
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Investigation of benzoyloximes as benzoylating reagents: benzoyl-Oxyma as a selective benzoylating reagent†
Satvika
Burugupalli
,
Sayali
Shah
,
Phillip L.
van der Peet
,
Seep
Arora
,
Jonathan M.
White
and
Spencer J.
Williams
*
School of Chemistry and Bio21 Molecular Science and Biotechnology Institute, University of Melbourne, Parkville, Victoria, Australia 3010. E-mail: sjwill@unimelb.edu.au
First published on 29th October 2015
Abstract
Hydroxybenzotriazole (HOBt) and HOBt-derived reagents have been classified as Class I explosives, with restrictions on their transportation and storage. We explored a range of benzoylated oxime-based reagents as alternatives to benzoyloxybenzotriazole (BBTZ) for the selective benzoylation of carbohydrate polyols. Benzoylated oximes derived from 2-hydroximino-malononitrile, ethyl 2-hydroximino-2-cyanoacetate (Oxyma), and tert-butyl 2-hydroximino-2-cyanoacetate were most effective for benzoylation of a simple primary alcohol, with yields approaching that obtained for BBTZ. When applied to carbohydrate diols, the most effective reagent was identified as benzoyl-Oxyma. Benzoyl-Oxyma is a highly crystalline, readily prepared alternative to BBTZ, useful in the selective benzoylation of carbohydrate polyols.
Introduction
Benzoic acid esters are widely used hydroxyl protecting groups for polyhydroxylated substrates such as nucleosides, sugars, and inositols owing to their ease of introduction and removal, and their stability to a wide range of reaction conditions.1,2 In the context of carbohydrate chemistry, benzoates can be used to alter stereoselectivity of glycosylations by providing anchimeric assistance,3 and to attenuate reactivity according to the armed/disarmed principle.4 The selective benzoylation of a polyol5 can be achieved by careful control of reaction conditions6 and through use of a range of selective benzoylating reagents including benzoyl cyanide,7,8N-benzoyltetrazole,9–11N-benzoylimidazole,9,10,12 Mitsunobu conditions,13 benzoyl chloride/collidine,14 and BzCl in combination with organotin reagents.15 However, depending on the substrate or reagent, various problems emerge including poor solubility for many polyol substrates at the low temperatures sometimes required, high toxicity, poor reactivity, and/or poor regioselectivity. Among the most reliable and widely-used selective benzoylating reagent is the hydroxybenzotriazole (HOBt; 1) derived benzoyloxybenzotriazole (BBTZ, 2), first introduced for this purpose by Kim and co-workers (Fig. 1).16 BBTZ reacts under mild conditions with outstanding regio- and chemoselectivity that surpasses that of most other benzoylating reagents.17 The intrinsically high regioselectivity of benzoylation with BBTZ has been harnessed in the use of BzOH with the HOBt-derived ammonium salt TBTU (3),18 which ostensibly proceeds through a BBTZ intermediate. | ||
| Fig. 1 Benzotriazole- and Oxyma-based reagents. | ||
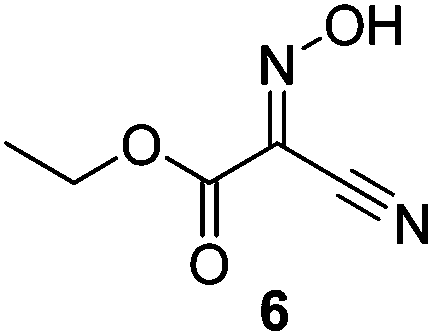
In recent years, the safety of HOBt has been called into question. HOBt and its monohydrate are classified as Class 1 explosives, with significant limitations on their transport and storage.19 Safety concerns extend to HOBt-derivatives such as TBTU (3),19 HDMA (4)20 and HDMB (5) (Fig. 1).20 Against this backdrop, various oxime-based alternatives have emerged as viable alternatives to HOBt as nucleophilic catalysts in peptide couplings. In particular ethyl 2-hydroximino-2-cyanoacetate (Oxyma, OxymaPure, 6) has been identified as a viable alternative to HOBt,21 and has been elaborated to the uronium-type coupling reagent COMU (7),20,22 which is effective for both peptide couplings20 and esterifications.18,23 Both Oxyma and COMU show a better safety profile than HOBt and HOBt-based reagents, including a lower heatflow associated with smaller increases in temperature and lower released pressures, equating to a lower risk of runaway thermal explosions.20,21 In this work we report the synthesis of a range of benzoylated oximes and investigation of their ability to act as selective benzoylating reagents. Benzoyl-Oxyma is identified as a readily synthesized, highly crystalline and storable alternative for BBTZ in the regio- and chemoselective acylation of carbohydrate alcohols.
