
Copper-catalyzed direct C–H fluoroalkenylation of heteroarenes†
Kevin
Rousée
,
Cédric
Schneider
,
Jean-Philippe
Bouillon
,
Vincent
Levacher
,
Christophe
Hoarau
*,
Samuel
Couve-Bonnaire
* and
Xavier
Pannecoucke
Normandie Univ, COBRA, UMR 6014 & FR 3038, Univ Rouen, INSA Rouen, CNRS, IRCOF, 1 rue Tesnière 76821, Mont-Saint-Aignan Cedex, France. E-mail: samuel.couve-bonnaire@insa-rouen.fr; christophe.hoarau@insa-rouen.fr
First published on 25th November 2015
Abstract
Copper-catalyzed direct C–H fluoroalkenylation of heterocycles using various gem-bromofluoroalkenes as electrophiles is reported. This efficient method offers step-economical, low-cost and stereocontrolled access to relevant heteroarylated monofluoroalkenes. The synthesis of fluorinated analogues of biomolecules and therapeutic agents for the treatment of Duchenne muscular dystrophy as application is reported.
Introduction
The importance of fluorinated compounds in agrochemicals,1,2 pharmaceuticals/medicinals,1,3 and materials science1,4 has triggered an explosion of research efforts in developing new and efficient methods to introduce a fluorinated functional group into organic molecules. Of particular relevance is the emergence of fluoroalkenes,5 versatile compounds that have found many applications as, for example, peptidomimetics,6 drugs7 and materials.8 In peptide synthesis and fine organic chemistry, fluoroalkenes are widely looked upon as stable isosteric and isoelectronic mimics of the amide bond,6 and bioisosteres in the structure/activity relationship studies.Within the readily available fluorinated building blocks for the construction of fluoroolefins, gem-bromofluoroalkenes are easily accessible9 and versatile reagents for the achievement of highly useful cross-coupling reactions.10 The development of catalytic direct C–H bond functionalization methodologies using transition metals as catalysts has received considerable attention avoiding thus the preparation of organometallic intermediates as coupling partners.11 For a realistic catalyst loading of these precious metals, less expensive transition elements such as copper have received significant attention. Indeed, since the breakthroughs made by Daugulis,12 Miura13 and Piguel,14 remarkable advances have been made in copper-catalyzed direct arylation,15 alkynylation16 and alkenylation17,18 of azoles from monohalogenoalkenes (Fig. 1, eqn (1) and (2)). However, no example of copper-catalyzed direct C–H halogenoalkenylation so far has been reported from gem-dihalogenoalkenes. Indeed, the latter have been used only for copper-catalyzed C–H alkynylation of heterocycles, the non-coupled second halogen being eliminated during the catalytic process (Fig. 1, eqn (3)).19 Recently, Cao has described an elegant metal-free base mediated nucleophilic vinylic substitution reaction between tetrasubstituted gem-difluoroalkenes and azoles (Fig. 1, eqn (4)).20a In this case, only tetrasubstituted gem-difluoroalkenes can be used as substrates, and the trisubstituted ones would directly lead to the dehydrofluorination process. This year, Loh have successfully engaged gem-difluoroalkenes in an innovative Rh(III)-catalyzed ortho-directed C–H activation, migratory insertion into the double bond and a defluorination sequence to produce fluoroalkenylated (hetero)aromatics.20b Our group has previously reported the first Pd-catalyzed direct C–H halogenoalkenylation of heterocycles demonstrating that gem-bromofluoroalkenes are suitable building blocks for this transformation (Fig. 1, eqn (5)).21 During this study, a combination of copper salt with a palladium catalyst led to a cooperative Pd(0)/Cu(I) catalysis which was found highly performant to achieve the direct C–H alkenylation of a broad range of 1,3-diazoles with any gem-bromofluoroalkenes as electrophiles. This efficiency was based upon the catalytic generation of an heteroaryl copper intermediate reacting as a transmetallating agent.22 In our ongoing project devoted to the production of fluorinated biomolecule analogues, we have recently turned our attention to the evaluation of the reactivity of the heteroaryl copper intermediate towards gem-bromofluoroalkenes in view of developing a novel palladium free copper-catalyzed direct C–H fluoroalkenylation of heterocycles offering step-economical, low-cost and stereocontrolled access to heteroarylated monofluoroalkenes (Fig. 1, eqn (6)). Moreover, taking into account that the direct fluoroalkenylation is very scarce in the literature, it could represent an efficient alternative to produce fluorinated fine chemicals. We reported herein this methodology giving access to a wide variety of trisubstituted monofluoroalkene5,23 derivatives including fluorinated analogues of therapeutic agents.
