
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Enantioselective synthesis of (−)-chloramphenicol via silver-catalysed asymmetric isocyanoacetate aldol reaction†
Allegra
Franchino
,
Pavol
Jakubec
and
Darren J.
Dixon
*
Chemistry Research Laboratory, Department of Chemistry, University of Oxford, 12 Mansfield Road, Oxford, OX1 3TA, UK. E-mail: darren.dixon@chem.ox.ac.uk
First published on 29th October 2015
Abstract
The highly enantio- and diastereoselective aldol reaction of isocyanoacetates catalysed by Ag2O and cinchona-derived amino phosphines applied to the synthesis of (−)- and (+)-chloramphenicol is described. The concise synthesis showcases the utility of this catalytic asymmetric methodology for the preparation of bioactive compounds possessing α-amino-β-hydroxy motifs.
Chiral vicinal amino alcohols represent a very important class of compounds, which are of interest to synthetic chemists not only as valuable building blocks and chiral auxiliaries, but also by virtue of their pharmacological properties.1 Bioactive vicinal amino alcohols of differing complexity include the broad spectrum antibiotics chloramphenicol2 (1) and thiamphenicol3 (2), the protease inhibitor for HIV treatment saquinavir4 (3) and the antihypertensive drug aliskiren5 (4, Fig. 1). Among others,1,6 a privileged access to these structures is offered by the aldol reaction of glycine equivalents,7 including isocyanoacetates,8 followed by reduction of the carboxylic group. Recently, our group developed a cooperative catalytic system consisting of a Lewis acid (Ag+) and a cinchona-derived amino phosphine ligand, bearing both Brønsted and Lewis basic sites, for the activation of isocyanoacetate pronucleophiles towards electrophiles, such as aldehydes,9 ketimines10 and ketones.11
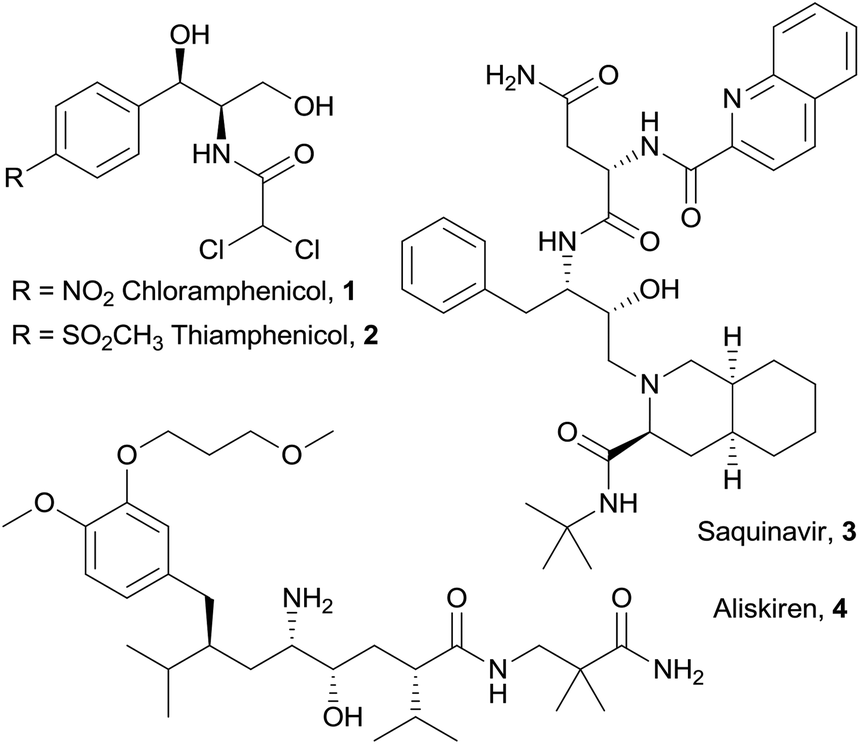 | ||
| Fig. 1 Selected pharmaceuticals containing vicinal amino alcohol fragments. | ||
As a direct demonstration of the utility of this asymmetric methodology, herein we report a short asymmetric synthesis of (−)-chloramphenicol,12 which is the first one relying on a catalytic enantio- and diastereoselective aldol reaction.13 According to our retrosynthetic plan, outlined in Scheme 1, (−)-chloramphenicol would be derived through standard chemical manipulations from the trans oxazoline (4S,5R)-6.14 It was envisioned that the latter could be obtained via the Ag-catalysed asymmetric isocyanoacetate aldol reaction (IAR) between a suitable isocyanoacetate ester157 and 4-nitrobenzaldehyde (8). Specifically, on the basis of our previous work9 it was anticipated that the use of cinchonine-derived amino phosphine L-1 as chiral ligand in the IAR would provide oxazoline 6 with the desired absolute configuration for the preparation of (−)-chloramphenicol (Table 1).