Results and discussion
Design and synthesis of oxime-based benzoylating reagents
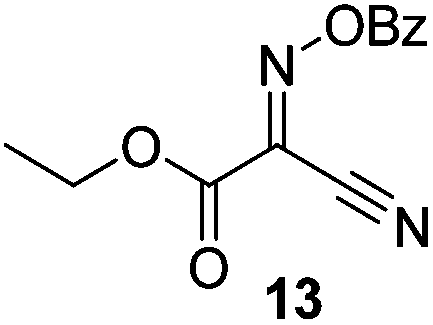
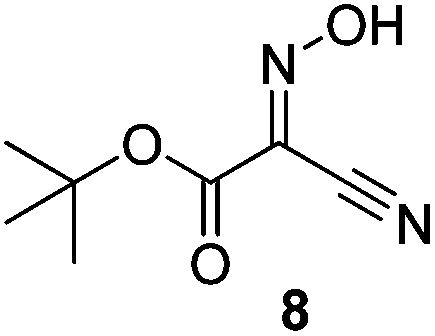
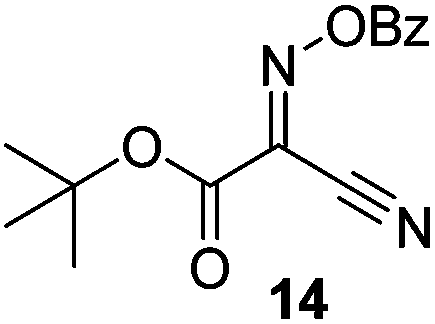
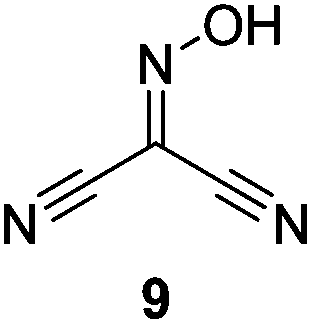
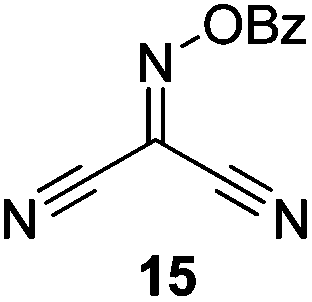
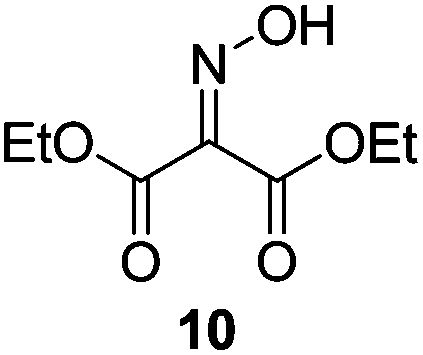
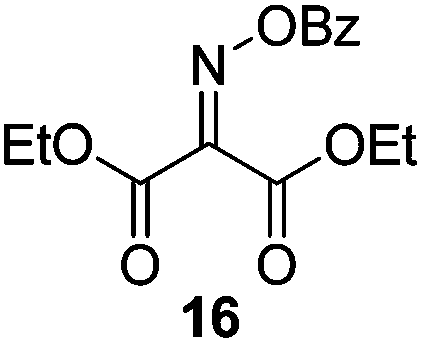
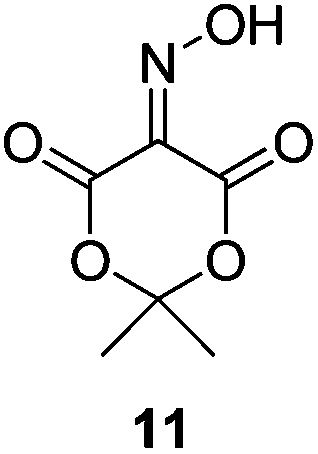
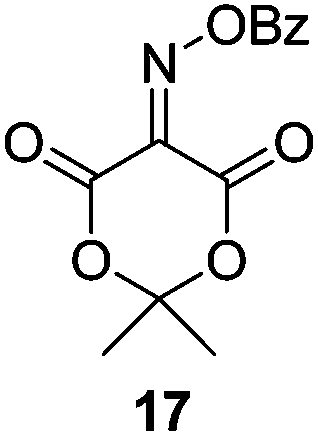
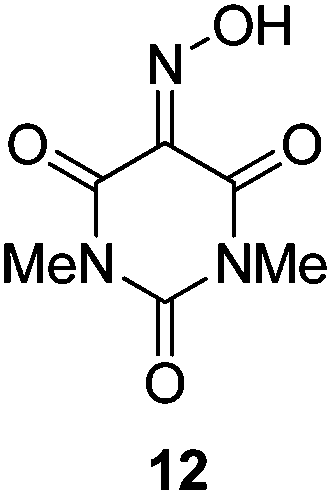
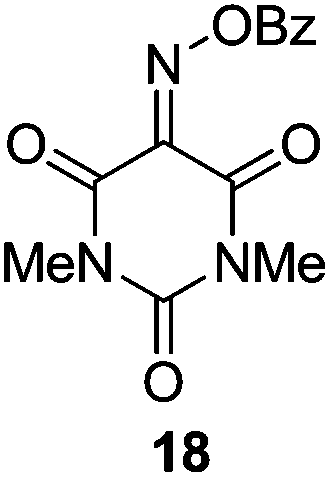
Initially we sought to explore a range of benzoylated oxime-based reagents varying in the nature of the electron-withdrawing groups adjacent to the oxime. It has been found that Oxyma (6) can undergo side-reactions at the ethoxylcarbonyl group.21,24 Therefore we also studied the tert-butyl ester 8 (derived from tert-butyl cyanoacetate)25 and the bis-nitrile 9 (derived from malonitrile)26 (Table 1). In order to complete the series we included the bis-ethoxycarbonyl derivative 10 (derived from diethyl malonate).27 Several analogues of COMU have been reported to provide improved peptide couplings, most notably based on oximes 11 (derived from Meldrum's acid)28 and 12 (derived from dimethylbarbituric acid),29 which were also included in our studies. Oximes 6, 10–12 or the corresponding silver salts of 8 and 9, were converted to benzoylated derivatives 13, 14, 15,2616,3017 and 18 by treatment with benzoic anhydride (Bz2O) in ether, or using BzCl and pyridine or Et3N. Among these examples, we highlight the exceptionally simple preparation of benzoyl-Oxyma (13): addition of Bz2O to a 0.3 M solution of Oxyma in Et2O resulted in the formation of a precipitate within 1 min. Filtration and drying provided the pure reagent. This result stands in contrast with the preparation of BBTZ (2): commercial HOBt containing 20% water can either be dried under vacuum (caution: explosion risk) or alternatively stirred with activated molecular sieves, prior to reaction with BzCl and Et3N.16 Following aqueous workup, the reagent may, with some difficulty, be crystallized from EtOAc/petrol, with the crystallization likely complicated by the known interconversion of O- and N-acyl isomers.31 In the case of the esters 13 and 14 derived from unsymmetrical oximes, the stereochemistry about the oxime double bond was defined as Z by single crystal X-ray diffraction analysis (Fig. 2). | ||
| Fig. 2 ORTEP representations of the molecular structures of benzoylated oximes (a) 13 and (b) 14, determined by single-crystal X-ray crystallography. Ellipsoids are at the 50% probability level. | ||
While simple oximes possess pKa values of 8.3–11.8,33 Oxyma (6) is characterized by the presence of strongly electron-withdrawing cyano and ethoxycarbonyl groups. The pKa value of Oxyma is reported to be 4.6,34 matching that reported for HOBt (1).35 In order to understand the leaving group ability of various oximes we initially sought to extract pKa data from the literature; however, owing to the range of different solvents used for these studies we were unable to directly compare the various oximes. Consequently, we measured the pKa values for the series of compounds by spectrophotometric titration of the oximes in a solution in 95% MeCN in water.32 As revealed in Table 1, the pKa values for the series of oximes fell within the range 3.96–4.39, and under these conditions the pKa value of HOBt was determined to be 4.25; based on their pKa values, the series of oximes possess similar intrinsic leaving group ability.
Investigation of the benzoylation of a simple primary alcohol
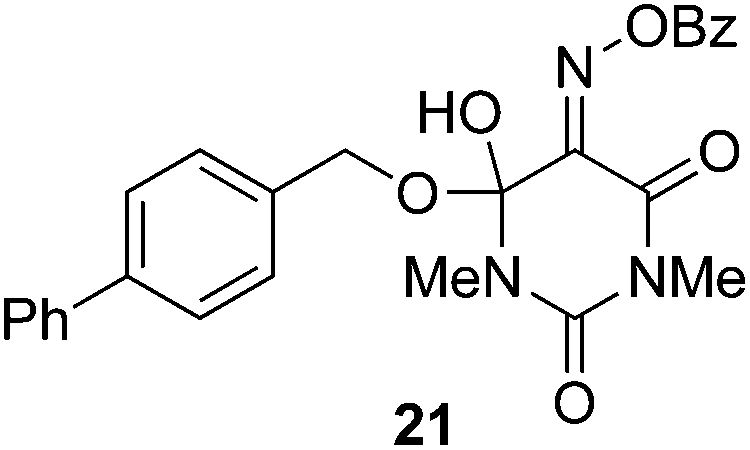
The effectiveness of the benzoylated oximes as benzoylating agents was investigated by studying their reaction with a simple alcohol, 4-phenylbenzyl alcohol 19 (Table 2). This substrate was chosen as it is crystalline, easily dried and readily detected owing to the presence of a chromaphore. Treatment of 19 with 1.1 equiv. of BBTZ (2) and Et3N at 0 °C and warming to room temperature overnight afforded the benzoate 20 in quantitative yield (entry 1), highlighting the impressive benzoylating ability of this reagent. Treatment with 1.1 equiv. of the nitrile-containing reagents 13–15 gave yields of 95–98% (entries 2–4). Significantly, we found that for optimal yields