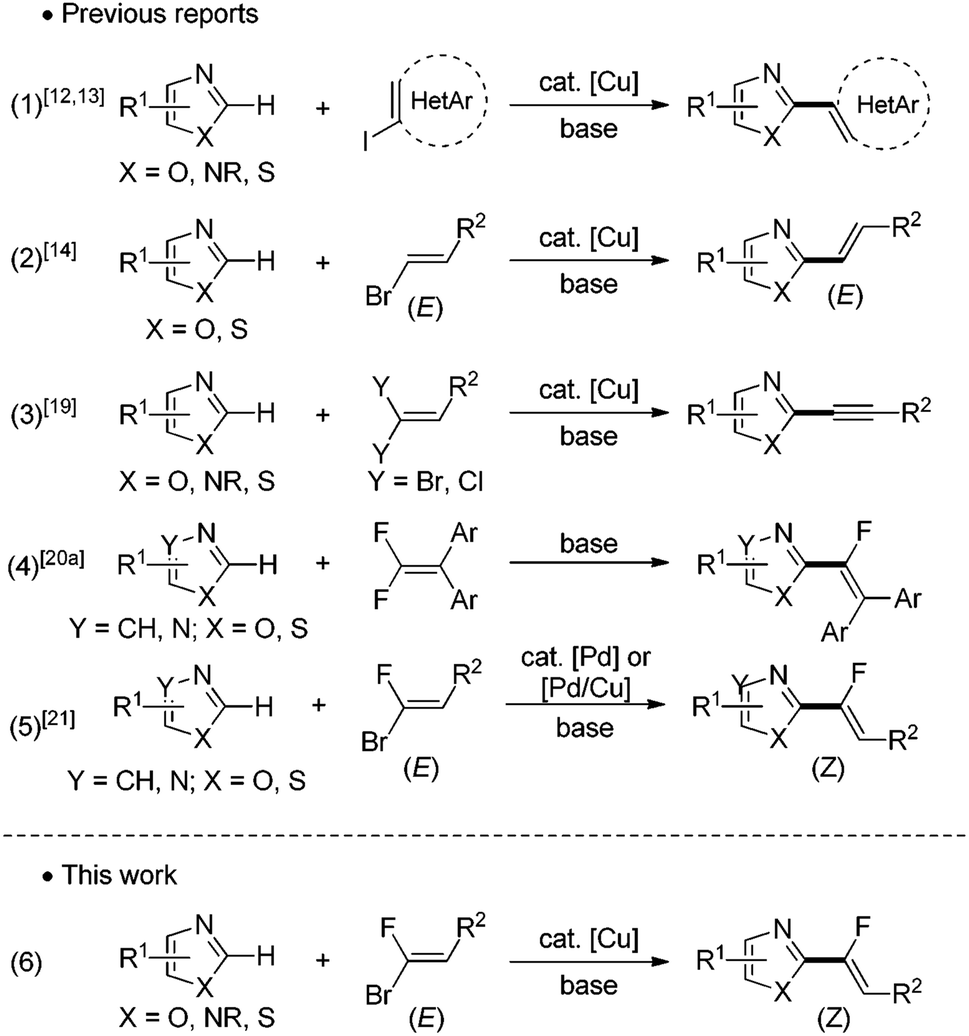 | ||
| Fig. 1 Direct functionalization of ubiquitous C–H bonds using halogenoaryls/alkenes and gem-dihalogenoalkenes. | ||
Results and discussion
We initiated our investigation by probing various reaction conditions for the direct C–H fluoroalkenylation of 5-phenyl-oxazole (2a) with easily accessible (E)-gem-bromofluoroalkene91A (Table 1). Indeed, whereas a set of experiments with our model substrate, phenyloxadiazole, under an optimized previously reported procedure was found unsuccessful without the palladium catalyst,242a proved to be a suitable substrate for direct fluoroalkenylation without a palladium source, providing 3Aa in 63% yield compared to 75% yield under bimetallic Pd/Cu catalysis (entries 1 and 2). Switching the nature of the copper source for CuI led to a slight enhancement of the yield (entry 3). Among the bases, t-BuOLi proved to be the most effective without formation of the alkynylated side-product via a dehydrofluorination process (entries 4 and 5). Subsequently, common ligands of copper have been screened, such as Phen and derivatives, diamine ligands or mono- and bidendate phosphines (entries 4–10), and surprisingly, dppe revealed considerable efficiency affording the desired product in almost quantitative yield (entry 10). Crucially, formation of 3Aa was not observed when copper was omitted from the reaction mixture (entry 11). We then performed the reaction with different copper sources (entries 12–14). The best performance of the reaction was thus attained by using CuI as the catalyst; however, we observed that the monofluoroalkenylation was slightly insensitive to the copper source (Cu(I) or Cu(II)).|
|
||||
|---|---|---|---|---|
| Entry | [Cu] | Ligand | Base | Yieldb (%) |
| a All reactions were performed using 1A (1.1 equiv.), 2a (0.2 mmol, 1.0 equiv.), copper source (10 mol%), ligand (20 mol%), and base (3 equiv.) in 1,4-dioxane (0.25 M) at 110 °C. b Yield based on the isolated product after flash chromatography. c In the presence of PdCl2(PPh3)2 (5 mol%). d L1 = 3,4,7,8-(Me)4-1,10-Phen. e L2 = trans-N,N′-dimethylcyclohexane-1,2-diamine. | ||||