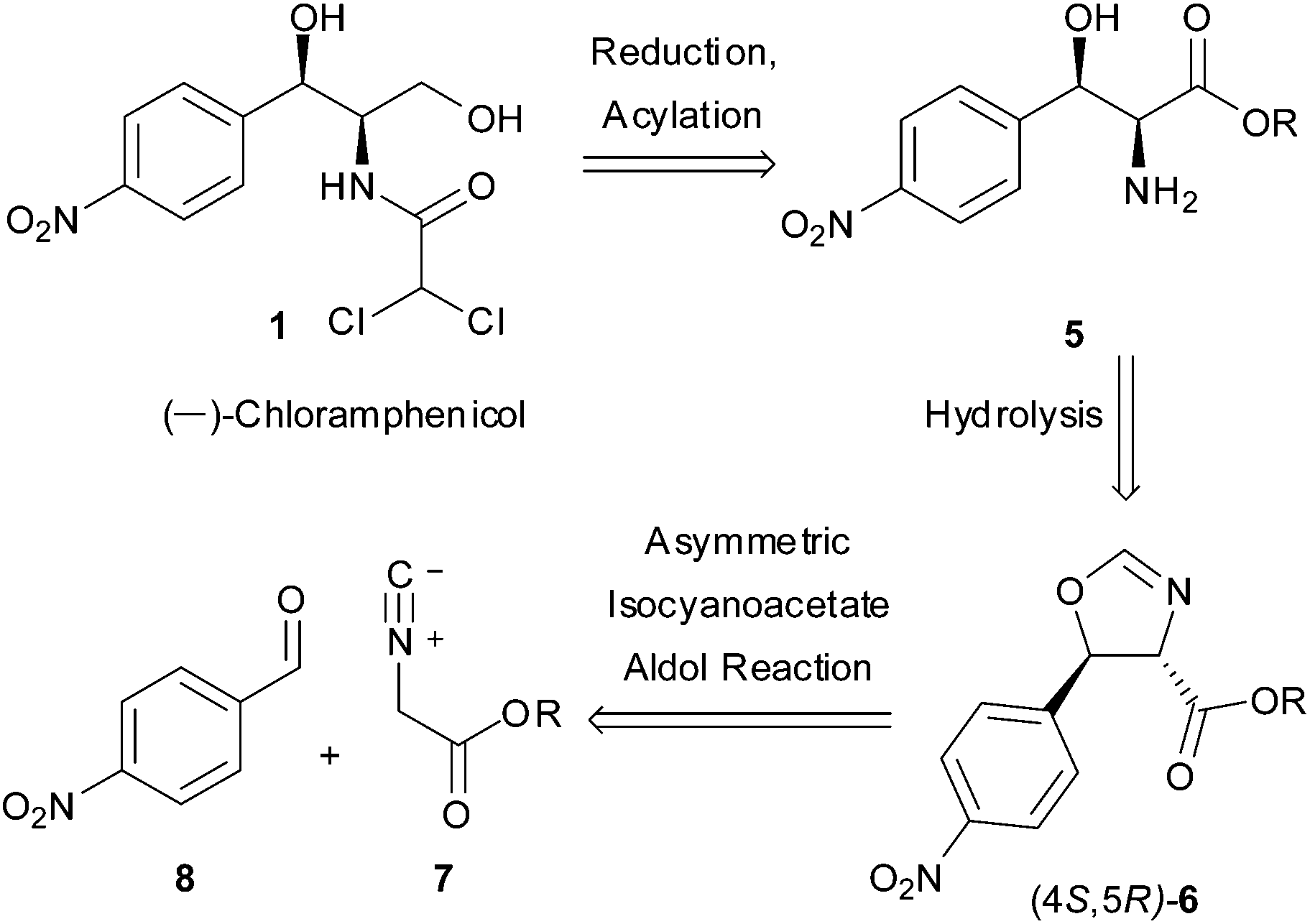 | ||
| Scheme 1 Retrosynthetic approach to (−)-chloramphenicol. | ||
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) a
a
|
|
||||||
|---|---|---|---|---|---|---|
| Entry | T (°C) | [7a] (M) | Time (h) | Yieldb (%) | d.r.c (trans:cis) |
e.e.d (%) |
| a Reaction performed on 0.25 mmol of 7a using 1.1 eq. of 8. Configuration of 6a assigned by analogy with previous work.9 b Isolated yield of trans diastereomer after FCC. c d.r. determined by 1H NMR analysis of the crude reaction mixture. d e.e. of trans diastereomer determined by HPLC on chiral stationary phase. e 0.50 mmol of 7a. f Stirred overnight, as TLC control after 3 hours indicated that the reaction was progressing. g Stirred overnight, as TLC control after 6 hours indicated that the reaction was progressing. | ||||||
| 1e | −20 | 0.3 | 21f | 45 | 91:9 |
44 |
| 2e | 20 | 0.3 | 0.25 | 59 | 91:9 |
53 |
| 3 | 20 | 0.05 | 0.5 | 73 | 91:9 |
69 |
| 4 | 20 | 0.01 | 0.5 | 70 | 91:9 |
78 |
| 5 | 20 | 0.0025 | 17g | 70 | 90:10 |
80 |
| 6 | 0 | 0.01 | 16f | 72 | 90:10 |
72 |
| 7 | 50 | 0.01 | 2 | 61 | 88:12 |
65 |
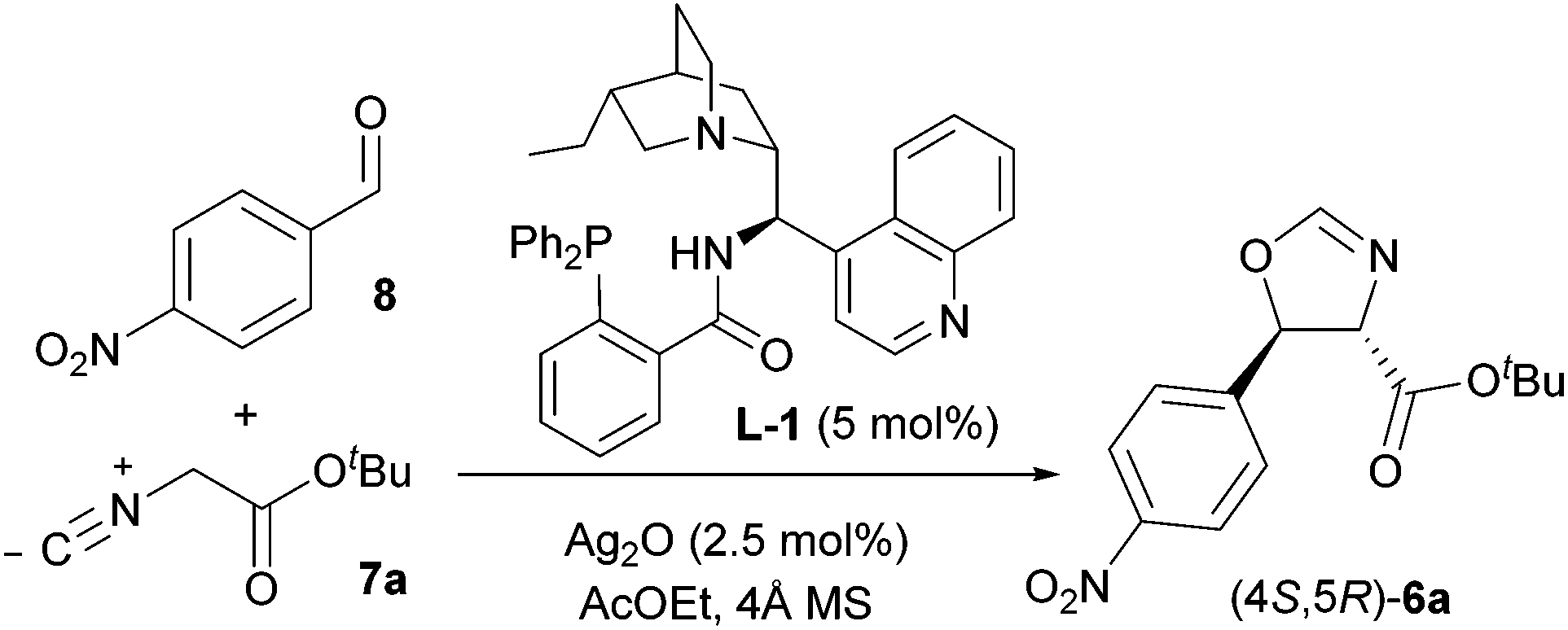
