reaction. Ester 6 and phosphonate 7 in turn could be prepared from the chiral starting materials, D-serine (9) and D-methyl lactate (10), respectively. The C2 and C6 stereochemistry is expected to arise from 9 and C3-hydroxyl stereochemistry envisaged from 10 using diastereoselective Luche reduction14 of keto functionality.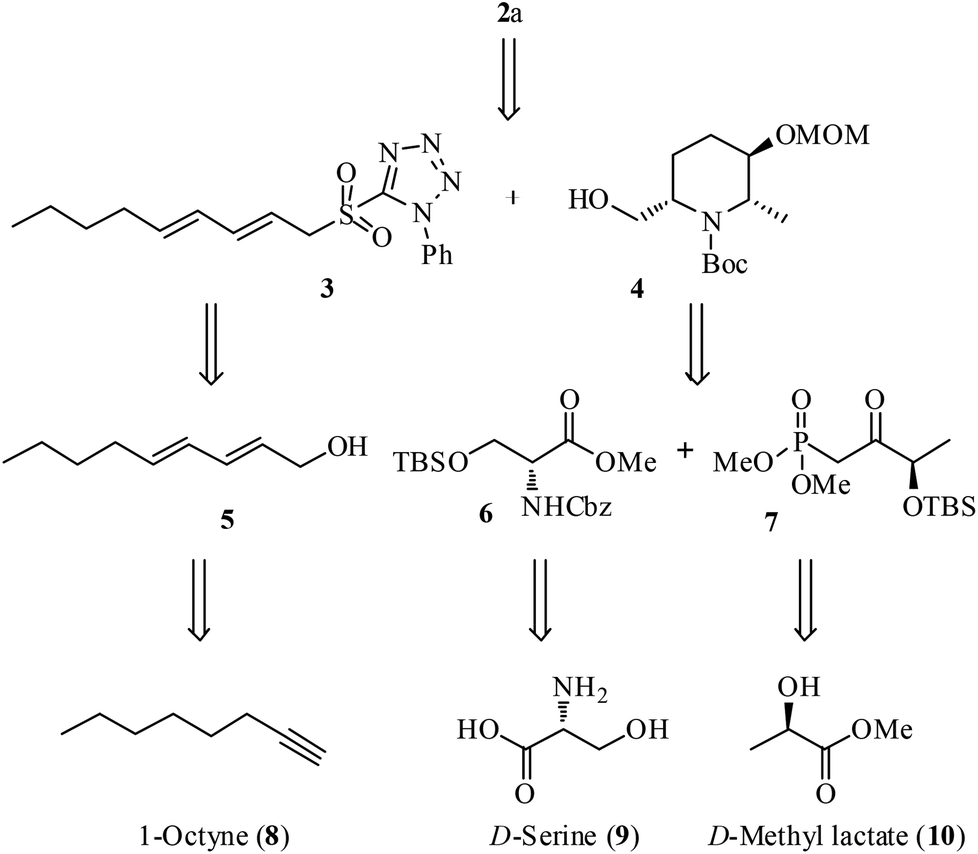 | ||
| Scheme 1 Retrosynthetic analysis of 2a. | ||
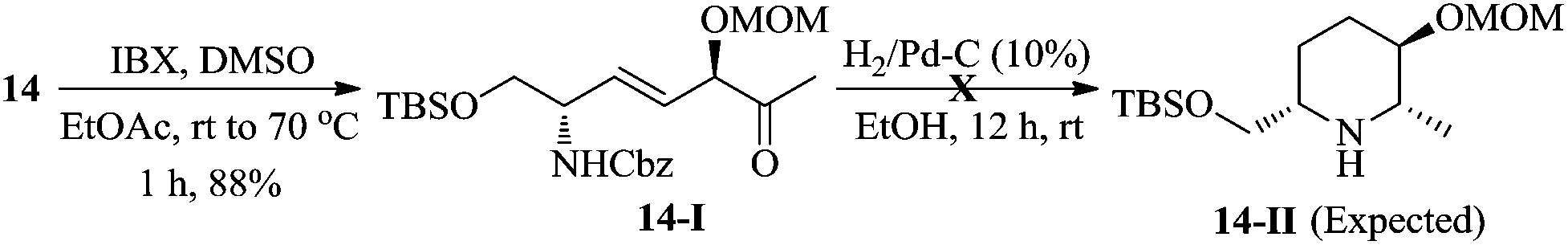
The ester intermediate 6 was prepared in three steps from D-serine (9) following the reported procedure.15 The desired β-keto phosphonate 7 was also smoothly obtained in two steps from D-methyl lactate (10) using a literature protocol.16 The synthesis of the piperidine fragment is outlined in Scheme 2. Initially, the ester 6 was subjected to DIBAL-H reduction to aldehyde followed by HWE olefination with β-keto phosphonate 7 under Ba(OH)2·8H2O in THF/H2O conditions to afford the enone 11 in 87% yield, a precursor for diastereoselective Luche reduction. Exposure of 11 to NaBH4 in MeOH in the presence of CeCl3·H2O at −78 °C provided the allylic alcohol 12 along with its minor diastereomer in 85% yield [dr >9![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, based on the diastereomers 17 & 17a, separated in the cyclization step]. At this stage, we were unable to separate these diastereomers either by column or by HPLC (in 1H NMR spectra, the signals were not separated to verify the diastereomeric ratio) and hence, were moved for further transformations as a mixture. Thus, the hydroxyl group of 12 was protected as methoxy methyl (MOM) ether 13 using MOMCl/diisopropylethyl amine in CH2Cl2 (89%). To obtain a free secondary hydroxyl group, a two-step protecting group manipulation was chosen. Deprotection of both the tert-butyldimethyl silyl groups of 13 under HF (40% in water) in CH3CN followed by selective protection of the resulting primary hydroxyl group as a TBS ether produced the required alcohol 14 in 81% yield over two steps. After the oxidation of alcohol 14 to the corresponding ketone 14-I, the attempt to form the piperidine ring 14-II through hydrogenation was unsuccessful.17