| 1c | CuBr | — | t-BuOLi | 75 |
| 2 | CuBr | — | t-BuOLi | 63 |
| 3 | CuI | — | t-BuOLi | 65 |
| 4 | CuI | Phen | t-BuOLi | 51 |
| 5 | CuI | Phen | K2CO3 | — |
| 6 | CuI | L1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) d d |
t-BuOLi | 51 |
| 7 | CuI | L2e |
t-BuOLi | 81 |
| 8 | CuI | PPh3 | t-BuOLi | 83 |
| 9 | CuI | PCy3·HBF4 | t-BuOLi | 51 |
| 10 | CuI | dppe | t-BuOLi | 96 |
| 11 | — | dppe | t-BuOLi | — |
| 12 | CuBr | dppe | t-BuOLi | 66 |
| 13 | CuCl2 | dppe | t-BuOLi | 73 |
| 14 | Cu(OTf)2 | dppe | t-BuOLi | 65 |
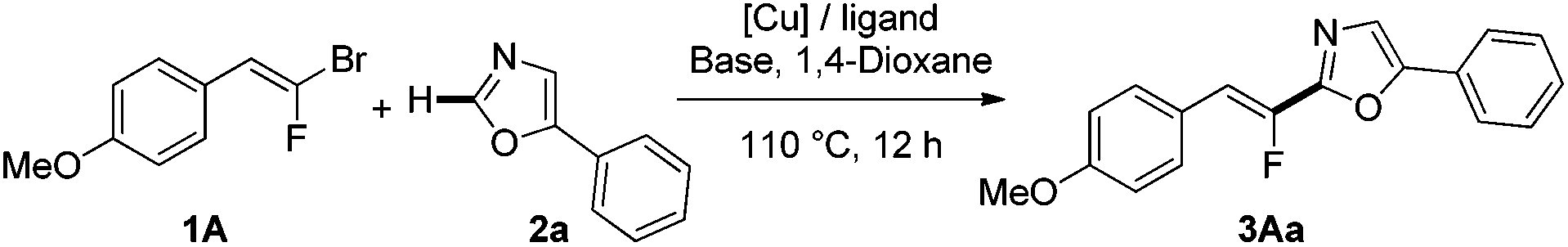
Under these optimized reaction conditions, the heteroarylated fluoroalkene 3Aa was produced in 96% isolated yield as a pure (Z)-isomer, demonstrating that the reaction proceeds with complete retention of the stereochemistry.
With the optimized conditions in hand, the establishment of the scope of the direct C–H fluoroalkenylation was undertaken on 5-phenyloxazole (2a) with various readily accessible (E)-gem-bromofluoroalkenes9 (Table 2). The latter flanked indifferently with electron-donating or electron-withdrawing groups on the aromatic unit at the ortho, meta and para positions, reacted at the C-2 position of the 5-phenyloxazole producing the desired product in moderate to excellent yields.
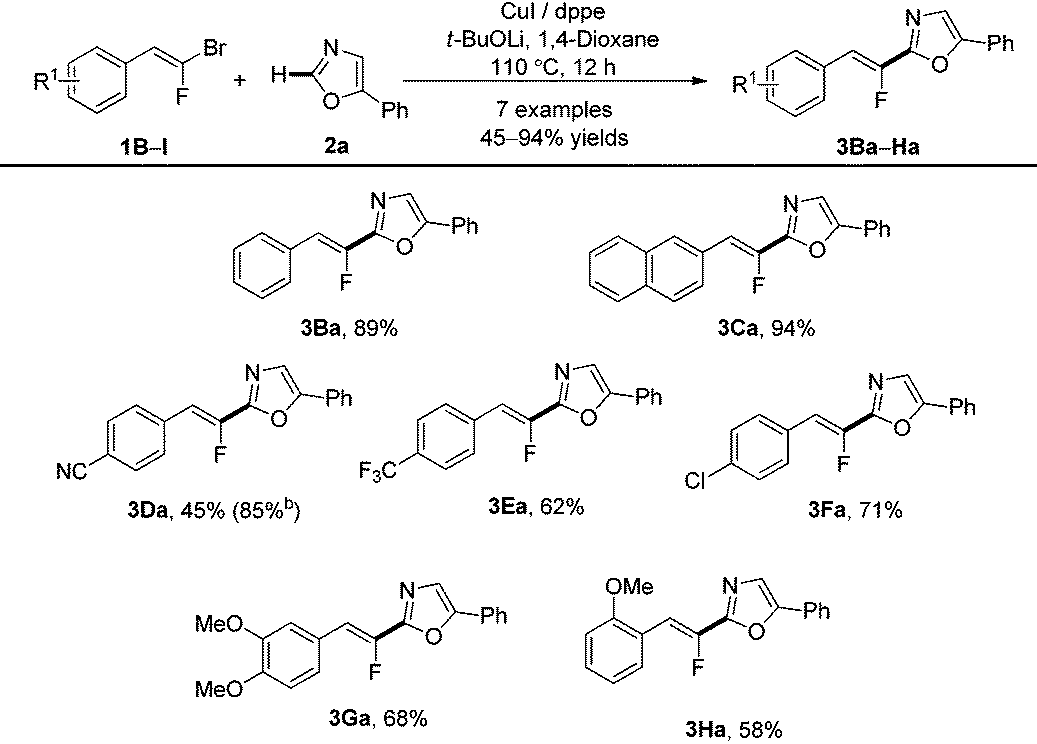
Interestingly, (E)-gem-bromofluoroalkenes bearing the cyano group (1D) or the chlorine atom (1F) on the aromatic ring, both valuable functional groups for further post-functionalizations, displayed good reactivity, even if, in the case of 3Da, the reaction yield was lower than that obtained under Pd/Cu catalysis.21
Alkylated or tetrasubstituted gem-bromofluoroalkenes proved to be unreactive under Cu-catalysis whereas the reaction occurred under Pd/Cu catalysis.21,25 The aromatic ring bearing an electron-withdrawing group at the ortho position proved to be unstable under these reaction conditions.25