Our investigation thus began by performing the reaction between 8 and tert-butyl isocyanoacetate 7a under the conditions that had already been optimised for a range of aldehydes, namely in AcOEt at −20 °C and 0.3 M concentration, employing 5 mol% L-1 and 2.5 mol% Ag2O. To our surprise, the desired trans oxazoline 6a was obtained with modest yield (45%) and low enantioselectivity (44% e.e., Table 1, entry 1), pointing out the need for optimisation of the IAR. Screening of several reaction parameters (concentration, temperature, solvent, nature of the isocyanoacetate ester group, structure of the ligand and Ag/ligand ratio) was therefore undertaken, and the main findings are reported below.
Adjustment of the temperature to 20 °C was beneficial both for yield (59%) and enantiocontrol (53% e.e., entry 2). At this temperature, dilution of the reaction mixture to 0.01 M isocyanoacetate concentration afforded the desired product 6a in 30 minutes with good yield and stereoselectivity (70% yield, 91:9 d.r., 78% e.e., entry 4). These conditions improved solubility, diminishing a competitive non-asymmetric background reaction catalysed by Ag2O only, which we hypothesized was responsible for the poor enantioselectivity observed at lower temperature and higher concentration. At the same time, dilution of the reaction mixture increased the yield of the product by reducing the amount of undesired double aldol side product. Further dilution to 0.0025 M resulted in slightly improved enantiocontrol (80% e.e., entry 5) over longer reaction time, but it was discarded for practical scale-up reasons. A temperature screen at 0.01 M (entries 6 and 7) revealed that 20 °C was the optimal temperature. A quick solvent survey confirmed AcOEt to be optimal (see ESI, Table S1†).
With the optimised reaction conditions established, the performance of isocyanoacetates with different ester groups was then investigated (Table 2). Methyl isocyanoacetate 7b (entry 2) performed better than its tert-butyl analogue 7a (entry 1), suggesting that excessive steric bulk hampered the transmission of stereochemical information. However, the presence of a benzyl or benzhydryl group was well-tolerated: from isocyanoacetates 7c and 7f the desired oxazolines could be obtained with the highest enantioselectivity (87% e.e., entries 3 and 7). Isocyanoacetates 7d and 7e, possessing 4-methoxybenzyl and 3,5-bis(trifluoromethyl)benzyl groups respectively, provided enantioenriched oxazolines with similar e.e. (86% and 84% e.e. respectively, entries 5 and 6), suggesting that electronic factors didn't play a major role in enantiocontrol.