:1, based on the diastereomers 17 & 17a, separated in the cyclization step]. At this stage, we were unable to separate these diastereomers either by column or by HPLC (in 1H NMR spectra, the signals were not separated to verify the diastereomeric ratio) and hence, were moved for further transformations as a mixture. Thus, the hydroxyl group of 12 was protected as methoxy methyl (MOM) ether 13 using MOMCl/diisopropylethyl amine in CH2Cl2 (89%). To obtain a free secondary hydroxyl group, a two-step protecting group manipulation was chosen. Deprotection of both the tert-butyldimethyl silyl groups of 13 under HF (40% in water) in CH3CN followed by selective protection of the resulting primary hydroxyl group as a TBS ether produced the required alcohol 14 in 81% yield over two steps. After the oxidation of alcohol 14 to the corresponding ketone 14-I, the attempt to form the piperidine ring 14-II through hydrogenation was unsuccessful.17
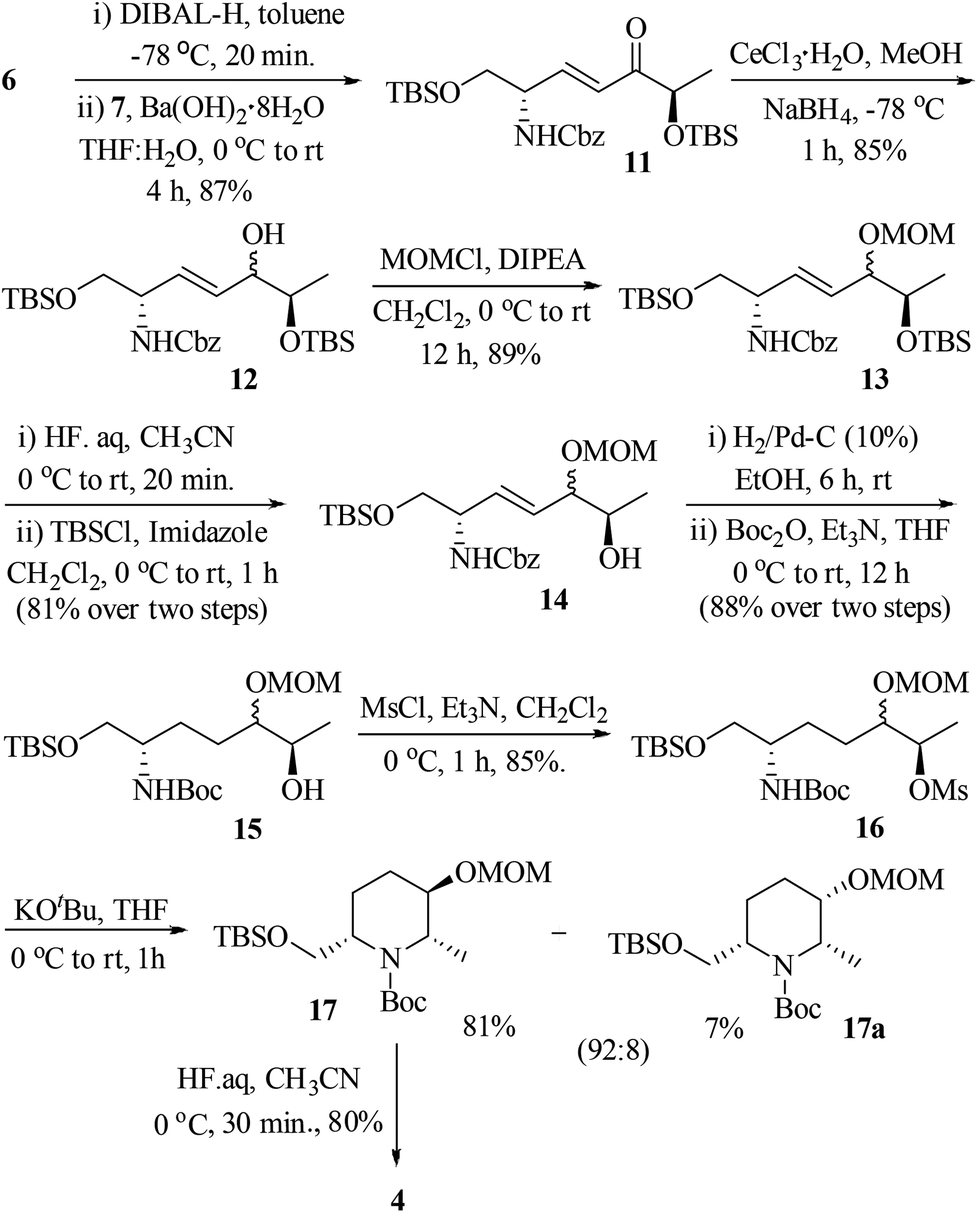 | ||
| Scheme 2 Synthesis of 4 from 6 and 7. | ||
Thus, an alternative sequence was followed. The hydrogenation reaction of 14 using 10% Pd/C in EtOH involves the olefin reduction as well as Cbz-deprotection to free amine, which was subsequently treated with di-tert-butyl-dicarbonate (Boc2O)/Et3N to obtain Boc-protected amino alcohol 15 in 88% yield. Treatment of 15 with methanesulfonyl chloride in the presence of triethyl amine in CH2Cl2 gave the mesylate 16 in 85% yield. Compound 16 was successfully converted into 2,3,6-trisubstituted piperidine via intramolecular carbamate N-alkylation (SN reaction) using potassium tert-butoxide in THF (88%).18 At this point, the diastereomers formed during the Luche reduction of 11 were separated by column chromatography (dr 92:8). The major isomer 17 was found to be the desired one and the minor isomer 17a was undesired, which were characterized by 2D COSY and NOESY experiments.19 The nOe cross correlations between H8(Me)/H7(H7′), H2/H9 and H3/H8(Me) for 17 (Fig. 2) support the desired diastereomer. In the case of 17a the nOe cross correlations observed between H8(Me)/H7(H7′), H7/H9, H8/H9, and H2/H3 support the undesired diastereomer (Fig. 2). Next, the TBS group of 17 was deprotected using HF (40% in water) in CH3CN to give the piperidinol 4 in 80% yield.
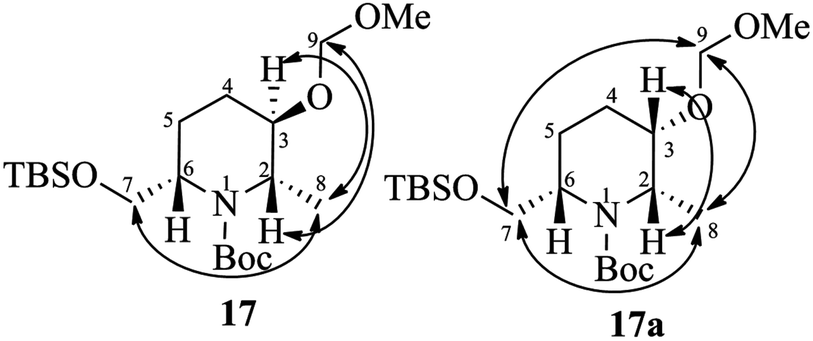 | ||
| Fig. 2 The characteristic nOe cross correlations of compounds 17 and 17a. | ||
The sulfone 3 required for Julia olefination was synthesised from dienol 5, obtained from 1-octyne (8).12 The Mitsunobu reaction20 of the alcohol 5 with 1-phenyl-1H-tetrazole-5-thiol to thio-tetrazole 18 (95% yield) followed by ammonium molybdate catalyzed oxidation21 using hydrogen peroxide in EtOH provided the sulfone 3 in 82% yield (Scheme 3).