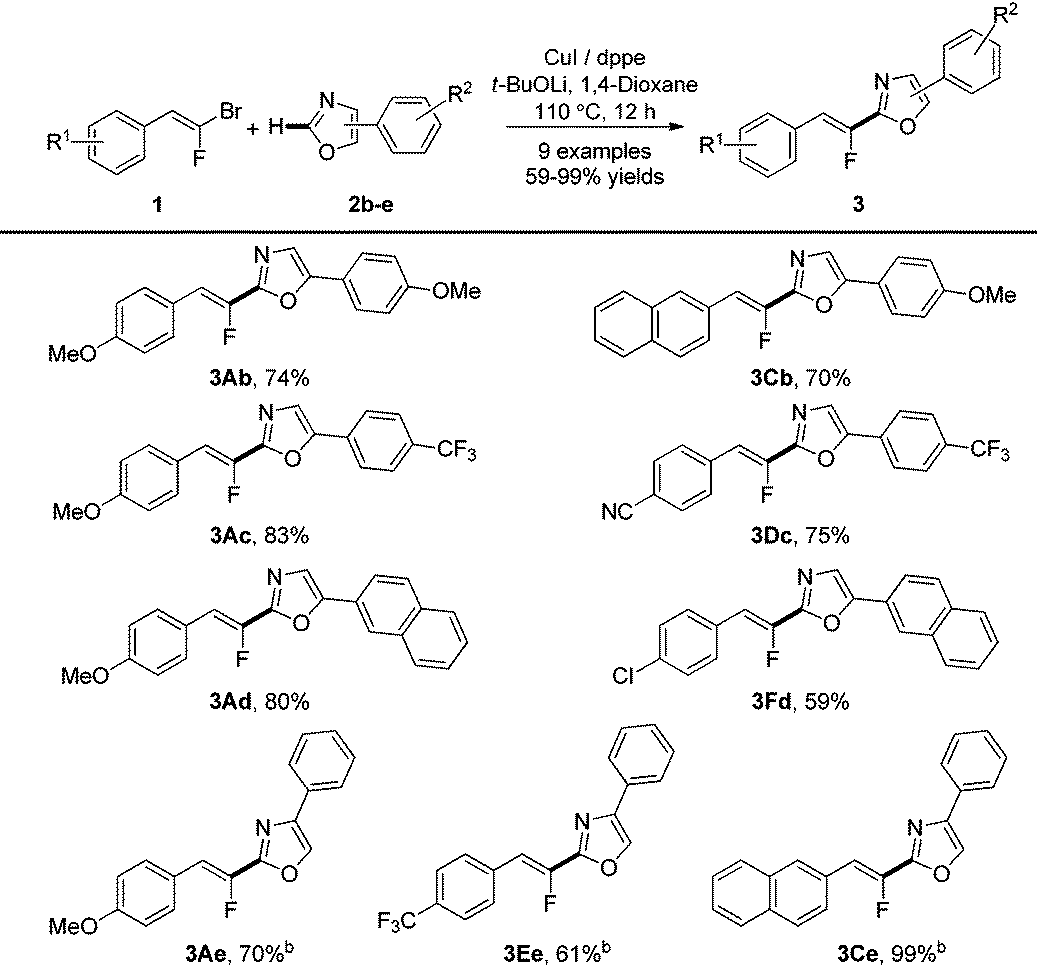
We next examined the substrate scope of various aryloxazoles (2b–e) with a panel of gem-bromofluoroalkenes in this transformation (Table 3). The reaction was efficient whatever the electronic nature of the substituents on the benzene ring of the 5-phenyloxazole 2b–d. It is noteworthy that the use of Phen instead of dppe as a ligand with 4-phenyloxazole 2e as the substrate was crucial to obtain trisubstituted Z-fluoroalkenes 3Ae, 3Ce and 3Ee in good yields.25
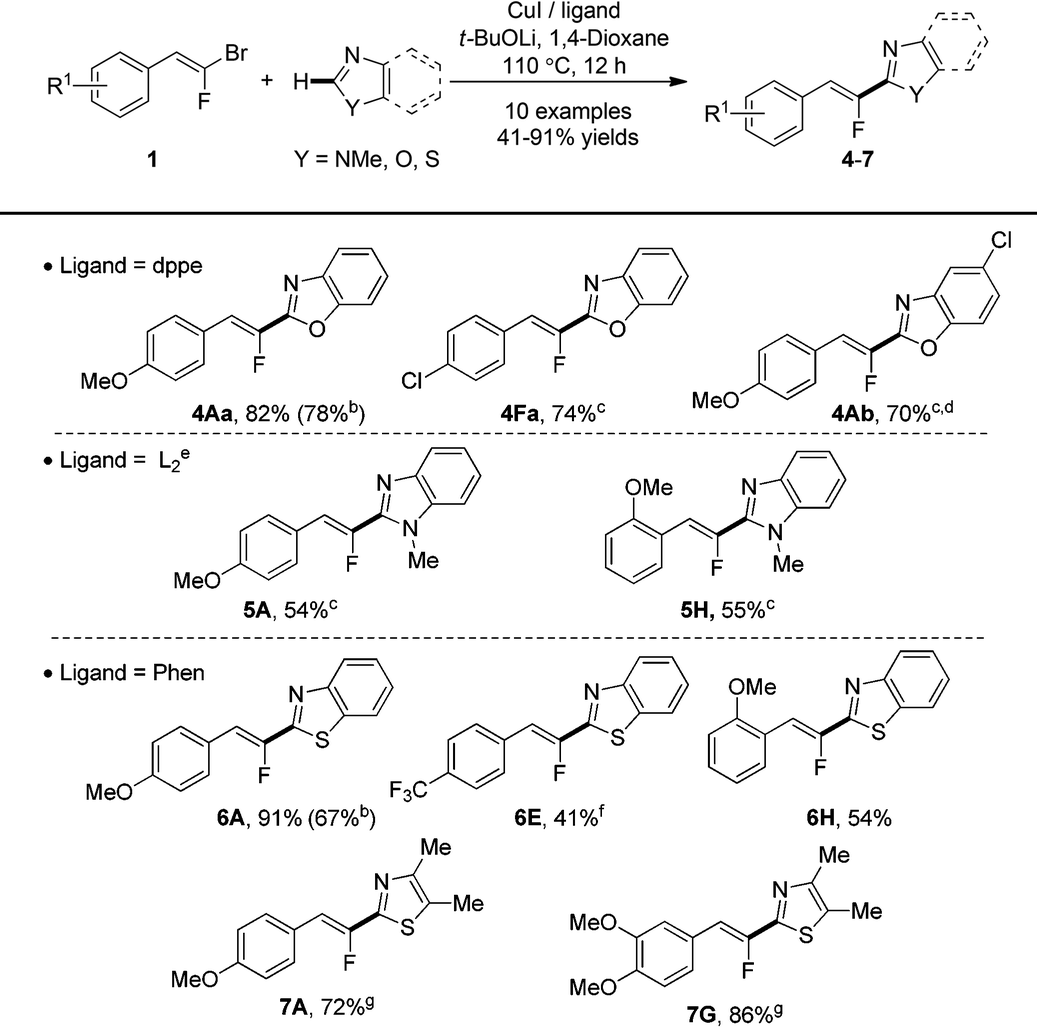
We then applied our reaction conditions to various relevant 1,3-diazoles (Table 4). A first set of experiments engaging benzoxazole 4a with the (E)-gem-bromofluoroalkenes 1A and 1F, as coupling partners was addressed. Through the expected monofluoroalkenes, 4Aa was obtained in good (82%) yield whereas the chlorinated benzoxazolylfluoroalkene 4Fa was produced in poor (20%) yield (19F NMR yield). Nevertheless, when the temperature was increased from 110 to 130 °C, full completion of benzoxazole 4a was attained to provide product 4Fa in 74% yield. The reaction performed with chlorinated benzoxazole 4b at the same 130 °C temperature delivered the expected benzoxazolylfluoroalkene 4Ab in good (70%) yield and importantly, the reaction could be scaled up from 0.2 mmol to 1 mmol without any loss of efficiency.
| a All reactions were performed using 1 (1.1 equiv.), heterocycle (0.2 mmol, 1.0 equiv.), CuI (10 mol%), ligand (20 mol%), and t-BuOLi (3 equiv.) in 1,4-dioxane (0.25 M) at 110 °C. Yields are based on the isolated product after flash chromatography. b Yield obtained under bimetallic Pd/Cu catalysis. c Reaction performed at 130 °C. d Reaction performed on a 1 mmol scale. e L2 = trans-N,N′-dimethylcyclohexane-1,2-diamine. f Reaction performed at 90 °C. g CuI (20 mol%) and Phen (40 mol%) were used. |
|---|
|