using benzoyl-Oxyma 13, use of dry Et3N was critical, as using undried Et3N only afforded an 80% yield. This sensitivity to trace moisture appears to be unique to the Oxyma-derived reagent as close to quantitative yields were obtained with the t-butyl ester 14, dicyanooxime 15 and BBTZ using undried Et3N, results that suggest that the carbonyl group of the Oxyma-derived 13 may be a site of unwanted reactivity. On the other hand the malonate-derived reagents 10–12 gave much poorer yields. The diethyl malonate derivative 10 provided just 50% after 5 d, with tlc evidence of both unreacted 10 and 19 (entry 5); the Meldrum's acid derivative 11 delivering only 2% (entry 6), accompanied by degradation of the reagent to materials that could not be identified; and the barbiturate 12 providing only a 15% yield (entry 7). In the last case, the major by-product was tentatively assigned as the addition product 21. Overall, these results reveal that despite similarity in the pKa value of the conjugate acid of the leaving group, significant differences in reactivity occur across the series.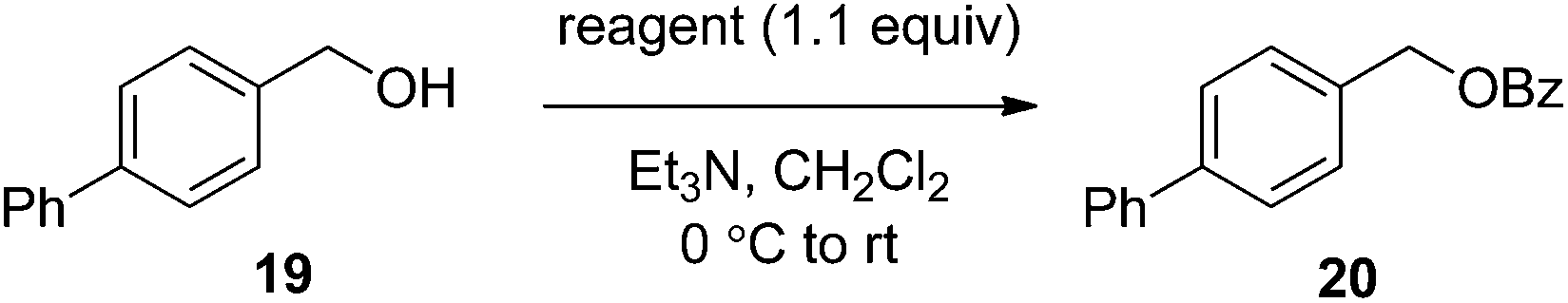
Selective benzoylation of carbohydrate alcohols
Having identified the nitrile-containing oximes 13–15 as the most effective benzoylating reagents for a simple alcohol, we next turned our attention to the selective acylation of carbohydrate diols. Typically, good acylation selectivity is observed for diols when one hydroxyl group has a cis-disposed vicinal oxygen, which is believed to result from the presence of an intramolecular hydrogen bond that enhances the reactivity of the hydroxyl group. Using the optimum protocol identified from Table 2, the benzoylation of galactoside 22 was studied (Table 3). Consistent with literature yields36 treatment of galactoside 22 with BBTZ provided a quantitative yield of the 3-benzoate 23 (entry 1). Among the three nitrile-based reagents, the best yield of 23 was obtained for the Oxyma-derived reagent 13 (90%, entry 2), which could be improved to a quantitative yield by using 1.4 equivalents of the reagent. We next explored acylation of the glucoside 24. BBTZ afforded 25 in an 87% yield (entry 5). The three nitrile-containing oximes 13–15 gave marginally lower yields, with the best result obtained for the Oxyma-derived reagent (83%, entry 6). Increasing the amount of the Oxyma-derived reagent to 1.4 equivalents provided 25 in an 89% yield, commensurate with that obtained using 1.1 equivalents of BBTZ. We therefore conclude that the nitrile-based reagents are effective for selective acylation of diols, with the Oxyma-derived reagent 13 the most promising. Although these reagents are slightly less effective than BBTZ, use of 1.4 equiv. of 13 provides similar yields to that obtained using 1.1 equiv. BBTZ.|
|
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Entry | Substrate | Reagent | Yielda (%) | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| a Reactions were performed using 1.1 equivalent of reagent, 1.2 equiv. dry Et3N in dry CH2Cl2, 0 °C to rt overnight. b Yield for reaction performed using 1.4 equiv. of benzoyl-Oxyma. | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 1 | 22 | BBTZ | 100 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 2 | 22 | Benzoyl-Oxyma (13) | 90 (100)b | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 3 | 22 | 14 | 90 (100)b | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 4 | 22 | 15 | 72 (90)b | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 5 | 24 | BBTZ (1) | 87 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 6 | 24 | Benzoyl-Oxyma (13) | 83 (89)b | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 7 | 24 | 14 | 75 (93)b | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 8 | 24 | 15 | 80 (93)b | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
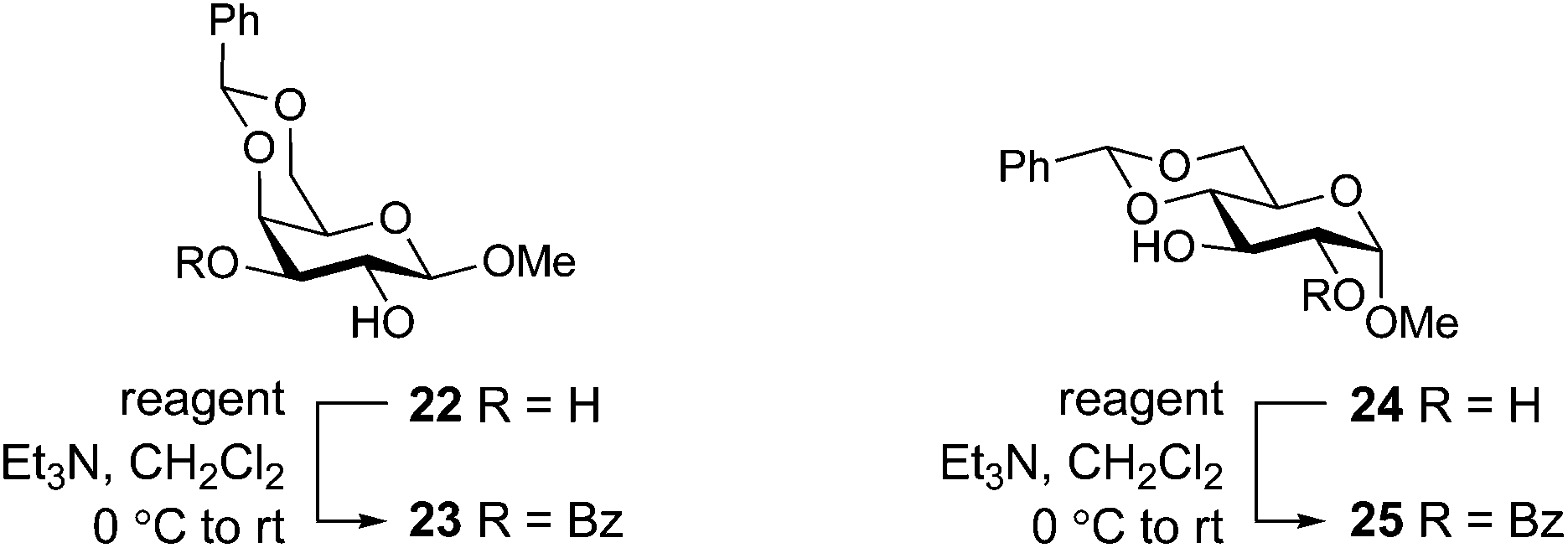
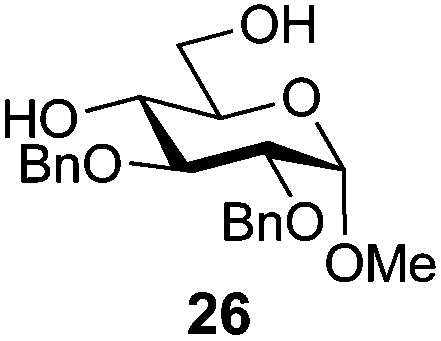
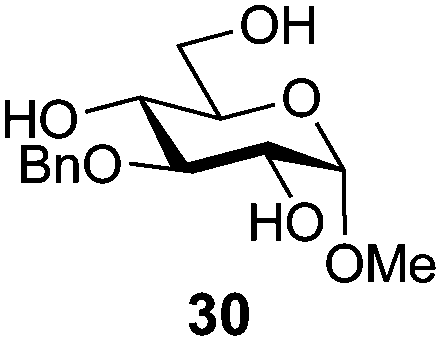
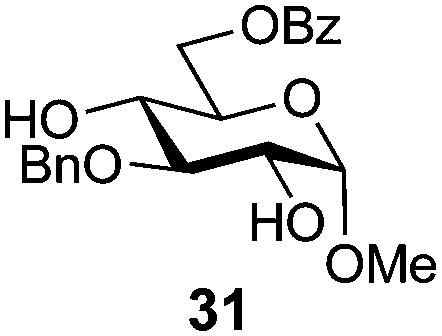
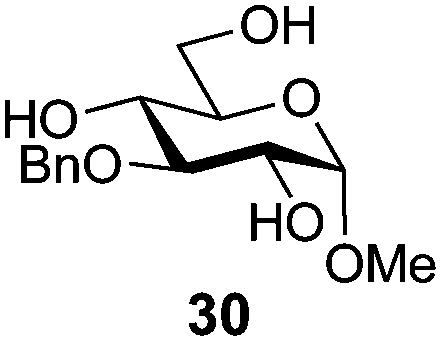
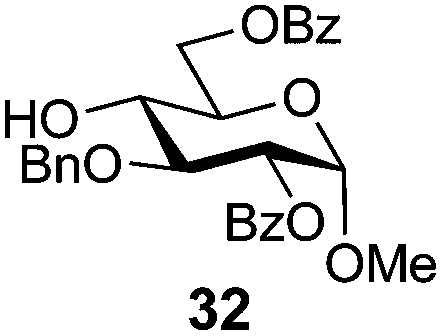
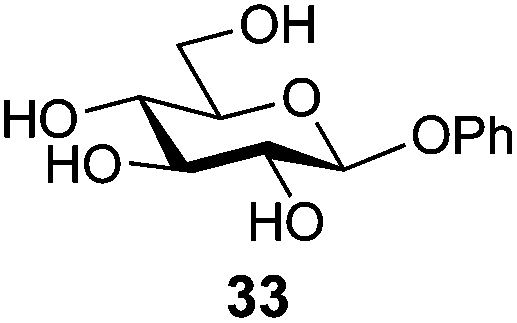
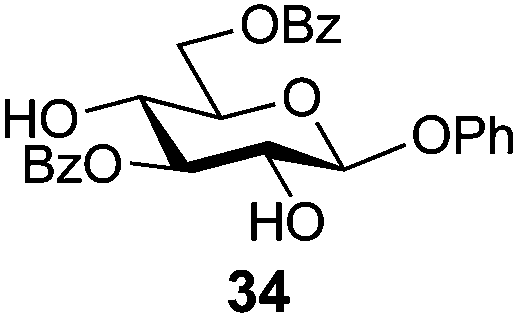