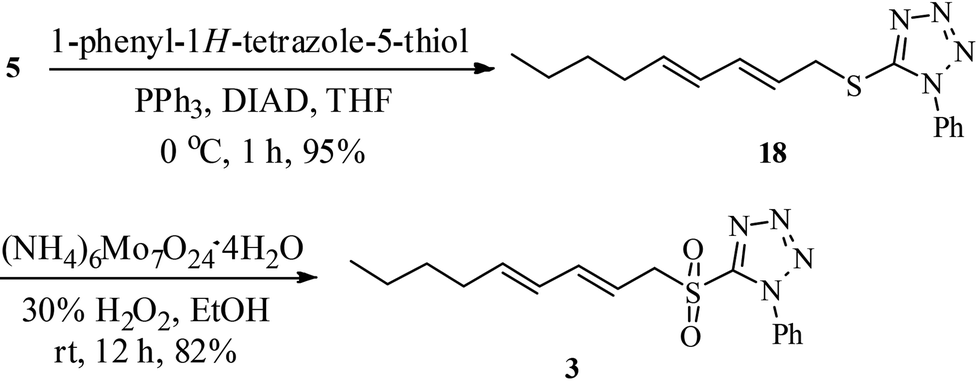 | ||
| Scheme 3 Synthesis of sulfone 3. | ||
The stage was set for the conversion of 4 to microcosamine A (2a) by connecting the side chain (Scheme 4). Accordingly, the alcohol 4 was oxidized with IBX (2-iodoxybenzoic acid) to the corresponding aldehyde followed by Julia–Kocienski olefination with the trienyl sulfone 3 by treating with KHDMS in the presence of 18-crown-6 in DME provided the trienyl-piperidine 19 exclusively as the Z-isomer in 72% yield over two steps.22 Removal of MOM and Boc groups was accomplished in one step by the treatment of 19 with 3 N HCl in MeOH to give the desired microcosamine A (2a) in 78% yield.
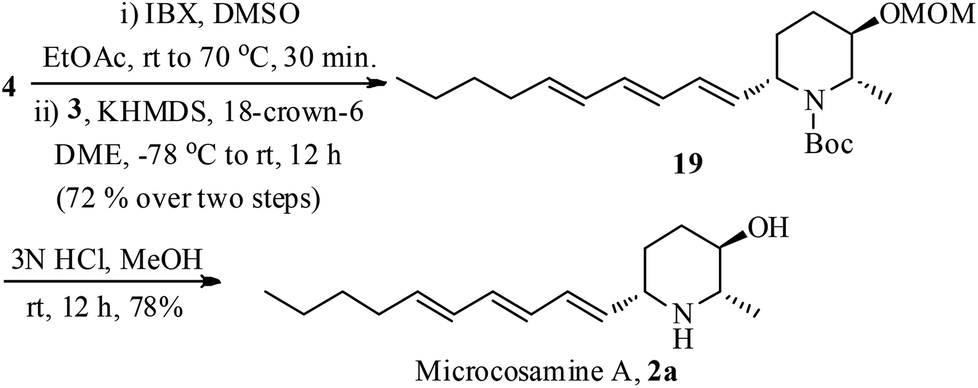 | ||
| Scheme 4 Synthesis of 2a from 4. | ||
The spectral data (1H, 13C NMR and mass) of our synthetic microcosamine A (2a) were in full agreement with those reported for the natural product (see Table S1 in the ESI†). The specific rotation of synthetic 2a {[α]20D: +5.6 (c 1.00, CH3OH)} was also comparable to the natural product {[α]20D: +4.0 (c 1.00, CH3OH)}. These results confirm the structure and absolute configuration of the natural product 2a.
Conclusions
In summary, the first asymmetric total synthesis of a natural piperidine alkaloid, microcosamine A, was accomplished using commercially available D-serine, D-methyl lactate (for piperidine unit) and 1-octyne (for triene-side chain) as starting materials. The key features of the strategy are the successful utilization of HWE-olefination and intramolecular carbamate N-cyclization for piperidine ring construction and Julia–Kocienski olefination for the installation of the side chain in the natural product synthesis. The approach is handy for the synthesis of other natural products and their analogues having different side chains.Experimental
General
Melting points were determined on a POLMON melting point apparatus and are uncorrected. NMR spectra were recorded in CDCl3 on 300 MHz, 400 MHz or 500 MHz spectrometers at ambient temperature. Chemical shifts δ were denoted with reference to TMS or solvent residual (CDCl3: δ 7.26 ppm for 1H and 77.0 ppm for 13C) peaks given in ppm (parts per million) and coupling constants J are measured in Hz (hertz). FTIR spectra were recorded on a Perkin-Elmer 683 infrared spectrophotometer in KBr or as neat. Optical rotations were measured on an Anton Paar MLP 200 modular circular digital polarimeter by using a 2 mL cell with a path length of 1 dm with MeOH or CHCl3 as the solvent. Low-resolution MS were collected on an Agilent Technologies LC-MSD trap SL spectrometer in positive ion mode. Technical-grade EtOAc and hexanes used for column chromatography were distilled before use. All the reagents and solvents were of reagent grade and used without further purification unless otherwise stated. THF, when used as a solvent for reactions, was freshly distilled from a sodium benzophenone ketyl radical. Progress of the reactions was monitored by thin-layer chromatography using silica plates (UV254, glass backed; Merck KGaA) and the spots were visualized under UV-light and/or after charring with ninhydrin or potassium permanganate or β-naphthol stain solutions. Column chromatography was performed over silica gel (60–120 mesh) or on alumina (aluminium oxide activated, neutral, 150 mesh) packed in glass columns, eluted with gradients of petroleum ether and ethyl acetate. Column fractions were concentrated under reduced pressure at temperatures not more than 40 °C. All the reactions were performed under N2 in oven dried glassware with magnetic stirring.To a stirred solution of (R)-dimethyl (3-((tert-butyldimethylsilyl)oxy)-2-oxobutyl)phosphonate167 (2.2 g, 7.19 mmol) in THF (30 mL), was added Ba(OH)2·8H2O (2.8 g, 8.99 mmol) at rt and stirred vigorously for 45 min. The reaction mixture was cooled to 0 °C before adding the above crude aldehyde in 15 mL of THF/H2O (20:1) and the mixture was allowed to warm to rt. After completion of the reaction (4 h), the reaction mixture was diluted with EtOAc (30 mL), the organic layer was washed with water (30 mL), and brine (30 mL), dried over Na2SO4 and concentrated under reduced pressure. The crude product was purified by flash column chromatography (neutral alumina, hexanes/EtOAc 9:1) to afford enone 11 (2.7 g, 87%) as a colorless oil. Rf = 0.4 (petroleum ether:EtOAc = 9:1); [α]20D = +9.5 (c 1.50, CHCl3); IR (neat): νmax 3446, 2954, 2931, 2858, 1705, 1631, 1255, 1116, 837 cm−1; 1H NMR (400 MHz, CDCl3): δ 7.41–7.28 (m, 5H, Ph), 6.93 (dd, J = 15.8, 4.7 Hz, 1H, CH![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH), 6.73 (d, J = 15.8 Hz, 1H, CHCH), 5.22–5.06 (m, 3H, CH2–Ph, –NH), 4.52–4.43 (br m, 1H, CH–N), 4.25 (q, J = 13.6, 6.8 Hz, 1H, CH–CH3), 3.76 (d, J = 3.7 Hz, 2H, CH2–O), 1.29 (d, J = 6.8 Hz, 3H, CH–CH3), 0.90 (s, 9H, tBu-Si), 0.85 (s, 9H, tBu-Si), 0.06 (s, 3H, Si(CH3)2), 0.05 (s, 3H, Si(CH3)2), 0.03 (s, 3H, Si(CH3)2), 0.02 (s, 3H, Si(CH3)2); 13C NMR (100 MHz, CDCl3): δ 201.0, 155.7, 145.2, 136.3, 128.5, 128.1, 128.0, 124.2, 74.3, 66.9, 64.6, 53.7, 25.7, 25.7, 20.8, 18.2, 18.0, −4.8, −4.9, −5.5, −5.5; MS (ESI): m/z 544 (M + Na)+; HRMS (ESI): m/z calcd for C27H47NO5Si2Na (M + Na)+, 544.2885; found 544.2890.