We then examined the selective C–H monofluoroalkenylation with N-methyl-benzimidazole 5 as a heterocycle under our optimized conditions and, unfortunately no desired product was obtained. A careful screening of bases, solvents and ligands at different reaction temperatures25 led to select trans-N,N′-dimethylcyclohexane-1,2-diamine as a ligand and a reaction temperature of 130 °C to produce fluoroalkenes 5A and 5H in optimized 54 and 55% yields, respectively.
Finally, the reaction was investigated in a thiazole series. In this case, the phenanthroline ligand was found highly performant to achieve the cross-coupling at 110 °C of para-, ortho- or disubstituted gem-bromofluoroalkenes 1A, 1G and 1H with benzothiazole 6 and 4,5-dimethythiazole 7 giving the fluoroalkenes in fair 54% to excellent 91% yields. However, the reaction performed with the trifluoromethylated (E)-gem-bromofluoroalkenes 1E provided the desired product 6E in poor 20% isolated yield mainly due to the degradation of the coupling partner 1E. Fortunately, the yield was significantly improved to 41% when operating at lower temperature, 90 °C. It has to be noted that the yields obtained for 4Aa and 6A under these experimental conditions were better than the yields obtained under bimetallic catalysis.21
Finally, we applied this copper-catalyzed fluoroalkenylation to the synthesis of relevant biomolecules as depicted in Scheme 1. Taking into account the similarities between the fluoroolefin moiety and the amide bond,6 we first synthesized a fluorinated analogue 4Ba, in 53% yield, of the antiasthmatic agent 8,26 starting from benzoxazole 4a and gem-bromofluoroalkene 1B. Then, we decided to apply our methodology as an alternative pathway to produce potential active molecules used in the treatment of Duchenne muscular dystrophy exemplified by the molecule 4Bc27 which was produced in quantitative yield by reacting 5-methoxybenzoxazole 4c with gem-bromofluoroalkene 1B (Scheme 1). Interestingly, a library of compounds may be readily produced for further SAR study introducing various substituents on both aromatic rings bearing heterocycle 4 or gem-bromofluoroalkene partner 1.