In view of the importance of selective hydroxyl functionalization in carbohydrate chemistry, we explored several additional examples using benzoyl-Oxyma (13) (Table 4). Selectivity for a primary versus a secondary alcohol was demonstrated through the benzoylation of the 4,6-diol 26 to afford the 6-benzoate 27 in 86% yield (entry 1), the 2,6-diol 28 to afford the 6-benzoate 29 in 50% yield (entry 2), and the 2,4,6-triol 30 to afford the 6-benzoate 31 in 93% yield (entry 3), in each case using 1.4 equiv. of 13. Two additional transformations are noteworthy. Treatment of 2,4,6-triol 30 with 2.8 equivalents of 13 afforded 2,6-dibenzoate 31 in 41%, with the greater reactivity of the 2-position relative to the 4-position a result of the cis-relationship with the anomeric oxygen. Finally, treatment of tetraol 33 with 2.2 equiv. of 13 directly provided the 3,6-dibenzoate 34 in 55% yield.
Conclusions
The classification of HOBt and related reagents as Class I explosives has provided an impetus for the development of new reagents that allow similar transformations with a lower risk of explosion. Oxime-based reagents, particularly those based on Oxyma, have emerged as effective alternatives for HOBt and have formed the basis of the development of COMU as a replacement for HBTU and HDMB. In this work we have explored a range of oxime-based alternatives to the selective acylating reagent BBTZ, leading to the identification of the nitrile-based benzoates 13–15 as the most promising representatives. In particular, benzoyl–Oxyma (13) can be considered a viable alternative to BBTZ, featuring a facile preparation, yields that approach those of BBTZ in various model reactions and a broad utility in the selective benzoylation of various carbohydrates.Experimental
General
Proton nuclear magnetic resonance spectra (1H NMR) and proton decoupled carbon nuclear magnetic resonance spectra (13C NMR) were obtained in deuterated chloroform, methanol-d4 (CD3OD) and DMSO-d6 with residual protonated solvent as internal standard. Abbreviations for multiplicity are s, singlet; d, doublet; t, triplet; q, quartet; p, pentet. Flash chromatography was carried out on silica gel 60 according to the procedure of Still et al.37 IR spectra were obtained as thin films or solids on a Fourier-transform attenuated total reflectance infrared spectrophotometer equipped with a diamond-coated zinc selenide sample accessory. Analytical thin layer chromatography (t.l.c.) was conducted on aluminium-backed 2 mm thick silica gel 60 GF254 and chromatograms were visualized with ceric ammonium molybdate (Hanessian's stain) or orcinol/FeCl3 (Bial's reagent). High resolution mass spectra (HRMS) were obtained by ionizing samples using electro-spray ionization (ESI) and a time-of-flight mass analyzer. Dry DMF was obtained by drying over 4 Å molecular sieves. Hexanes refers to petroleum ether, boiling range 40–60 °C. CH2Cl2 and THF were dried over alumina according to the method of Pangborn et al.38 Oxyma (ethyl 2-hydroximino-2-cyanoacetate, 6) was purchased from Sigma-Aldrich. The synthesis of various oximes and oxime benzoates has been reported previously: 8,259,2610,2711,2812,2915,26 and 16.30Ethyl 2-benzoyloximino-2-cyanoacetate (benzoyl-Oxyma, 13)
A mixture of benzoic anhydride (2.21 g, 8.44 mmol) and Oxyma (6) (1.00 g, 7.04 mmol) in dry diethyl ether (25 mL) was stirred at room temperature for 1 min. The resulting precipitate was filtered and washed with cold petroleum ether to afford 13 as a white crystalline powder (1.50 g, 90%), m.p. 99.3 °C (lit.39 100 °C); 1H NMR (CDCl3, 400 MHz) δ 1.43 (3 H, t, J 7.1 Hz, CH3), 4.50 (2 H, q, J 7.1 Hz, CH2), 7.54–8.21 (5 H, m, Ph); 13C NMR (CDCl3, 100 MHz) 14.1 (CH3), 64.6 (CH2), 107.1 (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N), 125.8 (C
N), 125.8 (C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N), 129.2, 130.6, 131.7, 135.2 (Ph), 157.0 (CO), 160.7 (CO); IR ν 2988.1, 2200.2, 1776.1, 1752.3, 1599.1, 1583.7, 1451.9, 1368.7, 1297.3, 1233.3, 1179.0, 1146.7, 1110.4, 1077.1, 1031.0, 996.2, 904.4, 837.5, 801.6, 764.9, 709.2, 666.5 cm−1; HRMS [M + Na]+ calcd for C12H10N2O4Na m/z 269.0533, found 269.0532.
N), 129.2, 130.6, 131.7, 135.2 (Ph), 157.0 (CO), 160.7 (CO); IR ν 2988.1, 2200.2, 1776.1, 1752.3, 1599.1, 1583.7, 1451.9, 1368.7, 1297.3, 1233.3, 1179.0, 1146.7, 1110.4, 1077.1, 1031.0, 996.2, 904.4, 837.5, 801.6, 764.9, 709.2, 666.5 cm−1; HRMS [M + Na]+ calcd for C12H10N2O4Na m/z 269.0533, found 269.0532.