a
|
|
|||||||
|---|---|---|---|---|---|---|---|
| Entry | R | 7 | Ligand | Time (min) | Yieldb (%) | d.r.c (trans:cis) |
e.e.d (%) |
| a Reaction performed on 0.25 mmol of 7 (0.01 M in AcOEt) using 1.1 eq. of 8. Configuration of 6 assigned by analogy with previous work.9 b Isolated yield of trans diastereomer after FCC. c d.r. determined by 1H NMR analysis of the crude reaction mixture. d e.e. of trans diastereomer determined by HPLC on chiral stationary phase. e Opposite enantiomer obtained. | |||||||
| 1 | (CH3)3C | 7a | L-1 | 30 | 70 | 91:9 |
78 |
| 2 | CH3 | 7b | L-1 | 100 | 80 | 88:12 |
82 |
| 3 | PhCH2 | 7c | L-1 | 180 | 61 | 90:10 |
87 |
| 4 | PhCH2 | 7c | L-2 | 80 | 64 | 90:10 |
87 |
| 5 | 4-(OCH3) C6H4CH2 | 7d | L-2 | 60 | 63 | 89:11 |
86 |
| 6 | 3,5-(CF3)2 C6H3CH2 | 7e | L-2 | 60 | 56 | 90:10 |
84 |
| 7 | Ph2CH | 7f | L-1 | 100 | 81 | 93:7 |
87 |
| 8 | Ph2CH | 7f | L-2 | 45 | 78 | 91:9 |
89 |
| 9 | Ph2CH | 7f | L-3 | 200 | 82 | 93:7 |
88e |
| 10 | Ph2CH | 7f | L-4 | 60 | 68 | 92:8 |
93e |
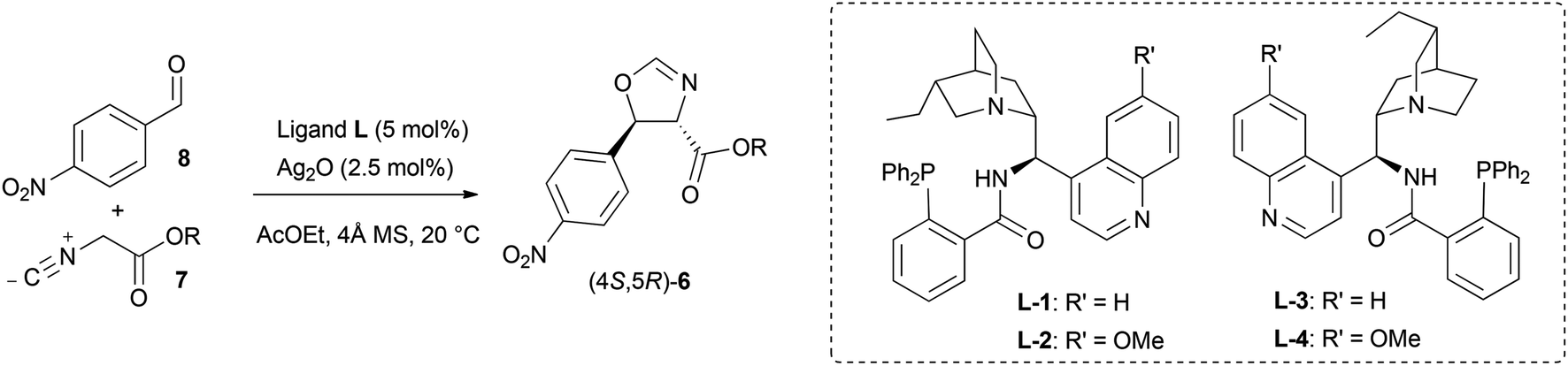
Next the effect of fine tuning of the ligand was taken into account by testing four amino phosphines prepared from 9-amino(9-deoxy) epicinchona alkaloids (Table 2). The use of quinidine-derived L-2 in the reaction between the benzhydryl isocyanoacetate 7f and 8 resulted in further improved enantiocontrol (89% e.e., entry 8), whereas no boost in enantioselectivity was observed starting from benzyl isocyanoacetate 7c (87% e.e., entry 4). The pseudoenantiomeric catalytic system comprising quinine-derived L-4 afforded the enantiomeric oxazoline (4R,5S)-6f in slightly lower yield (68%) and better stereocontrol (92:8 d.r., 93% e.e., entry 10).
Finally catalyst loading studies confirmed the ideal Ag/ligand ratio to be 1:1, specifically with 2.5 mol% Ag2O and 5 mol% L-2 (see ESI, Table S2†).