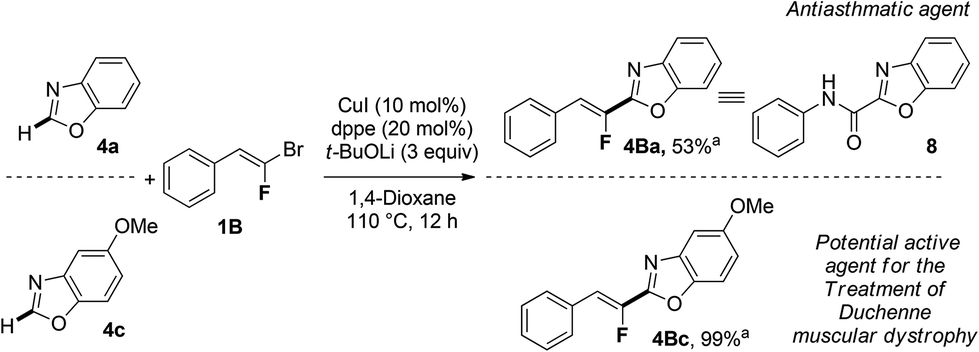 | ||
| Scheme 1 Synthesis of fluorinated therapeutic agents. aYields based on the isolated product after flash chromatography. | ||
Conclusions
In summary, an efficient Cu(I)/t-BuOLi catalyst has been employed for direct C–H fluoroalkenylation of 1,3-diazoles with readily available gem-bromofluoroalkenes as coupling partners. Although phenyloxadiazole remained unreactive as well as the use of alkylated gem-bromofluoroalkenes as electrophiles were unsuitable under these experimental conditions compared to the bimetallic Pd/Cu catalysis, the palladium free copper catalyzed fluoroalkenylation proved to be very efficient with gem-bromofluorostyrenes. Remarkably, a broad scope of 1,3-diazoles was accomplished modulating the nature of the ligand. Notably, the (benzo)oxazole, (benzo)thiazole and benzimidazole series were successfully coupled with various gem-bromofluoroalkenes using diarylphosphine, phenanthroline and diamine ligands. The methodology gave access to innovative and valuable heteroarylated fluoroalkenes 3–7 produced in fair to excellent yields. It was finally applied to the synthesis of valuable benzoxazolylfluoroalkenes 4Ba, a fluorinated analogue of an antiasthmatic agent, and 4Bc which is potentially active in the treatment of Duchenne muscular dystrophy.Acknowledgements
This work has been partially supported by INSA Rouen, Rouen University, CNRS EFRD, Labex SynOrg (ANR-11-LABX-0029).Notes and references
- (a) T. Hiyama in Organofluorine Compounds: Chemistry and Applications, edH. Yamamoto, Springer-Verlag, Berlin, 2000 Search PubMed; (b) Approximately 30–40% of agrochemicals and 20–25% of pharmaceuticals on the market are estimated to contain fluorine: A. M. Thayer, Chem. Eng. News, 2006, 84, 15 CrossRef.
- P. Jiechke, ChemBioChem, 2004, 5, 570 CrossRef PubMed.
- (a) J.-P. Bégué and D. Bonnet-Delphon, J. Fluorine Chem., 2006, 127, 992 CrossRef; (b) K. L. Kirk, J. Fluorine Chem., 2006, 127, 1013 CrossRef CAS; (c) K. Muller, C. Faeh and F. Diederich, Science, 2007, 317, 1881 CrossRef PubMed; (d) W. K. Hagmann, J. Med. Chem., 2008, 51, 4359 CrossRef CAS PubMed; (e) S. Purser, P. R. Moore, S. Swallow and V. Gouverneur, Chem. Soc. Rev., 2008, 37, 320 RSC; (f) J. Wang, M. Sánchez-Roselló, J. L. Aceña, C. del Pozo, A. E. Sorochinsky, S. Fustero, V. A. Soloshonok and H. Liu, Chem. Rev., 2014, 114, 2432 CrossRef CAS PubMed.
- M. Pagliaro and R. Ciriminna, J. Mater. Chem., 2005, 15, 4981 RSC.
- (a) G. Landelle, N. Bergeron, M.-O. Turcotte-Savard and J.-F. Paquin, Chem. Soc. Rev., 2011, 40, 2867 RSC; (b) H. Yanai and T. Taguchi, Eur. J. Org. Chem., 2011, 5939 CrossRef CAS; (c) S. Hara, Top. Curr. Chem., 2012, 327, 59 CrossRef CAS PubMed.