t-Butyl 2-benzoyloximino-2-cyanoacetate (14)
A solution of sodium nitrite (2.93 g, 42.5 mmol) in water (13 mL) was added to a solution of t-butyl cyanoacetate (2.00 g, 14.1 mmol) in acetic acid (6 mL) and water (6 mL) at 0 °C. The solution was stirred at room temperature for 12 h in the dark. A solution of AgNO3 (2.40 g, 14.1 mmol) in water (13 mL) was added and stirred for 30 min. The resulting yellow precipitate was collected by filtration, washed with cold petroleum ether, and dried under vacuum (3.57 g, 70%). BzCl (0.554 g, 3.94 mmol) was added to a stirred solution of the yellow precipitate (1.00 g, 3.58 mmol) in toluene (7.5 mL). A drop of pyridine was added to the solution resulting in the rapid formation of a white precipitate. Stirring was continued for another 30 min and then the precipitate was collected by filtration. The filtrate was concentrated and the residue recrystallized to afford 14 as a white crystalline solid (0.62 g, 65%), m.p. 72.5–73.5 °C (ether/petroleum ether); 1H NMR (CDCl3, 400 MHz) δ 1.63 (9 H, s), 7.55–8.21 (5 H, m, Ph); 13C NMR (CDCl3, 100 MHz) 27.0, 87.4, 107.4, 125.7, 128.9, 130.3, 132.6, 134.8, 155.2, 160.6; IR ν 3679.5, 2984.8, 2938.9, 2844.3, 2238.0, 1978.2, 1928.0, 1841.7, 1778.1, 1730.8, 1598.1, 1449.1, 1347.5, 1259.5, 1224.9, 1175.7, 1141.2, 1028.8, 989.8, 901.6, 843.0, 701.9, 665.7 cm−1; HRMS [M + Na]+ calcd for C14H14N2O4Na m/z 297.0846, found 297.0847.Benzoate ester of isonitroso Meldrum's acid (17)
Isonitroso Meldrum's acid 1128 (0.500 g, 2.89 mmol) was dissolved in CH2Cl2 (7 mL) and Et3N (0.44 mL, 3.16 mmol) was added, followed by addition of BzCl (0.34 mL, 2.9 mmol). The solution was stirred for 1 h, then diluted with CH2Cl2 (35 mL) and stirred for another 5 min. The solution was washed with water (2 × 20 mL), dried (MgSO4), filtered and concentrated under reduced pressure. Flash chromatography (5–10% EtOAc/petroleum ether) afforded 17 as a brown solid (1.50 g, 70%), m.p. 108.5–109 °C; 1H NMR (CDCl3, 400 MHz) δ 1.87 (6 H, s, CH3), 7.55–8.27 (5 H, m, Ph); 13C NMR (CDCl3, 100 MHz) 28.4, 46.0, 106.8, 126.2, 129.0, 129.2, 130.7, 131.0, 134.6, 135.1, 139.2, 150.9, 155.7, 161.3, 162.5; IR ν 3750.7, 2943.4, 1778.5, 1749.4, 1589.9, 1556.2, 1493.8, 1453.1, 1396.4, 1384.2, 1293.3, 1264.7, 1230.0, 1198.3, 1181.8, 1159.3, 1055.4, 1032.0, 1011.9, 982.3, 969.9, 949.0, 930.9, 891.0, 864.9, 798.8, 739.0, 701.8, 677.7, 667.1 cm−1; HRMS [M + Na]+ calcd for C13H11NO6Na m/z 300.0479, found 297.0478.Benzoate ester of isonitroso dimethylbarbituric acid (18)
Benzoic anhydride (0.70 g, 2.97 mmol) was added to a solution of isonitroso dimethylbarbituric acid 1229 (0.500 g, 2.70 mmol) in dry Et2O (12.5 mL) and the solution was stirred overnight at room temperature. The resulting precipitate was filtered and washed with cold petroleum ether. The product was obtained as a green solid (0.65 g, 83%), m.p. 218.5–219 °C; 1H NMR (CDCl3, 400 MHz) δ 3.45 (3 H, s, CH3), 3.47 (3 H, s, CH3), 7.54–8.33 (5 H, m, Ph); 13C NMR (CDCl3, 100 MHz) 29.0, 29.5, 31.0, 126.5, 129.1, 130.9, 134.9, 161.7; IR ν 3000.1, 1949.2, 1773.7, 1686.0, 1447.8, 1367.7, 1283.3, 1234.9, 1070.9, 1014.7, 994.2, 970.3, 924.2, 866.2, 793.4, 746.3, 716.5 cm−1; HRMS [M + Na]+ calcd for C13H11N3O5Na m/z 312.0591, found 312.0595.Protocol for benzoylation of 4-phenylbenzyl alcohol (19)
A mixture of 4-phenylbenzyl alcohol (19) (0.100 g, 0.542 mmol), 1.1 equivalents of the benzoylating reagent and Et3N (0.091 mL, 0.650 mmol) in dry CH2Cl2 (3.0 mL) at 0 °C was stirred overnight. The solution was diluted with CH2Cl2, washed with water (2 × 20 mL), dried (MgSO4), filtered and concentrated under reduced pressure. The residue was purified by flash chromatography (10–15% EtOAc/petroleum ether) to afford 4-phenylbenzyl benzoate 20 as a white solid, m.p. 62–63 °C (lit.40 61.5–62.5 °C); 1H NMR (CDCl3, 400 MHz) δ 5.42 (2 H, s, CH2), 7.34–8.12 (15 H, m, Ph); 13C NMR (CDCl3, 100 MHz) 66.6 (CH2), 127.2, 127.5, 127.6, 128.5, 128.9, 129.8, 130.2, 133.2, 135.1, 140.8, 141.3 (Ph), 166.6 (CO); HRMS [M + Na]+ calcd for C20H16O2Na m/z 311.1043, found 311.1042.
Characterization data for adduct of benzoate ester of isonitroso dimethylbarbituric acid and 4-phenylbenzyl alcohol, compound (21)
1H NMR (CDCl3, 400 MHz) δ 3.01 (3 H, s, Me), 3.05 (3 H, s, Me), 5.29 (1 H, d, J 12 Hz, CH2), 5.41 (1 H, d, J 12 Hz, CH2), 7.42–7.85 (14 H, m, 2 × Ph, C6H4); 13C NMR (CDCl3, 100 MHz) δ 25.54, 26.15, 69.32, 82.3, 126.4, 127.2, 127.3, 127.4, 127.6, 127.7, 128.5, 128.8, 128.9, 129.0, 129.4, 129.2, 129.4, 132.8, 134.1, 140.4, 142.2, 155.9, 162.3, 164.8, 166.3; HRMS [M + H]+ calcd for C26H23N3O6m/z 474.1620, found 474.1659.Methyl 3-O-benzoyl 4,6-O-benzylidene-β-D-galactopyranoside (23)
A mixture of methyl 4,6-O-benzylidene-β-D-galactopyranoside (22) (0.100 g, 0.354 mmol), benzoylating reagent (1.1 equivalent), and Et3N (0.060 mL, 0.42 mmol) in dry CH2Cl2 (3.0 mL) at 0 °C was stirred overnight. The solution was diluted with CH2Cl2, washed with water (2 × 20 mL), dried (MgSO4), filtered and concentrated under reduced pressure. Flash chromatography (75–85% EtOAc/petroleum ether) of the residue afforded the 3-benzoate 23 as a white solid, m.p. 163–164 °C (lit.36 m.p. 166–167 °C); [α]D +94 (c 1.0, CHCl3; lit.36 [α]D +95); 1H NMR (CDCl3, 400 MHz) δ 2.34 (1 H, d, J 2.4 Hz, OH), 3.60–3.62 (4 H, m, H5, CH3), 4.11 (1 H, dd, J 12.5, 1.7 Hz, H6), 4.18 (1 H, ddd, J 10.1, 7.8, 2.5 Hz, H2), 4.41–4.36 (2 H, m, H1,6), 4.51 (1 H, d, J 3.6 Hz, H4), 5.15 (1 H, dd, J 10.1, 3.7 Hz, H3), 5.53 (1 H, s, PhCH), 7.29–8.12 (10 H, m, Ph) 13C NMR (CDCl3, 100 MHz) 57.4 (CH3), 66.7, 68.9, 69.1, 73.7, 74.3, 100.9, 104.2 (C1), 126.2, 128.1, 128.5, 128.9, 129.8, 130.0, 133.4, 137.7 (Ph), 166.6 (CO); HRMS [M + H]+ calcd for C21H22O7Na m/z 409.1254, found 409.1254.