CH), 6.73 (d, J = 15.8 Hz, 1H, CHCH), 5.22–5.06 (m, 3H, CH2–Ph, –NH), 4.52–4.43 (br m, 1H, CH–N), 4.25 (q, J = 13.6, 6.8 Hz, 1H, CH–CH3), 3.76 (d, J = 3.7 Hz, 2H, CH2–O), 1.29 (d, J = 6.8 Hz, 3H, CH–CH3), 0.90 (s, 9H, tBu-Si), 0.85 (s, 9H, tBu-Si), 0.06 (s, 3H, Si(CH3)2), 0.05 (s, 3H, Si(CH3)2), 0.03 (s, 3H, Si(CH3)2), 0.02 (s, 3H, Si(CH3)2); 13C NMR (100 MHz, CDCl3): δ 201.0, 155.7, 145.2, 136.3, 128.5, 128.1, 128.0, 124.2, 74.3, 66.9, 64.6, 53.7, 25.7, 25.7, 20.8, 18.2, 18.0, −4.8, −4.9, −5.5, −5.5; MS (ESI): m/z 544 (M + Na)+; HRMS (ESI): m/z calcd for C27H47NO5Si2Na (M + Na)+, 544.2885; found 544.2890.
:2) to furnish 12 (2.1 g, 85%) as a colorless oil. Rf = 0.2 (petroleum ether:EtOAc = 9:1); [α]20D = −6.6 (c 1.10, CHCl3); IR (neat): νmax 3446, 2953, 2930, 1714, 1253, 773 cm−1; 1H NMR (500 MHz, CDCl3): δ 7.41–7.28 (m, 5H, Ph), 5.79 (dd, J = 15.6, 5.7 Hz, 1H, CHCH), 5.65 (dd, J = 15.6, 5.9 Hz, 1H, CHCH), 5.15–5.04 (m, 1H, –NH), 5.11 (s, 2H, CH2–Ph), 4.32–4.20 (br m, 1H, CH–N), 3.84–3.76 (m, 1H, CH–OH), 3.72 (dd, J = 10.1, 4.1 Hz, 1H, CH2–OTBS), 3.67–3.58 (m, 2H, CH2–OTBS, CH–OTBS), 1.10 (d, J = 5.8 Hz, 3H, CH3), 0.91 (s, 9H, tBu-Si), 0.89 (s, 9H, tBu-Si), 0.09 (s, 3H, Si(CH3)2), 0.08 (s, 3H, Si(CH3)2), 0.05 (s, 6H, Si(CH3)2); 13C NMR (125 MHz, CDCl3): δ 155.7, 136.5, 131.2, 130.9, 130.4, 128.4, 128.0, 76.5, 71.9, 66.6, 65.2, 53.7, 25.7, 25.8, 19.8, 18.2, 17.9, −4.2, −4.8, −5.4; MS (ESI): m/z 524 (M + H)+; HRMS (ESI): m/z calcd for C27H50NO5Si2 (M + H)+, 524.3222; found 524.3228.
:1) to obtain compound 13 as a colourless liquid (1.9 g, 89%). Rf = 0.4 (petroleum ether:EtOAc = 9:1); [α]20D = −15.4 (c 1.00, CHCl3); IR (neat): νmax 3446, 2954, 2857, 1725, 1254, 1106, 836 cm−1; 1H NMR (400 MHz, CDCl3): δ 7.43–7.27 (m, 5H, Ph), 5.71 (dd, J = 15.8, 5.6 Hz, 1H, CHCH), 5.57 (dd, J = 15.7, 6.9 Hz, 1H, CHCH), 5.16–5.03 (m, 3H, CH2–Ph, –NH), 4.64 (A of AB q, J = 6.5 Hz, 1H, OCH2O), 4.57 (B of AB q, J = 6.6 Hz, 1H, OCH2O), 4.35–4.21 (m, 1H, CH–N), 3.89 (t, J = 6.4 Hz, 1H, CHOMOM), 3.85–3.76 (m, 1H, CH–OTBS), 3.70 (dd, J = 10.0, 4.3 Hz, 1H, CH2–OTBS), 3.64 (dd, J = 9.6, 3.5 Hz, 1H, CH2–OTBS), 3.34 (s, 3H, OCH3), 1.06 (d, J = 6.1 Hz, 3H, CH3), 0.89 (s, 9H, tBu-Si), 0.87 (s, 9H, tBu-Si), 0.06 (s, 6H, Si(CH3)2), 0.03 (s, 3H, Si(CH3)2), 0.03 (s, 3H, Si(CH3)2). 13C NMR (100 MHz, CDCl3): δ 155.7, 136.5, 131.8, 128.5, 128.4, 128.0, 94.4, 80.3, 70.4, 66.6, 65.2, 55.3, 53.7, 25.8, 25.8, 19.4, 18.2, 18.1, −4.6, −4.7, −5.5, −5.5; MS (ESI): m/z 590 (M + Na)+; HRMS (ESI): m/z calcd for C29H53NO6Si2Na (M+ Na)+, 590.3303; found 590.3304.
:3) gave 14 (1.23 g, 81%) as a colorless oil. Rf = 0.3 (petroleum ether:EtOAc = 7:3); [α]20D = −46.2 (c 1.70, CHCl3); IR (neat): νmax 3445, 3332, 2953, 2931, 1716, 1255, 1102, 838 cm−1; 1H NMR (400 MHz, CDCl3) δ 7.39–7.30 (m, 5H, Ph), 5.75 (dd, J = 15.7, 5.5 Hz, 1H, CHCH), 5.49 (dd, J = 15.6, 8.0 Hz, 1H, CHCH), 5.11 (s, 2H, CH2–Ph), 4.70 (d, J = 6.1 Hz, 1H, OCH2O), 4.55 (d, J = 6.4 Hz, 1H, OCH2O), 4.28 (m, 1H, CH–N), 3.79 (t, J = 7.6 Hz, 1H, CHOMOM), 3.74–3.62 (m, 3H, CH2–OTBS, CH–OH), 3.38 (s, 3H, OCH3), 2.03 (br s, 1H, OH), 1.12 (d, J = 6.0 Hz, 3H, CH3), 0.88 (s, 9H, tBu-Si), 0.04 (s, 3H, Si(CH3)2), 0.04 (s, 3H, Si(CH3)2). 13C NMR (100 MHz, CDCl3) δ 155.7, 136.4, 134.0, 128.5, 128.1, 93.9, 81.7, 69.5, 66.7, 65.0, 55.6, 53.7, 25.8, 18.4, 18.2, −5.5, −5.5; MS (ESI): m/z 476 (M + Na)+; HRMS (ESI): m/z calcd for C23H39NO6SiNa (M + Na)+, 476.2438; found 476.2445.