- (a) K. Uneyama, Organofluorine Chemistry, Blackwell, Oxford, 2006 Search PubMed; (b) S. Couve-Bonnaire, D. Cahard and X. Pannecoucke, Org. Biomol. Chem., 2007, 5, 1151 RSC and references therein; (c) C. Pierry, S. Couve-Bonnaire, L. Guilhaudis, C. Neveu, A. Marotte, B. Lefranc, D. Cahard, I. Segalas-Milazzo, J. Leprince and X. Pannecoucke, ChemBioChem, 2013, 14, 1620 CrossRef CAS PubMed.
- Monofluoroalkenes as (a) Antitumoral: S. Osada, S. Sano, M. Ueyama, Y. Chuman, H. Kodama and K. Sakaguchi, Bioorg. Med. Chem., 2010, 18, 605 CrossRef CAS PubMed; (b) Antimicrobial: Y. Asahina, K. Iwase, F. Iinuma, M. Hosaka and T. Ishizaki, J. Med. Chem., 2005, 48, 3194 CrossRef CAS PubMed; (c) Anti-HIV: S. Oishi, H. Kamitani, Y. Kodera, K. Watanabe, K. Kobayashi, T. Narumi, K. Tomita, H. Ohno, T. Naito, R. Kodama, M. Matsuoka and N. Fujii, Org. Biomol. Chem., 2009, 7, 2872 RSC; (d) Antidiabetic: S. D. Edmondson, L. Wei, J. Xu, J. Shang, S. Xu, J. Pang, A. Chaudhary, D. C. Dean, H. He, B. Leiting, K. A. Lyons, R. A. Patel, S. B. Patel, G. Scapin, J. K. Wu, M. G. Beconi, N. A. Thornberry and A. E. Weber, Bioorg. Med. Chem. Lett., 2008, 18, 2409 CrossRef CAS PubMed.
- (a) F. Babudri, G. M. Farinola, F. Naso and R. Ragni, Chem. Commun., 2007, 1003 RSC; (b) F. Babudri, A. Cardone, G. M. Farinola, C. Martinelli, R. Mendichi, F. Naso and M. Striccoli, Eur. J. Org. Chem., 2008, 1977 CrossRef CAS.
- All gem-bromofluoroalkenes were prepared following our optimized olefination procedure: (a) X. Lei, G. Dutheuil, X. Pannecoucke and J.-C. Quirion, Org. Lett., 2004, 6, 2101 CrossRef CAS PubMed; (b) L. Zoute, G. Dutheuil, J.-C. Quirion, P. Jubault and X. Pannecoucke, Synthesis, 2006, 3409 CAS; (c) For other preparation see: D. J. Burton, Z.-Y. Yang and W. Qiu, Chem. Rev., 1996, 96, 1641 CrossRef CAS PubMed.
- (a) F. Legrand, K. Jouvin and G. Evano, Isr. J. Chem., 2010, 50, 588 CrossRef CAS; (b) G. Chelucci, Chem. Rev., 2012, 112, 1344 CrossRef CAS PubMed and references therein.
- For recent reviews see: (a) D. Alberico, M. E. Scott and M. Lautens, Chem. Rev., 2007, 107, 174 CrossRef CAS PubMed; (b) B. Seregin and V. Gevorgyan, Chem. Soc. Rev., 2007, 36, 1173 RSC; (c) K. Fagnou, Top. Curr. Chem., 2010, 292, 35 CrossRef CAS PubMed; (d) I. A. I. Mkhalid, J. H. Barnard, T. B. Marder, J. M. Murphy and J. F. Hartwig, Chem. Rev., 2010, 110, 890 CrossRef CAS PubMed; (e) D. A. Colby, R. G. Bergman and J. A. Ellman, Chem. Rev., 2010, 110, 624 CrossRef CAS PubMed; (f) C. L. Sun, B.-J. Li and Z.-J. Shi, Chem. Commun., 2010, 46, 677 RSC; (g) M. Wasa, K. M. Engle and J.-Q. Yu, Isr. J. Chem., 2010, 50, 605 CrossRef CAS PubMed; (h) C. Liu, H. Zhang, W. Sui and A. Lei, Chem. Rev., 2011, 111, 1780 CrossRef CAS PubMed; (i) P. Herrmann and T. Bach, Chem. Soc. Rev., 2011, 40, 2022 RSC; (j) L. Ackermann, Chem. Rev., 2011, 111, 1315 CrossRef CAS PubMed; (k) C. S. Yeung and V. M. Dong, Chem. Rev., 2011, 111, 1215 CrossRef CAS PubMed; (l) W. Shi, C. Liu and A. Lei, Chem. Soc. Rev., 2011, 40, 2761 RSC; (m) K. M. Engle, T.-S. Mei, M. Wasa and J.-Q. Yu, Acc. Chem. Res., 2012, 45, 788 CrossRef CAS PubMed; (n) J. Yamaguchi, A. D. Yamaguchi and K. Itami, Angew. Chem., Int. Ed., 2012, 51, 8960 CrossRef CAS PubMed; (o) T. Besset, T. Poisson and X. Pannecoucke, Chem. – Eur. J., 2014, 20, 16830 CrossRef CAS PubMed.