After having successfully improved yield and stereocontrol for the isocyanoacetate aldol reaction, oxazoline (4S,5R)-6f was prepared on 2.5 mmol scale with 72% yield and 89% e.e.,16 and then was readily elaborated to the target molecule (Scheme 2). Ring opening of 6f using thionyl chloride in methanol proceeded with 75% yield to afford amino alcohol 5, whose enantiomeric purity could be improved to 98% e.e. by a single recrystallisation from toluene (61% yield, first crop). The amino alcohol was then acylated with dichloroacetyl chloride to provide dichloroacetamide 9 in 83% yield. Finally, chemoselective reduction of the ester group with excess sodium borohydride delivered (−)-chloramphenicol in 80% yield and 99% e.e.17 (+)-Chloramphenicol was prepared in an analogous manner from oxazoline (4R,5S)-6f.17
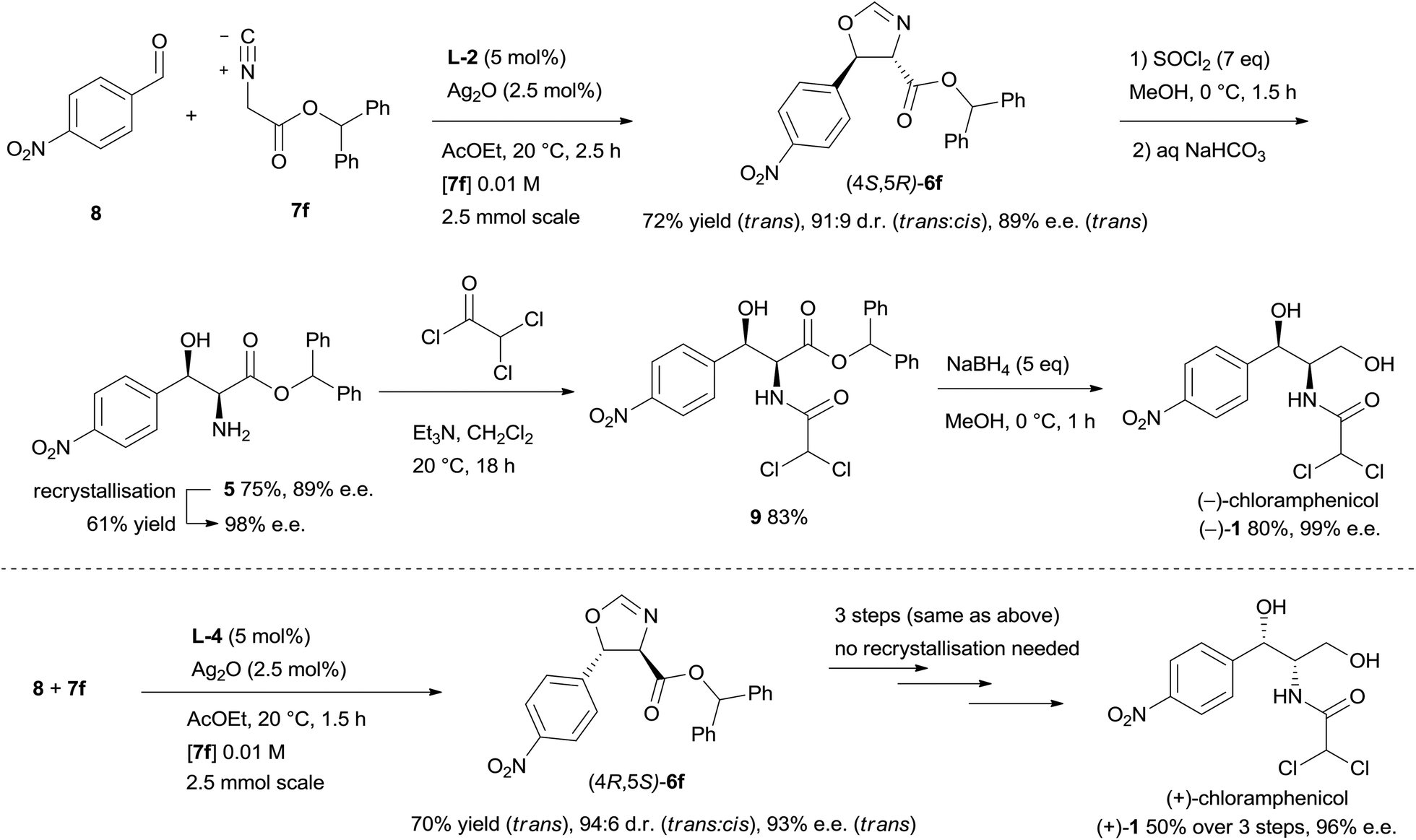 | ||
| Scheme 2 Synthesis of (−)- and (+)-chloramphenicol. | ||
In summary, a catalytic asymmetric synthesis of (−)-chloramphenicol has been accomplished, delivering the target molecule in 4 steps and 22% yield calculated from 4-nitrobenzaldehyde. The concise synthetic route relies on the enantio- and diastereoselective aldol reaction of isocyanoacetates catalysed by Ag2O and cinchona-derived amino phosphine ligands. Extensive screening of the reaction parameters has been undertaken to optimise the key step, eventually achieving the formation of the two contiguous stereocentres of the target molecule with good enantiocontrol. The present work demonstrates the utility of this asymmetric methodology for the preparation of bioactive molecules bearing an α-amino-β-hydroxy motif.