:2) to yield ketone 14-I as the product (175 mg, 88%). Rf = 0.7 (petroleum ether:EtOAc = 7:3); [α]20D = −28.8 (c 1.57, CHCl3); IR (neat): νmax 3441, 2940, 1716, 766 cm−1; 1H NMR (500 MHz, CDCl3): δ 7.38–7.29 (m, 5H, Ph), 5.91 (dd, J = 15.6, 5.5 Hz, 1H,CHCH), 5.61 (dd, J = 15.6, 6.6 Hz, 1H, CHCH), 5.15–5.07 (m, 1H, –NH), 5.09 (s, 2H, CH2–Ph), 4.68 (d, J = 6.4 Hz, 1H, OCH2O), 4.61 (d, J = 6.6 Hz, 1H, OCH2O), 4.52 (d, J = 6.6 Hz, 1H, CHOMOM), 4.34–4.26 (br m, 1H, CH–N), 3.70 (dd, J = 10.0, 4.3 Hz, 1H, CH2–OTBS), 3.65 (d, J = 7.2 Hz, 1H, CH2–OTBS), 3.35 (s, 3H, OCH3), 2.15 (s, 3H, COCH3), 0.86 (s, 9H, tBu-Si), 0.03 (s, 3H, Si(CH3)2), 0.02 (s, 3H, Si(CH3)2); 13C NMR (125 MHz, CDCl3) δ 206.1, 155.7, 136.3, 133.9, 128.4, 128.1, 125.7, 94.7, 82.0, 66.8, 64.9, 55.8, 53.6, 25.7, 18.2, −5.5; MS (ESI): m/z 452 (M + H)+; HRMS (ESI): m/z calcd for C23H37NO6SiNa (M+ Na)+, 474.2282; found 474.2286.
:3) gave 15 (817 mg, 88%) as a colorless oil. Rf = 0.5 (petroleum ether:EtOAc = 7:3); [α]20D = −19.5 (c 1.00, CHCl3); IR (neat): νmax 3451, 2955, 2932, 1709, 1103, 838 cm−1; 1H NMR (400 MHz, CDCl3) δ 4.71–4.66 (m, 2H, OCH2O), 3.71–3.63 (m, 1H, CH–OH), 3.59–3.52 (m, 2H, CH2–OTBS), 3.40 (s, 3H, OCH3), 3.29–3.22 (m, 1H, CH–N), 3.05–3.14 (m, 1H, CHOMOM), 1.70–1.57 (m, 2H, CH2–CH2), 1.50–1.38 (m, 11H, tBu in Boc, CH2–CH2), 1.13 (d, J = 6.4 Hz, 3H, CH3), 0.87 (s, 9H, tBu-Si), 0.03 (s, 6H, Si(CH3)2). 13C NMR (100 MHz, CDCl3) δ 155.5, 97.4, 85.4, 79.0, 69.1, 64.6, 55.7, 52.0, 28.3, 27.7, 27.1, 25.8, 18.8, 18.2, −5.4; MS (ESI): m/z 444 (M + Na)+; HRMS (ESI): m/z calcd for C20H44NO6Si (M + H)+, 422.2751; found 422.2761.
:3) to yield pale yellow oil 16 as the product (1 g, 85%). Rf = 0.6 (petroleum ether:EtOAc = 7:3); [α]20D = −6.9 (c 1.20, CHCl3); IR (neat): νmax 3392, 2933, 1712, 1361, 1176, 837 cm−1; 1H NMR (400 MHz, CDCl3) δ 4.87–4.59 (m, 3H, OCH2O, CHOMs), 3.69–3.49 (m, 4H, CH2–OTBS, CH–N, CHOMOM), 3.39 (s, 3H, OCH3), 3.02 (s, 3H, CH3 in Ms), 1.80–1.60 (m, 2H, CH2–CH2), 1.56–1.47 (m, 2H, CH2–CH2), 1.47–1.38 (m, 12H, tBu in Boc, CH3), 0.88 (s, 9H, tBu-Si), 0.04 (s, 6H, Si(CH3)2). 13C NMR (100 MHz, CDCl3) δ 155.6, 96.9, 79.6, 79.2, 79.1, 64.8, 55.9, 51.9, 38.4, 28.3, 26.9, 26.6, 25.8, 18.2, 16.8, −5.4; MS (ESI): m/z 522 (M + Na)+; HRMS (ESI): m/z calcd for C21H45NO8SSiNa (M + Na)+, 522.2527; found 522.2560.
:8) of diastereomers. Rf = 0.4 (petroleum ether:EtOAc = 9:1); 17: [α]20D = −8.7 (c 0.90, CHCl3); IR (neat): νmax 2954, 2932, 1691, 1367, 1099, 838 cm−1; 1H NMR (400 MHz, CDCl3) δ 4.70–4.61 (m, 2H, OCH2O), 4.25 (q, J = 6.8 Hz, 1H, 2-CH–N), 4.17–4.06 (m, 1H, 6-CH–N), 3.61–3.50 (m, 2H, CH2–OTBS, CHOMOM), 3.46 (dd, J = 9.3, 4.7 Hz, 1H, CH2–OTBS), 3.36 (s, 3H, OCH3), 1.97–1.56 (m, 4H, CH2–CH2), 1.45 (s, 9H, tBu in Boc), 1.07 (d, J = 7.1 Hz, 3H, CH3), 0.89 (s, 9H, tBu-Si), 0.06 (s, 3H, Si(CH3)2), 0.05 (s, 3H, Si(CH3)2). 13C NMR (100 MHz, CDCl3) δ 155.4, 94.7, 79.3, 73.5, 63.1, 55.3, 51.2, 50.0, 28.4, 25.8, 19.6, 19.3, 18.2, 17.3, −5.2, −5.4; MS (ESI): m/z 426 (M + Na)+; HRMS (ESI): m/z calcd for C20H42NO5Si (M + H)+, 404.2827; found 404.2858.
17a: [α]20D = −26.2 (c 1.01, CHCl3); 1H NMR (300 MHz, CDCl3) δ 4.62 (q, J = 7.0 Hz, 2H, OCH2O), 4.11 (qd, J = 7.0, 3.1 Hz, 1H, 2-CH–N), 3.87 (t, J = 9.5 Hz, 1H, CH2–OTBS), 3.75 (dd, J = 9.6, 4.2 Hz, 1H, CH2–OTBS), 3.69–3.55 (m, 2H, 6-CH–N, CHOMOM), 3.34 (s, 3H, OCH3), 2.00–1.83 (m, 2H, CH2–CH2), 1.81–1.69 (m, 2H, CH2–CH2), 1.44 (s, 9H, tBu in Boc), 1.22 (d, J = 7.1 Hz, 3H, CH3), 0.88 (s, 9H, tBu-Si), 0.04 (s, 6H, Si(CH3)2). 13C NMR (75 MHz, CDCl3) δ 155.7, 94.5, 79.2, 73.9, 62.9, 55.3, 53.3, 51.3, 28.4, 25.9, 22.9, 20.2, 18.2, 17.9, −5.2, −5.3.