Methyl 2-O-benzoyl 4,6-O-benzylidene-α-D-glucopyranoside (25)
A mixture of methyl 4,6-O-benzylidene-α-D-glucopyranoside (24) (0.100 g, 0.354 mmol), 1.4 equivalents of the benzoylating reagent and Et3N (0.060 mL, 0.415 mmol) in dry CH2Cl2 (3.0 mL) at 0 °C was stirred overnight. The solution was diluted with CH2Cl2, washed with water (2 × 20 mL), dried (MgSO4), filtered and concentrated under reduced pressure. Flash chromatography (25–35% EtOAc/petroleum ether) of the residue afforded the 2-benzoate 25 as a white solid, m.p. 169–170 °C (169–170 °C); [α]D +108 (c 1.0, CHCl3; lit.16 [α]D +107); 1H NMR (CDCl3, 400 MHz) δ 2.57 (1 H, s, OH), 3.40 (3 H, s, CH3), 3.63 (1 H, t, J 9.4 Hz, H6), 3.80 (1 H, t, J 10.3 Hz, H6), 3.91 (1 H, ddd, J 9.8, 9.8, 4.7 Hz, H5), 4.34 (2 H, m, H3,4), 5.04 (1 H, dd, J 9.5, 3.8 Hz, H2), 5.08 (1 H, d, J 3.8 Hz, H1), 5.58 (1 H, s, PhCH), 7.36–8.13 (10 H, m, Ph); 13C NMR (CDCl3, 100 MHz) 55.7 (CH3), 62.2, 69.0, 69.1, 74.2, 81.6, 97.9, 102.2 (C1), 126.4, 128.5, 128.6, 129.4, 130.1, 133.5, 137.1 (Ph), 166.3 (CO); HRMS [M + H]+ calcd for C21H22O7Na m/z 409.1257, found 409.1260.
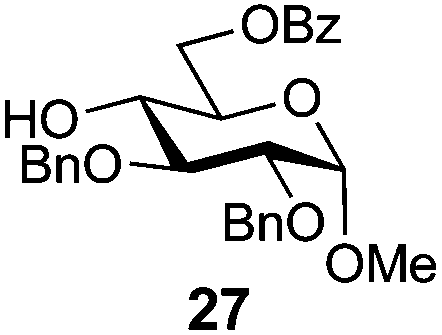
Methyl 2,3-di-O-benzyl-6-O-benzoyl-α-D-glucopyranoside (27)
A mixture of benzoyl-Oxyma 13 (0.092 g, 0.374 mmol), methyl 2,3-di-O-benzyl-α-D-glucopyranoside 2641 (0.100 g, 0.267 mmol) and Et3N (0.050 mL, 0.32 mmol) in dry CH2Cl2 (3.0 mL) at 0 °C was stirred overnight. The solution was diluted with CH2Cl2, washed with water (2 × 20 mL), dried over (MgSO4), filtered and concentrated under reduced pressure. Flash chromatography (30–40% EtOAc/petroleum ether) of the residue afforded 27 (0.11 g, 86%), m.p. 77–79 °C (lit.42 75–77 °C); [α]D +35 (c 0.25, CHCl3; lit.42 [α]D +25); 1H NMR (CDCl3, 400 MHz) δ 2.53 (1 H, d, J 2.9 Hz, OH), 3.40 (3 H, s, CH3), 3.53 (2 H, m, H4,5), 3.87 (2 H, m, H2,3), 4.51 (1 H, dd, J 12.1, 2.1 Hz, H6), 4.59–4.68 (3 H, m, H1,6,CH2), 4.77 (2 H, dd, J 11.6, 10.3 Hz, CH2Ph), 5.01 (1 H, d, J 11.3, CH2Ph), 7.28–8.02 (15 H, m, Ph); 13C NMR (CDCl3, 100 MHz) 55.3, 63.8, 69.5, 70.2, 73.3, 75.7, 79.7, 81.3, 98.2 (C1), 128.0, 128.1, 128.2, 128.4, 128.6, 128.7, 129.8, 133.2, 138.0, 138.7 (Ph), 166.8 (CO); HRMS [M + Na]+ calcd for C28H30O7Na m/z 501.1884, found 501.1886.
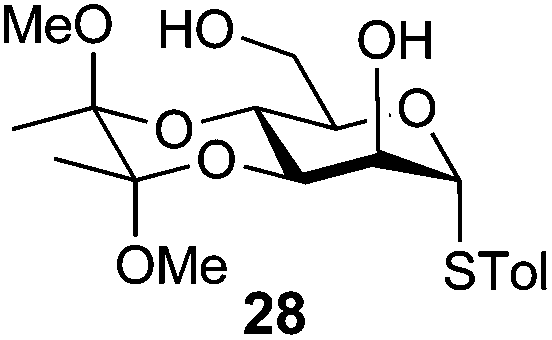
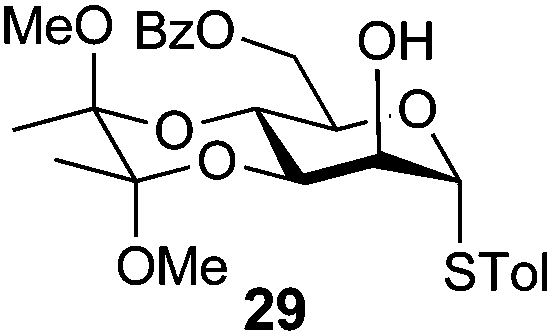
4-Methylphenyl 3,4-O-(2′,3′-dimethoxybutan-2′,3′-diyl)-6-O-benzoyl-1-thio-α-D-mannopyranoside (29)
A mixture of benzoyl-Oxyma 13 (0.036 g, 0.146 mmol), 3,4-di-O-(2,3-dimethoxybutane-2,3-diyl)-α-D-mannopyranoside 2843 (0.042 g, 0.105 mmol), and Et3N (0.020 mL, 0.126 mmol) in dry CH2Cl2 (3.0 mL) at 0 °C was stirred overnight. The solution was diluted with CH2Cl2, washed with water (2 × 20 mL), dried over (MgSO4), filtered and concentrated under reduced pressure. Flash chromatography (50–70% EtOAc/petroleum ether) of the residue afforded 29 as a syrup (0.030 g, 50%), [α]D +167.8 (c 0.25, CHCl3); 1H NMR (CDCl3, 400 MHz) δ 1.30 (3 H, s, CH3), 1.34 (3 H, s, CH3), 2.27 (3 H, s, CH3Ar), 3.17 (3 H, s, OCH3), 3.32 (3 H, s, OCH3), 4.07 (1 H, dd, J 9.1, 3.0 Hz, H2), 4.18–4.22 (2 H, m, H3,6), 4.49 (1 H, ddd, J 11.9, 6.1 Hz, H6), 4.55–4.65 (2 H, m, H4,5), 5.52 (1 H, s, H1), 6.98–8.01 (10 H, m, Ph); 13C NMR (CDCl3, 100 MHz) 14.1 (CH3), 17.7 (CH3), 17.8, 21.2, 29.8, 48.1, 48.3, 63.0, 64.1, 64.7, 67.1, 69.7, 72.7, 87.1, 100.2, 100.5, 107.1, 125.8, 128.4, 128.54, 129.2, 129.7, 129.8, 130.0, 130.0, 130.7, 132.7, 133.1, 133.3, 135.3, 138.2, 157.1 (Ph), 160.7, 165.9, 166.4 (CO); HRMS [M + H]+ calcd for C26H32O8SNa m/z 527.1701 found 527.1705.