The authors gratefully acknowledge the EPSRC (leadership fellowship to D.J.D. and postdoctoral fellowship to P.J.) and the People Programme (Marie Curie Actions) of the European Union's Seventh Framework Programme (A.F., FP7/2007-2013, REA grant agreement no. 316955).
Notes and references
- S. C. Bergmeier, Tetrahedron, 2000, 56, 2561 CrossRef CAS.
- (a) J. Ehrlich, Q. R. Bartz, R. M. Smith, D. A. Joslyn and P. R. Burkholder, Science, 1947, 106, 417 CAS; (b) M. C. Rebstock, H. M. Crooks, J. Controulis and Q. R. Bartz, J. Am. Chem. Soc., 1949, 71, 2458 CrossRef CAS.
- R. A. Cutler, R. J. Stenger and C. M. Suter, J. Am. Chem. Soc., 1952, 74, 5475 CrossRef CAS.
- E. de Clercq, J. Clin. Virol., 2004, 30, 115 CrossRef CAS PubMed.
- J. M. Wood, J. Maibaum, J. Rahuel, M. G. Grütter, N.-C. Cohen, V. Rasetti, H. Rüger, R. Göschke, S. Stutz, W. Fuhrer, W. Schilling, P. Rigollier, Y. Yamaguchi, F. Cumin, H.-P. Baum, C. R. Schnell, P. Herold, R. Mah, C. Jensen, E. O'Brien, A. Stanton and M. P. Bedigian, Biochem. Biophys. Res. Commun., 2003, 308, 698 CrossRef CAS PubMed.
- O. K. Karjalainen and A. M. P. Koskinen, Org. Biomol. Chem., 2012, 10, 4311 CAS.
- (a) J. Patel, G. Clavé, P.-Y. Renard and X. Franck, Angew. Chem., Int. Ed., 2008, 47, 4224 CrossRef CAS PubMed and references therein; (b) A. Jakubowska and K. Kulig, Curr. Org. Synth., 2013, 10, 547 CAS.
- (a) S. Kohta and S. Halder, Synlett, 2010, 337 Search PubMed; (b) A. V. Gulevich, A. G. Zhdanko, R. V. A. Orru and V. G. Nenajdenko, Chem. Rev., 2010, 110, 5235 CrossRef CAS PubMed; (c) S. Chakrabarty, S. Choudhari, A. Doshi, F.-Q. Liu, R. Mohan, M. P. Ravindra, D. Shah, X. Yang and F. F. Fleming, Adv. Synth. Catal., 2014, 356, 2135 CrossRef CAS PubMed; (d) V. P. Boyarskiy, N. A. Bokach, K. V. Luzyanin and V. Y. Kukushkin, Chem. Rev., 2015, 115, 2698 CrossRef CAS PubMed.
- F. Sladojevich, A. Trabocchi, A. Guarna and D. J. Dixon, J. Am. Chem. Soc., 2011, 133, 1710 CrossRef CAS PubMed.
- I. Ortín and D. J. Dixon, Angew. Chem., Int. Ed., 2014, 53, 3462 CrossRef PubMed.
- R. de la Campa, I. Ortín and D. J. Dixon, Angew. Chem., Int. Ed., 2015, 54, 4895 CrossRef CAS PubMed.