:1) gave 4 (229 mg, 80%) as a colorless oil. Rf = 0.2 (petroleum ether:EtOAc = 6:4); [α]20D = −5.4 (c 1.10, CHCl3); IR (neat): νmax 3444, 2973, 2936, 1667, 1686, 1369, 1042 cm−1; 1H NMR (300 MHz, CDCl3) δ 4.67 (s, 2H, OCH2O), 4.29 (q, J = 6.9 Hz, 2H, CH–N), 3.68–3.52 (m, 3H, CH2–OH, CHOMOM), 3.37 (s, 3H, OCH3), 2.06–1.94 (m, 1H, CH2–CH2), 1.84–1.49 (m, 3H, CH2–CH2), 1.46 (s, 9H, tBu in Boc), 1.15 (d, J = 7.2 Hz, 3H, CH3). 13C NMR (75 MHz, CDCl3) δ 156.9, 94.9, 80.0, 73.5, 65.6, 55.4, 51.1, 50.5, 28.4, 19.7, 19.5, 18.4; MS (ESI): m/z 312 (M + Na)+; HRMS (ESI): m/z calcd for C14H27NO5Na (M + Na)+, 312.1781; found 312.1808.
:1) to give 18 (1.01 g, 94%) as a colorless oil. Rf = 0.4 (petroleum ether:EtOAc = 9:1); 1H NMR (300 MHz, CDCl3) δ 7.68–7.43 (m, 5H, Ar), 6.31 (dd, J = 15.0, 10.4 Hz, 1H, 3-CH), 5.97 (dd, J = 15.2, 10.4 Hz, 1H, 4-CH), 5.81–5.55 (m, 2H, 2-CH, 5-CH), 4.06 (d, J = 7.6 Hz, 2H, 1-CH2), 2.05 (q, J = 6.7 Hz, 2H, 6-CH2), 1.43–1.20 (m, 4H, CH2–CH2), 0.87 (t, J = 7.0 Hz, 3H, –CH3); 13C NMR (75 MHz, CDCl3) δ 153.8, 137.0, 135.8, 133.6, 130.0, 129.6, 128.7, 123.7, 122.8, 35.7, 32.2, 31.1, 22.1, 13.8; MS (ESI): m/z 323 (M + Na)+; HRMS (ESI): m/z calcd for C16H21N4S (M + H)+, 301.1481; found 301.1482.
:1) gave 3 (453 mg, 82%) as a colorless oil. Rf = 0.4 (petroleum ether:EtOAc = 9:1); IR (neat): νmax 2958, 2930, 2871, 1723, 1498, 1349, 1153, 763 cm−1; 1H NMR (500 MHz, CDCl3) δ 7.69–7.50 (m, 5H, Ar), 6.35 (dd, J = 15.2, 10.5 Hz, 1H, 3-CH), 6.01 (dd, J = 15.2, 10.5 Hz, 1H, 4-CH), 5.86–5.78 (m, 1H, 2-CH), 5.48 (dt, J = 15.3, 7.6 Hz, 1H, 5-CH), 4.41 (d, J = 7.6 Hz, 2H, 1-CH2), 2.09 (q, J = 7.0 Hz, 2H, 6-CH2), 1.40–1.24 (m, 4H, CH2–CH2), 0.89 (t, J = 7.2 Hz, 3H, –CH3); 13C NMR (125 MHz, CDCl3) δ 153.0, 142.4, 139.9, 132.9, 131.3, 129.5, 128.3, 125.2, 111.8, 60.0, 32.2, 30.9, 22.1, 13.8; MS (ESI): m/z 355 (M + Na)+; HRMS (ESI): m/z calcd for C16H21N4O2S (M + H)+, 333.1380; found 333.1404.
To a solution of sulfone 3 (82 mg, 0.24 mmol) and 18-crown-6 (64 mg, 0.24 mmol) in dry DME (5 mL) was added dropwise KHMDS (1 M in THF, 0.2 mL, 0.2 mmol) at −78 °C under a nitrogen atmosphere. After being stirred for 30 min, a solution of the above prepared aldehyde in dry DME (3 mL) was added slowly to the reaction mixture and stirred for 2 h at −78 °C before warming to rt and stirred overnight. The reaction mixture was poured into aqueous saturated NH4Cl solution (5 mL) and extracted with ethyl acetate (2 × 5 mL). The combined organic layer was washed with brine (5 mL), dried over Na2SO4, and the solvent was removed under reduced pressure. Flash chromatography of the crude over neutral alumina (5% EtOAc in hexanes) gave 19 (58 mg, 72%) as a colorless oil. Rf = 0.3 (petroleum ether:EtOAc = 9:1); [α]20D = −18.6 (c = 1.30, CHCl3); IR (neat): νmax 2930, 1688, 1365, 1038 cm−1; 1H NMR (300 MHz, CDCl3) δ 6.20–5.98 (m, 4H, CH–CHCH–CH), 5.76–5.62 (m, 2H, –CH2–CH, CH–CHN), 4.77 (br t, J = 5.1 Hz, 1H, 6-CH–N), 4.66 (q, J = 7.0 Hz, 2H, OCH2O), 4.31 (q, J = 7.0 Hz, 1H, 2-CH–N), 3.64–3.55 (m, 1H, CHOMOM), 3.36 (s, 3H, OCH3), 2.19–2.00 (m, 2H, CH–CH2), 1.90–1.54 (m, 3H, CH2–CH2), 1.45 (s, 9H, tBu in Boc), 1.50–1.22 (m, 5H, CH2–CH2), 1.11 (d, J = 7.2 Hz, 3H, CH–CH3), 0.88 (t, J = 7.0 Hz, 3H, CH2–CH3). 13C NMR (75 MHz, CDCl3) δ 155.4, 135.4, 134.7, 132.5, 130.3, 130.1, 130.1, 94.7, 79.4, 73.3, 55.3, 50.5, 50.4, 32.4, 31.4, 28.4, 22.1, 21.6, 19.8, 19.3, 13.8; MS (ESI): m/z 416 (M + Na)+; HRMS (ESI): m/z calcd for C23H40NO4 (M + H)+, 394.2952; found 394.2976.