Methyl 3-O-benzyl-6-O-benzoyl-α-D-glucopyranoside (31)
A mixture of benzoyl-Oxyma 13 (0.121 g, 0.491 mmol), methyl 3-O-benzyl-α-D-glucopyranoside 3044 (0.100 g, 0.351 mmol) and Et3N (0.060 mL, 0.421 mmol) in dry CH2Cl2 (3.0 mL) at 0 °C was stirred overnight. The solution was diluted with CH2Cl2 washed with water (2 × 20 mL), dried over (MgSO4), filtered and concentrated under reduced pressure. Flash chromatography (40–50% EtOAc/petroleum ether) of the residue afforded 31 as a white solid (0.13 g, 55%), m.p. 70–71 °C; [α]D +88.0 (c 1.0, CHCl3); 1H NMR (CDCl3, 400 MHz) δ 2.27 (1 H, d, J 8.3 Hz, OH), 2.83 (1 H, d, J 3.2 Hz, OH), 3.45 (3 H, s, CH3), 3.54 (1 H, ddd, J 9.1, 9.1, 2.3 Hz, H4), 3.63 (1 H, dd, J 8.4, 8.4 Hz, H3), 3.69 (1 H, ddd, J 9.1, 3.8 Hz, H2), 3.87 (1 H, ddd, J 9.8, 4.7, 2.2 Hz, H5), 4.52 (1 H, dd, J 12.1, 2.2 Hz, H6), 4.67 (1 H, dd, J 12.1, 4.7 Hz, H6), 4.77–4.83 (2 H, m, H1, CH2), 4.98 (1 H, d, J 11.4, CH2), 7.29–8.08 (10 H, m, Ph); 13C NMR (CDCl3, 100 MHz) 55.4 (CH3), 63.7, 69.9, 69.9, 72.7, 75.3, 82.5, 99.6 (C1), 120.3, 128.1, 128.5, 128.7, 129.8, 129.8, 133.3, 138.5 (Ph), 167.0 (CO); HRMS [M + H]+ calcd for C21H24O7 Na m/z 411.1414, found 411.1412.
Methyl 3-O-benzyl-2,6-di-O-benzoyl-α-D-glucopyranoside (32)
A mixture of benzoyl-Oxyma 13 (0.194 g, 0.787 mmol), of methyl 3-O-benzyl-α-D-glucopyranoside 3044 (0.080 g, 0.281 mmol) and Et3N (0.05 mL, 0.337 mmol) in dry CH2Cl2 (3.0 mL) at 0 °C was stirred overnight. The solution was diluted with CH2Cl2, washed with water (2 × 20 mL), dried over (MgSO4), filtered and concentrated under reduced pressure. Flash chromatography (30–40% EtOAc/petroleum ether) of the residue afforded 32 as a syrup (0.0600 g, 41%), [α]D +100.4 (c 0.5, CHCl3); 1H NMR (CDCl3, 400 MHz) δ 3.42 (3 H, s, CH3), 3.72 (1 H, t, J 9.1 Hz, H4), 3.98 (1 H, ddd, J 10.1, 4.4, 2.2 Hz, H5), 4.08 (1 H, t, J 9.0 Hz, H3), 4.56 (1 H, dd, J 12.1, 2.2 Hz, H6), 4.73–4.79 (2 H, m, H2,6), 4.88 (1 H, d, J 11.3 Hz, CH2Ph), 5.07–5.12 (2 H, m, H1,CH2Ph), 7.48–8.12 (10 H, m, Ph); 13C NMR (CDCl3, 100 MHz) 55.4 (CH3), 63.6, 69.7, 70.4, 73.9, 75.5, 79.6, 82.5, 97.5, 99.6 (C1), 128.0, 128.1, 128.5, 128.6, 128.7, 129.7, 129.8, 129.9, 129.9, 130.2, 133.3, 133.4, 133.5, 138.1 (Ph), 160.0, 167.1 (CO); HRMS [M + H]+ calcd for C28H28O8Na m/z 515.1676 found 515.1668.
Phenyl 3,6-di-O-benzoyl-β-D-glucopyranoside (34)
A mixture of benzoyl-Oxyma 13 (0.210 g, 0.853 mmol), phenyl β-D-glucopyranoside 33 (0.100 g, 0.390 mmol) and Et3N (0.060 mL, 0.468 mmol) in dry CH2Cl2 (3.0 mL) at 0 °C was stirred overnight. The solution was diluted with CH2Cl2, washed with water (2 × 20 mL), dried (MgSO4), filtered and concentrated under reduced pressure. Flash chromatography (2–5% acetone/CH2Cl2) of the residue afforded 34 as a white solid (0.11 g, 55%), m.p. 144 °C; [α]D +2.7 (c 0.5, CHCl3); 1H NMR (CDCl3, 400 MHz) δ 2.81 (1 H, s, OH), 3.49 (1 H, s, OH), 3.83 (1 H, t, J 9.3 Hz, H4), 3.90 (1 H, ddd, J 9.7, 6.1, 2.3 Hz, H5), 4.00 (1 H, dd, J 9.1, 8.0 Hz, H2), 4.63 (1 H, dd, J 12.0, 6.1 Hz, H6), 4.74 (1 H, dd, J 12.0, 2.3 Hz, H6), 5.07 (1 H, d, J 7.7 Hz, H1), 5.33 (1 H, t, J 9.1 Hz, H3), 7.07–8.08 (15 H, m, Ph); 13C NMR (CDCl3, 100 MHz) 63.0, 69.7, 72.2, 74.6, 78.6 (C2,3,4,5), 101.0 (C1), 117.0, 123.2, 128.5, 128.6, 129.3, 129.6, 129.7, 129.9, 130.1, 133.4, 133.7, 157.0 (Ph), 166.9, 167.6 (CO); HRMS [M + H]+ calcd for C26H24O8Na m/z 487.1363, found 487.1361.
X-ray crystallography
Crystals of 13 and 14 were mounted in low temperature oil then flash cooled using an Oxford low temperature device. Intensity data were collected at 130 K on an Oxford SuperNova X-ray diffractometer with CCD detector using Cu-Kα (α = 1.54184 Å) radiation. Data were reduced and corrected for absorption. The structures were solved by direct methods and difference Fourier synthesis using the SHELX suite of programs45 as implemented within the WINGX software.46 Thermal ellipsoid plots were generated using the program ORTEP-3.Crystal data for 13: C12H10N2O4M = 246.22, T = 130.0(1) K, λ = 1.54184, monoclinic, space group P21/n, a = 11.8867(1), b = 8.0556(1), c = 12.3853(1) Å, β = 99.064(1), V = 1171.14(2) Å3, Z = 4, Dc = 1.396 g cm−3, μ(Cu-Kα) 0.904 mm−1, F(000) = 512, crystal size 0.49 × 0.44 × 0.32 mm. 8000 reflections measured, 2434 independent reflections (Rint = 0.0170), the final R was 0.0342 [I > 2σ(I) 2347 data] and wR(F2) (all data) was 0.0966. CCDC deposition: 1406156.
Crystal data for 14: C14H14N2O4M = 274.27, T = 130.0(1) K, λ = 1.54184 Å, monoclinic, space group P21/n, a = 5.9020(1), b = 21.5230(3), c = 11.2041(2) Å, β = 102.073(2)°, V = 1391.76(4) Å3, Z = 4, Dc = 1.309 g cm−3, μ(Cu-Kα) 0.813 mm−1, F(000) = 576, crystal size 0.50 × 0.15 × 0.06 mm. 9619 reflections measured, 2897 independent reflections (Rint = 0.0299), the final R was 0.0392 [I > 2σ(I) 2605 data] and wR(F2) (all data) was 0.1018. CCDC deposition: 1406157.
Spectrophotometric titrations
Dissociation constants (pKa values) of the oximes or HOBt were measured spectrophotometrically using a Cary-50 Bio UV/Vis spectrophotometer in 95% (v/v) MeCN-water at wavelengths of 220–315 nm. Buffer solutions across the pH range of 2–10 were prepared by adding 0.2 M NaOH to a mixture of 0.04 M phosphoric acid, acetic acid, and boric acid. HOBt or oximes at 0.1 mM concentration were prepared in the buffer solutions and the absorbance measured. Each cuvette contained 50 μl of 1 mM stock solution of oxime or HOBt in MeCN, 100 μl of 40 mM buffer, and 1.85 mL of MeCN were used to obtain a final concentration of 0.025 mM of the reagent. Absorbances were measured at the wavelength where the difference between the absorbance of the oxime or HOBt, and its conjugate base was maximized. Titration curves are provided in the ESI.†Acknowledgements
We thank the Australian Research Council (ARC) for financial support. SJW is an ARC-funded Future Fellow. SS thanks the Department of State Development, Business and Innovation of Victoria for a Victoria-India Doctoral Scholarship and the Australia India Institute.References
- P. Wuts, Greene's Protective Groups in Organic Synthesis, John Wiley & Sons, New York, 5th edn, 2014 Search PubMed.