- (a) H.-Q. Do and O. Daugulis, J. Am. Chem. Soc., 2007, 129, 12404 CrossRef CAS PubMed; (b) H.-Q. Do, R. K. M. Khan and O. Daugulis, J. Am. Chem. Soc., 2008, 130, 15185 CrossRef CAS PubMed.
- (a) M. Yamashita, K. Hirano, T. Satoh and M. Miura, Org. Lett., 2009, 11, 2337 CrossRef CAS PubMed; (b) T. Yoshizumi, H. Tsurugi, T. Satoh and M. Miura, Tetrahedron Lett., 2008, 49, 1598 CrossRef CAS.
- F. Besselièvre, S. Piguel, F. Mahuteau-Betzer and D. S. Grierson, Org. Lett., 2008, 10, 4029 CrossRef PubMed.
- (a) A. A. Kulkarni and O. Daugulis, Synthesis, 2009, 4087 CAS; (b) O. Daugulis, H.-Q. Do and D. Shabashov, Acc. Chem. Res., 2009, 41, 1074 CrossRef PubMed; (c) O. Daugulis, Top. Curr. Chem., 2010, 292, 57 CrossRef CAS PubMed; (d) K. Hirano and M. Miura, Synlett, 2011, 294 CAS; (e) K. Hirano and M. Miura, Chem. Commun., 2012, 48, 10704 RSC; (f) K. Hirano and M. Miura, Top. Catal., 2014, 57, 878 CrossRef CAS.
- (a) F. Besselièvre and S. Piguel, Angew. Chem., Int. Ed., 2009, 48, 9553 CrossRef PubMed; (b) N. Matsuyama, M. Kitahara, K. Hirano, T. Satoh and M. Miura, Org. Lett., 2010, 12, 2358 CrossRef CAS PubMed; (c) T. Kawano, N. Matsuyama, K. Hirano, T. Satoh and M. Miura, J. Org. Chem., 2010, 75, 1764 CrossRef CAS PubMed.
- For recent reviews of C–H alkenylation see: (a) S. Messaouidi, J. –D. Brion and M. Alami, Eur. J. Org. Chem., 2010, 6495 CrossRef; (b) R. Rossi, F. Bellina and M. Lessi, Synthesis, 2010, 4131 CrossRef CAS.
- (a) Q. Liao, L. Zhang, S. Li and C. Xi, Org. Lett., 2011, 13, 228 CrossRef CAS PubMed; (b) B. Das, G. C. Reddy, P. Balasubramanyam and N. Salvanna, Tetrahedron, 2012, 68, 300 CrossRef CAS; (c) W. Zhang, Y. Tian, N. Zhao, Y. Wang, J. Li and Z. Wang, Tetrahedron, 2014, 70, 6120 CrossRef CAS.
- (a) B. P. Berciano, S. Lebrequier, F. Besselièvre and S. Piguel, Org. Lett., 2010, 12, 4038 CrossRef PubMed; (b) G. C. Reddy, P. Balasubramanyam, N. Salvanna and B. Das, Eur. J. Org. Chem., 2012, 471 CrossRef CAS; (c) K. Jouvin, A. Coste, A. Bayle, F. Legrand, G. Karthikeyan, K. Tadiparthi and G. Evano, Organometallics, 2012, 31, 7933 CrossRef CAS; (d) L. Ackermann, C. Kornhaass and Z. Yingjun, Org. Lett., 2012, 14, 1824 CrossRef CAS PubMed.
- (a) X. Zhang, Y. Lin, J. Zhang and S. Cao, RSC Adv., 2015, 5, 7905 RSC; (b) P. Tian, C. Feng and T.-P. Loh, Nat. Commun., 2015, 6, 7472 CrossRef CAS PubMed.
- C. Schneider, D. Masi, S. Couve-Bonnaire, X. Pannecoucke and C. Hoarau, Angew. Chem., Int. Ed., 2013, 52, 3246 CrossRef CAS PubMed.
- F. Bellina, S. Cauteruccio and R. Rossi, Curr. Org. Chem., 2008, 12, 774 CrossRef CAS.
- K. Rousée, C. Schneider, S. Couve-Bonnaire, X. Pannecoucke, V. Levacher and C. Hoarau, Chem. – Eur. J., 2014, 20, 15000 CrossRef PubMed.
- Reaction of 1A with phenyloxadiazole in 1,4-dioxane with t-BuOLi as a base gave 83% yield under PdCl2(PPh3)2/CuBr catalysis, 0% under CuBr catalysis and 20% under CuI catalysis.
- See the ESI† for more details.
- B. Loev, R. E. Brown, F. –C. Huang and H. Jones, Patent, EP0127066 (A2), USV Pharmaceutical Corporation, 1984 Search PubMed.
- (a) S. P. Wren, G. M. Wynne, F. X. Wilson and S. D. Poignant, Patent, WO2010/112093 (A1), BioMarin IGA, Ltd, 2010 Search PubMed; (b) S. P. Wren, G. M. Wynne and F. X. Wilson, Patent, WO2010/112092 (A1), BioMarin IGA, Ltd, 2010 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures, optimization tables, characterization data and NMR spectra of new compounds. See DOI: 10.1039/c5ob02213d |
| This journal is © The Royal Society of Chemistry 2016 |