:MeOH = 95:5); [α]20D = +5.6 (c 1.00, CH3OH); IR (neat): νmax 3445, 2924, 2854, 1660, 1127, 473 cm−1; 1H NMR (500 MHz, CDCl3) δ 6.22–6.11 (m, 2H, CH–CHCH–CH), 6.11–5.98 (m, 2H, CH–CHCH–CH), 5.73–5.66 (m, 1H, –CH2–CH), 5.60 (dd, J = 15.2, 7.1 Hz, 1H, CH–CHN), 3.23–3.14 (m, 2H, 6-CH–N, CHOH), 2.57–2.50 (m, 1H, 2-CH–N), 2.25 (br s, 1H, OH), 2.11–2.04 (m, 3H, CH–CH2, CH2–CH2), 1.77–1.71 (m, 1H, CH2–CH2), 1.40–1.28 (m, 6H, CH2–CH2), 1.20 (d, J = 6.1 Hz, 3H, CH–CH3), 0.89 (t, J = 7.1 Hz, 3H, CH2–CH3); 13C NMR (125 MHz, CDCl3) δ 135.6, 135.0, 132.9, 130.4, 130.1, 129.8, 73.6, 58.6, 58.3, 33.9, 32.4, 31.9, 31.4, 22.2, 18.9, 13.9; MS (ESI): m/z 250 (M + H)+; HRMS (ESI): m/z calcd for C16H28NO (M + H)+, 250.2165; found 250.2180.
Acknowledgements
The authors thank the Council of Scientific and Industrial Research (CSIR)-New Delhi for the award of research fellowship to BL and KW, and for financial support as part of XII-five year plan project under title ORIGIN (CSC-0108).Notes and references
- For selected reviews of piperidine alkaloids, see: (a) H. Makabe, Stud. Nat. Prod. Chem., 2014, 42, 353–371 CrossRef CAS; (b) I. Ojima and D. M. Iula, Alkaloids: Chemical and Biological Perspectives, Elsevier, Oxford, UK, 1999, vol. 13, p. 371–412 Search PubMed; (c) O. Plunkett and M. Sainsbury, in Rodd's Chemistry of Carbon Compounds, ed. M. Sainsbury, Elsevier, Amsterdam, 2nd edn, 1998, pp. 365–421, part F/part G (partial) Search PubMed; (d) M. J. Schneider, Alkaloids: Chemical and Biological Perspectives, Elsevier, Oxford, UK, 1996, vol. 10, pp. 155–299 Search PubMed; (e) S. R. Angle and J. G. Breitenbucher, Stud. Nat. Prod. Chem., 1995, 16, 453–502 CrossRef CAS; (f) G. M. Strunz and J. A. Findlay, in The Alkaloids, ed. A. Brossi, Academic, New York, NY, 1985, vol. 26, pp. 89–183 Search PubMed; (g) G. B. Fodor and B. Colasanti, in Alkaloids: Chemical and Biological Perspectives, ed. S. W. Pelletier, Wiley, New York, NY, 1985, vol. 3, pp. 1–90 Search PubMed; (h) T. H. Jones and M. S. Blum, in Alkaloids: Chemical and Biological Perspectives, ed. S. W. Pelletier, Wiley, New York, 1983, ch. 2, vol. 1, pp. 33–84 Search PubMed.
- Representative references, for isolation, see: (a) C. Viegas Jr., V. da S. Bolzani, M. Furlan, E. J. Barreiro, M. C. M. Young, D. Tomazela and M. N. Eberlin, J. Nat. Prod., 2004, 67, 908–910 CrossRef PubMed; (b) G. Kusano, S. Orihara, D. Tsukamoto, M. Shibano, M. Coskun, A. Guvenc and C. S. Erdurak, Chem. Pharm. Bull., 2002, 50, 185–192 CrossRef CAS PubMed; (c) R. J. Highet, J. Org. Chem., 1964, 29, 471–474 CrossRef CAS; (d) W. Y. Rice and J. L. Coke, J. Org. Chem., 1966, 31, 1010–1012 CrossRef CAS. For activity, see: (e) P. Sansores-Peraza, M. Rosado-allado, W. Brito-Loeza, G. J. Mena-Rejon and L. Quijano, Fitoterapia, 2000, 71, 690–692 CrossRef CAS PubMed; (f) S. L. Astudillo, S. K. Jurgens, G. SchmedaHirschmann, G. A. Griffith, D. H. Holt and P. R. Jenkins, Planta Med., 1999, 65, 161–162 CrossRef PubMed; (g) G. R. Cook, L. G. Beholz and J. R. Stille, J. Org. Chem., 1994, 59, 3575–3584 CrossRef CAS; (h) A. M. Aguinaldo and R. W. Read, Phytochemistry, 1990, 29, 2309–2313 CrossRef CAS; (i) A. Ahmad, K. A. Khan, V. U. Ahmad and S. Qazi, Planta Med., 1986, 4, 285–288 CrossRef PubMed; (j) G. B. Fodor and B. Colasanti, in Alkaloids: Chemical and Biological Perspectives, ed. S. W. Pelletier, Wiley, New York, 1985, ch. 1, vol. 3, pp. 1–90 Search PubMed; (k) G. Fodor, J.-P. Fumeaux and V. Sankaran, Synthesis, 1972, 464–472 CrossRef CAS; (l) P. Bourinet and A. Quevauviller, Compt. Rend. Soc. Biol., 1968, 162, 1138–1140 Search PubMed; (m) P. Bourinet and A. Quevauviller, Ann. Pharm. Fr., 1968, 26, 787–796 Search PubMed.
- Selected recent references for synthesis, see: (a) F. V. D. Pijl, F. L. V. Delft and F. P. J. T. Rutjes, Eur. J. Org. Chem., 2015, 4811–4829 CrossRef; (b) K.-J. Xiao, Y. Wang, Y.-H. Huang, X.-G. Wang and P.-Q. Huang, J. Org. Chem., 2013, 78, 8305–8311 CrossRef CAS PubMed; (c) M. A. Wijdeven, J. Willemsen and F. P. J. T. Rutjes, Eur. J. Org. Chem., 2010, 2831–2844 CrossRef CAS; (d) S. D. Koulocheri, E. N. Pitsinos and S. A. Haroutounian, Curr. Org. Chem., 2008, 12, 1454–1467 CrossRef CAS; (e) L.-X. Liu, Y.-P. Ruan, Z.-Q. Guo and P.-Q. Huang, J. Org. Chem., 2004, 69, 6001 CrossRef CAS PubMed; (f) D. Maa and N. Ma, Tetrahedron Lett., 2003, 44, 3963–3965 CrossRef; (g) R. Singh and S. K. Ghosh, Tetrahedron Lett., 2002, 43, 7711–7715 CrossRef CAS; (h) H. Zhai, M. Parvez and T. G. Back, J. Org. Chem., 2007, 72, 3853–3858 CrossRef CAS PubMed; (i) G. Kim and N. Kim, Tetrahedron Lett., 2007, 48, 4481–4483 CrossRef CAS; (j) S. Yu, X. Pu, T. Cheng, R. Wang and D. Ma, Org. Lett., 2006, 8, 3179–3182 CrossRef CAS PubMed; (k) X. Pu and D. Ma, J. Org. Chem., 2006, 71, 6562–6572 CrossRef CAS PubMed; (l) B. B. Snider and B. J. Neubert, Org. Lett., 2005, 7, 2715–2718 CrossRef CAS PubMed; (m) J. Lofstedt, H. Pettersson-Fasth and J.-E. Backvall, Tetrahedron, 2000, 56, 2225–2230 CrossRef CAS; (n) N. Toyooka, Y. Yoshida, Y. Yotsui and T. Momose, J. Org. Chem., 1999, 64, 4914–4919 CrossRef CAS PubMed.