- For racemic syntheses of chloramphenicol see: (a) J. Controulis, M. C. Rebstock and H. M. Crooks, J. Am. Chem. Soc., 1949, 71, 2463 CrossRef CAS; (b) L. M. Long and H. D. Troutman, J. Am. Chem. Soc., 1949, 71, 2469 CrossRef CAS; (c) L. M. Long and H. D. Troutman, J. Am. Chem. Soc., 1949, 71, 2473 CrossRef CAS; (d) G. Ehrhart, W. Siedel and H. Nahm, Chem. Ber., 1957, 90, 2088 CrossRef CAS; (e) B. G. Hazra, V. S. Pore and S. P. Maybhate, Synth. Commun., 1997, 27, 1857 CrossRef CAS. For asymmetric syntheses of (−)-chloramphenicol using chiral auxiliaries see: (f) U. Schöllkopf and T. Beulshausen, Liebigs Ann. Chem., 1989, 223 CrossRef; (g) S. Hajra, A. Karmakar, T. Maji and A. K. Medda, Tetrahedron, 2006, 62, 8959 CrossRef CAS; (h) Q. Li, H. Zhang, C. Li and P. Xu, Chin. J. Chem., 2013, 31, 149 CrossRef. For asymmetric syntheses of (−)-chloramphenicol starting from enantiopure aminoacids see: (i) C. J. Easton, C. A. Hutton, M. C. Merrett and E. R. T. Tiekink, Tetrahedron, 1996, 52, 7025 CrossRef CAS; (j) G. Veeresa and A. Datta, Tetrahedron Lett., 1998, 39, 8503 CrossRef CAS. For asymmetric syntheses of (−)-chloramphenicol based on Sharpless epoxidation see: (k) A. V. R. Rao, S. P. Rao and M. N. Bhanu, J. Chem. Soc., Chem. Commun., 1992, 859 RSC; (l) B.-L. Lou, Y.-Z. Zhang and L.-X. Dai, Chem. Ind., 1993, 7, 249 Search PubMed; (m) G. Bhaskar, V. S. Kumar and B. V. Rao, Tetrahedron: Asymmetry, 2004, 15, 1279 CrossRef CAS; (n) S. George, S. V. Narina and A. Sudalai, Tetrahedron, 2006, 62, 10202 CrossRef CAS. For asymmetric syntheses of (−)-chloramphenicol based on Sharpless dihydroxylation see: (o) J. N. Park, S. Y. Ko and H. Y. Koh, Tetrahedron Lett., 2000, 41, 5553 CrossRef CAS; (p) J. Boruwa, J. C. Borah, S. Gogoi and N. C. Barua, Tetrahedron Lett., 2005, 46, 1743 CrossRef CAS. For other asymmetric syntheses of (−)-chloramphenicol see: (q) R. Chênevert and S. Thiboutot, Synthesis, 1989, 444 CrossRef (enzymatic kinetic resolution); (r) C. Loncaric and W. D. Wulff, Org. Lett., 2001, 3, 3675 CrossRef CAS PubMed (catalytic enantioselective aziridination).
- For an asymmetric synthesis of chloramphenicol relying on a diazaborolidine-mediated aldol reaction, proceeding via a chiral boron enolate, see: E. J. Corey and S. Choi, Tetrahedron Lett., 2000, 41, 2765 CrossRef CAS.
- For previous work on the synthesis of oxazolines and their hydrolysis to vicinal amino alcohols, see: (a) Y. Ito, M. Sawamura and T. Hayashi, J. Am. Chem. Soc., 1986, 108, 6405 CrossRef CAS; (b) D. Hoppe and U. Schöllkopf, Liebigs Ann. Chem., 1972, 763, 1 CrossRef CAS; (c) U. Schöllkopf and K.-H. Scheunemann, Liebigs Ann. Chem., 1980, 1348 CrossRef.
- The preparation of isocyanoacetates 7a–f from commercial reagents over three steps is detailed in the ESI.†.
- Removal of 4 Å molecular sieves had no detrimental effect on the reaction.
- The observed slight upgrade in e.e. between 9 and 1 is linked to the chromatographic purification process.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures, characterisation data, copies of 1H and 13C NMR spectra, and HPLC traces. See DOI: 10.1039/c5ob02141c |
| This journal is © The Royal Society of Chemistry 2016 |