- R. V. Stick and S. J. Williams, Carbohydrates: The Essential Molecules of Life, Elsevier, Oxford, United Kingdom, 2009 Search PubMed.
- D. Crich, Z. Dai and S. Gastaldi, J. Org. Chem., 1999, 64, 5224–5229 CrossRef CAS.
- D. R. Mootoo, P. Konradsson, U. Udodong and B. Fraser-Reid, J. Am. Chem. Soc., 1988, 110, 5583–5584 CrossRef CAS.
- A. H. Haines, in Adv. Carbohydr. Chem. Biochem, ed. R. S. Tipson and H. Derek, Academic Press, 1976, vol. 33, pp. 11–109 Search PubMed.
- J. M. Williams and A. C. Richardson, Tetrahedron, 1967, 23, 1369–1378 CrossRef CAS.
- A. Holý and M. Souček, Tetrahedron Lett., 1971, 12, 185–188 CrossRef.
- S. A. Abbas and A. H. Haines, Carbohydr. Res., 1975, 39, 358–363 CrossRef CAS.
- F. A. Carey and K. O. Hodgson, Carbohydr. Res., 1970, 12, 463–465 CrossRef CAS.
- G. J. F. Chittenden, Carbohydr. Res., 1971, 16, 495–496 CrossRef CAS.
- J. Stawinski, T. Hozumi and S. A. Narang, J. Chem. Soc., Chem. Commun., 1976, 243–244, 10.1039/C39760000243.
- N. L. Holder and B. Fraser-Reid, Synthesis, 1972, 83 CrossRef CAS.
- O. Mitsunobu, J. Kimura, K.-i. Iiizumi and N. Yanagida, Bull. Chem. Soc. Jpn., 1976, 49, 510–513 CrossRef CAS.
- K. Ishihara, H. Kurihara and H. Yamamoto, J. Org. Chem., 1993, 58, 3791–3793 CrossRef CAS.
- S. David and S. Hanessian, Tetrahedron, 1985, 41, 643–663 CrossRef CAS.
- S. Kim, H. Chang and W. J. Kim, J. Org. Chem., 1985, 50, 1751–1752 CrossRef CAS.
- B. L. Wilkinson, Aust. J. Chem., 2013, 66, 910–912 CrossRef CAS.
- J. D. Twibanire and T. B. Grindley, Org. Lett., 2011, 13, 2988–2991 CrossRef CAS PubMed.
- K. D. Wehrstedt, P. A. Wandrey and D. Heitkamp, J. Hazard. Mater., 2005, 126, 1–7 CrossRef CAS PubMed.
- A. El-Faham, R. S. Funosas, R. Prohens and F. Albericio, Chem. – Eur. J., 2009, 15, 9404–9416 CrossRef CAS PubMed.
- R. Subiros-Funosas, R. Prohens, R. Barbas, A. El-Faham and F. Albericio, Chem. – Eur. J., 2009, 15, 9394–9403 CrossRef CAS PubMed.
- R. Subirós-Funosas, S. N. Khattab, L. Nieto-Rodríguez, A. El-Faham and F. Albericio, Aldrichimica Acta, 2013, 46 Search PubMed.
- B. Cao, X. Chen, Y. Yamaryo-Botte, M. B. Richardson, K. L. Martin, G. N. Khairallah, T. W. Rupasinghe, R. M. O'Flaherty, R. A. O'Hair, J. E. Ralton, P. K. Crellin, R. L. Coppel, M. J. McConville and S. J. Williams, J. Org. Chem., 2013, 78, 2175–2190 CrossRef CAS PubMed.
- D. Dev, N. B. Palakurthy, N. Kumar and B. Mandal, Tetrahedron Lett., 2013, 54, 4397–4400 CrossRef CAS.
- G. Duguay, J.-P. Guémas, J.-C. Meslin, J.-P. Pradère, F. Reliquet, A. Reliquet, C. Tea-Gokou, H. Quiniou and C. Rabiller, J. Heterocycl. Chem., 1980, 17, 767–770 CrossRef CAS.
- J. Perrocheau, R. Carrié and J.-P. Fleury, Can. J. Chem., 1994, 72, 2458–2467 CrossRef CAS.
- D. A. May and T. D. Lash, J. Org. Chem., 1992, 57, 4820–4828 CrossRef CAS.
- A. El-Faham, R. Subirós-Funosas and F. Albericio, Eur. J. Org. Chem., 2010, 3641–3649 CrossRef CAS.
- Y. E. Jad, S. N. Khattab, B. G. de la Torre, T. Govender, H. G. Kruger, A. El-Faham and F. Albericio, Org. Biomol. Chem., 2014, 12, 8379–8385 CAS.
- A. I. Mishchenko, A. V. Prosyanik, P. N. Belov, V. A. Romanchenko, E. G. Belova and V. I. Markov, Chem. Heterocycl. Compd., 1984, 20, 270–274 CrossRef.
- B. D. Brink, J. R. DeFrancisco, J. A. Hillner and B. R. Linton, J. Org. Chem., 2011, 76, 5258–5263 CrossRef CAS PubMed.
- M. F. Fathallah and S. N. Khattab, J. Chem. Soc. Pak., 2011, 33, 324–332 Search PubMed.
- M. Charton, in The Chemistry of Hydroxylamines, Oximes and Hydroxamic Acids, ed. Z. Rappoport and J. F. Liebman, John Wiley & Sons, Ltd, 2008, pp. 553–608, DOI:10.1002/9780470741962.ch12.
- M. Itoh, Bull. Chem. Soc. Jpn., 1973, 46, 2219–2222 CrossRef CAS.
- I. E. Pop, B. P. Deprez and A. L. Tartar, J. Org. Chem., 1997, 62, 2594–2603 CrossRef CAS PubMed.
- I. F. Pelyvás, T. K. Lindhorst, H. Streicher and J. Thiem, Synthesis, 1991, 1015–1018 CrossRef.
- W. C. Still, M. Kahn and A. M. Mitra, J. Org. Chem., 1978, 43, 2923–2925 CrossRef CAS.
- A. B. Pangborn, M. A. Giardello, R. H. Grubbs, R. K. Rosen and F. J. Timmers, Organometallics, 1996, 15, 1518–1520 CrossRef CAS.
- J.-M. Biehler, J.-P. Fleury, J. Perchais and A. Regent, Tetrahedron Lett., 1968, 9, 4227–4230 CrossRef.
- M. Sawada, Y. Tsuno and Y. Yukawa, Bull. Chem. Soc. Jpn., 1972, 45, 1206–1209 CrossRef CAS.
- D. J. Bell and J. Lorber, J. Chem. Soc., 1940, 453–455, 10.1039/JR9400000453.
- H. C. Jarrell and W. A. Szarek, Can. J. Chem., 1978, 56, 144–146 CrossRef CAS.
- Y. Zhang, C. Meng, L. Jin, X. Chen, F. Wang and H. Cao, Chem. Commun., 2015, 51, 11654–11657 RSC.
- J. M. Küster and I. Dyong, Liebigs Ann. Chem., 1975, 1975, 2179–2189 CrossRef.
- G. Sheldrick, Acta Crystallogr., Sect. A: Fundam. Crystallogr., 2008, 64, 112–122 CrossRef CAS PubMed.
- L. J. Farrugia, J. Appl. Crystallogr., 1999, 32, 837–838 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1406156 and 1406157. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5ob02092a |
| This journal is © The Royal Society of Chemistry 2016 |