- (a) N. Lindquist, N. Shigematsu and L. Pannell, J. Nat. Prod., 2000, 63, 1290–1291 CrossRef CAS PubMed; (b) M. McCrea-Hendrick and C. J. Nichols, Synth. Commun., 2009, 39, 3611–3620 CrossRef CAS.
- P. C. Still, B. Yi, T. F. G. Cestari, L. Pan, R. E. Pavlovicz, H. B. Chai, T. N. Ninh, C. Li, D. D. Soejarto, D. B. McKay and A. D. Kinghorn, J. Nat. Prod., 2013, 76, 243–249 CrossRef CAS PubMed.
- S. X. Feng, L. D. Lin, H. H. Xu and X. Y. Wei, J. Asian Nat. Prod. Res., 2008, 10, 1155–1158 CrossRef CAS PubMed.
- Y. Saitoh, Y. Moriyama, H. Hirota, T. Takahashi and Q. Khuong-Huu, Bull. Chem. Soc. Jpn., 1981, 54, 488 CrossRef CAS.
- (a) C. R. Reddy, M. D. Reddy and U. Dilipkumar, Eur. J. Org. Chem., 2014, 6310–6313 CrossRef CAS; (b) C. R. Reddy, B. Latha and N. N. Rao, Tetrahedron, 2012, 68, 145–151 CrossRef CAS; (c) C. R. Reddy and B. Latha, Tetrahedron: Asymmetry, 2011, 22, 1849–1854 CrossRef CAS.
- (a) L. J. Dorr, in Flora of China, ed. Z. Y. Wu, P. H. Raven and D. Y. Hong, Science Press, Beijing, and Missouri Botanical Garden Press, St. Louis, MO, 2007, vol. 12, pp. 251–258 Search PubMed; (b) T. Ya, M. G. Gilbert and L. J. Dorr, Tiliaceae, eFlora China 12, 2007 Search PubMed; (c) State Administration of Traditional Chinese Medicine “Chinese Herbal Medicine”, the Editorial Committee of Codification, Chinese Herbal Medicine, Shanghai Science and Technology Press, Shanghai, 1999, vol. 5, pp. 324–326 Search PubMed.
- (a) J. Luo, L. Zhang, M. F. Roberts and J. D. Phillipson, Acta Pharm. Sin., 2009, 44, 150–153 CAS; (b) K. A. N. P. Bandara, V. Kumar, U. Jacobsson and L.-P. Molleyres, Phytochemistry, 2000, 54, 29–32 CrossRef CAS PubMed; (c) A. M. Aguinaldo and R. W. Read, Phytochemistry, 1990, 29, 2309–2313 CrossRef CAS.
- (a) G. E. Keck, K. A. Savin and M. A. Weglarz, J. Org. Chem., 1995, 60, 3194–3204 CrossRef CAS; (b) P. J. Kocienski, Phosphorus Sulfur, 1985, 24, 97–127 CrossRef; (c) M. Julia, J. Launay, S. Verpeaux and J. Verpeaux, Tetrahedron Lett., 1982, 23, 2465–2472 CrossRef CAS; (d) P. J. Kocienski, B. Lythgoe and S. Ruston, J. Chem. Soc., Perkin Trans. 1, 1978, 829–834 RSC; (e) M. Julia and J. M. Paris, Tetrahedron Lett., 1973, 14, 4833–4836 CrossRef.
- Experimental procedures for preparation of the conjugated alcohol 5 from 1-octyne 8 are described in the ESI (Scheme S1†). For reference, see: (a) S. D. Rychnovsky and J. Kim, J. Org. Chem., 1994, 59, 2659–2660 CrossRef CAS; (b) U. Kazmaier, Tetrahedron, 1998, 54, 1491–1496 CrossRef CAS.
- (a) I. Paterson, K. S. Yeung and J. B. Smaill, Synlett, 1993, 774–776 CrossRef CAS; (b) W. S. Wadsworth and W. D. Emmons, J. Am. Chem. Soc., 1961, 83, 1733–1738 CrossRef CAS; (c) L. Horner, H. M. R. Hoffmann and H. G. Wippel, Ber., 1958, 91, 61–63 CrossRef CAS.
- (a) A. L. Gemal and J. L. Luche, J. Am. Chem. Soc., 1981, 103, 5454–5459 CrossRef CAS; (b) J. L. Luche, J. Am. Chem. Soc., 1978, 100, 2226–2227 CrossRef CAS.
- (a) H. Tamamura, Y. Koh, S. Ueda, Y. Sasaki, T. Yamasaki, M. Aoki, K. Maeda, Y. Watai, H. Arikuni, A. Otaka, H. Mitsuya and N. Fujii, J. Med. Chem., 2003, 46, 1764–1768 CrossRef CAS PubMed; (b) E. J. Jacobsen, et al. , J. Med. Chem., 1999, 42, 1525–1536 CrossRef CAS PubMed; (c) A. W. Konradi, et al. , J. Am. Chem. Soc., 1994, 116, 1316–1323 CrossRef CAS.
- M. Rodriquez, I. Bruno, E. Cini, M. Marchetti, M. Taddei and L. Gomez-Paloma, J. Org. Chem., 2006, 71, 103–107 CrossRef CAS PubMed.
- Oxidation of compound 14 under IBX conditions smoothly gave ketone 14-I in 88% yield. Further, hydrogenation of compound 14-I in the presence of H2/Pd–C (10 wt%) in EtOH gave the complex mixture of diastereomeric products 14-II along with the starting material.
. - (a) A. Biela, F. Oulaïdi, E. Gallienne, M. Gorecki, J. Frelek and O. R. Martin, Tetrahedron, 2013, 69, 3348–3354 CrossRef CAS; (b) B. Chandrasekhar, J. P. Rao, B. V. Rao and P. Naresh, Tetrahedron Lett., 2011, 52, 5921–5925 CrossRef CAS; (c) C. Alegret, X. Ginesta and A. Riera, Eur. J. Org. Chem., 2008, 1789–1796 CrossRef CAS.
- The 2D COSY and NOESY spectra of compounds 17 and 17a are included in the ESI.†.
- O. Mitsunobu, Synthesis, 1981, 1–28 CrossRef CAS.
- H. S. Schultz, H. B. Freyermuth and S. R. Buc, J. Org. Chem., 1963, 28, 1140–1142 CrossRef CAS.
- (a) J. Pospisil, Tetrahedron Lett., 2011, 52, 2348–2352 CrossRef CAS; (b) Y. Nakatani, J. Oshita, K. Ishigami, H. Watandbe and T. Kitahara, Tetrahedron, 2006, 62, 160–165 CrossRef CAS; (c) P. R. Blakemorea, W. J. Colea, P. J. Kocienski and A. Morley, Synlett, 1998, 26–28 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5ob02085a |
| This journal is © The Royal Society of Chemistry 2016 |